
Abstract
Tumor-associated carcinoembryonic antigen (CEA) is a natural target for vaccines against colorectal cancers. Our previous experience with a DNA vaccine with scFv6.C4, a CEA surrogate, showed a CEA-specific immune response with 40% of tumor-free mice after challenge with B16F10-CEA and 47% with MC38-CEA cells. These percentages increased to 63% after using FrC as an adjuvant. To further enhance the vaccine efficacy, we tested GM-CSF and IFNγ as adjuvants. C57BL/6J-CEA2682 mice were immunized 4 times with uP-PS/scFv6.C4, uP-PS/scFv6.C4 + uP-IFNγ, or uP-PS/scFv6.C4 + uP-GMCSF. After one week, the mice were challenged with MC38-CEA, and tumor growth was monitored over 100 days. Immunization with scFv6.C4 and scFv6.C4 + GM-CSF resulted in a gradual increase in the anti-CEA antibody titer, while scFv6.C4 + IFNγ immunization led to a rapid and sustained increase in the titer. The addition of IFNγ also induced higher CD4 + and CD8 + responses. When challenged, almost 80% of the scFv6.C4 + IFNγ-vaccinated mice did not develop tumors, while the others had a significant tumor growth delay. The probability of being tumor-free was 2700% higher using scFv6.C4 + IFNγ than scFv6.C4. The addition of GM-CSF had no additional effect on tumor protection. DNA immunization with scFv6.C4 + IFNγ, but not GM-CSF, increased the antitumor effect via readily sustained specific humoral and cytotoxic responses to CEA.
Similar content being viewed by others


Introduction
Colorectal cancer is among the three most common cancers expected to be diagnosed in the United States in 2020, accounting for 9% of estimated cancer deaths in both men and women in this year. Although this number has been falling over the last 10 years due mostly to preventive diagnosis, therapeutic options are still insufficient to cover all pathophysiological variations [1].
Our previous results with DNA vaccines against the main tumor-associated antigen (TAA) of colorectal cancers, carcinoembryonic antigen (CEA), demonstrated CEA-specific humoral and cellular immune responses [2, 3]. The immunization was performed with a CEA surrogate, scFv6.C4, originated from the anti-idiotypic mAB 6.C4 variable heavy and light chain sequences [4,5,6]. Immunization of CEA-expressing transgenic mice (C57BL/6J-CEA2682) with scFv6.C4 prevented tumor growth in 40% of the mice after challenge with CEA-expressing tumor B16F10-CEA cells [2] and 47% of those with MC38-CEA cells [3]. The addition of tetanus toxin fragment C (FrC) as an adjuvant increased the percentage of tumor-free animals to 63%, with induction of strong and specific humoral and cellular immune responses to CEA [3, 7]. Although the protection provided by the FrC adjuvant is significant, for transfer of this vaccine to a clinical trial in the future, the effectiveness of this vaccine must be improved because other factors related to differences between animal models and humans can reduce the effectiveness of vaccines.
Granulocyte-macrophage colony-stimulating factor (GM-CSF) is the major stimulator for the growth and differentiation of monocytes and granulocyte progenitors and for the proliferation and survival of dendritic cells and other antigen-presenting cells (APCs) [8,9,10]. This molecule plays important roles in lymphocyte cytotoxicity, antigen presentation, antibody-mediated cytotoxicity, and cell-mediated immunity, as well as the production of various cytokines, such as IL-1, TNF-α, and IL-6 [11]. Due to these activities, GM-CSF has been extensively tested against CEA-expressing cancers in preclinical [12,13,14,15,16,17,18] and clinical [19,20,21,22,23] trials.
Interferon gamma (IFNγ) is a cytokine produced by T lymphocytes, NK cells, and NKT cells and is known to have antiviral, antitumor, and immunoregulatory activities [24]. This cytokine regulates the differentiation of macrophages, MHC expression in APCs, and Th1-CD4 + and cytotoxic CD8 + cell development, in addition to having an important role in immunoglobulin class switching [24]. In the antitumor response, IFNγ exerts antiproliferative and antimetabolic activity, promotes tumor cell apoptosis and inhibits angiogenesis as well as innate and adaptive immune responses against tumors [25, 26]. In a preclinical trial against CEA-expressing tumors, IFNγ could substantially increase immune responses, mainly by inducing the Th1 cytotoxic cellular response [27].
In the present study, adjuvant effects of GM-CSF and IFNγ on a DNA vaccination with a scFv6.C4-expressing vector, uP-PS/scFv6.C4, were assessed in C57BL/6J-CEA2682 mice. Humoral and cellular responses were evaluated before and after tumor challenge with MC38-CEA cells.
Materials and methods
Research Ethics Committee approval
Animal procedures were performed in full compliance with guidelines of the UNIFESP Institutional Research Ethics Committee (http://www.unifesp.br/reitoria/ceua) only after approval by this committee (Approval number: CEUA 703012).
Construction of vectors
scFv6.C4 and GM-CSF expression vectors were named uP-PS/scFv6.C4 and uP-GM-CSF, respectively, and were described previously [2, 28]. For generation of the IFNγ-expressing vector uP-IFNγ, pUC57-IFNγ plasmid (GenScript, Piscataway, NJ, USA) containing mouse IFNγ cDNA (NM_008337.4) was treated with BamHI and ApaI enzymes to release the 489 bp IFNγ fragment, which was inserted into the uP plasmid vector [28] previously treated with the same enzymes. The identity of the vectors was confirmed by Sanger sequencing.
Cell culture
The murine colon adenocarcinoma cell lines MC38 [29], MC38-CEA [3], and MC38-scFv6.c4 [3] were maintained in DMEM (Dulbecco’s modified Eagle’s medium; Thermo Fisher Scientific; Waltham, MA, USA) buffered with sodium bicarbonate (24 mM) plus 2-[4-(2-hydroxyethyl)-1-piperazinyl] ethanesulfonic acid (HEPES; 10 mM) and supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen; Grand Island, NY, USA), L-glutamine (2 mM), penicillin (100 units/ml) and streptomycin (100 μg/ml) in a humidified atmosphere (5% CO2, 37 °C).
