
Abstract
Nanocomposite Fe–Al and Cu–Fe–Al oxide catalysts are studied in the CO oxidation reaction. It is shown that the introduction of copper leads to a more than two orders of magnitude increase in the reaction rate. The most active catalyst is a nanocomposite containing 5 wt % CuO, 78 wt % Fe2O3, and 17 wt % Al2O3. According to X-ray diffraction analysis, the catalysts consist of hematite nanoparticles, α-Fe2O3, and amorphous alumina. Copper is in a highly dispersed state to form CuO and CuFeOx clusters. Results of in situ X‑ray absorption spectroscopy studies suggest that the reduction of copper from Cu2+ to Cu1+ and Cu0 in a CO stream begins at a temperature of 200–250°C; at 400–600°C, copper is mostly in the metallic state; the Fe3O4 → FeO → Fe reduction begins at 400–500°С. In a stoichiometric mixture of CO and O2, Fe3+ does not undergo reduction; the reduction of copper oxide to metal occurs at temperatures above 400°C. In the presence of excess oxygen in a temperature range of 100–600°C, no changes in the chemical state of the catalysts are detected. The mechanism of CO oxidation in the presence of Cu–Fe–Al nanocomposites is proposed.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
INTRODUCTION
In recent years, a steady tendency of developing and implementing catalytic fuel combustion technologies for both the production of thermal energy and the recycling of various industrial wastes has been observed [1–6]. The use of catalysts leads to a significant decrease in the material consumption and the heating plant dimensions and a more than 20-fold increase in the specific load per reactor volume [5]. Catalysts make it possible to decrease the combustion temperature from 1200–1600 to 350–750°C and thereby provide a significant decrease in the atmospheric emissions of nitrogen oxides. At the same time, the combustion efficiency considerably increases; therefore, low-calorie fuel (peat, brown coal, wood waste, etc.) can be used. Fuel combustion in a fluidized catalyst bed is thought of as the most promising approach. In this case, at the first stage, a solid fuel undergoes gasification to form a CO-containing gas. After that, CO undergoes oxidation to CO2 on the surface of the catalyst particles. Activity in the CO oxidation is a key factor determining the quality of catalysts for this process [7]. In addition, to provide a stable on-stream behavior of a catalyst in a fluidized bed mode, the catalyst should be shaped into spherical particles exhibiting a high mechanical strength and abrasion resistance. The use of catalysts based on noble metals for this process is not reasonable [8]. This is attributed to the high cost of these catalysts and severe on-stream conditions in a fluidized bed mode, which lead to the irreversible loss of noble metals due to the abrasion of the catalyst granules [9]. Catalyst systems based on mixed chromium-containing oxides (CuCr2O4, MgCr2O4) are effectively used as catalysts for fuel combustion in a fluidized bed [10]. However, their use leads to the emission of toxic chromium-containing compounds. Therefore, the development of new environmentally friendly and cheap catalyst systems for fuel combustion in a fluidized bed remains an urgent task.
Recently, it has been shown [11, 12] that nanocomposites based on iron and aluminum oxides exhibit a high catalytic activity in the CO oxidation. The addition of copper oxide leads to a considerable increase in this activity. The presence of alumina in the composition of catalysts makes it possible to use them to prepare strong spherical granules by the oil-drop granulation method [6]. In this case, the addition of pseudoboehmite at the oil-drop granulation stage not only contributes to the formation of high-strength uniform granules, but also makes it possible to stabilize the phase state of the active component. In this study, nanocomposite Cu–Fe–Al oxide catalysts containing different amounts of copper were studied by in situ X‑ray absorption spectroscopy methods, namely, extended X-ray absorption fine structure (EXAFS) and X-ray absorption near-edge structure (XANES) spectroscopy. Testing in the in situ mode revealed how the state of iron and copper cations changes under the action of reaction conditions. Comparison of the derived data with the results of kinetic studies revealed the nature of the catalytic action of nanocomposite Cu–Fe–Al oxide catalysts in the CO oxidation.
EXPERIMENTAL
The catalysts were prepared by fusing Cu(NO3)2 ⋅ 3H2O, Fe(NO3)3 ⋅ 9H2O, and Al(NO3)3 ⋅ 9H2O nitrates. During synthesis, the salts were mechanically mixed in required ratios; after that, the resulting mixture was heated to obtain a homogeneous melt of salt hydrates and held at a temperature of 200°C until the complete removal of water. The resulting powder was sequentially calcined in air at a temperature of 400°C for 1 h and at 700°C for 1 h. The catalysts are designated as CuxFeyAlz, where x, y, and z correspond to the content (wt %) of CuO, Fe2O3, and Al2O3, respectively.
