
Abstract
Body mass index (BMI) is a crucial health indicator for obesity. With the progression of socio-economic status and alterations in lifestyle, an increasing number of global populations are at risk of obesity. Given the complexity and severity of neurological diseases, early identification of risk factors is vital for the diagnosis and prognosis of such diseases. In this study, we employed Mendelian randomization (MR) analysis utilizing the most comprehensive genome-wide association study (GWAS) data to date. We selected single nucleotide polymorphisms (SNPs) that are unaffected by confounding factors and reverse causality as instrumental variables. These variables were used to evaluate the genetic and causal relationships between Body Mass Index (BMI) and various neurological diseases, including Parkinson’s Disease (PD), Alzheimer's Disease (AD), Amyotrophic Lateral Sclerosis (ALS), Multiple Sclerosis (MS), Ischemic Stroke (IS), and Epilepsy (EP). The Inverse Variance Weighted (IVW) analysis indicated that there was no significant causal relationship between Body Mass Index (BMI) indicators and PD (P-value = 0.511), AD (P-value = 0.076), ALS (P-value = 0.641), EP (P-value = 0.380). However, a causal relationship was found between BMI indicators and MS (P-value = 0.035), and IS (P-value = 0.000), with the BMI index positively correlated with the risk of both diseases. The Cochran’s Q test for MR-IVW showed no heterogeneity in the MR analysis results between the BMI index and the neurological diseases (P > 0.05). The Egger intercept test for pleiotropy revealed no horizontal pleiotropy detected in any of the neurological diseases studied (P > 0.05). It was found that there was no causal relationship between BMI and PD, AD, ALS, EP, and a genetic causal association with MS, and IS. Meanwhile, the increase in BMI can lead to a higher risk of MS and IS, which reveals the critical role of obesity as a risk factor for specific neurological diseases in the pathogenesis of the diseases.
Similar content being viewed by others

Introduction
Body Mass Index (BMI), a prevalent metric for assessing obesity, has been extensively employed for its simplicity, convenience, and high sensitivity in detecting fat accumulation in clinical settings1,2. With the ongoing evolution of economic standards and shifts in dietary habits and lifestyles, a significant number of children and adults are confronted with the risk of obesity3. An increasing body of research has investigated the health implications of elevated BMI, revealing that a high BMI not only elevates the risk of various diseases but also augments mortality rates4,5,6. Numerous studies have corroborated the association between a high BMI and an increased likelihood of developing specific conditions, such as type 2 diabetes, hypertension, and coronary heart disease7,8. Moreover, a high BMI contributes to the onset of musculoskeletal disorders, encompassing rheumatoid arthritis and psoriatic arthritis, as well as neoplastic growths9,10.
Neurological diseases encompass a wide array of lesions that affect the human nervous system. These include but are not limited to, cerebrovascular disorders, neurodegenerative conditions, demyelinating lesions, infectious neurological diseases, oncological disorders, metabolic neurological diseases, and hereditary neurological illnesses. As society ages, neurological diseases have emerged as a leading cause of mortality and disability among humans. Among them, Parkinson’s Disease (PD) and Alzheimer’s Disease (AD) are the two most common neurodegenerative diseases. The etiology of Parkinson’s disease is not yet clear and is believed to be related to age, gender, smoking, alcohol consumption, vitamin D exposure, pesticide use, uric acid levels, and cranial brain injuries11,12,13,14,15. Its main pathological basis is the concentration of striatal dopamine and the presence of neuronal Lewy bodies12,13. Alzheimer’s disease is considered to be related to age, infection, and environmental factors16,17, and its main pathology includes the deposition of β-amyloid protein, phosphorylation of tau protein, inflammatory mechanisms, and mitochondrial dysfunction18. Amyotrophic Lateral Sclerosis (ALS) is a neurodegenerative disease affecting both upper and lower motor neurons. The etiology and pathogenesis of ALS remain largely unclear. Pathological studies have reported the aggregation and accumulation of ubiquitinated protein inclusion bodies within motor neurons19. Multiple Sclerosis (MS), is an immune-mediated demyelinating disease of the central nervous system, distinguished by its spatial and temporal multiplicity. To date, the etiology of MS remains elusive, though it is postulated to be linked to immune responses triggered by infections, including those caused by Epstein–Barr Virus (EBV), human herpesvirus type 6, and measles virus, among others20. Ischemic stroke (IS), a prevalent cerebrovascular condition, has seen an increasing trend in recent years. It is widely believed to be closely associated with factors such as advancing age, hypertension, hyperlipidemia, diabetes mellitus, smoking, alcohol consumption, cardiac diseases, and vascular inflammatory conditions21,22. Additonally, epilepsy (EP), is characterized by clinical syndromes arising from abnormally synchronized neuronal discharges triggered by diverse factors. The etiology of EP encompasses genetic predispositions, structural damage, or functional irregularities within the central nervous system, including traumatic brain injuries, cerebrovascular diseases, brain tumors, and central nervous system infections. Research has indicated that this condition correlates with elements such as age and metabolic processes23.
Mendelian randomization (MR) is an analytical method, recently proposed, for assessing the causal relationship between exposure and outcome factors. It utilizes genetic instrumental variables, such as single nucleotide polymorphisms (SNPs), which are randomly assigned and not subject to any causality. This method controls for confounders while maintaining the constancy of the genes and their independence from disease progression. MR studies avoid the effects of reverse causality and can provide a scientific evaluation of the genetics of the relationship between exposure and outcome24,25. This study explored the genetic association between BMI and various neurological diseases, aiming to provide some ideas and evidence for disease prevention and treatment.
Materials and methods
Study design
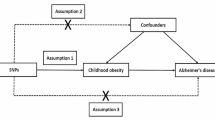
In this investigation, single nucleotide polymorphisms (SNPs) correlated with exposure and outcome factors within the Genome-Wide Association Study (GWAS) dataset were employed as instrumental variables to probe the genetic causality linking them. The study adhered to the tripartite hypothetical prerequisites for MR studies: (1) All instrumental variables exhibited a robust correlation with exposure factors, with underperforming instrumental variables omitted (P < 5 × 10−8, F-statistics > 10); (2) Instrumental variables bore no association with potential confounders, a premise corroborated by an exhaustive review of the literature and pertinent statistical methodologies; (3) All instrumental variables were stipulated to impact the outcome exclusively via BMI levels, precluding alternative pathways26,27. The data selected for this study was sourced from open-access public databases. An illustration of the study design is depicted in Fig. 1.