The human colorectal carcinoma cell line HCT-8 [30] was maintained in RPMI-1640 (Thermo Fisher Scientific) buffered with sodium bicarbonate (24 mM) and HEPES (10 mM) and supplemented with 10% heat-inactivated fetal bovine serum (Invitrogen), L-glutamine (2 mM), penicillin (100 units/ml) and streptomycin (100 μg/ml) in a humidified atmosphere (5% CO2, 37 °C). The supplemented medium was named RPMIc.
Preventive DNA immunization and tumor cell challenge
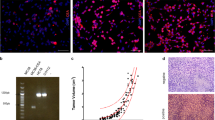
CEA-expressing transgenic mice (CEA2682; kindly donated by Dr. Wolfgang Zimmermann; University of Munich, Munich, Germany) [31] were separated into three groups: (i) the scFv6.C4-immunized group: 70 μg uP-PS/scFv6.C4 in 70 μl PBS was injected into each quadriceps muscle (n = 17); (ii) the scFv6.C4 + IFNγ -immunized group: 70 μg uP-PS/scFv6.C4 and 70 μg uP-IFNγ in 70 μl PBS were injected into each quadriceps muscle (n = 10); (iii) the scFv6.C4 + GM-CSF immunized group: 70 μg uP-PS/scFv6.C4 and 70 μg uP-GM-CSF in 70 μl PBS were injected into each quadriceps muscle (n = 10). All injections were performed slowly using insulin syringes, and 6 electric pulses (100 V; 40 ms duration per pulse; 1 s interval) were applied through 10 mm tweezer electrodes, which were placed around the DNA injection site (Electroporator ECM830, BTX, Harvard Apparatus, Holliston, MA, USA). Three subsequent immunizations were performed at two-week intervals. Blood samples were collected one week after each immunization to quantify AB3 antibody production (Fig. 1a).

a Immunization scheme. The black arrows indicate electroporation with uP-PS/scFv 6.C4 plus adjuvant vectors. Blood samples were collected 7 days after each immunization, as indicated by red arrows. The tumor challenge was performed 7 days after the last immunization, as indicated by the blue arrow. The animals were euthanized between days 90 and 130 for cellular and humoral response evaluation. b CEA-specific AB3 antibodies in mice immunized with scFv6.C4 + -IFNγ and scFv6.C4 + GM-CSF were detected by ELISA. Blood samples were diluted (1:50) for the assays. Sera from the mice immunized with the scFv6.C4 vector were used as controls. Data are expressed as the mean ± SD of OD490 nm of each group (scFv6.C4 + IFNγ (n = 10), scFv6.C4 + uP-GM-CSF (n = 10), and scFv6.C4 (n = 17)); 1 = p = 0.030 compared to scFv6.C4 + IFNγ preimmune sera; 2 = p = 0.004 compared to the first immunization with scFv6.C4; 3 = p = 0.004 compared to the first immunization with scFv6.C4 + GM-CSF; 4 = p < 0.001 compared to the scFv6.C4 and scFv6.C4 + GM-CSF second immunizations; 5 = p = 0.041 compared to the scFv6.C4 third immunization; 6 = p = 0.026 compared to the scFv6.C4 + GM-CSF third immunization. c Tumor-free animals estimated by Cox regression. Mice with tumors smaller than 500 mm3 were considered survivors. Nonimmunized and scFv6.C4-immunized animals were used as controls (scFv6.C4 + -IFNγ (n = 10), scFv6.C4 + GM-CSF (n = 10)). The animals were observed for at least 100 days; however, since day 65, no changes in the survival rate were observed.
One week after the last immunization, the mice were challenged by s.c. injection of 1 × 105 MC38-CEA cells into the left flank. For 100 days, the tumor size was periodically measured with a caliper. Tumor volume (in mm3) was estimated using the following equation:
Quantification of CEA‑specific antibody response
Sera from the mice were assessed for anti-CEA antibodies (AB3 = AB1′) by ELISA as we described previously [3]. Briefly, 96-well plates were sensitized with 1 μg/ml CEA (Abcam; Cambridge, UK) at 37 °C for 1 h. The plates were blocked with 1% bovine serum albumin (BSA; Sigma-Aldrich; St. Louis, MO, USA) and 2% nonfat milk in PBS (room temperature; 1 h). Then, mouse sera (1:50) were incubated overnight at 4 °C. Biotinylated rabbit anti-mouse IgG (Dako; Carpinteria, CA, USA) was incubated for 1 h at 37 °C, followed by horseradish peroxidase-streptavidin (Dako) incubation in a dark chamber at room temperature for 30 min. Finally, the reaction was revealed by 50 mM citrate-phosphate buffer (51.4 mM Na2HPO4; 24.3 mM acetic acid; pH 5.0) containing ortho phenylenediamine (OPD; Sigma-Aldrich; 3 mg/ml) and 0.03% H2O2. The reactions were stopped with 2 N H2SO4, and the products were read at 492 nm in a microplate spectrophotometer (Spectra Max M2e, Molecular Devices; Sunnyvale, CA, USA). Each sample was run in triplicate.