The activity of the catalysts in the CO oxidation was studied using two units: a flow unit (in a pulsed mode) and a flow–circulation unit. Testing of the catalysts in a pulsed mode was conducted on a Chemosorb instrument (OOO SOLO, Russia), which consisted of a reactor, a gas supply system, and an automatic control and data acquisition system. To this end, 0.113 mL of a catalyst (bed height of 10 mm) was placed in a 13.6-mL U-shaped quartz reactor; a steady stream of an oxidizing gas—a mixture of helium (80 vol %) and oxygen (20 vol %)—was continuously passed through the reactor at a flow rate of 30 mL/min. During the test, CO was fed in a pulsed mode into the reactor placed in an oven; the pulse duration was 0.75 s; the amount of CO fed per pulse was 0.1 mL. The O2 : CO molar ratio in the stream was unstable at a level of no less than 3.5 owing to the pulsed oxidation technique. The change in the CO concentration at the outlet of the reactor was determined using a thermal conductivity detector. During testing, the temperature in the reactor was varied in a range of 100–600°C at a rate of 5°C/min. The measure of catalytic activity was temperature T50 at which a 50% CO conversion was achieved. The absolute error in determining the activity was 5°C. Testing of the catalysts in a flow–circulation mode was conducted on a PTsKU-1 unit (OOO SOLO, Russia), which consisted of a gas stream preparation system, a reactor, a condensing separator, and an automatic control and data acquisition system. During testing, a weighed portion of the test sample was placed in a 20‑mL reactor, which was connected to a circulation pump in a flow–circulation circuit. A stream of the feed reaction mixture—1 vol % CO and 1 vol % O2 in N2 —was fed into the circuit. The flow rate was varied in a range of 100–1000 mL/min. Carbon monoxide concentration was determined using a TEST-1 multicomponent gas analyzer (BONER, Russia) equipped with CO and CO2 optical sensors. Carbon monoxide conversion rate (W) was calculated by the formula
where V0 is the total flow rate of the reaction mixture (mL/min), mcat is the catalyst weight, and \(C_{{{\text{CO}}}}^{0}\) and CCO is the CO concentration (%) at the inlet and outlet of the reactor. Specific reaction rate at a given concentration of the key component was determined using typical kinetic tests [13], which are conducted to determine linear regression coefficients of the equation
In fact, by varying the total flow rate of the gas mixture at the inlet, specific steady-state oxidation rates at CO concentrations achieved at the outlet were determined. These rates can be used to calculate CO oxidation rate W * at a 50% conversion using the determined A and B coefficients.
The phase composition of the catalysts was determined by X-ray powder diffraction (XRD). Diffraction patterns were recorded on a D8 Advance diffractometer (Bruker, Germany) using monochromatic CuKα radiation (λ = 1.5418 Å). The size of domains—coherent scattering region (CSR)—was determined by the Scherrer formula.
The specific surface area of the catalysts (Ssp) was determined by the Brunauer–Emmett–Teller method using nitrogen adsorption isotherms measured at the liquid nitrogen temperature. The studies were performed using an ASAP 2400 automated system (Micromeritics Instrument Corp., United States).
Fe and Cu K-edge X-ray absorption spectra were recorded at the Structural Materials Science End-Station of the Kurchatov specialized synchrotron radiation source (Kurchatov Centre for Synchrotron Radiation and Nanotechnology, Moscow). The electron beam energy in the storage ring was 2.5 GeV; the average beam current was 100 mA. Synchrotron radiation (SR) was generated using a bending magnet. To monochromatize SR, a silicon single crystal in the form of a monoblock with a cut-out channel was used; double reflection of SR from the Si(111) planes provided an energy resolution of ΔЕ/Е = 2 × 10−4. To record spectra, the test sample was irradiated with an X-ray beam; the intensity of the incident and transmitted radiation was detected using ionization chambers filled with a nitrogen–argon mixture. Signals were digitized using Keithley 6487 picoammeters (Keithley Instruments, United States). To calibrate the monochromator, before measuring each set of Fe and Cu K-edge X-ray absorption spectra, the X-ray absorption spectra of the reference samples—iron or copper foils, respectively, were recorded. The position of the absorption edge E0 was determined from the maximum of the first derivative of the spectrum with respect to the energy near the K-absorption edge, which was 7112.0 eV for iron and 8979.0 eV for copper [14].
For in situ studies, a powder of the test catalysts was mixed with hexagonal boron nitride in a ratio of 1 : 15; the resulting mixture was compressed into a 1-mm-thick pellet. To record X-ray absorption spectra in the in situ mode, the pellet was placed in a high-temperature chamber through which a mixture of gases of a given composition was blown at atmospheric pressure. The design of the chamber made it possible to heat the catalyst and the gas mixture to a temperature of 600°C [15]. In situ X-ray absorption spectra were recorded in a stream of 5% CO in N2 and in a stream of a CO–O2 mixture at a molar ratio of CO : O2 = 2 : 1 and 1 : 1 under stepwise heating from room temperature to 600°C. The total gas flow rate was 200 cm3/min.
The experimental spectra were processed using standard procedures for subtracting the background and normalizing to the magnitude of the K-absorption edge jump using the DEMETER software package [16]. The Fourier transform of EXAFS spectra χ(k) was performed in a wavenumber range of 2.0–11.0 Å−1 with weight function k2. For a correct interpretation of the EXAFS data, a simulation of the spectra was conducted. To this end, a preliminary model was constructed using crystallographic data [17]. After that, the theoretical curve of EXAFS oscillations, which is the sum of single and multiple scattering paths taking into account several coordination spheres, was calculated. The minimization of the functional of discrepancy between the theoretical and experimental spectra using the FEFF6 software [18] made it possible to refine the parameters of the used model and determine coordination numbers, radii of coordination spheres, Debye–Waller factors, and the chemical shift.
