
Abstract
Heterogeneous catalysts xRu/Ce0.75Zr0.25O2 (x = 1, 5 wt.%) are prepared by the sorption-hydrolytic precipitation. It is shown that these catalysts are active in the methanation of carbon dioxide. The composition and structural features of these compounds are studied by a complex of methods such as powder XRD, high-resolution electron microscopy, chemisorption, and X-ray photoelectron spectroscopy (XPS). It is established that catalysts obtained by the sorption-hydrolytic method contain the active component in a highly dispersed state. The catalyst with 1 wt.% Ru contains ruthenium compounds in the form of atomic clusters, while that with 5 wt.% Ru contains, along with ultrafine forms, also crystallized ruthenium-containing particles. It is shown that the initial catalysts contain oxide ruthenium compounds that are reduced to the metallic state under the methanation reaction conditions. In situ studies by powder XRD, XPS studies, temperature-programmed reduction in the hydrogen atmosphere (H2-TPR) show that metal ruthenium particles, which were obtained by the activation treatment of the catalysts as a result of heating in a hydrogen-enriched flow, promote the process of the partial reduction of the support material particles due to the spillover effect.

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
INTRODUCTION
Various techniques of carbon dioxide processing have been rapidly developed nowadays due to the deterioration of global environmental problems caused by anthropogenic greenhouse gas emissions. The catalytic reaction of CO2 hydrogenation or methanation (Sabatier process) attracts interest as a process of carbon dioxide recycling to obtain methane utilized as a valuable energy carrier.
The modern “power-to-gas” (P2G) technique used for converting electrical energy into chemically bound energy also involves the methanation catalytic process. In this technique, electrolysis is used to convert the excess electricity into hydrogen which reacts with carbon dioxide and transforms into methane [1-3]. The advantages of keeping excess electricity in the form of methane include its long-term storage and easy transportation.
The progress of these technologies stimulated the development of effective heterogeneous catalysts for CO2 methanation. It was shown that the most efficient catalysts are based on noble metals such as ruthenium [4-6], rhodium [7-9], palladium [10, 11] or on less expensive transition metals such as nickel, iron, cobalt, etc. [12-14]. Ruthenium-containing catalysts exhibit the highest rates of methane conversion in the low-temperature region [15]. Supported ruthenium catalysts based on easily reducible oxide supports such as cerium dioxide CeO2 or mixed cerium-zirconium oxides Ce1–xZrxO2 achieve high activity at low reaction temperatures and low active component contents [16-19]. It is assumed that their high catalytic activity is due to the participation of the support surface in the catalytic process. In methanation reactions, the catalysts operate in a hydrogen-enriched reaction atmosphere under reducing conditions, and before the use they are reduced in a hydrogen-containing flow at the temperatures of their subsequent operation. It is assumed that the reduced CeO2 or Ce1–xZrxO2 surface contains sites of CO2 activation [16, 20].
The easiest and most commonly approach to the structural studies of catalysts includes the diagnostics of materials in the initial state, after activation treatments, and after reaction tests. However, ex situ studies alone cannot reveal the relationship between the composition and structure of a catalyst and its catalytic characteristics, whereas structural diagnostics of catalytic materials under conditions most closely similar to the conditions of their activation and operation can reveal the nature of active sites.
The purpose of this work is to study the structural features of Ru/Ce1–xZrxO2 ruthenium-containing catalysts prepared by the sorption-hydrolytic precipitation and to reveal the changes in the structure of catalysts during their activation by continuous heating in a hydrogen-enriched gaseous environment. We conducted powder XRD and electron microscopy ex situ studies of the catalysts in the initial state and after CO2 methanation. Also, transformations of the phase composition and structure of the catalyst components were studied by in situ powder XRD. Changes in the surface composition of the catalysts were studied by pseudo in situ XPS. The reduction process was investigated by hydrogen temperature-programmed reduction (H2–TPR).
EXPERIMENTAL
Catalyst preparation. The material for the catalyst support was the Ce0.75Zr0.25O2 commercial mixed oxide (Ekoalliance). The samples of xRu/Ce0.75Zr0.25O2 catalysts (x = 1 wt.%, 5 wt.%) were prepared by the sorption-hydrolytic precipitation. It was shown in [21-23] that this method allows for a reproducible production of catalysts based on platinum group metals (Pt, Ru, Rh) with high dispersed supported particles. The ruthenium chloride hydrate RuCl3·H2O (Krastsvetmet) was used as the ruthenium precursor. Firstly, a hydrochloric acid solution of ruthenium chloride with the molar ratio HCl:RuCl3 = 3:1 was prepared to obtain hexachlororuthenic acid H3RuCl6. Then an oxidizing agent (H2O2) was added to the solution, and the obtained solution was boiled . Next, hydrazine dichloride was added to the solution in a molar ratio of N2H6Cl2:Ru = 3:1. The resulting solution was evaporated to dryness and then dissolved in water to a volume of 10 mL. A weighted amount of powder support was added to the solution using a magnetic stirrer, and then a Na2CO3 solution (1 mol) was added dropwise to the molar ratio Na:Cl = 1:1. The suspension was stirred for 10 min at 25 °C and then for 30 min at 80 °C. 10% from the initial amount of sodium carbonate were added. The obtained precipitate was filtered and dried at 80 °C. The final heat treatment of the samples was carried out in air at 420 °C for 1 h. The material of the support sample Ce0.75Zr0.25O2 is referred to below as CZ, the catalyst samples with different ruthenium contents xRu/Ce0.75Zr0.25O2 are referred to as xRu/CZ.
According to X-ray fluorescence analysis, the elemental composition of all the obtained samples corresponded to the required one within the determination error.