Immunocytochemistry of HCT-8, MC38, and MC38-CEA cells
Immunocytochemistry was performed as we described previously [2, 3]. Briefly, on day 0, cells were seeded onto 13 mm coverslips and incubated overnight in a humidified atmosphere (5% CO2; 37 °C). On day 1, media were aspirated, and the cells were fixed in 4% paraformaldehyde (1 h), incubated with NH4Cl (50 mM; 15 min), permeabilized with 0.5% Triton X-100 (10 min), blocked with 10% BSA (Sigma-Aldrich) and 8% nonfat milk (1 h), and incubated with 1:50 diluted sera from uP-PS/scFv6.C4-, uP-PS/scFv6.C4 plus uP-IFNγ- or uP-PS/scFv6.C4 plus uP-GM-CSF-immunized animals in a humidified chamber (4 °C; overnight). Nonimmunized mouse sera were used as a negative control, while monoclonal antibody 1F5H2 [6] (final concentration: 10 μg/ml) was used as a positive control. On day 2, the coverslips were incubated with biotinylated rabbit anti-mouse IgG antibody (Dako; 1:100; 1 h), followed by Alexa 594-streptavidin (Invitrogen; 1:1000; 1 h) and 4′,6-diamidino-2-phenylindole (DAPI) for cell nuclei labeling (Invitrogen; 1:1000; 15 min). Finally, coverslips were rinsed with distilled water and mounted with Fluoromount aid (Sigma-Aldrich), and images were acquired using a fluorescence microscope (Olympus BX51, Tokyo, Japan).
In vitro cellular proliferation assay
An in vitro cellular proliferation assay was performed as we described previously [3]. Briefly, 90-110 days after MC38-CEA injection, the mice were euthanized to collect spleens, and 2 × 107 splenocytes suspended in PBS were labeled with 2.0 μM carboxyfluorescein diacetate succinimidyl ester (CFSE) (Cell-Trace™; CFSE Cell Proliferation Kit; Invitrogen) (37 °C; 5 min). In a U-bottom shape 96-well plate, 5×105 cells in 200 μl of RPMIc were seeded per well. The cells were stimulated with CEA (2 μg/ml; Abcam), mAB 6.C4 (10 μg/ml) [5], or concanavalin A (2.5 μg/ml; ConA; Sigma-Aldrich) as a positive control. Unstimulated cells were used as a negative control. All experiments were carried out in triplicate. After 6 days of incubation in a humidified atmosphere (5% CO2; 37 °C), the cells were washed with MACS buffer (PBS containing 2 mM EDTA and 0.5% BSA; pH 7.2) and labeled in a dark chamber (4 °C; 45 min) with anti-CD4-APC (1:100) or anti-CD8-PE (1:100) antibodies (BD Biosciences; San Jose, CA, USA). The samples were quantified by flow cytometry (FACSCanto II, BD Biosciences) and analyzed by FlowJo software (Tree Star; Ashland, OR, USA). The gating strategy used to determine the lymphocyte population was based on the SSC-A and FSC-A parameters, followed by determination of the CD8 + (SSC-A, PE) and CD4 + (SSC-A, APC) subpopulations. The proliferated cell gate (CFSE low) was established in unstimulated samples from the same animal and applied to the stimulated subpopulations. The percentage of CFSE-low cells was normalized by subtracting the percentage observed in unstimulated samples of the same animal. The samples stimulated with ConA were used for experimental validation.
Cytotoxicity assay
The cytotoxicity assay was performed as we described previously [3]. Briefly, 5–7 × 107 splenocytes were labeled with the anti-CD8-PE (BD Biosciences) antibody and sorted by flow cytometry (FACSAria II, BD Biosciences). Concomitantly, 2 × 104 tumor cells (MC38, MC38-CEA or MC38-scFv6.C4) were plated in duplicate in 96-U-well plates with sorted CD8 + T cells in three different targets: effect ratios: 1:5 (10 × 104 T CD8 + ), 1:2 (4 × 104 T CD8 + ), and 1:1 (2 × 104 T CD8 + ). RPMIc was added to a final volume of 200 μl. The cells were incubated in a humidified chamber (5% CO2; 16 h). Wells containing only tumor cells were used as controls. The LDH activity assay (In Vitro Toxicology Assay Kit, Lactic Dehydrogenase based; Sigma-Aldrich) was performed in 50 μl of medium following the manufacturer’s instructions. The lysis percentage was calculated using the following formula: % cell lysis = 100 x [(A - B)/(C- B)], where A: OD690nm tumor plus target, B: OD690nm target spontaneous release (background), and C: OD690nm target maximus lysis.
Statistical analysis
Statistical analyses were performed using IBM SPSS Statistics software (v. 21; New York, NY, USA), and graphs were plotted using GraphPad Prism software (v. 5.0; La Jolla, CA, USA). The anti-CEA antibody titers were analyzed by GEE (general estimating equations) using linear distribution, defined according to the QIC parameter, followed by Bonferroni tests. Survival analysis was performed by Cox regression, in which the observed outcome was the time necessary to reach 0.5 cm3 tumor volume. The reported hazard ratio (HR) corresponds to the mean of all observed times. The effect of the different groups of animals on the proliferation and cytotoxicity assays was assessed by GLzM using linear distribution, defined according to the AIC parameter, followed by Bonferroni tests. The sample size was chosen based upon experiences from previous experiments, and the numbers of samples per group are indicated in the legends. The significance was 5%. Data are presented as the mean and standard deviation.
Results
Humoral responses and protection against tumor challenge induced by DNA vaccinations
CEA2682 mice were immunized four times in alternate weeks by i.m. injection of uP/PS-scFv6.C4 + uP-IFNγ or uP/PS-scFv6.C4 + uP-GM-CSF plasmid solutions followed by electroporation, according to our previous experience [2, 3, 32]. Mice immunized with uP/PS-scFv6.C4 alone were used as controls. Blood samples were collected 7 days after each immunization to determine the AB3-CEA antibody titers (Fig. 1a).