RESULTS AND DISCUSSION
Catalytic Properties of Nanocomposites
Nanocomposite Fe–Al oxide catalysts were studied in detail earlier [11, 12]. According to XRD and transmission electron microscopy, the Fe–Al catalysts consist of Fe2O3 nanoparticles with a hematite structure and amorphous Al2O3. Aluminum cations (Al3+) are partially dissolved in the Fe2O3 lattice to form a (Fe,Al)2O3 solid solution. It was found that the activity of catalysts in the CO oxidation reaction depends on their chemical composition. The highest activity is exhibited by a catalyst containing 82 wt % Fe2O3 and 18 wt % Al2O3 [11]. For this catalyst (Fe82Al18), the light-off temperature T50 measured in a flow reactor is 275°C. Earlier, it was shown that the introduction of copper contributes to an increase in the activity of the catalyst [11]. To elucidate the nature of this effect, catalysts with an identical Fe2O3 : Al2O3 ratio of 82 : 18 and a CuO content of 3, 5, 8, and 10 wt % were synthesized (Table 1). The results of catalytic tests in a flow reactor showed that the addition of copper leads to a decrease in the light-off temperature (Fig. 1). The minimum light-off temperature (190°C) is observed in the case of CO oxidation in the presence of a catalyst containing 5 wt % CuO (Cu5Fe78Al17).

Dependence of the light-off temperature (T50) in the CO oxidation in the presence of the Cu–Fe–Al nanocomposites on the CuO content.
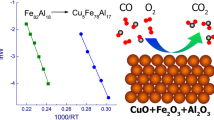
Detailed kinetic studies of the most active Fe–Al and Cu–Fe–Al nanocomposites in a flow–circulation reactor showed that, at 250°C in the presence of the Cu5Fe78Al17 catalyst, the CO oxidation rate is 260 times higher than that provided by the Fe82Al18 catalyst (Table 2). Carbon monoxide oxidation rate hardly depends on oxygen concentration in the gas phase; that is, in the presence of either of the catalysts, the order with respect to oxygen is close to zero, while the order with respect to CO is 1.10–1.11. The temperature dependences of the CO oxidation rate at a 50% conversion in the Arrhenius coordinates for the studied catalysts are shown in Fig. 2. The dependence of ln W * on 1/T is linear; therefore, the apparent activation energy value (Ea) can be determined. For the Cu5Fe78Al17 catalyst, the activation energy is 88 kJ/mol. For the Fe82Al18 catalyst, a significantly higher Ea value, 102 kJ/mol, is observed. This fact suggests that the introduction of copper leads to a change in the nature of active sites.

Dependence of the logarithm of the CO oxidation rate constant on the reciprocal of temperature for (1) the Fe82Al18 and (2) Cu5Fe78Al17 catalysts.
Structure and Chemical Composition of Cu–Fe–Al Nanocomposites
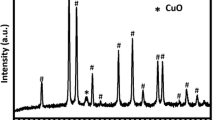
According to Fig. 3, diffraction patterns of the Cu–Fe–Al catalysts with a copper oxide content of 3–8 wt %, as in the case of the Fe–Al catalysts, exhibit peaks corresponding to Fe2O3 with a hematite structure, space group (sp. gr.) \(R\bar {3}c\) (PDF 330664). Additional reflections corresponding to crystalline copper-containing phases were not detected. An increase in the copper oxide content to 10 wt % leads to the appearance of additional low-intensity peaks in the diffraction pattern, which correspond to mixed oxide (Cu,Al,Fe)3O4 with a spinel structure (sp. gr. Fd3m) and a lattice parameter of 8.333Å, which is between the values characteristic of CuFe2O4 (8.400 Å, ICSD no. 39132) and AlFe2O4 (8.204 Å, ICSD no. 86571).

Diffraction patterns of the Cu–Fe–Al nanocomposite catalysts containing different amounts of copper: (1) α-Fe2O3 (PDF 330664), (2) Fe82Al18, (3) Cu3Fe79Al18, (4) Cu5Fe78Al17, (5) Cu8Fe75Al17, and (6) Cu10Fe74Al16.