Diagram of the design of the two-sample Mendelian randomization study.
Data sources
Data on BMI indicators were sourced from the IEU database, which contains genome-wide association summary data28,29 (available at https://gwas.mrcieu.ac.uk/datasets/ieu-b-4816/). The cohort comprised 999,998 male and female participants of European descent, with measurements for 7,191,606 SNPs. Ethical approval and informed consent were previously obtained in the original study, as documented in the relevant literature. The initial ethical guidelines adhered to the Declaration of Helsinki. All indicators about age, gender mismatch, non-European ancestry, and duplicate samples underwent stringent quality control. Subsequently, the identified SNPs were utilized for further analysis following covariate harmonization.
Summary genome-wide association data30,31,32,33,34,35 for multiple neurologic diseases were obtained from the IEU database (https://gwas.mrcieu.ac.uk/datasets/ieu-b-7/, https://gwas.mrcieu.ac.uk/datasets/ieu-b-2/, https://gwas.mrcieu.ac.uk/datasets/ieu-b-18, https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST90013429/, https://gwas.mrcieu.ac.uk/datasets/ebi-a-GCST0058), which included 33,674 PD cases, and 449,056 controls, 21,982 AD cases and 41,944 controls, 47,429 MS cases and 68,374 controls, 22,040 amyotrophic lateral sclerosis cases and 62,644 controls, 34,217 IS cases and 40,611 controls,15,212 EP cases and 29,677 controls. The participants with PD, AD, MS, and IS were of European origin, and those with amyotrophic lateral sclerosis and EP were a mixed multiracial and multiregional population. We have attributed and controlled the quality of all disease data above and obtained relevant genetic variations (nSNPs; PD = 12,858,066, AD = 10,528,610, MS = 6,304,359, ALS = 10,181,076, IS = 7,537,579, EP = 4,880,492). On this basis, SNPs related to BMI indicators were further extracted. All participants, sample size, genetic variation, and quality control information can be searched and obtained through the IEU database website.
Selection of instrumental variables
By identifying SNPs that are significantly associated with the BMI index (p < 5 × 10−8), and subsequently conducting cluster analysis based on these SNPs (r2 < 0.001, cluster distance = 10,000 kb), we excluded SNPs with similar effects. Additionally, SNPs that were directly causally linked to the outcomes (PD, AD, MS, ALS, IS, EP; P < 5 × 10−8) were also excluded. By reviewing the literature, further exclusion of SNPs related to confounding factors was conducted, such as risk factors associated with PD: age, gender, coffee consumption, pesticides, smoking, alcohol consumption, uric acid levels, stroke, cardiovascular diseases, encephalitis, head injuries, and thyroid diseases11,36,37,38,39,40,41,42,43,44. Risk factors associated with AD: gender, age, smoking, diet, traumatic brain injury, cardiovascular diseases, diabetes, stroke, and hearing impairment45,46,47,48,49,50,51. Risk factors associated with MS: age, gender, smoking, birth season, EBV infection, and VitD52,53,54,55. Risk factors associated with ALS: gender, age, smoking, physical activity, alcohol consumption, environmental factors, viral exposure, and cyanide toxicity56,57,58,59,60,61. Risk factors associated with IS: gender, age, smoking, alcohol consumption, income, diabetes, and heart disease62,63,64,65. Risk factors associated with EP: brain infections, metabolic disorders, immunity, head trauma, stroke, brain tumors, hypoxia, and drug or alcohol poisoning66,67,68,69,70,71,72,73,74. To mitigate the impact of weak instrumental variables on the study’s outcomes, we selected only those instrumental variables with a statistic F value greater than 10. Furthermore, to ensure that SNPs influenced outcome factors exclusively through exposure factors, we omitted SNPs of moderate allelic frequency. In instances where particular SNPs were unavailable, we utilized an online platform to search for and obtain information on suitable proxy SNPs (https://ldlink.nci.nih.gov/).
MR analysis
The association between BMI and PD, AD, MS, ALS, IS, and EP, was investigated through a genetic study. This study employed a two-sample MR analysis, utilizing the “TwoSampleMR” package in R language software, version 4.2.1. The primary analytical approach was the Inverse Variance Weighting (IVW) method, complemented by additional techniques such as MR Egger, Weighted Median (WM), Simple Mode, and Weighted Mode. IVW is a technique that aggregates multiple random variables to minimize the variance of their sum. It assigns weights to each variable inversely proportional to its variance, facilitating the combination of results from independent studies using the Wald ratio method. The Wald ratio method calculates exposure-outcome effect values for each SNP, providing a robust causality estimate between exposure and outcome, and is regarded as the principal analytical method75,76. The Egger regression method facilitates multivariate assessment of outcomes through its intercept. The MR-PRESSO (MR Pleiotropy Residual Sum and Outlier) method calculates the median of a distribution function derived from ranking SNP effect sizes based on their weights. This method is primarily used when at least 50% of the data originates from valid instrumental variables, resulting in more robust estimates. Compared to IVW, both simple and weighted modal assessments are less efficacious, rendering them supplementary and complementary analyses.
Sensitivity analysis
We performed sensitivity analysis on the results of the causal relationship between BMI indicators and PD, AD, MS, ALS, IS, and EP, which mainly included horizontal pleiotropy, heterogeneity test, and Leave-one-out method77,78,79. The MR-Egger method was used to test whether the results had horizontal pleiotropy, and when the regression intercept was not zero and P > 0.05, it indicated that there was no sign of horizontal pleiotropy. The Cochran Q test was used to test for the presence of heterogeneity among individual SNPs by assessing the P-value, and when P > 0.05, it indicated the absence of heterogeneity, in which case the fixed-effects IVW method would be used, otherwise the random-effects IVW method would be used. The leave-one-out method was used to assess whether the combined effect of the remaining SNPs after the removal of one SNP was consistent with the main effect; if it was consistent, it indicated that the individual SNPs removed did not have an excessive effect on the MR analysis, then the result was considered to be reliable, and vice versa.
Ethical approval and consent to participate
This study used only publicly available GWAS summary statistics and therefore did not require ethical approval. The relevant GWAS researchers have obtained their respective ethical approvals.