The first scFv6.C4 + IFNγ immunization induced a significant increase in the AB3 titer (0.415 ± 0.052 vs. preimmune 0.185 ± 0.015, p = 0.030), and this level was maintained during the remaining immunizations (day 21: 0.408 ± 0.038; day 35: 0.390 ± 0.018; day 49: 0.442 ± 0.017, p = 1.000 in relation to day 7) (Fig. 1b). Compared to those of the mice immunized with scFv6.C4 only, the humoral responses after scFv6.C4 + IFNγ immunization were notably higher after the first immunization (0.415 ± 0.052 vs. 0.169 ± 0.012, p = 0.004). Although the magnitude of the difference decreased after the second (0.408 ± 0.038 vs. 0.182 ± 0.009, p < 0.001) and third (0.390 ± 0.018 vs. 0.286 ± 0.018, p = 0.041) immunizations, the titers were still significantly higher. Nevertheless, after the last immunization, the anti-CEA titer reached a similar level in the scFv6.C4 + IFNγ and scFv6.C4 groups (0.422 ± 0.017 vs. 0.379 ± 0.024, p = 1.000).
Unlike IFNγ treatment, the addition of the uP-GM-CSF adjuvant vector did not improve the AB3 anti-CEA antibody titer significantly compared to that in the control uP/PS-scFv6.C4 group after the first (0.130 ± 0.010 vs. 0.169 ± 0.012, p = 1.000), second (0.190 ± 0.015 vs. 0.182 ± 0.009, p = 1.000) and third (0.257 ± 0.026 vs. 0.286 ± 0.018, p = 1.000) immunizations (Fig. 1b). However, after the last immunization, the AB3 titer of the scFv6.C4 + GM-CSF group reached a level close to that of the scFv6.C4-IFNγ group (0.357 ± 0.61 vs. 0.422 ± 0.017, p = 1.000).
For evaluation of the effectiveness of preventive DNA vaccines with scFv6.C4 + IFNγ and scFv6.C4 + GM-CSF vectors, 1 × 105 MC38-CEA cells were s.c. injected into the left flank of mice one week after the last immunization (Fig. 1a), and tumor growth was measured periodically. In the scFv6.C4 + IFNγ-immunized group, 78% of the vaccinated animals remained free of tumors over 100 days of observation compared to 40% of the scFv6.C4 + GM-CSF-immunized mice and 47% of those immunized with scFv6.C4 only. All nonimmunized mice developed tumors 40 days after the challenge.
Cox regression survival analysis demonstrated the probability of the scFv6.C4 + INFγ-immunized mice being free of tumors was 58 times higher than that of the nonimmunized mice (95% CI HR 7.43–453.87; p < 0.001) and 2.7 times higher than that of the group immunized with scFv6.C4 (CI95% HR: 0.58–12.44; p = 0.204). The scFv6.C4 + GM-CSF-immunized animals had an average 6 times greater chance of being tumor-free than the nonimmunized animals (95% CI: 1.66–21.15; p = 0.006) but did not have improved survival compared to the scFv6.C4-treated group (95% CI: 0.25–1.97, p = 0.500) (Fig. 1c). These results showed that the IFNγ adjuvant vaccine regimen, but not GM-CSF, strongly potentializes scFv6.C4 immunization, remarkably delays tumor growth, and increases animal survival, and it can even completely inhibit tumor growth. A summary of the tumor cell challenge experiments is shown in Table 1.
The AB3 antibody affinity to CEA was evaluated by immunocytochemistry of the CEA-expressing human colorectal cell line HCT-8 and murine cell line MC38-CEA using mouse sera obtained from preimmunization, post-last immunization, and post-tumor challenge as primary antibodies. The MC38 cell line was used as a negative control. Sera from the nonimmunized and scFv6.C4-immunized animals were used for comparison (Fig. 2). Immune reactions with sera from the scFv6.C4-IFNγ and scFv6.C4-GM-CSF-immunized mice were strong in the CEA-expressing cell lines, whereas the sera from preimmunized and nonimmunized mice showed no reaction. These results corroborate AB3-specific antibody responses described previously [2, 3].

Blood samples were collected before the first immunization, after the last immunizations and after the tumor challenge. MC38 and HCT-8 cells were used as negative and positive controls, respectively. Samples obtained from the nonimmunized and scFv6.C4-immunized animals were used for comparison. CEA staining is shown in red, and DAPI nuclear staining is shown in blue. Representative images of each group are shown. Bar = 100 µm.
Cellular responses induced by scFv6.C4+IFNγ DNA vaccination
As the scFv6.C4 + IFNγ vaccination was able to increase animal survival, specific T- and B-cell proliferative activities of animals of this group were assessed and compared to those of the nonimmunized and scFv6.C4-immunized mice. Splenocytes from the vaccinated and MC38-CEA-challenged mice were harvested and stimulated with CEA or mAB 6.C4 (Fig. 3). ConA (2.5 μg/ml) and no stimulation were used as positive and negative controls, respectively. Splenocytes from the mice with and without tumors after tumor challenge were analyzed separately.

Splenocytes extracted from the MC38-CEA-challenged animals were labeled with 2.0 µM CSFE and stimulated with CEA (a, b) or mAB 6. C4 (c, d) for 6 days. CD4 + (a, c) and CD8 + (b, d) CFSE-low cells were counted by flow cytometry. The nonimmunized and scFv6.C4-immunized animals were used for comparison. Data are expressed as the mean ± SD (scFv6.C4 + IFNγ (n = 4), scFv6.C4 (n = 8), and nonimmunized (n = 5)). *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001.
Under CEA stimulation, the CD4 + T-cell proliferation rate of the scFv6.C4 + IFNγ-immunized animals that did not develop any tumors (3.52 ± 0.37%) was significantly higher than that of the scFv6.C4 animals that were also tumor-free (2.06 ± 0.53% p = 0.013, Fig. 3a). No differences in CD4 + proliferation were observed between the animals that developed tumors (Fig. 3a). However, CD8 + proliferation of the scFv6.C4 + IFNγ-immunized animals that did not develop tumors (2.12 ± 0.47%) was lower than that of the scFv6.C4-immunized animals (8.52 ± 0.66% p < 0.001, Fig. 3b). In the animals with tumors, the CD8 + response was similar between the scFv6.C4 + IFNγ (1.15 ± 0.54%) and scFv6.C4 (1.29 ± 0.41%) groups and was lower than the nonimmunized mouse response (8.59 ± 0.42%, p < 0.001 in relation to both groups, Fig. 3b).