To obtain more detailed information, the catalysts were studied by the XANES and EXAFS methods. Both methods are structure-sensitive. The use of XANES makes it possible to determine the chemical state of atoms; the use of EXAFS, provides information about the structure of the local environment of atoms. Unlike diffraction methods, EXAFS makes it possible to study the structure of nanomaterials and amorphous substances. Figure 4a shows the copper K‑edge XANES spectra of Cu–Fe–Al catalysts with a CuO content of 3–10 wt %. The Cu K-edge XANES spectra of all the studied samples are represented by a single intense peak at the absorption edge (1s → 4p transition). Comparison of the position of the Cu K‑edge and the shape of the spectra of metallic copper, Cu2O, CuO, CuAl2O4, CuFe2O4, and the catalysts (Fig. 4b) suggests that that copper is present in the catalysts mostly in the Cu2+ state. In fact, the shape of the Cu K-edge XANES spectra significantly depends on the chemical state of copper. In the K-edge absorption spectra of metallic copper, the actual absorption edge Е0 is lied at 8979 eV. Copper oxidation leads to a shift of the edge toward higher energies, which indicates the presence of a positive charge on the copper atoms. The value of the shift depends on the charge state of copper. The absorption edge in the spectrum of Cu2O, where copper is in the Cu1+ state, is shifted toward higher photon energies (~8980 eV); in addition, the spectrum exhibits an intense shoulder at 8981 eV [19]. The appearance of a shoulder at the absorption edge is attributed to the 1s → 4p transition arising from the transfer of outer-shell electrons to an inner-shell vacancy, the so-called “shakedown” electronic transitions [20, 21]. The Cu2+ atoms have a partially filled 3d electron shell. The X-ray absorption spectra of these compounds are characterized by the presence of a low-intensity peak in the pre-edge region of the spectrum and a shoulder at the absorption edge. The prepeak corresponds to the 1s → 3d dipole-forbidden quadrupole transitions arising from the hybridization of the p- and d-orbitals of copper [22]. The absorption edge of CuO and CuFe2O4 is observed at 8983 and 8989 eV, respectively.

Cu K-edge XANES spectra of (a) the Cu–Fe–Al nanocomposite catalysts and (b) bulk copper oxides and copper foil; the inset shows the pre-edge region of the XANES spectra of (1) CuFe2O4, (2) CuAl2O4, and (3) CuO.
The prepeak can be considered as an identifier of the Cu2+ state, because in compounds containing copper in the Cu1+ state, the 1s → 3d transition is completely forbidden due to the filled 3d electron shell, and a prepeak is not recorded in the absorption spectra of Cu(I) compounds. The intensity of the pre-edge peak increases with the deviation of the copper atom from the centrosymmetric position owing to an increase in the mixing of 3d and 4p orbitals and, therefore, depends on the filling of the 3d orbitals, coordination geometry, and local symmetry. This prepeak is present in the spectra of CuO, CuAl2O4, and CuFe2O4 (Fig. 4b). The prepeak intensity is higher for copper atoms in a tetrahedral coordination (in compounds of the CuAl2O4 type) and significantly lower for copper atoms in a centrosymmetric square-planar or octahedral environment (in compounds of the CuO and CuFe2O4 type) [23].
A significant shoulder is present in the low-energy region of the absorption edge of the spectra of CuO and CuAl2O4. This feature is attributed to the dipole-allowed 1s → 4p transition, which arises from the transfer of outer-shell electrons into the unbound state of the continuous spectrum and shakedown transitions. These transitions depend on the centrosymmetry of the environment of the absorbing atom and are sensitive to the nature of the ligand. A fairly intense shoulder is observed in the spectrum of CuO owing to the square-planar coordination of copper atoms, whereas the spectrum of CuAl2O4 exhibits a low-intensity shoulder, which can be attributed to the octahedral symmetry of the environment of the copper atoms. The spectrum of CuFe2O4 is characterized by an abruptly increasing absorption edge with an intense peak at 8998 eV [24, 25]. Unlike CuAl2O4, the spectrum of CuFe2O4 exhibits hardly any shoulder on the low-energy slope of the absorption edge and splitting of the main absorption peak; this fact indicates a high degree of inversion of the spinel structure [23, 24].
Thus, comparison of the spectra of the nanocomposites (Fig. 4a) with the spectra of the reference compounds (Fig. 4b) suggests that copper is present in the catalysts mostly in the Cu2+ state, because the absorption edge Е0 lies in a range of 8985–8987 eV, which is close to Е0 for CuFe2O4. The spectra of the nanocomposites are structurally similar to the spectra of CuFe2O4. The presence of a small shoulder at the absorption edge for the nanocomposites with a copper oxide content of 3–8 wt % can indicate the presence of a certain amount of copper cations with a local environment close to that of bulk CuO.
To obtain quantitative information, the XANES spectra of the catalysts were approximated by a linear combination of spectra of the reference compounds using the DEMETER software package [16]. This method is referred to as linear combination fitting (LCF); it is commonly used to analyze XANES spectra [11]. A theoretical substantiation of the LCF method is the additivity of the nature of X-ray radiation absorption by various components that constitute the test material. Hence, the absorption spectrum of a test substance is the sum of the absorption spectra of the components that constitute the test substance. Weight coefficients in the linear combination of spectra of the reference compounds are selected by the least square method. The LCF analysis revealed that the appearance of a shoulder at the absorption edge in the spectra of the Cu–Fe–Al catalysts is associated with the presence of a certain amount of copper in the composition of CuO; copper in the metallic state and Cu2O are not detected. As an example, Fig. 5 shows the result of approximation of the XANES spectrum of the Cu3Fe79Al18 catalyst by a linear combination of spectra of CuO (43 wt %) and CuFe2O4 (57 wt %). The figure shows (1) an experimental spectrum of the Cu3Fe79Al18 catalyst and (2) a model spectrum theoretically constructed by varying the weight coefficients of each individual spectrum of (3) bulk copper oxide CuFe2O4 and (4) CuO to achieve the best agreement between the theoretical and experimental spectra. Upon the addition of the spectra of metallic copper or Cu2O to a linear combination to construct a model spectrum, their weight coefficients vanished. Therefore, these substances were excluded from consideration as components of the Cu–Fe–Al catalysts.