Studying the catalytic properties. The experiments were carried out in a flow-through U-shaped quartz reactor (inner diameter = 3 mm) at the atmospheric pressure in the 200-450 °C temperature range (catalyst weight: 125 mg, fraction: 0.25×0.5 mm, gas mixture flow rate: 30000 cm3/gcath). The composition of the reaction mixture (vol.%) was 4CO2 + 16H2 + + 80Ar (balance). The temperature was controlled using a chromel-alumel thermocouple inserted in the catalyst bed. Before the catalytic tests, the catalysts were heated in He up to 170 °C and reduced in an H2/Ar flow for 1 h at 400 °C. The inlet and outlet reaction mixture was analyzed using a Khromos GKh-1000 gas chromatograph (Russia) equipped with a thermal conductivity detector (column - CaA molecular sieves) and a flame ionization detector (PorapakQ column) with a methanator, which determines CO, CO2, CH4 contents up to 1 ppm. The deviations of CO2 and H2 concentration in the initial mixture from the required ones did not exceed ±0.5 vol.% and 1 vol.%, respectively. Equilibrium compositions were calculated using the HSC 7.0 program under the assumption that they contained only gaseous substances (CH4, CO, CO2, H2, H2O).
Studying the samples by physical and chemical methods. The ex situ powder XRD studies were conducted on a STOE STADI MP diffractometer (STOE, Germany) in the transmission mode using MoKα radiation (λ = 0.7093 Å). The diffractometer was equipped with a linear position-sensitive MYTHEN2 1K detector (Dectris AG, Switzerland). The scanning was performed in the 2θ range of 3-35° with a step of 0.015°. The qualitative powder XRD analysis was carried out using the ICDD PDF-4+ database. The average coherent scattering region (CSR) of crystal phases was determined from peak broadening using the Scherrer equation while taking into account the instrumental broadening measured from the XRD pattern of a NIST SRM 660c reference sample (LaB6).
The in situ powder XRD studies were conducted on a Bruker D8 Advance diffractometer (Bruker, Germany; CuKα radiation, λ = 1.5418 Å) equipped with a LynxEye linear detector (2θ range = 2.9°). The measurements were carried out in the 2θ range 10-80° with a step of 0.05° using an XRK-900 high-temperature reactor chamber (Anton Paar, Austria) in the temperature range 25-400 °C under atmospheric pressure. The heating rate was 10 °C/min. The flow rate of the gaseous reaction mixture was controlled by an ALBORG gas control system. The supplied reaction mixture contained 10 vol.% of H2 in He.
The hydrogen temperature-programmed reduction was performed on a ChemBET Pulsar TPR/TPD analyzer (Quantachrome Inst., USA). The TPR profile was recorded in a flow of 10% H2 in Ar from 30 °C to 900 °C. The heating rate was 10 °C/min. TPR curves were normalized to the catalyst mass.
Transmission electron microscopy studies were conducted in the direct resolution mode (HRTEM) and scanning electron microscopy studies were conducted in the dark-field mode (STEM) using a high-angle annular dark-field (HAADF) detector for recording electrons scattered at large angles. Experiments were performed on a Themis Z microscope (Thermo Fisher Scientific, Netherlands) equipped with a two-fold astigmatism corrector at an accelerating voltage of 200 kV. Local elemental analysis was carried out using the energy-dispersive X-ray spectroscopy (EDS) data obtained on a Super-X EDX spectrometer (Thermo Fisher Scientific, Netherlands).
The pseudo in situ XPS research were conducted on a SPECS Surface Nano Analysis GmbH spectrometer (Germany) equipped with a PHOIBOS-150-MCD-9 hemispherical analyzer. The spectra were recorded using non-monochromatic AlKα radiation (hν = 1486.61 eV). The catalyst samples were studied in the initial state and after a reduction treatment in hydrogen at temperature of 400 °C and under a pressure of 1000 mbar for 30 min. The spectrometer was equipped with a special high-pressure cell allowing the sample to be processed and transferred into the analyzer chamber without contacting the air. The spectrometer was calibrated for the Ce3d3/2-u′′′ line (Eb = 916.7 eV). Relative contents of elements were determined from the integrated line intensities while taking into account photoionization cross sections of the corresponding terms [24]. After performing the Shirley background correction, the experimental curve was fitted by a number of lines corresponding to the electron photoemission of atoms in different chemical environments. The data were processed using the CasaXPS package [25].
The average size of ruthenium particles in the catalyst samples was determined by CO pulse adsorption (CO chemisorption). The samples were placed in a quartz reactor and reduced in a H2 flow at 350 °C for 30 min. Complete reduction of the samples was determined from the hydrogen absorption. Then the temperature was diminished to 20 °C and the required amount of carbon monoxide was supplied in pulses, and the moment of its appearance at the reactor exit was registered. From the data obtained, the surface area and size of ruthenium particles were calculated under the assumptions of their spherical shape and the adsorption of one CO molecule by one metal atom.
RESULTS AND DISCUSSION
The prepared xRu/CZ catalysts demonstrated high activity in the reaction of CO2 methanation. Fig. 1 shows temperature dependences of the concentrations of reaction products (CH4 and CO). After having reached the maximum, the methane concentration increases with increasing temperature and then decreases down to equilibrium values. The concentration of the CO reaction byproduct also increases with increasing temperature (not higher than 0.1 vol.%). Above 360 °C, the CO concentration approaches equilibrium values. Higher than equilibrium concentrations of CO below 340 °C indicate that CO is an intermediate product of CO2 methanation. The 5Ru/CZ catalyst exhibited a higher conversion of CO2 into methane at a significantly lower temperature and a higher selectivity to methane. Thus, the catalytic activity was improved significantly by increasing the amount of the active component from 1 wt.% to 5 wt.%. This result agrees with the data reported earlier on the catalytic activity of Ru/CeO2 supported catalysts in the reaction of CO2 methanation for ruthenium contents increasing from 1 wt.% to 2.5-4 wt.% [26].