Under mAB 6.C4 stimulation, CD4 + proliferation rates were similar among the scFv6.C4 + IFNγ, scFv6.C4, and nonimmunized animals (Fig. 3c). CD8 + proliferation rates were similar between the tumor-free animals from the scFv6.C4 + IFNγ (0.77 ± 0.46%) and scFv6.C4 (0.11 ± 0.66%) groups (Fig. 3d). For the animals that developed tumors, the scFv6.C4 CD8 + proliferation rate (5.65 ± 0.42%) was higher than that of the scFv6.C4 + IFNγ (0.54 ± 0.54%, p < 0.001) and nonimmunized (1.02 ± 0.38%, p < 0.001, Fig. 3d) groups.
To evaluate the CTL immune response, we used sorted CD8 + cells from tumor-challenged animals (with and without tumors after challenge) for cytotoxicity assays with a different target: effector ratios using MC38-CEA or MC38-scFv6.C4 as target cells and MC38 cells as a negative control (Fig. 4). At a 1:5 ratio, ∼4% of MC38 cells were lysed by CD8 + cells from naïve mice (Fig. 4a), and this was considered nonspecific basal activity.

MC38-CEA (b, e) and MC38-scFv6.C4 (c, f)-specific CTL lysis was carried out using CD8 + cells sorted by FACS from tumor-challenged animals that developed tumors (Tumor) or animals that did not develop tumors (No tumor). The cells were incubated for 16 h together with tumor target cells (target tumor: CD8 + ) at ratios of 1:5, 1:2, and 1:1 before LDH activity assessment. The nonimmunized, naïve and scFv6.C4-immunized animals were used for comparison. MC38 cells were used as negative controls (a, d). The naïve group represents the nonimmunized and nonchallenged animals. Data are expressed as the mean ± SD (scFv6. C4 + IFNγ (n = 4), scFv6.C4 (n = 4), nonimmunized (n = 6), and naïve (n = 4)). *** p ≤ 0.001.
Specific lysis of MC38-CEA was observed in the scFv6.C4 + IFNγ-immunized and tumor-free mice (14.98 ± 1.03%, Fig. 4b) and was much higher than that of the tumor-free animals from the scFv6.C4 (5.12 ± 1.45%; p < 0.001) and naïve (8.93 ± 1.45%; p = 0.002) groups. However, nonspecific lysis of MC38 cells was also observed by CD8 + cells from the scFv6.C4-IFNγ group (15.55 ± 1.06%, Fig. 4a) and was higher than the nonspecific lysis from the scFv6.C4 (4.66 ± 1.06%; p < 0.001) and naïve animals (3.68 ± 0.95%; p < 0.001). No specific lysis of MC38-scFv6.C4 was observed by CTL from the immunized groups compared to the naïve group (Fig. 4c).
In the group of animals that developed tumors, specific lysis of MC38- CEA cells of the scFv6.C4 + IFNγ group (11.87 ± 0.89, Fig. 4e) was higher than that of the scFv6.C4 group (8.11 ± 0.90% p = 0.009), but the mean value was similar to that of the nonimmunized animal group (12.80 ± 0.65; p = 1.000). No differences on MC38 (Fig. 4d) and MC38-scFv6.C4 (Fig. 4f) lysis were observed.
Discussion
CEA is a potential target for antitumor vaccines, since its expression increases substantially in patients with some types of cancer, mainly those from the gastrointestinal tract [33, 34]. Our group constructed the CEA surrogate scFv6.C4 for a DNA vaccine to promote anti-CEA humoral and cellular responses after immunization; in the CEA-expressing transgenic mice, the scFv6.C4 vaccine prevented 40–50% of tumor growth after challenge with tumor cells expressing CEA [2, 3]. This result is a major advance and proof-of-concept of the scFv6.C4-based DNA vaccine, but for use in humans, this vaccine still requires further improvement.
Therefore, we used FrC from Clostridium tetani as an adjuvant to the scFv6.C4 DNA vaccine [3] because it has a promiscuous universal epitope for MHC class II that can promote better antigen presentation and development of immune responses [35, 36]. Although a significant increase of 40% in the survival rate was observed, in our opinion, there is still a window for further improvement based on the other adjuvants. Therefore, in this study, we focused on the use of the endogenous factors GM-CSF and IFNγ rather than exogenous antigens to stimulate the immune response system.
Surprisingly, the immunization of CEA2682 mice with scFv6.C4 associated with IFNγ resulted in strong protection, increasing the tumor-free rate from 47% (only with scFv6.C4) to 78% after tumor challenge (Fig. 1c). In other words, the use of IFNγ in the scFv6.C4 DNA vaccine increased the chance of being free of tumors by ∼270% (Table 1). Taking into account that all nonimmunized mice died within 40 days after tumor injection and that the follow-up time after tumor challenge was 100 days, this improvement is a remarkable achievement.
IFN-γ is a Th1 cytokine involved in the induction and repression of immunoglobulin isotypes. In mice, IFN-γ promotes the production of IgG2a and IgG3, but it inhibits IgG1, IgM, and IgE [37]. A similar phenomenon was observed with human cells in the presence of IFN-γ [38], showing the modulatory effect of IFN-γ on antibody production. These effects of IFN-γ on anti-CEA antibody production were observed in our vaccination regimen; after the first immunization, the IgG antibody titer increased immediately, and it seemed to have peaked at this immunization because the titer did not change significantly with additional immunizations (Fig. 1b). The specificity of antibodies raised after immunization was observed in the immunocytochemistry assay using MC38-CEA and the human colorectal cell line HCT-8 (Fig. 2). As the use of scFv6.C4 in our vaccine regimen is to disrupt the tolerance to CEA, allowing its own immunity to recognize CEA as a nonself-antigen, the high antibody titer against CEA is a very important parameter to evaluate CEA recognition and, consequently, vaccination efficacy.