Approximation of the Cu K-edge XANES spectrum of the Cu3Fe79Al18 catalyst by a linear combination of spectra of CuO and CuFe2O4: (1) the experimental spectrum of the Cu3Fe79Al18 catalyst, (2) the model spectrum (dashed curve), (3) the spectrum of CuFe2O4, and (4) the spectrum of CuO.
The EXAFS analysis of the Cu–Fe–Al catalysts provides more detailed information on the local environment of copper atoms, in particular, interatomic distances and coordination numbers of each coordination sphere. Figure 6a shows Fourier transform modules (FTM) of the EXAFS spectra of metallic copper, CuO, Cu2O, CuAl2O4, and CuFe2O4; FTM of the EXAFS spectra of the nanocomposite catalysts and the results of optimization of their structural models are shown in Fig. 6b. The determined values of parameters of the local environment of Cu atoms (interatomic distances and coordination numbers) are listed in Table 3. It should be noted that the form of the Fourier transform of the EXAFS signal resembles the atomic pair radial distribution function; therefore, sometimes it is incorrectly interpreted. The difference between Fourier transform and radial distribution function lies in the fact that the former contains information about both pair and polyatomic distribution functions; the positions of the peaks do not correspond to the real interatomic distances owing to a phase shift, and the shape of the peaks is heavily distorted owing to a nontrivial dependence of the backscattering amplitude on wave number k. Fourier transform can give only an approximate idea of the geometric structure; it can be completely interpreted only with the use of simulation.

Fourier transforms of EXAFS spectra of (a) copper compounds and (b) Cu–Fe–Al nanocomposite catalysts. The solid and dashed lines show the Fourier transforms of the experimental and model spectra, respectively.
Structural information is derived from EXAFS spectra using a method based on comparison of theoretical and experimental EXAFS functions. The functions of amplitudes and phases are calculated theoretically for an assumed structure of atoms in the nearest environment of the absorbing atom. According to these data, a theoretical EXAFS spectrum is constructed and the difference between the theoretical and experimental EXAFS spectra is minimized by varying the following parameters: coordination numbers, interatomic distances, Debye–Waller factors, and ionization threshold E0. The “fitting” of the model spectrum to the experimental spectrum makes it possible to largely determine the degree of correspondence between the proposed local structures and the real structures.
The simulation results shown in Fig. 6b suggest that the first peak, which lies at about 1.5 Å in the Fourier transform curve of the spectra of the catalysts, corresponds to the first coordination sphere and characterizes the Cu–O interatomic distances; the second peak in the region of 2.5 Å corresponds to the Cu–Cu and Cu–Fe interatomic distances. It is well known that a decrease in the size of a nanoparticle leads to a change in the ratio of atoms localized in the bulk and on the surface of the particle. This factor provides a change in the coordination number compared with the value characteristic of bulky standard compounds. It is evident from Fig. 6b that the intensity of the peak at 2.5 Å gradually increases with an increase in the copper content in the catalysts. According to the simulation results, the coordination number of the Cu and Fe scattering atoms (at a distance of ~2.9 and ~3.5 Å, respectively) and the Cu–O and Cu–Cu interatomic distances also monotonically increase with an increase in the copper content (Table 3). This change in the coordination numbers and interatomic distances indicates an increase in the CuFeOx and CuO cluster sizes. The results confirm the above assumption that copper atoms are present in the catalysts in the composition of CuO and CuFeOx clusters, whose small sizes hindered their detection by XRD.
In Situ XANES/EXAFS Studies
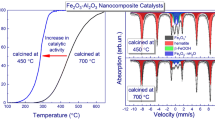
To determine the nature of the active sites, three sets of in situ tests were conducted; during the tests, Cu and Fe K-edge absorption spectra were recorded under stepwise heating of the Cu5Fe78Al17 catalyst from room temperature to 600°C in a stream of pure CO and a CO–O2 gas mixture at a ratio of CO : O2 = 2 : 1 and 1 : 1. The studies were conducted using the Cu5Fe78Al17 catalyst, which exhibited the highest activity in the CO oxidation (Fig. 1). Figure 7 shows a set of Cu and Fe K-edge XANES spectra recorded in situ during the treatment of the catalyst in a pure CO stream. Table 4 shows results of simulation of the absorption spectra by the LCF method. The shape of the Cu and Fe K-edge XANES spectra gradually changes with an increase in temperature. The Fe K‑edge XANES spectrum of the feed catalyst is similar to the spectrum of α-Fe2O3, where the iron atoms are in the Fe3+ state. The spectrum exhibits a broad pre-edge peak at 7114 eV and an intense peak at the absorption edge at 7133 eV, which is characteristic of Fe3+ cations in an octahedral oxygen environment [26, 27]. After heating of the catalyst to 400°C in a CO stream, the spectrum exhibits a small shoulder at the absorption edge, the intensity of which increases with an increase in temperature. The LCF analysis revealed that, at a temperature of about 400°C, the Fe2O3 → Fe3O4 transformation takes place; that is, the Fe3+ iron cations undergo partial reduction to Fe2+. Simultaneously with this process, the further reduction of Fe3O4 to FeO and metallic iron occurs; however, Fe3O4 does not undergo complete reduction; even at 600°C, about 25% of the iron is in the spinel structure of Fe3O4. Similar results were obtained earlier by in situ XRD studies of the reduction of the Cu5Fe78Al17 catalyst in a CO stream [28]. The Fe3O4 → Fe reduction was detected at 500°C; a small amount of FeO was detected at 650–700°C. This discrepancy of XANES and XRD results can be associated with the formation of X-ray amorphous phases, to which the XRD method is insensitive. Thus, the Fe3O4 → FeO → Fe reduction in CO occurs at 400–500°С.