Results
Association estimation of SNPs for genome-wide significance of BMI and multiple neurological diseases
In accordance with the instrumental variable screening criteria employed in this study, the 7,191,606 SNPs associated with the BMI index and the 12,858,066 associated with PD, the 10,528,610 associated with AD, the 6,304,359 associated with MS, the 10,181,076 associated with ALS, the 7,537,579 associated with IS, and the EP related 4,880,492 SNPs were analyzed separately for coordination. A total of 42 SNPs associated with PD, 42 SNPs associated with AD, 39 SNPs associated with MS, 42 SNPs associated with ALS, 42 SNPs associated with IS, and 31 SNPs associated with EP, all exhibiting genome-wide significance for BMI indicators.
Results of MR analysis
The results of IVW analysis suggested that there was no clear genetic causality between BMI index and PD (β = − 0.044 95% Cl − 0.175–0.087; P-value = 0.511), AD (β = − 0.037; 95% Cl − 0.078–0.004; P-value = 0.076 ), ALS (β = − 0.008; 95% Cl − 0.039–0.023; P-value = 0.641), epilepsy (β = − 0.008 95% Cl − 0.026–0.010; P-value = 0.380). Nevertheless, there was a positive genetic causality between BMI index and MS (β = 0.052; 95% Cl 0.003–0.101; P-value = 0.035), IS (β = 0.034; 95% Cl 0.016–0.052; P-value = 0.000) and BMI was positively associated with the risk of developing both. The results of MR Egger's analysis suggested that there was no causal relationship between BMI and PD, AD, MS, ALS, IS, and EP (P > 0.05). The results of the weighted median method indicated that there was a causal relationship between BMI and MS, IS (P < 0.05). Simple model results implied a relationship between BMI and IS (P < 0.05). The results of the weighted model revealed that no genetic causality existed between BMI and PD, AD, MS, ALS, IS, and EP (P > 0.05). The results are shown in Table 1. Scatter plots were used to visualize the results, in which the regression lines for IVW, weighted median, MR-Egger, simple mode, and weighted mode are shown in Fig. 2. The forest plot demonstrated that among the neurological diseases studied, only the red lines of MS and IS indicators were located on the “0” side, suggesting that the BMI indicators MS and IS had a significant effect (Fig. 3).

Scatter plots of the causal effect of BMI metrics with multiple neurological disorders ((a) PD, (b) AD, (c) MS, (d) ALS, (e) IS, (f) EP). The horizontal coordinate on the plot is the effect of SNP on BMI indicators (black dots are beta values, horizontal crosses are standard errors, and the vertical coordinate is the effect of SNP on diseases (black dots are beta values, vertical crosses are standard errors), and the slopes of the straight lines represent the causal effect of each method, respectively. Regression lines for inverse variance weighted (IVW), weighted median, MR-Egger, simple, and weighted models are shown.

Forest plots of causal effects of BMI metrics with multiple neurological diseases ((a) PD, (b) AD, (c) MS, (d) ALS, (e) IS, (f) EP). Red and black dots and lines represented causal estimates of the risk of multiple neurological disorders by BMI metrics.
Sensitivity analysis of MR results
The results of Cochran's Q test in MR-IVW indicated that there was no significant heterogeneity between the results of MR analysis of BMI index and various neurological diseases (P > 0.05). The findings from the MR Egger’s test were in line with those from the Cochran's Q test. The funnel plots (Fig. 4) showed that the SNPs for MS and IS were relatively evenly distributed on both sides of the straight line in multiple neurological diseases. Additionally, the Egger intercept test indicated that no significant pleiotropic effects were detected in any of the neurological diseases examined (P > 0.05). MR presso test further corroborated this finding (Table 2).

Funnel plot of causal effects of BMI metrics with multiple neurological diseases ((a) PD, (b) AD, (c) MS, (d) ALS, (e) IS, (f) EP), the blue vertical line is the causality estimation for all SNPs, and whether all black dots are symmetrically distributed on both sides of the line is used as a measure of the reliability of the MR results.
By visualizing and expressing the results of the IVW method through the Leave-one-out method and eliminating individual SNPs sequentially, it becomes evident that the distribution of the remaining SNPs associated with the investigated neurological disorders remains proximal to the red-marked position on the graph. The aggregate findings remain largely unperturbed, thereby corroborating the stability and reliability of the IVW analytical results, as depicted in Fig. 5.

Leave-one-out method to exclude the effect of individual SNPs on the overall results ((a) PD, (b) AD, (c) MS, (d) ALS, (e) IS, (f) EP). The black dots in the lines in the figure indicate the results of the IVW method after excluding a particular SNP, and the red dots indicate the estimation of the results of the IVW method for all SNPs.
Discussion
In this study, we examined the genetic associations between BMI indicators and various neurological diseases for the first time. All data were obtained from genome-wide association data. Following rigorous quality control, we identified SNPs associated with BMI and extracted SNPs linked to various neurological disorders (PD, AD, MS, ALS, IS, EP). Based on these findings, we further analyzed and categorized SNPs related to both BMI and the aforementioned diseases, yielding millions of quantitative genetic variants. By conducting MR and sensitivity analyses, the results suggested that there was a genetically causal relationship between BMI and MS (P value = 0.035) and IS (P value = 0.000) and that BMI was positively associated with the risk of developing both. There was no clear genetic causal relationship between BMI and PD (P value = 0.511), AD (P value = 0.076), ALS (P value = 0.641), and epilepsy (P value = 0.380). Sensitivity analysis suggested no heterogeneity and horizontal pleiotropy in the results of MR analysis between BMI index and the neurological diseases studied (P > 0.05).