In addition, the anti-CEA antibody itself has the potential to neutralize CEA-expressing tumor growth by antibody-dependent cell-mediated cytotoxicity (ADCC). At least two humanized anti-CEA antibodies have been used to treat colorectal and lung cancers [39,40,41], showing high efficiency in neutralizing tumors.
Furthermore, IFNγ promotes the development of CD4+ and CD8+ cells and the differentiation of macrophages, which are very important activities for vaccination against cancer [24]. Figure 3 shows a specific proliferative activity of CD4 + T cells under CEA stimulation in the scFv6.C4 + IFNγ-immunized group without tumor. Some CTL activities were also observed in several groups, but they did not show specificity. Therefore, the high protection against the tumor in the scFv6.C4 + IFNγ group is probably due to the rapid increase and high titer of anti-CEA antibody and the CD4 + T-cell response.
Curiously, in both assays, cells with CEA stimulation or cells expressing CEA responded much better to cell proliferation and CTL activity, respectively, than those with mAB 6.C4 or scFv6.C4. This change of specificity from scFv6.C4 to CEA was previously observed with the scFv6.C4 vaccination [3]. As we commented there, we understand that immunization with scFv6.C4 should elicit immune responses against sequences specific to scFv6.C4 and to the sequences common to both CEA and scFv6.C4 because of their similarities [3]. However, as CEA2682 transgenic mice constitutively express CEA, we hypothesized that endogenous CEA may have favored the selection of clones that are more reactive to CEA than scFv6.C4.
One of the most relevant concerns in using antigen surrogates for vaccines is the possibility of inducing autoimmune disease and attacking own cells and tissues. In our previous study [3] and here, we did not observe any notable healthy alterations during any experimentation period. As the reaction between antibodies and antigens is concentration dependent, we understand that CEA expressed in CEA 2682 mouse cells and tissues was not sufficient to trigger autoimmune reactions. Such observations were also observed by others during vaccine studies [2, 17, 42, 43].
CD8 + T cells from mice with tumors showed significant proliferation after stimulation with CEA (in the nonimmunized group) and mAB 6.C4 (the scFv6.C4 group) (Fig. 3). Injection of highly CEA-expressing MC38-CEA tumor cells in nonimmunized mice should stimulate immunity against CEA. This phenomenon explains the rationality of using tumor extracts for immunization [44, 45]. CD8 + T-cell proliferative activity with mAB 6.C4 in the scFv6.C4-vaccinated group showed that the organism was working to eliminate tumors (Fig. 3), but the CTL activities were not enough (Fig. 4).
However, the immunization with scFv6.C4 plus GM-CSF worsened protection, decreasing the survival rate by approximately 40% (Fig. 1c). As we discussed above, the humoral response is a very important step for DNA vaccines that use a tumor-associated antigen surrogate. Therefore, the lower and slower anti-CEA antibody increase after immunization is likely the main cause of this low protection (Fig. 1b). Strong immune staining was shown by immunocytochemistry of sera from the scFv6.C4 + GM-CSF group, and because of the low survival rate, we continued this study with only the IFNγ group.
In conclusion, preventive DNA vaccination of CEA-expressing transgenic mice with uP-PS/scFv6.C4 plus uP-IFNγ, but not with uP-GM-CSF, resulted in strong humoral and CD4 + cell responses, which were sufficient to maintain almost 80% of mice free of tumors over 100 days after challenge with MC38-CEA cells. The adjuvant activity of IFNγ on tumor growth is likely to be associated with prompt and sustained specific humoral responses and stimulation of CD4 + cell proliferation. The use of IFNγ as an adjuvant has proven to be safe and tolerable in colon and pancreatic cancer vaccines [46, 47]. Therefore, its use as an adjuvant with scFv.6C4 for vaccination of future CEA-expressing cancer patients is promising.
The increased protection against tumors shown here with IFNγ and with FrC in the previous study [3] raise the possibility that the use of these two adjuvants together can develop even greater immunity and protection, but this hypothesis has to be validated in the future.
Change history
20 March 2023
A Correction to this paper has been published: https://doi.org/10.1038/s41434-023-00396-z
References
Siegel RL, Miller KD, Jemal A. Cancer statistics, 2020. CA Cancer J Clin. 2020;70:7–30.
Denapoli PMA, Zanetti BF, Dos Santos AA, de Moraes JZ, Han SW. Preventive DNA vaccination against CEA-expressing tumors with anti-idiotypic scFv6.C4 DNA in CEA-expressing transgenic mice. Cancer Immunol Immunother. 2016;66:333–42.
Zanetti BF, Ferreira CP, de Vasconcelos JRC, Han SW. scFv6.C4 DNA vaccine with fragment C of Tetanus toxin increases protective immunity against CEA-expressing tumor. Gene Ther. 2019;26:441–54.
Pignatari GC, Takeshita D, Parise CB, Soares FA, de Moraes JZ, Han SW. Carcinoembryonic antigen (CEA) mimicry by an anti-idiotypic scFv isolated from anti-Id 6.C4 hybridoma. J Biotechnol. 2007;127:615–25.
de Moraes JZ, Carneiro CR, Buchegger F, Mach JP, Lopes JD. Induction of an immune response through the idiotypic network with monoclonal anti-idiotype antibodies in the carcinoembryonic antigen system. J Cell Biochem. 1992;50:324–35.
de Moraes JZ, Gesztesi JL, Westermann P, Le Doussal JM, Lopes JD, Mach JP. Anti-idiotypic monoclonal antibody AB3, reacting with the primary antigen (CEA), can localize in human colon-carcinoma xenografts as efficiently as AB1. Int J Cancer. 1994;57:586–91.
Geethadevi A, Jadhav K, Kumar G, Parashar D scFv6.C4 DNA vaccine with fragment C of tetanus toxin increases protective immunity against CEA-expressing tumor. Gene Ther. 2020;26:441–54. https://doi.org/10.1038/s41434-020-0161-9.