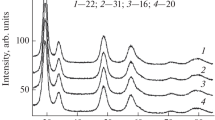
The initial and tested catalysts were studied by powder XRD. The main contribution to the diffraction patterns is due to the oxide support material. The XRD patterns of all the samples contain strong reflections characteristic of a mixed cerium–zirconium oxide with a cubic fluorite-type structure (Fig. 2). As it was shown earlier in a study of this commercial oxide support [27], some weak asymmetry of its diffraction peaks at large angles is due to the heterogeneity of the material′s phase composition. The composition of the oxide support contains the Ce0.94Zr0.06O2 cerium enriched solid solution with the average CSR size DXRD = 10.0 nm and the Ce0.2Zr0.8O2 highly dispersed (DXRD ≤ 3 nm) zirconium-enriched solid solution. The determined lattice parameter a = 5.398(1) Å corresponds to the composition of the Ce0.94Zr0.06O2 solid solution characterized by the highest content.
It was established that the 1Ru/CZ catalyst before and after the catalytic tests contains no crystals of ruthenium-containing phases detectable by powder XRD. The XRD patterns show no additional reflections from ruthenium-containing phases. The catalyst with a higher content of metallic 5Ru/CZ contains the RuO2 ruthenium(IV) oxide crystal phase with rutile structure (PDF#00-040-1290, P42/mnm space group, a = b = 4.499 Å, c = 3.107 Å) in the initial state and the Ru0 metallic ruthenium phase (PDF#00-060-0663, P63/mmc space group, a = b = 2.705 Å, c = 4.281 Å) after the catalytic tests. Within the definition error (0.002 Å), the lattice parameters obtained for the RuO2, Ru0 crystal phases coincide with those of the ICDD PDF-4+ database. The obtained data indicate that the reducing conditions of the activation process and subsequent CO2 methanation lead to the formation of Ru0 metal particles. The determined average CSR sizes of the detected phases are listed in Table 1. We also determined the average sizes of catalyst ruthenium-containing particles using CO chemisorption of samples that were preliminary reduced to obtain CO-binding Ru0 metal particles (Table 1). The chemisorption method is much more sensitive to ultrafine compounds than the powder XRD method. The fact that the size of ruthenium particles is significantly smaller than the CSR size according to the chemisorption data indicates that the 5Ru/CZ catalyst contains highly dispersed ruthenium compounds that are not detected by powder XRD. A smaller fraction of highly dispersed ruthenium particles in the tested 5Ru/CZ catalyst indicates their segregation and sintering under the conditions of the catalytic reaction. The analysis of the data obtained (Table 1) also showed that the 1Ru/CZ catalyst before and after catalytic tests contains ultrafine ruthenium compounds that are not detected by powder XRD.

Temperature dependence of obtained CH4 (a) and CO (b) concentrations as a result of CO2 methanation on the 1Ru/CZ and 5Ru/CZ catalysts.

Powder XRD patterns of initial (a) and tested (b) catalysts 1Ru/CZ (1) and 5Ru/CZ (2).
The analysis of STEM data and element distribution maps in the chosen region according to the EDX data revealed uniformly distributed atomic-scale ruthenium compounds over the 1Ru/CZ catalyst (Fig. 3(A.4)), in agreement with the chemisorption data indicating a highly dispersed state of supported particles. The 5Ru/CZ sample contains large (10-20 nm) ruthenium-containing particles and a small number of atomic-scale ruthenium species (Fig. 3(B.4)).
The study of the samples after catalytic tests showed that the ultrafine state of ruthenium is preserved in the 1Ru/CZ catalyst. The 5Ru/CZ sample also has ultrafine ruthenium compounds and segregated ruthenium-containing particles with a size of 10-20 nm (Fig. 4).

STEM images of initial 1Ru/CZ (A.1) and 5Ru/CZ (B.1) catalysts and element distribution maps in the analyzed regions according to the EDX data for the samples 1Ru/CZ (A.2, A.3, A.4) and 5Ru/CZ (B.2, B.3, B.4).

STEM images of the tested 1Ru/CZ (A.1) and 5Ru/CZ (B.1) catalysts and element distribution maps in the analyzed regions according to the EDX data for the samples 1Ru/CZ (A.2, A.3, A.4) and 5Ru/CZ (B.2, B.3, B.4).
The HRTEM data (Fig. 5) indicate that the large ruthenium-containing particles detected in the 5Ru/CZ catalyst before and after the catalytic tests correspond to the ruthenium oxide RuO2 and the metallic ruthenium Ru0, respectively. The result agrees with the powder XRD data.

HRTEM images showing the presence of RuO2 crystallites in the initial 5Ru/CZ catalyst (a) and Ru0 crystallites in the tested 5Ru/CZ catalyst (b).
During the catalytic CO2 methanation, the catalyst operates under reducing conditions in a hydrogen-rich gaseous environment. Before being used in the methanation reaction, the catalysts were activated by heating them up to 400-450 °C in a hydrogen-containing atmosphere. In the present work, we conducted in situ powder XRD studies to reveal changes in the composition and structural organization of the activated catalysts. In the XRD experiments, the catalyst samples and the support material were continuously heated in the 25-400 °C temperature range in a hydrogen-containing flow and then continuously cooled in the same environment. Fig. 6 shows the powder XRD patterns of the 5Ru/CZ sample measured upon continuous heating. The initial RuO2 crystal phase is detected up to 125 °C. At 175 °C, reflections of the RuO2 phase disappear and those of metallic ruthenium Ru0 appear, in agreement with the characteristic onset of RuO2 reduction in hydrogen at 170-200 °C [26].