In this study, we identified a genetic causal relationship between BMI indicators and MS as well as IS, with a positive correlation to the risk of morbidity. This finding aligns with previous research. MR studies in recent years have indicated that an increased BMI elevates the risk of MS, confirming obesity's causal role in the etiology of MS80,81,82. The main pathological mechanisms are unclear, and studies have shown that this may be related to the increased systemic organismal inflammatory response caused by obesity, which has a promoting effect on the development of autoimmune responses83,84,85. Morales et al.86 demonstrated significant improvements in inflammation, demyelination, and axonal damage significantly in mice, suggesting that intermittent fasting (IF) has neuroprotective and anti-inflammatory effects, which may be related to the modulation of intestinal bacterial abundance degree and metabolite changes. It has been discovered that BMI can influence MS by affecting specific hormone levels in the body, including lipocalin and leptin. Notably, leptin, as an essential immune system regulator, can effectively prevent the migration of immune cells and reactions to the central nervous system via receptor blockade, improving the progression of experimental autoimmune encephalomyelitis (EAE)87,88. Additionally, it has been associated with macrophage phagocytosis and the regulation of pro-inflammatory factor release89,90. Further investigation into additional mechanisms is necessary. Our findings also implied a causal relationship between BMI indicators and IS, consistent with most previous research. A retrospective study from 2015 revealed that obesity could increase the risk of IS, possibly due to the impact on blood pressure, blood glucose, and other relevant indicators91. Recent research has shown that a high BMI in adolescents correlates with an elevated risk of youthful stroke92. This may result from visceral fat deposition caused by a high BMI, triggering inflammatory responses and oxidative stress, impairing vascular endothelial function, and thus inducing thrombosis93,94. Moreover, fat deposition can affect liver and muscle metabolism in humans, leading to insulin resistance and consequent elevations in blood glucose levels, damaging the vascular endothelium95. A small number of studies suggest that obese patients might improve their prognosis after IS by enhancing the early-stage anti-inflammatory response96. Rodríguez and Liu97,98 propose that a high BMI might increase the survival rate of IS patients, potentially due to better metabolic reserves. More clinical studies and experimental validation are required in the future to clarify its mechanism.
The current study failed to establish a link between BMI and PD. Earlier investigations in this domain have yielded conflicting results. Wang et al.99 and others posited that elevated BMI does not elevate the risk of PD, while Yoon et al.100 found that women with lower BMI exhibited higher mortality rates compared to men with PD. Conversely, certain studies have presented opposing conclusions. A MR study previously indicated a causal relationship between reduced BMI and PD101. A meta-analysis suggested that being overweight or obese might act as a risk factor for PD, potentially triggering the onset of the condition102. Wills et al.103 concluded that no correlation exists between BMI and PD mortality. The precise mechanism underlying these observations remains elusive. No causal relationship was identified between BMI and AD in this study. Research in this area is currently inconclusive. Findings suggest that higher adult BMI or obesity is not linked to the progression of AD51,104. Nordestgaard et al.105 suggested that low BMI is not a risk factor for AD and is not associated with an increased risk of developing AD. However, contradictory findings propose that higher BMI correlates with a decreased risk of AD among individuals sharing the same genetic susceptibility106. It has also been postulated that both high and low BMI could augment the risk of AD development105. This may be attributed to obesity-induced inflammatory responses that further compromise endothelial function and the blood–brain barrier, consequently affecting cognition107,108. Moreover, research has uncovered that high BMI in premenopausal women can escalate the risk of AD, which might be associated with hormonal levels, inflammation, and other metabolic factors. Another study confirmed that after menopause, high BMI can reduce the risk of AD in women. The researchers suggested that this may be due to changes in body composition and metabolism in postmenopausal women109,110. Therefore, further research is necessary to establish a clear association and mechanism between the two variables. The current study found no correlation between BMI and ALS. There has been inconsistency regarding the results of studies in this area. A MR study demonstrated that genetically increasing or decreasing BMI had no causal effect on the risk of developing ALS111. Conversely, a prospective study indicated that higher BMI and weight gain were associated with a lower risk of ALS after 50 years. O'Reilly discovered that a lower pre-morbid BMI increased the risk of ALS112. Military personnel and athletes, particularly football players, appear to have a higher risk of ALS. This may be attributed to muscle damage caused by strenuous exercise, free radical production, or other unknown factors. However, this does not imply that exercise is detrimental113,114. Early epidemiology has identified a decrease in body mass index early in the onset of ALS115,116, but this merely reflects the symptoms and prognosis of the disease after its onset, and the effect of pre-morbid BMI on the disease requires further investigation. This study did not find a causal link between BMI and EP. Experts hold varying views on this matter. Some scholars have argued that there was no significant difference in exercise participation rates between epileptic and non-epileptic patients117,118, refuting the impact of obesity on EP development. A recent prospective study concluded an association between obesity and idiopathic generalized EP and a family history of EP, but not drug-resistant EP119. A MR analysis investigated the relationship between obesity, institutional distribution, and EP subtypes and found that obesity, a risk factor for EP, can lead to an increased risk of spasmodic seizures in adolescents, while a high BMI raised the risk of cataplectic seizures in children120. Early studies identified the possibility of co-morbidity between EP and obesity, suggesting a risk of obesity-induced EP121,122,123. Indeed, the early discovery of the serotonin 5ht2c receptor is not only involved in organismal obesity124 but has been recognized as a potential target for antiepileptic drug action125. Therefore, we hypothesized that the reason for the association between high BMI and EP may be related to neurological dysfunction caused by mutations in some specific genes. However, our study did not show positive results, which may be due to factors such as sample size and ethnicity of origin. The exact mechanism needs to be further clarified and studied.
In the current investigation, we utilized the GWAS database to offer a precise assessment of the genetic linkage between exposure and outcomes. This was achieved by identifying instrumental SNPs that exhibit robust associations with both exposure and outcome factors. Our approach circumvented the influences of confounding variables and the reverse causation inherent in transcriptional processes. Additionally, we employed a database derived from an open and dependable source24,25. Therefore, the use of the MR method is feasible and has good value in providing a more reliable genetic assessment of the association between BMI and various neurological disorders.
Limitations
There are several limitations to this study. First, most of the data involved in this study were patients of European ancestry, which biases the results somewhat and needs to be validated by including more ancestries and ethnicities in the future. Secondly, numerous risk factors are associated with neurological disorders. Despite our efforts to remove all factors that can be accessed, we still cannot fully cover them, which may have a certain impact on the study. Third, BMI is a simple index that measures the relationship between weight and height, but it does not distinguish the proportion of fat and muscle in body weight, nor does it reflect important health factors such as body fat distribution or insulin sensitivity. In future studies, other indicators such as waist circumference, waist-to-hip ratio, body fat percentage, and bioelectrical impedance analysis should be included to prevent bias in results from a single indicator.
Conclusion
In summary, this study investigated the genetic associations between BMI and various neurological diseases using MR analysis. The findings indicated that there was no causal relationship between BMI and PD, AD, ALS, and EP. The study found that there was a genetic association between BMI and MS as well as IS, and a higher BMI increased the risk of MS and IS. The findings underscore the significant role of obesity as a risk factor for certain neurological disorders, influencing their onset and progression. These insights are anticipated to offer a foundational framework for the prevention, treatment, and prognosis of these conditions.
Data availability
The data for this study is sourced from public databases, and relevant data and materials are available from corresponding GWAS databases.