Coleman DL, Chodakewitz JA, Bartiss AH, Mellors JW. Granulocyte-macrophage colony-stimulating factor enhances selective effector functions of tissue-derived macrophages. Blood. 1988;72:573–8.
Inaba K, Inaba M, Romani N, Aya H, Deguchi M, Ikehara S, et al. Generation of large numbers of dendritic cells from mouse bone marrow cultures supplemented with granulocyte/macrophage colony-stimulating factor. J Exp Med. 1992;176:1693–702.
Steinman RM, Witmer-Pack M, Inaba K. Dendritic cells: antigen presentation, accessory function and clinical relevance. Adv Exp Med Biol. 1993;329:1–9.
Clive KS, Tyler JA, Clifton GT, Holmes JP, Mittendorf EA, Ponniah S, et al. Use of GM-CSF as an adjuvant with cancer vaccines: beneficial or detrimental? Expert Rev Vaccines. 2010;9:519–25.
Kass E, Panicali DL, Mazzara G, Schlom J, Greiner JW Granulocyte / macrophage-colony stimulating factor produced by recombinant avian poxviruses enriches the regional lymph nodes with antigen-presenting cells and acts as an immunoadjuvant granulocyte / macrophage-colony stimulating factor produced by recomb. Cancer Res. 2001;27:206–14.
Aarts WM, Schlom J, Hodge JW. Vector-based vaccine / cytokine combination therapy to enhance induction of immune responses to a self-antigen and antitumor activity. Am Assoc Cancer Res. 2002;62:5770–7.
Lima J, Jenkins C, Guerrero A, Triozzi PL, Shaw DR, Strong TV. A DNA vaccine encoding genetic fusions of carcinoembryonic antigen (CEA) and granulocyte/macrophage colony-stimulating factor (GM-CSF). Vaccine. 2005;23:1273–83.
Hallermalm K, Johansson S, Bråve A, Ek M, Engström G, Boberg A, et al. Pre-clinical evaluation of a CEA DNA prime/protein boost vaccination strategy against colorectal cancer. Scand J Immunol. 2007;66:43–51.
Schwegler C, Dorn-Beineke A, Nittka S, Stocking C, Neumaier M. Monoclonal anti-idiotype antibody 6G6.C4 fused to GM-CSF is capable of breaking tolerance to carcinoembryonic antigen (CEA) in CEA-transgenic mice. Cancer Res. 2005;65:1925–33.
Hodge JW, Poole DJ, Aarts WM, Yafal AG, Gritz L, Schlom J. Modified vaccinia virus ankara recombinants are as potent as vaccinia recombinants in diversified prime and boost vaccine regimens to elicit therapeutic antitumor responses. Cancer Res. 2003;63:7942–9.
Luo Y, O’Hagan D, Zhou H, Singh M, Ulmer J, Reisfeld RA, et al. Plasmid DNA encoding human carcinoembryonic antigen (CEA) adsorbed onto cationic microparticles induces protective immunity against colon cancer in CEA-transgenic mice. Vaccine. 2003;21:1938–47.
Samanci A, Yi Q, Fagerberg J, Strigard K, Smith G, Ruden U, et al. Pharmacological administration of granulocyte/macrophage-colony-stimulating factor is of significant importance for the induction of a strong humoral and cellular response in patients immunized with recombinant carcinoembryonic antigen. Cancer Immunol Immunother. 1998;47:131–42.
Ullenhag GJ, Frödin JE, Jeddi-Tehrani M, Strigård K, Eriksson E, Samanci A, et al. Durable carcinoembryonic antigen (CEA)-specific humoral and cellular immune responses in colorectal carcinoma patients vaccinated with recombinant CEA and granulocyte/macrophage colony-stimulating factor. Clin Cancer Res. 2004;10:3273–81.
Duggan MC, Jochems C, Donahue RN, Richards J, Karpa V, Foust E, et al. A phase I study of recombinant (r) vaccinia-CEA(6D)-TRICOM and rFowlpox-CEA(6D)-TRICOM vaccines with GM-CSF and IFN-α-2b in patients with CEA-expressing carcinomas. Cancer Immunol Immunother. 2016;65:1353–64.
Marshall JL, Gulley JL, Arlen PM, Beetham PK, Tsang KY, Slack R, et al. Phase I study of sequential vaccinations with fowlpox-CEA(6D)-TRICOM alone and sequentially with vaccinia-CEA(6D)-TRICOM, with and without granulocyte-macrophage colony-stimulating factor, in patients with carcinoembryonic antigen-expressing carcinomas. J Clin Oncol. 2005;23:720–31.
Geynisman DM, Zha Y, Kunnavakkam R, Aklilu M, Catenacci DV, Polite BN, et al. A randomized pilot phase I study of modified carcinoembryonic antigen (CEA) peptide (CAP1-6D)/montanide/GM-CSF-vaccine in patients with pancreatic adenocarcinoma. J Immunother cancer. 2013;1:8.
Farrar M, Schreiber R. The molecular cell biology of interferon-gamma and its receptor. Annu Rev Immunol. 1993;11:571–611.
Ikeda H, Old LJ, Schreiber RD. The roles of IFNγ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002;13:95–109.
Miller CHT, Maher SG, Young HA. Clinical use of Interferon- γ. Ann N Y Acad Sci. 2009;79:69–79.
Song K, Chang Y, Prud’homme GJ. Regulation of T-helper-1 versus T-helper-2 activity and enhancement of tumor immunity by combined DNA-based vaccination and nonviral cytokine gene transfer. Gene Ther. 2000;7:481–92.
Sacramento CB, Cantagalli VD, Grings M, Carvalho LP, Baptista-Silva JCC, Beutel A, et al. Granulocyte-macrophage colony-stimulating factor gene based therapy for acute limb ischemia in a mouse model. J Gene Med. 2009;11:345–53.