It was established that the appearance of reflections from the Ru0 crystal phase coincides with the displacement of reflections from the Ce0.94Zr0.06O2 mixed oxide phase towards smaller reflection angles. We also analyzed changes in the unit cell parameters (UCPs) of the Ce0.94Zr0.06O2 mixed oxide. The expected UCP change caused by the lattice thermal expansion was determined using the lattice thermal expansion coefficient (TEC) for the isostructural CeO2 phase (α = 11.5·10−6 K–1) [28]. As can be seen in Fig. 7a, the increased UCP value correlates with the expected crystal lattice thermal expansion upon heating up to 125 °C, but then the UCP value increases abruptly relative to the expected one. Starting from 175 °C, the UCP changes linearly, but its values are much larger than the expected ones. Upon subsequent cooling, the UCP decreases linearly, in agreement with the lattice thermal contraction, and significantly exceeds its initial value after reaching the room temperature. The initial Ce0.94Zr0.06O2 UCP value is registered only after the sample contacts the air. The recorded abrupt UCP increase exceeds the thermal expansion value and indicates that the oxide support is partially reduced. Thus, reduction of CeO2 based oxides is detected by XRD from the UCP growth due to the larger size of Ce3+ cations and the formation of oxygen vacancies [29]. According to [30], the reduction of the cerium dioxide surface begins at ~500 °C, while the UCP increase not related to the thermal expansion was recorded in the experiment at 175 °C. A similar experiment was conducted for the pure support material in the temperature range of 25-450 °C. It was shown that the UCP change of individual Ce0.94Zr0.06O2 oxide corresponds to the lattice thermal expansion up to 400 °C, a slight increase is observed at a temperature of 450 °C, which is close to the point of cerium dioxide reduction [31] (Fig. 7b). Presumably, partial support reduction recorded in the catalyst at lower temperatures is due to the presence of ruthenium.

In situ powder XRD patterns of the 5Ru/CZ catalyst recorded upon continuous heating in H2/He.
Thus, an abrupt change of the Ce0.94Zr0.06O2 UCP value and formation of crystalline metallic ruthenium Ru0 are observed virtually simultaneously upon heating up to 175 °C. Apparently, the resulting Ru0 particles induce the reduction of the support surface due to the hydrogen spillover effect whereby activated adsorbed light hydrogen particles diffuse from the surface of Ru0 metal particles to the support surface, thus causing its reduction. This effect is characteristic for supported metal catalysts [32]. It is believed that partial reduction of the support surface increases the activity of catalysts in the carbon oxide methanation; the oxygen-deficient centers on the support surface participate in the activation of carbon oxide molecules [33-36].
Comparative studies of the reduction of the individual CZ support and the 5Ru/CZ catalyst were performed using hydrogen temperature-programmed reduction (H2–TPR) (Fig. 8).
The TPR-H2 profile of the individual support contains two broadened peaks with maxima at 545 °C and 815 °C, in agreement with the reported data on the two-stage reduction of CeO2 based oxides associated with a Ce4+ to Ce3+ transition [37]. The first and second (high-temperature) peaks corresponds to the surface and bulk reduction of Ce1–xZrxO2 particles, respectively. In the volume of Ce1–xZrxO2 particles, oxygen is stronger connected to Ce cations than to insufficiently coordinated surface oxygen, so the reduction begins at a higher temperature. A weak peak at 275 °C is apparently due to the reduction of molecular forms of oxygen on the surface of mixed oxide particles.

Temperature dependence of UCP for the Ce0.94Zr0.06O2 phase in the composition of the 5Ru/CZ catalyst (a) and for the Ce0.94Zr0.06O2 phase in the composition of the pure CZ support (b) upon heating in reduction conditions (1) and subsequent cooling (2). For comparison, linear UCP dependence due to thermal expansion is shown (dashed lines).

H2–TPR profiles of the pure CZ support (1) and the 5Ru/CZ catalyst (2).
The TPR–H2 profile of the 5Ru/CZ catalyst contains two narrow maxima in the low-temperature region and two broad ones in the high-temperature region. The reported data [23, 38, 39] suggest that the narrow peaks at 75-200 °C corresponds to the reduced ruthenium oxide RuO2. The splitting into two peaks may be caused by different particle sizes: the agglomerated ones are reduced at higher temperatures than highly dispersed ones [40]. This conclusion completely agrees with the electron microscopy data (Fig. 4, 5) and with the conclusion based on the comparison of powder XRD and chemisorption data suggesting the coexistence of crystallized and ultradispersed ruthenium compounds, which are not detected by powder XRD (Table 1). We also conducted a detailed quantitative analysis of the catalyst′s reduction temperature profile in hydrogen (Table 2). The amount of absorbed hydrogen, as determined from the integral intensity of the peaks at 110 °C and 140 °C, virtually coincides with that required for the reduction of all available ruthenium, in accordance with the catalyst composition and in the assumption that the initial state is 4+ (~0.99 mmol/gcat).
The amount of absorbed hydrogen observed at 270 °C on the H2–TPR catalyst curve significantly exceeds the value obtained for the support′s peak at the same temperatures. Apparently, a wider and more intense peak at 270 °C corresponds to one stage of the reduction of mixed oxide particles, namely, the reduction of Ce4+ into Ce3+ in surface Ce1–xZrxO2 layers, while the maximum at 800 °C corresponds to the reduction of Ce4+ into Ce3+ in the volume of mixed oxide particles [40-44]. Note that the maximum on the TPR–H2 catalyst curve characteristic for the surface reduction of Ce1–xZrxO2 particles is strongly shifted towards lower temperatures compared with that on the individual support curve (Fig. 8). Thus, the obtained TPR data agree with the in situ powder XRD data indicating that the reduction of the Ce1–xZrxO2 surface in the catalyst begins at a lower temperature. First, under reduction conditions, Ru0 particles are formed, and then H2 molecules dissociate from them and migrate to the nearest surface of the Ce1–xZrxO2 support, thus causing its partial reduction.