References
Ryder, J. R., Kaizer, A. M., Rudser, K. D., Daniels, S. R. & Kelly, A. S. Utility of body mass index in identifying excess adiposity in youth across the obesity spectrum. J. Pediatr. 177, 255-261.e2 (2016).
Freedman, D. S. et al. Relation of BMI to fat and fat-free mass among children and adolescents. Int J Obes. 29, 1–8 (2005).
Blundell, J. E., Dulloo, A. G., Salvador, J., Frühbeck, G., EASO SAB Working Group on BMI. Beyond BMI–phenotyping the obesities. Obes. Facts. 7(5), 322–328 (2014).
Abdullah, A. et al. The number of years lived with obesity and the risk of all-cause and cause-specific mortality. Int. J. Epidemiol. 40(4), 985–996 (2011).
Cirulli, E. T. et al. Profound perturbation of the metabolome in obesity is associated with health risk. Cell Metab. 29(2), 488-500.e2 (2019).
Flegal, K. M., Kit, B. K., Orpana, H. & Graubard, B. I. Association of all-cause mortality with overweight and obesity using standard body mass index categories: A systematic review and meta-analysis. JAMA 309(1), 71–82 (2013).
Hu, H. et al. Duration and degree of weight change and risk of incident diabetes: Japan epidemiology collaboration on occupational health study. Prev. Med. 96, 118–123 (2017).
Reis, J. P. et al. Excess body mass index- and waist circumference-years and incident cardiovascular disease: The CARDIA study. Obesity (Silver Spring) 23(4), 879–885 (2015).
Liu, Y., Hazlewood, G. S., Kaplan, G. G., Eksteen, B. & Barnabe, C. Impact of obesity on remission and disease activity in rheumatoid arthritis: A systematic review and meta-analysis. Arthr. Care Res. (Hoboken) 69(2), 157–165 (2017).
Lauby-Secretan, B. et al. Body fatness and cancer: Viewpoint of the IARC working group. N. Engl. J. Med. 375(8), 794–798 (2016).
Ascherio, A. & Schwarzschild, M. A. The epidemiology of Parkinson’s disease: Risk factors and prevention. Lancet Neurol. 15(12), 1257–1272. https://doi.org/10.1016/S1474-4422(16)30230-7 (2016).
Hernan, M. A. et al. Cigarette smoking and the incidence of Parkinson’s disease in two prospective studies. Ann. Neurol. 50, 780–786 (2001).
Hernan, M. A., Chen, H., Schwarzschild, M. A. & Ascherio, A. Alcohol consumption and the incidence of Parkinson’s disease. Ann. Neurol. 54, 170–175 (2003).
Baldi, I. et al. Association between Parkinson’s disease and exposure to pesticides in southwestern France. Neuroepidemiology 22, 305–310 (2003).
Parkinson Study Group SURE-PD3 Investigators, Schwarzschild, M. A. et al. Effect of urate-elevating inosine on early parkinson disease progression: The SURE-PD3 randomized clinical trial. JAMA 326(10), 926–939 (2021).
Bjorklund, A. & Dunnett, S. B. Dopamine neuron systems in the brain: An update. Trends Neurosci. 30, 194–202 (2007).
Hornykiewicz, O. The discovery of dopamine deficiency in the Parkinsonian brain. J. Neural Transm. 70, 9–15 (2006).
Serý, O., Povová, J., Míšek, I., Pešák, L. & Janout, V. Molecular mechanisms of neuropathological changes in Alzheimer’s disease: A review. Folia Neuropathol. 51(1), 1–9 (2013).
Hardiman, O. et al. Amyotrophic lateral sclerosis [published correction appears in nat rev dis primers. 2017 Oct 20;3:17085]. Nat. Rev. Dis. Prim. 3, 17071 (2017).
Compston, A. & Coles, A. Multiple sclerosis. Lancet 372(9648), 1502–1517 (2008).
Feske, S. K. Ischemic stroke. Am. J. Med. 134(12), 1457–1464 (2021).
Shi, Y. et al. Risk factors for ischemic stroke: Differences between cerebral small vessel and large artery atherosclerosis aetiologies. Folia Neuropathol. 59(4), 378–385 (2021).
Shorvon, S. D. The etiologic classification of epilepsy. Epilepsia 52(6), 1052–1057 (2011).
Lawlor, D. A., Harbord, R. M., Sterne, J. A. C., Timpson, N. & Davey Smith, G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 27, 1133–1163 (2008).
Evans, D. M. & Davey Smith, G. Mendelian randomization: New applications in the coming age of hypothesis-free causality. Annu. Rev. Genom. Hum. Genet. 16, 327–350 (2015).
Birney, E. Mendelian randomization. Cold Spring Harb. Perspect. Med. 12(4), a041302 (2022).
Sekula, P., Del Greco, M. F., Pattaro, C. & Köttgen, A. Mendelian randomization as an approach to assess causality using observational data. J. Am. Soc. Nephrol. 27(11), 3253–3265 (2016).
Howe, L. J. et al. Within-sibship genome-wide association analyses decrease bias in estimates of direct genetic effects. Nat. Genet. 54(5), 581–592 (2022).
Howe, L. J., Evans, D. M., Hemani, G., Davey Smith, G. & Davies, N. M. Evaluating indirect genetic effects of siblings using singletons. PLoS Genet 18(7), e1010247 (2022).
Nalls, M. A. et al. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: A meta-analysis of genome-wide association studies. Lancet Neurol. 18(12), 1091–1102 (2019).
Kunkle, B. W. et al. Genetic meta-analysis of diagnosed Alzheimer’s disease identifies new risk loci and implicates Aβ, tau, immunity and lipid processing [published correction appears in Nat Genet. 2019 Sep;51(9):1423-1424]. Nat. Genet. 51(3), 414–430 (2019).
International Multiple Sclerosis Genetics Consortium. Multiple sclerosis genomic map implicates peripheral immune cells and microglia in susceptibility. Science 365(6460), eaav7188 (2019).
Iacoangeli, A. et al. Genome-wide meta-analysis finds the ACSL5-ZDHHC6 locus is associated with ALS and links weight loss to the disease genetics. Cell Rep. 33(4), 108323 (2020).
Malik, R. et al. Multiancestry genome-wide association study of 520,000 subjects identifies 32 loci associated with stroke and stroke subtypes [published correction appears in Nat Genet. 2019 Jul;51(7):1192-1193]. Nat. Genet. 50(4), 524–537 (2018).