(a) Fe and (b) Cu K-edge XANES spectra of the Cu5Fe78Al17 catalyst recorded in situ during heating in a CO stream.
The Cu K-edge XANES spectrum of the feed Cu5Fe78Al17 catalyst is a superposition of the spectra of CuO and CuFe2O4. During the heating of the catalyst to 250°C in a CO stream, an increase in the CuO content and the appearance of metallic copper at 250°C are observed. The reduction of copper from Cu2+ to Cu1+ and Cu0 begins at a temperature of about 200–250°C; at 400–600°C, copper is mostly in the metallic state (Table 4).
Figure 8 shows the FTM of Fe and Cu K-absorption edge EXAFS spectra of the Cu5Fe78Al17 catalyst. The FTM curve of the Fe K-absorption edges of the feed catalyst exhibits two peaks at 1.5 and 2.5 Å, which correspond to the Fe–O and Fe–Fe distances, respectively, in Fe2O3. It is clearly seen from Fig. 8a that the intensity of the second peak, which corresponds to the Fe–Fe distances in Fe2O3, monotonically decreases with an increase in temperature from room temperature to 500°C. This fact suggests that the filling of the cationic positions of iron decreases owing to thermal and structural disordering. At 600°C, the appearance of a peak at 2.25 Å corresponding to the Fe–Fe interatomic distance in the body-centered cubic lattice of metallic iron is observed. This means that the reduction of iron oxide to metal occurs at temperatures above 500°C. For the Cu K-absorption edge, the curves of the feed catalyst exhibit an intense peak at 1.5 Å, which is characteristic of the Cu–O distance (Fig. 8b). With an increase in temperature, the intensity of this peak abruptly decreases. After heating to 250°C, an additional peak at about 2.2 Å appears; it corresponds to the Cu–Cu distance in metallic copper; this peak becomes particularly pronounced at a temperature of 300°C. Thus, the EXAFS results support the earlier conclusion that fine copper oxide is reduced to metal in a CO stream at a temperature of about 300°C.

Fourier transform moduli of (a) Fe and (b) Cu EXAFS spectra of the Cu5Fe78Al17 catalyst recorded in situ during heating in a CO stream.
In situ Cu and Fe K-edge XANES spectra recorded for the Cu5Fe78Al17 catalyst treated in a stream of a CO–O2 gas mixture at a CO : O2 ratio of 2 : 1 are shown in Fig. 9. A shift of the Fe K-absorption edge is not observed up to 600°C, while above 400°C the Cu K-edge XANES spectra change similarly to the spectra recorded in studying the catalyst in a pure CO stream, yet with a shift by 200°C to the high temperature region. Hence, the presence of oxygen in the gas mixture hinders the reduction. It should be noted that, using XANES spectra, it is difficult to determine whether copper is in the Cu0 or Cu1+ state. More detailed information can be obtained using the EXAFS method.

(a) Fe and (b) Cu K-edge XANES spectra of the Cu5Fe78Al17 catalyst recorded in situ during heating in a stream of the reaction mixture (CO : O2 = 2 : 1).
Figure 10 shows the respective FTM curves of the Fe and Cu K-edge EXAFS spectra. The FTM curves of the copper EXAFS spectra at 500 and 600°C clearly exhibit peaks at 1.5 and 2.25 Å, which correspond to the Cu–O distances in copper oxide and the Cu–Cu distances in metallic copper. Hence, during CO oxidation at 500–600°C, copper undergoes reduction; the catalyst contains copper in both the oxidized and metallic states. The FTM curves of the iron K‑edge spectra exhibit two intense peaks at 1.5 and 2.5 Å, which are characteristic of the Fe–O and Fe–Fe interatomic distances, respectively, in Fe2O3. With an increase in temperature, the peak intensity gradually decreases by 2.5 Å. This fact suggests that, during heating to 500–600°C in a stream of a CO–O2 mixture, as in the case of reduction in a CO stream, cation vacancies are formed in the Cu5Fe78Al17 catalyst. However, even at 500–600°C, no additional peaks corresponding to the Fe–Fe interatomic distance in metallic iron are observed in the region of 2.25 Å in the FTM curves. That is, in the presence of oxygen in the gas mixture, iron is not reduced to the metallic state.

Fourier transform moduli of (a) Fe and (b) Cu EXAFS spectra of the Cu5Fe78Al17 catalyst recorded in situ during heating in a stream of the reaction mixture (CO : O2 = 2 : 1).