We conducted similar studies of structural changes in the 1Ru/CZ catalyst with a low metal content under similar conditions. The XRD patterns of the 1Ru/CZ catalyst show no reflections from ruthenium-containing phases over the entire temperature range (Fig. 9). However, at 200 °C the reflections from the Ce0.94Zr0.06O2 phase are shifted towards smaller angles.

In situ powder XRD patterns of the 1Ru/CZ catalyst recorded upon continuous heating and subsequent cooling in H2/He.
It was revealed that the increase of the Ce0.94Zr0.06O2 UCP corresponds to the expected one up to 100 °C and exceeds it significantly above 200 °C (Fig. 10). Upon cooling, the UCP decreases linearly in accordance with the lattice thermal contraction, but still exceeds the initial value after reaching the room temperature. Similarly to the previous case, such an abrupt UCP increase, exceeding the thermal expansion, indicates a partial reduction of the oxide support. Apparently, highly dispersed metallic ruthenium Ru0 particles are formed in the catalyst and induce the reduction of the support surface due to hydrogen spillover.
The formation Ru0 metal particles in the 1Ru/CZ catalyst upon heating in hydrogen was confirmed by pseudo in situ XPS studies. Fig. 11a shows the Ru 3d spectra of the 1Ru/CZ catalyst in the initial state and after treatment in an H2 reducing atmosphere at 400 °C. Note that the Ru 3d spectra partially overlap with the C 1s spectrum. The Ru 3d spectrum is a Ru 3d5/2–Ru 3d3/2 doublet. The ratio of integral component intensities is 3:2, the spin-orbital splitting has a value of 4.17 eV. Characteristic Ru 3d5/2 binding energies fall within a region of 279.8-280.3 eV for metallic ruthenium Ru0 and within 280.5-281.4 eV for the Ru4+ oxidized ruthenium in the form of the RuO2 oxide [45-48]. It was established that ruthenium in the initial 1Ru/CZ catalyst exists in the oxidized state, and the Ru 3d5/2 binding energy is 281.2 eV. After the treatment in hydrogen, the binding energy of the Ru 3d5/2 peak decreases down to 280.2 eV, indicating the reduction of ruthenium (Fig. 11). Thus, it was determined that the treatment in hydrogen completely reduces the ruthenium oxide compounds to the metallic Ru0 state.

Temperature dependence of UCP values for Ce0.94Zr0.06O2 in the composition of the 1Ru/CZ catalyst upon heating (1) under reduction conditions and subsequent cooling (2). For comparison, linear UCP dependence due to thermal expansion is shown (dashed line).

Ru 3d–C 1s (A) and Ce 3d spectra of the 1Ru/CZ catalyst before (1) and after the catalyst reduction in hydrogen (2).
Fig. 11b also shows Ce 3d spectra of the 1Ru/CZ catalyst before and after the reduction in hydrogen. As a result of spin-orbital interaction, the Ce 3d level splits into two sublevels Ce 3d5/2 and Ce 3d3/2 and forms a doublet with a 3:2 ratio of integrated line intensities. In turn, each component of the doublet is split into three Ce4+ lines (v/u, v″/u″, v′′′/u′′′) and two Ce3+ lines (v′/u′, v0/u0) [46, 47]. Fitting the spectra by individual components showed that the fraction of the Ce3+ cation in the initial catalyst is 24% and increases significantly (up to 43%) as a result of the reduction treatment in hydrogen. The XPS data confirm that the reduction treatment of the 1Ru/CZ catalyst leads to the formation of metallic ruthenium and causes partial reduction of the surface oxide support associated with a significant increase of the fraction of Ce3+ cations.
It was shown in a number of studies that ruthenium catalysts, containing cerium dioxide or CeO2 based mixed oxides as the support material, exhibit a higher activity in the methanation of carbon oxides than systems with supports based on inert materials [16, 49, 50]. It is believed that the enhanced catalytic properties are due to the fact that the easily reduced support surface participates in the catalytic reaction. The oxygen-deficient centers on the oxide support surface can act as activation centers for CO2 molecules so that the reaction proceeds in the region of the metal/support interface according to the formate mechanism [16, 17, 50]. The presented results of in situ studies of Ru/Ce1–xZrxO2 catalysts in the process of their activation in a hydrogen-containing reaction environment confirm that the surface of the Ce1–xZrxO2 oxide support is partially reduced by hydrogen atoms diffusing from the surface of supported Ru0 particles.
CONCLUSIONS
Comprehensive studies of the catalytic properties and structural features of supported xRu/Ce0.75Zr0.25O2 ruthenium catalysts of CO2 methanation were conducted. Catalysts with different contents of the active component (x = 1 wt.%, 5 wt.%) were prepared by the sorption-hydrolytic precipitation. According to the catalytical tests, the samples exhibit good activity in the reaction of CO2 methanation. It was established that the initial and the tested catalysts contain oxidized (Ru4+) and metallic (Ru0) ruthenium, respectively. The 1Ru/Ce0.75Zr0.25O2 catalyst with 1 wt.% Ru contains ruthenium compounds in an ultradispersed state. Increasing the ruthenium content up to 5 wt.% makes enlarges the active component particles. The catalyst with 5 wt.% Ru contains, in addition to ultrafine forms, crystallized ruthenium-containing particles. The in situ studies of the changes in the composition and structure of the catalysts during their activation treatment showed that the formation of metallic ruthenium particles under reduction conditions promotes partial reduction of the oxide support. The results demonstrate that the Ce1–xZrxO2 oxide support can participate in redox processes. It is believed that the high activity of supported metal catalysts in methanation reactions is due to the formation of oxygen-deficient centers in the region of the metal/support interface.
The studies were conducted using the equipment of the Shared Research Center “National Center of Investigation of Catalysts”.