International League Against Epilepsy Consortium on Complex Epilepsies. Genome-wide mega-analysis identifies 16 loci and highlights diverse biological mechanisms in the common epilepsies. Nat. Commun. 9(1), 5269 (2018).
Ben-Shlomo, Y. et al. The epidemiology of Parkinson’s disease. Lancet 403(10423), 283–292. https://doi.org/10.1016/S0140-6736(23)01419-8 (2024).
Domenighetti, C. et al. Mendelian randomisation study of smoking, alcohol, and coffee drinking in relation to Parkinson’s disease. J. Parkinsons Dis. 12(1), 267–282. https://doi.org/10.3233/JPD-21285 (2022).
Seifar, F., Dinasarapu, A. R. & Jinnah, H. A. Uric acid in Parkinson’s disease: What is the connection?. Mov. Disord. 37(11), 2173–2183 (2022).
Fang, S. et al. Parkinson’s disease and ischemic stroke: A bidirectional Mendelian randomization study. Transl. Stroke Res. 13(4), 528–532 (2022).
Potashkin, J. et al. Understanding the links between cardiovascular disease and Parkinson’s disease. Mov. Disord. 35(1), 55–74 (2020).
Xu, X., Fu, Z. & Le, W. Exercise and Parkinson’s disease. Int. Rev. Neurobiol. 147, 45–74 (2019).
Smeyne, R. J., Noyce, A. J., Byrne, M., Savica, R. & Marras, C. Infection and risk of Parkinson’s disease. J. Parkinsons Dis. 11(1), 31–43 (2021).
Brett, B. L., Gardner, R. C., Godbout, J., Dams-O’Connor, K. & Keene, C. D. Traumatic brain injury and risk of neurodegenerative disorder. Biol. Psychiatry. 91(5), 498–507 (2022).
Xu, J. et al. Genetic correlation between thyroid hormones and Parkinson’s disease. Clin. Exp. Immunol. 208(3), 372–379 (2022).
Nebel, R. A. et al. Understanding the impact of sex and gender in Alzheimer’s disease: A call to action. Alzheimers Dement. 14(9), 1171–1183 (2018).
Durazzo, T. C., Mattsson, N., Weiner, M. W., Alzheimer’s Disease Neuroimaging Initiative. Smoking and increased Alzheimer’s disease risk: A review of potential mechanisms. Alzheimers Dement. 10(3 Suppl), S122–S145 (2014).
Ramos-Cejudo, J. et al. Traumatic brain injury and Alzheimer’s disease: The cerebrovascular link. EBioMedicine 28, 21–30 (2018).
Rajna, Z. et al. Cardiovascular brain impulses in Alzheimer’s disease [published correction appears in Brain. 2021 Nov 29;144(10):e86]. Brain 144(7), 2214–2226 (2021).
Pugazhenthi, S., Qin, L. & Reddy, P. H. Common neurodegenerative pathways in obesity, diabetes, and Alzheimer’s disease. Biochim. Biophys. Acta Mol. Basis Dis. 1863(5), 1037–1045 (2017).
Sriram, S., Mehkri, Y., Quintin, S. & Lucke-Wold, B. Shared pathophysiology: Understanding stroke and Alzheimer’s disease. Clin. Neurol. Neurosurg. 218, 107306 (2022).
Ralli, M. et al. Hearing loss and Alzheimer’s disease: A review. Int. Tinnitus J. 23(2), 79–85 (2019).
Zeydan, B. & Kantarci, O. H. Impact of age on multiple sclerosis disease activity and progression. Curr. Neurol. Neurosci. Rep. 20(7), 24 (2020).
Nabizadeh, F. et al. Seasonal and monthly variation in multiple sclerosis relapses: A systematic review and meta-analysis. Acta Neurol. Belg. 122(6), 1447–1456 (2022).
Soldan, S. S. & Lieberman, P. M. Epstein–Barr virus and multiple sclerosis. Nat. Rev. Microbiol. 21(1), 51–64 (2023).
Feige, J. et al. Vitamin D supplementation in multiple sclerosis: A critical analysis of potentials and threats. Nutrients 12(3), 783 (2020).
Talbott, E. O., Malek, A. M. & Lacomis, D. The epidemiology of amyotrophic lateral sclerosis. Handb Clin Neurol. 138, 225–238 (2016).
Zhu, Y. et al. Mixed comparison of different exercise interventions for function, respiratory, fatigue, and quality of life in adults with amyotrophic lateral sclerosis: Systematic review and network meta-analysis. Front. Aging Neurosci. 14, 919059 (2022).
Ralli, M., Lambiase, A., Artico, M., de Vincentiis, M. & Greco, A. Amyotrophic lateral sclerosis: Autoimmune pathogenic mechanisms, clinical features, and therapeutic perspectives. Isr. Med. Assoc. J. 21(7), 438–443 (2019).
Motataianu, A., Serban, G., Barcutean, L. & Balasa, R. Oxidative stress in amyotrophic lateral sclerosis: Synergy of genetic and environmental factors. Int. J. Mol. Sci. 23(16), 9339 (2022).
Anzilotti, S. et al. Chronic exposure to l-BMAA cyanotoxin induces cytoplasmic TDP-43 accumulation and glial activation, reproducing an amyotrophic lateral sclerosis-like phenotype in mice. Biomed. Pharmacother. 167, 115503 (2023).
Levine, K. S. et al. Virus exposure and neurodegenerative disease risk across national biobanks. Neuron 111(7), 1086-1093.e2 (2023).
Rexrode, K. M. et al. The impact of sex and gender on stroke. Circ. Res. 130(4), 512–528 (2022).
GBD 2019 Stroke Collaborators. Global, regional, and national burden of stroke and its risk factors, 1990–2019: A systematic analysis for the global burden of disease study 2019. Lancet Neurol. 20(10), 795–820 (2021).
Tran, P. & Tran, L. Stroke and heart attack symptom recognition in older US adults by cognitive impairment status. Neuroepidemiology 55(3), 245–252 (2021).
Lau, L. H., Lew, J., Borschmann, K., Thijs, V. & Ekinci, E. I. Prevalence of diabetes and its effects on stroke outcomes: A meta-analysis and literature review. J. Diabetes Investig. 10(3), 780–792 (2019).
Vezzani, A. et al. Infections, inflammation and epilepsy. Acta Neuropathol. 131(2), 211–234 (2016).
Reddy, C. & Saini, A. G. Metabolic epilepsy. Indian J. Pediatr. 88(10), 1025–1032 (2021).