Thus, the XANES and EXAFS analysis results show that, in the presence of oxygen in the reaction mixture, the Cu2+ → Cu1+ reduction temperature is shifted to the high temperature region: reduction begins at ~400°C; after that, at 500°C, the formation of metallic copper begins. It should be emphasized that copper is present in two states, namely, Cu1+ and Cu0. At the same time, the oxidation state of the iron remains unchanged even at 600°C (Fig. 9).
Finally, in situ XANES studies of the catalyst during CO oxidation in the presence of excess oxygen (CO : O2 ratio of 1 : 1) were conducted. It was found that the shape of the Cu and Fe K-edge XANES spectra remains unchanged during heating to 600°C; that is, the states of iron and copper do not change under the reaction conditions (spectra are not shown). Hence, it can be assumed that, during CO oxidation in the presence of the Cu–Fe–Al nanocomposite catalysts, the Fe3+ → Fe2+ and Cu2+ → Cu1+ partial reduction with CO molecules occurs; however, in the presence of oxygen, a rapid reverse oxidation to Fe3+ and Cu2+ occurs. The oxidation rate is higher than the reduction rate; this feature is characteristic of the Mars–van Krevelen redox mechanism. This finding is in good agreement with the results of kinetic studies indicating the zero order of the reaction with respect to oxygen and the first order of the reaction with respect to CO during CO oxidation in the presence of the Fe–Al and Cu–Fe–Al nanocomposites (Table 2). Carbon monoxide oxidation in the presence of copper-containing nanocomposites occurs mostly on copper oxide: CO is adsorbed on the surface of CuO clusters and then reacts with lattice oxygen to form СО2 and a Cu1+—oxygen vacancy; the subsequent adsorption of oxygen leads to the filling of the vacancies and the oxidation of Cu1+ to Cu2+. Carbon monoxide oxidation in the presence of Cu–Fe–Al catalysts can occur via another route: CO is adsorbed on Fe3+ cations and then reacts with lattice oxygen Fe3+–[O2−]. However, the rate of the alternative route of CO oxidation is 260 times lower than the rate of CO oxidation on copper oxide (Table 2). This fact suggests that it is CuO and CuFeOx clusters that are active sites in the CO oxidation reaction in the presence of Cu–Fe–Al nanocomposites.
CONCLUSIONS
Two sets of Fe–Al and Cu–Fe–Al oxide nanocomposites promising for the development of catalysts for solid fuel combustion in a fluidized bed have been synthesized. They have been tested in the CO oxidation. It has been shown that the introduction of copper leads to a significant increase in the activity of the catalysts in the CO oxidation. The most active catalyst is a nanocomposite containing 5 wt % CuO, 78 wt % Fe2O3, and 17 wt % Al2O3. According to XRD, the catalysts consist of hematite nanoparticles, α-Fe2O3, and amorphous alumina. Copper is present in a highly dispersed state to form CuO and CuFeOx clusters. An increase in the copper content to 10 wt % leads to the formation of a CuxAlyFe3 – x – yO4 spinel, which is accompanied by a decrease in the catalyst activity. The results of in situ X-ray absorption spectroscopy studies have shown that CO oxidation in the presence of Cu–Fe–Al occurs mostly on the copper oxide clusters by the redox mechanism: CO is adsorbed on the surface of the copper oxide clusters and then reacts with lattice oxygen to form CO2 and a Cu1+—oxygen vacancy; the subsequent adsorption of oxygen leads to the filling of the vacancies and the oxidation of Cu1+ to Cu2+. The rate of CO oxidation on iron oxide is about 260 times lower than the rate of CO oxidation on copper oxide. Iron oxide and alumina are structural promoters that provide a high specific surface area of the catalysts and stability of the fine copper oxide particles.
REFERENCES
Prasad, R., Kennedy, L.A., and Ruckenstein, E., Catal. Rev., 1984, vol. 26, p. 1.
Ismagilov, Z.R. and Kerzhentsev, M.A., Catal. Today, 1999, vol. 47, p. 339.
Simonov, A.D., Yazykov, N.A., Vedyakin, P.I., Lavrov, G.A., and Parmon, V.N., Catal. Today, 2000, vol. 60, p. 139.
Simonov, A.D., Fedorov, N.A., Dubinin, Yu.V., Yazykov, N.A., Yakovlev, V.A., and Parmon, V.N., Catal. Ind., 2013, vol. 5, p. 42.
Parmon, V.N., Simonov, A.D., Sadykov, V.A., and Tikhov, S.F., Combust., Explos. Shock Waves, 2015, vol. 51, p. 143.
Fedorov, A.V., Yazykov, N.A., Bulavchenko, O.A., Saraev, A.A., Kaichev, V.V., and Yakovlev, V.A., Nanomaterials, 2020, vol. 10, p. 1002.
Fedorov, A.V., Saraev, A.A., Kremneva, A.M., Selivanova, A.V., Vorokhta, M., Šmíd, B., Bulavchenko, O.A., Yakovlev, V.A., and Kaichev, V.V., ChemCatChem, 2020, vol. 12, p. 4911.
Kamal, M.S., Razzak, S.A., and Hossain, M.M., Atmos. Environ., 2016, vol. 140, p. 117.