REFERENCES
C. Bassano, P. Deiana, L. Lietti, and C. G. Visconti. P2G movable modular plant operation on synthetic methane production from CO2 and hydrogen from renewables sources. Fuel, 2019, 253, 1071-1079. https://doi.org/10.1016/j.fuel.2019.05.074
C. Mebrahtu, F. Krebs, S. Abate, S. Perathoner, G. Centi, and R. Palkovits. CO2 methanation: Principles and challenges. In: Studies in Surface Science and Catalysis, Vol. 178 / Eds. S. Albonetti, S. Perathoner, and E. A. Quadrelli. Elsevier, 2019, 85-103. https://doi.org/10.1016/b978-0-444-64127-4.00005-7
S. Clegg and P. Mancarella. Integrated modeling and assessment of the operational impact of power-to-gas (P2G) on electrical and gas transmission networks. IEEE Trans. Sustainable Energy, 2015, 6(4), 1234-1244. https://doi.org/10.1109/tste.2015.2424885
P. Frontera, A. Macario, M. Ferraro, and P. Antonucci. Supported catalysts for CO2 methanation: A review. Catalysts, 2017, 7(12), 59. https://doi.org/10.3390/catal7020059
S. Sharma, Z. Hu, P. Zhang, E. W. McFarland, and H. Metiu. CO2 methanation on Ru-doped ceria. J. Catal., 2011, 278(2), 297-309. https://doi.org/10.1016/j.jcat.2010.12.015
G. Garbarino, D. Bellotti, P. Riani, L. Magistri, and G. Busca. Methanation of carbon dioxide on Ru/Al2O3 and Ni/Al2O3 catalysts at atmospheric pressure: Catalysts activation, behaviour and stability. Int. J. Hydrogen Energy, 2015, 40(30), 9171-9182. https://doi.org/10.1016/j.ijhydene.2015.05.059
A. Karelovic and P. Ruiz. Mechanistic study of low temperature CO2 methanation over Rh/TiO2 catalysts. J. Catal., 2013, 301, 141-153. https://doi.org/10.1016/j.jcat.2013.02.009
A. Karelovic and P. Ruiz. CO2 hydrogenation at low temperature over Rh/γ-Al2O3 catalysts: Effect of the metal particle size on catalytic performances and reaction mechanism. Appl. Catal., B, 2012, 113/114, 237-249. https://doi.org/10.1016/j.apcatb.2011.11.043
I. A. Fisher and A. T. Bell. A comparative study of CO and CO2 hydrogenation over Rh/SiO2. J. Catal., 1996, 162(1), 54-65. https://doi.org/10.1006/jcat.1996.0259
J.-N. Park and E. W. McFarland. A highly dispersed Pd–Mg/SiO2 catalyst active for methanation of CO2. J. Catal., 2009, 266(1), 92-97. https://doi.org/10.1016/j.jcat.2009.05.018
J. Martins, N. Batail, S. Silva, S. Rafik-Clement, A. Karelovic, D. P. Debecker, A. Chaumonnot, and D. Uzio. CO2 hydrogenation with shape-controlled Pd nanoparticles embedded in mesoporous silica: Elucidating stability and selectivity issues. Catal. Commun., 2015, 58, 11-15. https://doi.org/10.1016/j.catcom.2014.08.027
W. Wang, S. Wang, X. Ma, and J. Gong. Recent advances in catalytic hydrogenation of carbon dioxide. Chem. Soc. Rev., 2011, 40(7), 3703. https://doi.org/10.1039/c1cs15008a
J. Ashok, S. Pati, P. Hongmanorom, Z. Tianxi, C. Junmei, and S. Kawi. A review of recent catalyst advances in CO2 methanation processes. Catal. Today, 2020, 356, 471-489. https://doi.org/10.1016/j.cattod.2020.07.023
W. K. Fan and M. Tahir. Recent trends in developments of active metals and heterogenous materials for catalytic CO2 hydrogenation to renewable methane: A review. J. Environ. Chem. Eng., 2021, 9(4), 105460. https://doi.org/10.1016/j.jece.2021.105460
W. J. Lee, C. Li, H. Prajitno, J. Yoo, J. Patel, Y. Yang, and S. Lim. Recent trend in thermal catalytic low temperature CO2 methanation: A critical review. Catal. Today, 2021, 368, 2-19. https://doi.org/10.1016/j.cattod.2020.02.017
F. Wang, S. He, H. Chen, B. Wang, L. Zheng, M. Wei, D. G. Evans, and X. Duan. Active site dependent reaction mechanism over Ru/CeO2 catalyst toward CO2 methanation. J. Am. Chem. Soc., 2016, 138(19), 6298-6305. https://doi.org/10.1021/jacs.6b02762
C. Wang, Y. Lu, Y. Zhang, H. Fu, S. Sun, F. Li, Z. Duan, Z. Liu, C. Wu, Y. Wang, H. Sun, and Z. Yan. Ru-based catalysts for efficient CO2 methanation: Synergistic catalysis between oxygen vacancies and basic sites. Nano Res., 2023, 16(10), 12153-12164. https://doi.org/10.1007/s12274-023-5592-3
S. Sharma, K. B. Sravan Kumar, Y. M. Chandnani, V. S. Phani Kumar, B. P. Gangwar, A. Singhal, and P. A. Deshpande. Mechanistic insights into CO2 methanation over Ru-substituted CeO2. J. Phys. Chem. C, 2016, 120(26), 14101-14112. https://doi.org/10.1021/acs.jpcc.6b03224
T. Li, S. Wang, D. Gao, and S. Wang. Effect of support calcination temperature on the catalytic properties of Ru/Ce0.8Zr0.2O2 for methanation of carbon dioxide. J. Fuel Chem. Technol., 2014, 42(12), 1440-1446. https://doi.org/10.1016/s1872-5813(15)60001-9