Flammer, J., Neziraj, T., Rüegg, S. & Pröbstel, A. K. Immune mechanisms in epileptogenesis: Update on diagnosis and treatment of autoimmune epilepsy syndromes. Drugs 83(2), 135–158 (2023).
Ruggles, K. H., Hansotia, P. L. & Ahmann, P. A. Head trauma and epilepsy. Wis. Med. J. 87(2), 16–18 (1988).
Galovic, M. et al. Seizures and epilepsy after stroke: Epidemiology, biomarkers and management. Drugs Aging 38(4), 285–299 (2021).
Rajneesh, K. F. & Binder, D. K. Tumor-associated epilepsy. Neurosurg. Focus 27(2), E4 (2009).
Dong, B. et al. Relationship between right-to-left shunt, hypoxia, and epilepsy. Epilepsia Open 8(2), 456–465 (2023).
Wanleenuwat, P., Suntharampillai, N. & Iwanowski, P. Antibiotic-induced epileptic seizures: Mechanisms of action and clinical considerations. Seizure 81, 167–174 (2020).
Devetag, F., Mandich, G., Zaiotti, G. & Toffolo, G. G. Alcoholic epilepsy: Review of a series and proposed classification and etiopathogenesis. Ital. J. Neurol. Sci. 4(3), 275–284 (1983).
Grover, S., Del Greco, M. F., Stein, C. M. & Ziegler, A. Mendelian randomization. Methods Mol. Biol. 1666, 581–628 (2017).
Rees, J. M. B., Foley, C. N. & Burgess, S. Factorial Mendelian randomization: Using genetic variants to assess interactions. Int. J. Epidemiol. 49(4), 1147–1158 (2020).
Verbanck, M., Chen, C. Y., Neale, B. & Do, R. Detection of widespread horizontal pleiotropy in causal relationships inferred from mendelian randomization between complex traits and diseases. Nat. Genet. 50, 693–698 (2018).
Bowden, J. et al. Assessing the suitability of summary data for two sample mendelian randomization analyses using MR-Egger regression: the role of the I2 statistic. Int. J. Epidemiol. 45, 1961–1974 (2016).
Burgess, S. & Thompson, S. G. Interpreting findings from Mendelian randomization using the MR-Egger method [published correction appears in Eur J Epidemiol. 2017 Jun 29]. Eur. J. Epidemiol. 32(5), 377–389 (2017).
Mokry, L. E. et al. Obesity and multiple sclerosis: A Mendelian randomization study. PLoS Med. 13(6), e1002053 (2016).
Almramhi, M. M. et al. The role of body fat in multiple sclerosis susceptibility and severity: A Mendelian randomisation study. Mult. Scler. 28(11), 1673–1684 (2022).
Larsson, S. C. & Burgess, S. Causal role of high body mass index in multiple chronic diseases: A systematic review and meta-analysis of Mendelian randomization studies. BMC Med. 19(1), 320 (2021).
Visser, M., Bouter, L. M., McQuillan, G. M., Wener, M. H. & Harris, T. B. Low-grade systemic inflammation in overweight children. Pediatrics. 107, e13 (2001).
Timpson, N. J. et al. C-reactive protein levels and body mass index: Elucidating direction of causation through reciprocal Mendelian randomization. Int. J. Obes. 35, 300–308 (2011).
Würtz, P. et al. Metabolic signatures of adiposity in young adults: Mendelian randomization analysis and effects of weight change. PLoS Med. 11, e1001765 (2014).
Morales-Suarez-Varela, M., Collado Sánchez, E., Peraita-Costa, I., Llopis-Morales, A. & Soriano, J. M. Intermittent fasting and the possible benefits in obesity, diabetes, and multiple sclerosis: A systematic review of randomized clinical trials. Nutrients 13(9), 3179 (2021).
Lang, K. & Ratke, J. Leptin and adiponectin: New players in the fifield of tumor cell and leukocyte migration. Cell Commun. Sign. 7, 27 (2009).
Hsuchou, H. et al. Saturable leptin transport across the BBB persists in EAE mice. J. Mol. Neurosci. 51(2), 364–370 (2013).
Dayakar, A., Chandrasekaran, S., Veronica, J. & Maurya, R. Leptin induces the phagocytosis and protective immune response in leishmania donovani infected THP-1 cell line and human PBMCs. Exp. Parasitol. 160, 54–59 (2016).
Mancuso, P. et al. Ablation of the leptin receptor in myeloid cells impairs pulmonary clearance of streptococcus pneumoniae and alveolar macrophage bactericidal function. Am. J. Physiol. Lung Cell Mol. Physiol. 315(1), L78-86 (2018).
Mitchell, A. B. et al. Obesity increases risk of ischemic stroke in young adults. Stroke 46, 1690–1692 (2015).
Bardugo, A. et al. Body mass index in 1.9 million adolescents and stroke in young adulthood. Stroke 52, 2043–2052 (2021).
George, M. G., Tong, X. & Bowman, B. A. Prevalence of cardiovascular risk factors and strokes in younger adults. JAMA Neurol. 74, 695–703 (2017).
Koliaki, C., Liatis, S. & Kokkinos, A. Obesity and cardiovascular disease: Revisiting an old relationship. Metabolism 92, 98–107 (2019).
Jaakonmäki, N. et al. Obesity and the risk of cryptogenic ischemic stroke in young adults. J. Stroke Cerebrovasc. Dis. 31(5), 106380 (2022).
Lau, W. B. et al. Role of adipokines in cardiovascular disease. Circ. J. 81(7), 920–928 (2017).
Rodríguez-Castro, E. et al. Obesity paradox in ischemic stroke: Clinical and molecular insights. Transl. Stroke Res. 10(6), 639–649 (2019).
Liu, Z. et al. Adiposity and outcome after ischemic stroke: Obesity paradox for mortality and obesity parabola for favorable functional outcomes. Stroke 52(1), 144–151 (2021).
Wang, Y. L. et al. Body mass index and risk of Parkinson’s disease: A dose-response meta-analysis of prospective studies. PLoS One 10, e0131778 (2015).
Yoon, S. Y. et al. Initial BMI and weight loss over time predict mortality in Parkinson disease. J. Am. Med. Dir. Assoc. 23(10), 1719.e1-1719.e7 (2022).
Noyce, A. J. et al. Estimating the causal inflfluence of body mass index on risk of Parkinson disease: A Mendelian randomisation study. PLoS Med. 14, e1002314 (2017).