Corbett TH, Griswold DPJ, Roberts BJ, Peckham JC, Schabel FMJ. Tumor induction relationships in development of transplantable cancers of the colon in mice for chemotherapy assays, with a note on carcinogen structure. Cancer Res. 1975;35:2434–9.
Tompkins WA, Watrach AM, Schmale JD, Schultz RM, Harris JA. Cultural and antigenic properties of newly established cell strains derived from adenocarcinomas of the human colon and rectum. J Natl Cancer Inst. 1974;52:1101–10.
Eades-Perner AM, van der Putten H, Hirth A, Thompson J, Neumaier M, von Kleist S, et al. Mice transgenic for the human carcinoembryonic antigen gene maintain its spatiotemporal expression pattern. Cancer Res. 1994;54:4169–76.
Parise CB, Lisboa B, Takeshita D, Sacramento CB, de Moraes JZ, Han SW. Humoral immune response after genetic immunization is consistently improved by electroporation. Vaccine. 2008;26:3812–7.
Goldstein MJ, Mitchell EP. Carcinoembryonic antigen in the staging and follow-up of patients with colorectal cancer. Cancer Invest. England. 2005;23:338–51.
Duffy MJ. Carcinoembryonic antigen as a marker for colorectal cancer: is it clinically useful? Clin Chem. 2001;47:624–30.
Panina-Bordignon P, Tan A, Termijtelen A, Corradin G, Lanzavecchia A. Universally immunogenic T cell epitopes:promiscuous binding to human MHC class II and promiscuous recognition by T cells. Eur J Immunol. 1989;19:2237–42.
Demotz S, Lanzavecchia A, Eisel U, Niemann H, Widmann C, Corradin G. Delineation of several DR-restricted tetanus toxin T cell epitopes. J Immunol. 1989;142:394–402.
Finkelman FD, Katona IM, Mosmann TR, Coffman RL. IFN-gamma regulates the isotypes of Ig secreted during in vivo humoral immune responses. J Immunol. 1988;140:1022–7.
Kawano Y, Noma T, Yata J. Regulation of human IgG subclass production by cytokines. IFN-gamma and IL-6 act antagonistically in the induction of human IgG1 but additively in the induction of IgG2. J Immunol. 1994;153:4948–58.
Conaghan PJ, Ashraf SQ, Tytherleigh MG, Wilding JL, Tchilian E, Bicknell D, et al. Targeted killing of colorectal cancer cell lines by a humanised IgG1 monoclonal antibody that binds to membrane-bound carcinoembryonic antigen. Br J Cancer. 2008;98:1217–25.
Blumenthal RD, Osorio L, Hayes MK, Horak ID, Hansen HJ, Goldenberg DM. Carcinoembryonic antigen antibody inhibits lung metastasis and augments chemotherapy in a human colonic carcinoma xenograft. Cancer Immunol Immunother. 2005;54:315–27.
Sharkey RM, Juweid M, Shevitz J, Behr T, Dunn R, Swayne LC, et al. Evaluation of a complementarity-determining region-grafted (humanized) anti-carcinoembryonic antigen monoclonal antibody in preclinical and clinical studies. Cancer Res. 1995;55:5935s–5945s.
Greiner JW, Zeytin H, Anver MR, Schlom J. Vaccine-based therapy directed against carcinoembryonic antigen demonstrates antitumor activity on spontaneous intestinal tumors in the absence of autoimmunity. Cancer Res. 2002;62:6944–51.
Saha A, Chatterjee SK, Foon KA, Primus FJ, Sreedharan S, Mohanty K, et al. Dendritic cells pulsed with an anti-idiotype antibody mimicking carcinoembryonic antigen (CEA) can reverse immunological tolerance to CEA and induce antitumor immunity in CEA transgenic mice. Cancer Res. 2004;7:4995–5003.
Chiang CL-L, Benencia F, Coukos G. Whole tumor antigen vaccines. Semin Immunol. 2010;22:132–43.
Joshi VB, Geary SM, Gross BP, Wongrakpanich A, Norian LA, Salem AK. Tumor lysate-loaded biodegradable microparticles as cancer vaccines. Expert Rev Vaccines. 2014;13:9–15.
Tempero MA, Sivinski C, Steplewski Z, Harvey E, Klassen L, Kay HD. Phase II trial of interferon gamma and monoclonal antibody 17-1A in pancreatic cancer: biologic and clinical effects. J Clin Oncol. 1990;8:2019–26.
Wiesenfeld M, O’Connell MJ, Wieand HS, Gonchoroff NJ, Donohue JH, Fitzgibbons RJJ, et al. Controlled clinical trial of interferon-gamma as postoperative surgical adjuvant therapy for colon cancer. J Clin Oncol. 1995;13:2324–9.
Acknowledgements
This work was supported by the São Paulo Research Foundation (FAPESP; Grant Numbers: 2012/21861-1 and # 2013/17224-9). BFZ was a recipient of a FAPESP scholarship (2012/21861-1).
Author information
Authors and Affiliations
Contributions
BFZ: conception and design of the study, acquisition of data, analysis and interpretation of the data, drafting the article; CPF: acquisition of data, analysis, and interpretation of data; JRCV: analysis and interpretation of the data; SWH: conception and design of the study, drafting the article, revising it critically for important intellectual content, and final approval of the version to be submitted.
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
The original online version of this article was revised: Fig. 2 has been corrected.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zanetti, B.F., Ferreira, C.P., Vasconcelos, J.R.C. et al. Adjuvant properties of IFN-γ and GM-CSF in the scFv6.C4 DNA vaccine against CEA-expressing tumors. Gene Ther 30, 41–50 (2023). https://doi.org/10.1038/s41434-021-00270-w
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41434-021-00270-w
- Springer Nature Limited
This article is cited by
-
Engineering and design of promising T-cell-based multi-epitope vaccine candidates against leishmaniasis
Scientific Reports (2023)