Ismagilov, Z.R., Shkrabina, R.A., and Koryabkina, N.A., Catal. Today, 1999, vol. 47, p. 51.
Arendarskii, D.A., Ismagilov, Z.R., and Barannik, G.B., Catal. Lett., 1992, vol. 15, p. 347.
Fedorov, A.V., Tsapina, A.M., Bulavchenko, O.A., Saraev, A.A., Odegova, G.V., Ermakov, D.Yu., Zubavichus, Y.V., Yakovlev, V.A., and Kaichev, V.V., Catal. Lett., 2018, vol. 148, p. 3715.
Bulavchenko, O.A., Pochtar’, A.A., Gerasimov, E.Yu., Fedorov, A.V., Chesalov, Yu.A., Saraev, A.A., Yakovlev, V.A., and Kaichev, V.V., Appl. Catal., A, 2020, vol. 590, p. 117 364.
Pakharukov, I.Yu., Bobrov, N.N., and Parmon, V.N., Catal. Ind., 2009, vol. 1, p. 38.
Bearden, J.A. and Burr, A.F., Rev. Mod. Phys., 1967, vol. 39, p. 125.
Veligzhanin, A.A., Zubavichus, Ya.V., Chernyshov, A.A., Trigub, A.L., Khlebnikov, A.S., Nizovskii, A.I., Khudorozhkov, A.K., Beck, I.E., and Bukhtiyarov, V.I., J. Struc. Chem., 2010, vol. 51, p. S20.
Ravel, B. and Newville, M., J. Synchrotron Radiat., 2005, vol. 12, p. 537.
ISCD (Inorganic Crystal Structure Database) https://icsd.products.fiz-karlsruhe.de/
Rehr, J.J., Mustre de Leon, J., Zabinsky, S.I., and Albers, R.C., J. Am. Chem. Soc., 1991, vol. 113, p. 5135.
Kim, J.Y., Rodriguez, J.A., Hanson, J.C., Frenkel, A., and Lee, P.L., J. Phys. Chem. B, 2003, vol. 107, p. 9195.
Kosug, N., Yokoyama, T., Asakuna, K., and Kuroda, H., Chem. Phys., 1984, vol. 91, p. 249.
Bair, R.A. and Goddard, W.A., Phys. Rev. B, 1980, vol. 22, p. 2767.
Hahn, J.E., Scott, R.A., Hodgson, K.O., Doniach, S., Desjardins, S.E., and Solomon, E.I., Chem. Phys. Lett., 1982, vol. 88, p. 595.
Shimizu, K.-I., Maeshima, H., Yoshida, H., Satsuma, A., and Hattori, T., Phys. Chem. Chem. Phys., 2001, vol. 3, p. 862.
Krishnan, V., Selvan, R.K., Augustin, C.O., Gedanken, A., and Bertagnolli, H., J. Phys. Chem. C, 2007, vol. 111, p. 16 724.
Caddeo, F., Loche, D., Casula, M.F., and Corrias, A., Sci. Rep., 2018, vol. 8, p. 797.
Piquer, C., Roca, A.G., Laguna-Marco, M.A., Boada, R., Guglieri, C., and Chaboy, J., J. Phys. Chem. C, 2014, vol. 118, p. 1332.
Okube, M., Oshiumi, T., Nagase, T., Miyawaki, R., Yoshiasa, A., Sasaki, S., and Sugiyama, K., J. Synchrotron Radiat., 2018, vol. 25, p. 1694.
Bulavchenko, O.A., Vinokurov, Z.S., Saraev, A.A., Tsapina, A.M., Trigub, A.L., Gerasimov, E.Y., Gladky, A.Y., Fedorov, A.V., Yakovlev, V.A., and Kaichev, V.V., Inorg. Chem., 2019, vol. 58, p. 4842
ACKNOWLEDGMENTS
The kinetic and XRD studies were performed using the equipment of the Center for collective use “National Center for Catalyst Research.” The authors thank E. Khramov and A. Trigub for their assistance in the EXAFS and XANES measurements.
Funding
This work was supported by the Russian Science Foundation (project no. 17-73-20157).
Author information
Authors and Affiliations
Corresponding author
Additional information
Translated by M. Timoshinina
Abbreviations: EXAFS, extended X-ray absorption fine structure spectroscopy; XANES, X-ray absorption near-edge structure spectroscopy; LCF, linear combination fitting; FTM, Fourier transform moduli; XRD, X-ray diffraction analysis; CSR, coherent scattering region; W, CO conversion rate; V0, total flow rate of the reaction mixture (mL/min); mcat, catalyst weight; \(C_{{{\text{CO}}}}^{0}\) and CCO, CO concentration (%) at the inlet and outlet of the reactor; Ssp, specific surface area; Ea, apparent activation energy; SR, synchrotron radiation; sp. gr., space group.
Rights and permissions
About this article

Cite this article
Kremneva, A.M., Fedorov, A.V., Saraev, A.A. et al. In Situ X-Ray Absorption Spectroscopy Studies of Carbon Monoxide Oxidation in the Presence of Nanocomposite Cu–Fe–Al Oxide Catalysts. Kinet Catal 62, 160–171 (2021). https://doi.org/10.1134/S0023158421010079
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1134/S0023158421010079