H. T. T. Nguyen, Y. Kumabe, S. Ueda, K. Kan, M. Ohtani, and K. Kobiro. Highly durable Ru catalysts supported on CeO2 nanocomposites for CO2 methanation. Appl. Catal., A, 2019, 577, 35-43. https://doi.org/10.1016/j.apcata.2019.03.011
P. A. Simonov, T. B. Shoynkhorova, P. V. Snytnikov, D. I. Potemkin, V. D. Belyaev, and V. A. Sobyanin. Sposob prigotovleniya katalizatora (Method for Preparing a Catalyst). Patent RU 2653360 C1, 2017. [In Russian]
T. B. Shoynkhorova, P. A. Simonov, D. I. Potemkin, P. V. Snytnikov, V. D. Belyaev, A. V. Ishchenko, D. A. Svintsitskiy, and V. A. Sobyanin. Highly dispersed Rh-, Pt-, Ru/Ce0.75Zr0.25O2– catalysts prepared by sorption-hydrolytic deposition for diesel fuel reforming to syngas. Appl. Catal., B, 2018, 237, 237-244. https://doi.org/10.1016/j.apcatb.2018.06.003
A. M. Gorlova, P. A. Simonov, O. A. Stonkus, V. P. Pakharukova, P. V. Snytnikov, and D. I. Potemkin. Pt/Ce0.75Zr0.25O2–x catalysts for water gas shift reaction: Morphology and catalytic properties. Kinet. Catal., 2021, 62(6), 812-819. https://doi.org/10.1134/s0023158421060057
J. H. Scofield. Hartree-Slater subshell photoionization cross-sections at 1254 and 1487 eV. J. Electron Spectros. Relat. Phenom., 1976, 8(2), 129-137. https://doi.org/10.1016/0368-2048(76)80015-1
N. Fairley, V. Fernandez, M. Richard-Plouet, C. Guillot-Deudon, J. Walton, E. Smith, D. Flahaut, M. Greiner, M. Biesinger, S. Tougaard, D. Morgan, and J. Baltrusaitis. Systematic and collaborative approach to problem solving using X-ray photoelectron spectroscopy. Appl. Surf. Sci. Adv., 2021, 5, 100112. https://doi.org/10.1016/j.apsadv.2021.100112
S. López-Rodríguez, A. Davó-Quiñonero, E. Bailón-García, D. Lozano-Castelló, and A. Bueno-López. Effect of Ru loading on Ru/CeO2 catalysts for CO2 methanation. Mol. Catal., 2021, 515, 111911. https://doi.org/10.1016/j.mcat.2021.111911
V. P. Pakharukova, D. I. Potemkin, O. A. Stonkus, N. A. Kharchenko, A. A. Saraev, and A. M. Gorlova. Investigation of the structure and interface features of Ni/Ce1–xZrxO2 catalysts for CO and CO2 methanation. J. Phys. Chem. C, 2021, 125(37), 20538-20550. https://doi.org/10.1021/acs.jpcc.1c05529
M. Mogensen. Physical, chemical and electrochemical properties of pure and doped ceria. Solid State Ionics, 2000, 129(1-4), 63-94. https://doi.org/10.1016/s0167-2738(99)00318-5
D. Marrocchelli, S. R. Bishop, H. L. Tuller, and B. Yildiz. Understanding chemical expansion in non-stoichiometric oxides: Ceria and zirconia case studies. Adv. Funct. Mater., 2012, 22(9), 1958-1965. https://doi.org/10.1002/adfm.201102648
B. Guo, S. Li, and Y. Tian. Performance of cerium oxides from different preparation methods for desulfurizing hot coal gas. ACS Omega, 2019, 4(5), 9301-9305. https://doi.org/10.1021/acsomega.9b00247
F. Giordano, A. Trovarelli, C. de Leitenburg, and M. Giona. A model for the temperature-programmed reduction of low and high surface area ceria. J. Catal., 2000, 193(2), 273-282. https://doi.org/10.1006/jcat.2000.2900
T. Sakpal and L. Lefferts. Structure-dependent activity of CeO2 supported Ru catalysts for CO2 methanation. J. Catal., 2018, 367, 171-180. https://doi.org/10.1016/j.jcat.2018.08.027
M. V. Konishcheva, D. I. Potemkin, S. D. Badmaev, P. V. Snytnikov, E. A. Paukshtis, V. A. Sobyanin, and V. N. Parmon. On the mechanism of CO and CO2 methanation over Ni/CeO2 catalysts. Top. Catal., 2016, 59(15/16), 1424-1430. https://doi.org/10.1007/s11244-016-0650-7
N. M. Martin, P. Velin, M. Skoglundh, M. Bauer, and P.-A. Carlsson. Catalytic hydrogenation of CO2 to methane over supported Pd, Rh and Ni catalysts. Catal. Sci. Technol., 2017, 7(5), 1086-1094. https://doi.org/10.1039/c6cy02536f
E. Rombi, M. G. Cutrufello, L. Atzori, R. Monaci, A. Ardu, D. Gazzoli, P. Deiana, and I. Ferino. CO methanation on Ni–Ce mixed oxides prepared by hard template method. Appl. Catal., A, 2016, 515, 144-153. https://doi.org/10.1016/j.apcata.2016.02.002
L. Znak, K. Stołecki, and J. Zieliński. The effect of cerium, lanthanum and zirconium on nickel/alumina catalysts for the hydrogenation of carbon oxides. Catal. Today, 2005, 101(2), 65-71. https://doi.org/10.1016/j.cattod.2005.01.003
A. Jan, J. Shin, J. Ahn, S. Yang, K. J. Yoon, J.-W. Son, H. Kim, J.-H. Lee, and H.-I. Ji. Promotion of Pt/CeO2 catalyst by hydrogen treatment for low-temperature CO oxidation. RSC Adv., 2019, 9(46), 27002-27012. https://doi.org/10.1039/c9ra05965b