Chen, J. et al. Meta-analysis: overweight, obesity, and Parkinson’s disease [published correction appears in Int J Endocrinol. 2014;2014:306402]. Int. J. Endocrinol. 2014, 203930 (2014).
Wills, A. M. et al. Association between change in body mass index, unifified Parkinson’s disease rating scale scores, and survival among persons with Parkinson disease: Secondary analysis of longitudinal data from NINDS exploratory trials in Parkinson disease long-term study 1. JAMA Neurol. 73, 321e328 (2016).
Zhou, Y., Sun, X. & Zhou, M. Body shape and Alzheimer’s disease: A Mendelian randomization analysis. Front. Neurosci. 13, 1084 (2019).
Nordestgaard, L. T., Tybjærg-Hansen, A., Nordestgaard, B. G. & Frikke-Schmidt, R. Body mass index and risk of Alzheimer’s disease: A Mendelian randomization study of 399,536 individuals. J. Clin. Endocrinol. Metab. 102(7), 2310–2320 (2017).
Yuan, S. et al. Body mass index, genetic susceptibility, and Alzheimer’s disease: A longitudinal study based on 475,813 participants from the UK Biobank. J. Transl. Med. 20(1), 417 (2022).
Li, X. et al. Life course adiposity and Alzheimer’s disease: A Mendelian randomization study. J. Alzheimers Dis. 82(2), 503–512 (2021).
Gregor, M. F. & Hotamisligil, G. S. Inflammatory mechanisms in obesity. Annu. Rev. Immunol. 29, 415–445 (2011).
Wharton, W. et al. Short-term hormone therapy with transdermal estradiol improves cognition for postmenopausal women with Alzheimer’s disease: Results of a randomized controlled trial. J. Alzheimers Dis. 26(3), 495–505 (2011).
Luchsinger, J. A., Patel, B., Tang, M. X., Schupf, N. & Mayeux, R. Measures of adiposity and dementia risk in elderly persons. Arch. Neurol. 64(3), 392–398 (2007).
Zeng, P., Yu, X. & Xu, H. Association between premorbid body mass index and amyotrophic lateral sclerosis: Causal inference through genetic approaches. Front. Neurol. 10, 543 (2019).
O’Reilly, É. J. et al. Premorbid body mass index and risk of amyotrophic lateral sclerosis. Amyotroph Lateral Scler. Frontotemporal Degener. 14(3), 205–211 (2013).
Mariosa, D. et al. Body mass index and amyotrophic lateral sclerosis: A study of US military veterans. Am. J. Epidemiol. 185(5), 362–371 (2017).
Daneshvar, D. H. et al. Incidence of and mortality from amyotrophic lateral sclerosis in national football league athletes. JAMA Netw. Open. 4(12), e2138801 (2021).
Armon, C. & Traynor, B. J. High BMI is associated with low ALS risk: What does it mean?. Neurology 93(5), 189–191 (2019).
Kiernan, M. C. et al. Amyotrophic lateral sclerosis. Lancet 377(9769), 942–955 (2011).
Elliott, J. O., Lu, B., Moore, J. L., McAuley, J. W. & Long, L. Exercise, diet, health behaviors, and risk factors among persons with epilepsy based on the California Health Interview Survey, 2005. Epilepsy Behav. 13, 307–315 (2008).
Gordon, K. E., Dooley, J. M. & Brna, P. M. Epilepsy and activity: A population-based study. Epilepsia 51, 2254–2259 (2010).
Ladino, L. D., Hernández-Ronquillo, L. & Téllez-Zenteno, J. F. Obesity and its association with generalised epilepsy, idiopathic syndrome, and family history of epilepsy. Epileptic Disord. 16(3), 343–353 (2014).
Zhou, K., Yang, H., Chen, R., Wang, W. & Qu, Z. Causal relationship among obesity and body fat distribution and epilepsy subtypes. Front. Neurol. 13, 984824 (2022).
Gilliam, F. G., Mendiratta, A., Pack, A. M. & Bazil, C. W. Epilepsy and common comorbidities: Improving the outpatient epilepsy encounter. Epilept Disord. 7, 27–33 (2005).
Daniels, Z. S., Nick, T. G., Liu, C., Cassedy, A. & Glauser, T. A. Obesity is a common comorbidity for pediatric patients with untreated, newly diagnosed epilepsy. Neurology 73, 658–664 (2009).
Hinnell, C. et al. Health status and health-related behaviors in epilepsy compared to other chronic conditions-a national population-based study. Epilepsia 51, 853–861 (2010).
Palacios, J. M., Pazos, A. & Hoyer, D. A short history of the 5-HT2C receptor: From the choroid plexus to depression, obesity and addiction treatment. Psychopharmacology (Berl) 234(9–10), 1395–1418 (2017).
Isaac, M. Serotonergic 5-HT2C receptors as a potential therapeutic target for the design antiepileptic drugs. Curr. Top Med. Chem. 5(1), 59–67 (2005).
Acknowledgements
We thank all participants and associated GWAS databases for their generosity and contributions.
Funding
This study was funded by the National Natural Foundation of China under the National Science Foundation Program (Grant Nos. 81874389 and 82274493).
Author information
Authors and Affiliations
Contributions
X.W. designed the study. H.C. and Z.C. wrote the analysis plan, performed the data analysis, and interpreted the results. H.C. wrote the first draft of the manuscript and received critical feedback and revisions from J.Z. and D.X., and all authors have read and approved the final manuscript. X.W., H.C. contributed equally to this manuscript.
Corresponding author
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Open Access This article is licensed under a Creative Commons Attribution 4.0 International License, which permits use, sharing, adaptation, distribution and reproduction in any medium or format, as long as you give appropriate credit to the original author(s) and the source, provide a link to the Creative Commons licence, and indicate if changes were made. The images or other third party material in this article are included in the article's Creative Commons licence, unless indicated otherwise in a credit line to the material. If material is not included in the article's Creative Commons licence and your intended use is not permitted by statutory regulation or exceeds the permitted use, you will need to obtain permission directly from the copyright holder. To view a copy of this licence, visit http://creativecommons.org/licenses/by/4.0/.
About this article

Cite this article
Wang, X., Chen, H., Chang, Z. et al. Genetic causal role of body mass index in multiple neurological diseases. Sci Rep 14, 7256 (2024). https://doi.org/10.1038/s41598-024-57260-2
Received:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41598-024-57260-2
- Springer Nature Limited