J. Okal, M. Zawadzki, P. Kraszkiewicz, and K. Adamska. Ru/CeO2 catalysts for combustion of mixture of light hydrocarbons: Effect of preparation method and metal salt precursors. Appl. Catal., A, 2018, 549, 161-169. https://doi.org/10.1016/j.apcata.2017.09.036
N. Hamzah, N. M. Nordin, A. H. A. Nadzri, Y. A. Nik, M. B. Kassim, and M. A. Yarmo. Enhanced activity of Ru/TiO2 catalyst using bisupport, bentonite-TiO2 for hydrogenolysis of glycerol in aqueous media. Appl. Catal., A, 2012, 419/420, 133-141. https://doi.org/10.1016/j.apcata.2012.01.020
J. Okal, M. Zawadzki, P. Kraszkiewicz, and K. Adamska. Ru/CeO2 catalysts for combustion of mixture of light hydrocarbons: Effect of preparation method and metal salt precursors. Appl. Catal., A, 2018, 549, 161-169. https://doi.org/10.1016/j.apcata.2017.09.036
Z. Hu, X. Liu, D. Meng, Y. Guo, Y. Guo, and G. Lu. Effect of ceria crystal plane on the physicochemical and catalytic properties of Pd/ceria for CO and propane oxidation. ACS Catal., 2016, 6(4), 2265-2279. https://doi.org/10.1021/acscatal.5b02617
Z. Ma, S. Zhao, X. Pei, X. Xiong, and B. Hu. New insights into the support morphology-dependent ammonia synthesis activity of Ru/CeO2 catalysts. Catal. Sci. Technol., 2017, 7(1), 191-199. https://doi.org/10.1039/c6cy02089e
Y. Guo, S. Mei, K. Yuan, D.-J. Wang, H.-C. Liu, C.-H. Yan, and Y.-W. Zhang. Low-temperature CO2 methanation over CeO2-supported Ru single atoms, nanoclusters, and nanoparticles competitively tuned by strong metal–support interactions and H-spillover effect. ACS Catal., 2018, 8(7), 6203-6215. https://doi.org/10.1021/acscatal.7b04469
P. H. Ho, G. Sanghez de Luna, A. Poggi, M. Nota, E. Rodríguez-Castellón, G. Fornasari, A. Vaccari, and P. Benito. Ru–CeO2 and Ni–CeO2 coated on open-cell metallic foams by electrodeposition for the CO2 methanation. Ind. Eng. Chem. Res., 2021, 60(18), 6730-6741. https://doi.org/10.1021/acs.iecr.0c06024
X. Wang, J. C. Hanson, A. I. Frenkel, J.-Y. Kim, and J. A. Rodriguez. Time-resolved studies for the mechanism of reduction of copper oxides with carbon monoxide: Complex behavior of lattice oxygen and the formation of suboxides. J. Phys. Chem. B, 2004, 108(36), 13667-13673. https://doi.org/10.1021/jp040366o
H. Borchert, Y. V. Frolova, V. V. Kaichev, I. P. Prosvirin, G. M. Alikina, A. I. Lukashevich, V. I. Zaikovskii, E. M. Moroz, S. N. Trukhan, V. P. Ivanov, E. A. Paukshtis, V. I. Bukhtiyarov, and V. A. Sadykov. Electronic and chemical properties of nanostructured cerium dioxide doped with praseodymium. J. Phys. Chem. B, 2005, 109(12), 5728-5738. https://doi.org/10.1021/jp045828c
S. Y. Christou, M. C. Álvarez-Galván, J. L. G. Fierro, and A. M. Efstathiou. Suppression of the oxygen storage and release kinetics in Ce0.5Zr0.5O2 induced by P, Ca and Zn chemical poisoning. Appl. Catal., B, 2011, 106(1/2), 103-113 https://doi.org/10.1016/j.apcatb.2011.05.013
D. Alders, F. C. Voogt, T. Hibma, and G. A. Sawatzky. Nonlocal screening effects in 2p X-ray photoemission spectroscopy of NiO (100). Phys. Rev. B, 1996, 54(11), 7716-7719. https://doi.org/10.1103/physrevb.54.7716
J. A. H. Dreyer, P. Li, L. Zhang, G. K. Beh, R. Zhang, P. H.-L. Sit, and W. Y. Teoh. Influence of the oxide support reducibility on the CO2 methanation over Ru-based catalysts. Appl. Catal., B, 2017, 219, 715-726. https://doi.org/10.1016/j.apcatb.2017.08.011
S. Tada, O. J. Ochieng, R. Kikuchi, T. Haneda, and H. Kameyama. Promotion of CO2 methanation activity and CH4 selectivity at low temperatures over Ru/CeO2/Al2O3 catalysts. Int. J. Hydrogen Energy, 2014, 39(19), 10090-10100. https://doi.org/10.1016/j.ijhydene.2014.04.133
Funding
This work was funded by the Russian Science Foundation (project No. 21-73-20075).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
The authors of this work declare that they have no conflicts of interest.
Additional information
Russian Text © The Author(s), 2024, published in Zhurnal Strukturnoi Khimii, 2024, Vol. 65, No. 7, 128266.https://doi.org/10.26902/JSC_id128266
Publisher’s Note. Pleiades Publishing remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Kharchenko, N.A., Pakharukova, V.P., Stonkus, O.A. et al. Ex Situ and in Situ Studies of the Structural Features of Ruthenium-Containing Ru/Ce0.75Zr0.25O2 Catalysts of CO2 Methanation. J Struct Chem 65, 1277–1291 (2024). https://doi.org/10.1134/S0022476624070011
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0022476624070011