
Abstract
Inflammasomes are essential components of the innate immune system and its defense against infections, whereas the dysregulation of inflammasome activation has a detrimental effect on human health. The activation of inflammasomes is subjected to tight regulation to maintain immune homeostasis, yet the underlying mechanism remains elusive. Here, we identify USP3 as a direct deubiquitinating enzyme (DUB) for ASC, the central adapter mediating the assembly and activation of most inflammasomes. USP3 removes the K48-linked ubiquitination on ASC and strengthens its stability by blocking proteasomal degradation. Additionally, USP3 promotes inflammasome activation, and this function was confirmed in mouse models of aluminum (Alum)-induced peritonitis, F. novicida infection and flagellin-induced pneumonia in vivo. Our work unveils that USP3 functions as a key regulator of ASC ubiquitination and maintains the physiological role of ASC in mediating inflammasome activation, and we propose a new mechanism by which the ubiquitination of ASC regulates inflammasome activation.
Similar content being viewed by others


Introduction
Inflammasomes are multimeric complexes that defend against pathogenic microorganisms and environmental stress by the activation of inflammatory caspases and secretion of proinflammatory cytokines, including interleukin-1β (IL-1β) and IL-18 [1,2,3]. Although inflammasomes are critical components of the innate immune system and its defense against diverse organismal insults, inappropriate activation of inflammasomes has detrimental effects on human health and leads to various diseases, e.g., gout, atherosclerosis, diabetes, and Alzheimer’s disease [4,5,6]. Therefore, it is of great value to manipulate inflammasome activation to maintain immune homeostasis.
To date, several pattern recognition receptors (PRRs) have been shown to form inflammasomes, including NLRP1, NLRP3, NLRC4, Pyrin, and absent-in-melanoma (AIM2), among which the NLRP3 inflammasome is the best studied [2, 7]. The NLRP3 inflammasome is composed of four types of proteins: a ligand-sensor NLRP3, an adapter apoptosis-associated speck-like protein containing a caspase-recruitment domain (ASC), an effector pro-caspase-1, and the regulatory protein NIMA-related kinase 7 (NEK7) [2]. Activation of the NLRP3 inflammasome involves two steps: priming and activation. The priming step is typically mediated by PRRs as a response to microbial components and induces the upregulation of pro-IL1B and NLRP3 [2]. Then, in response to diverse activating stimuli, such as extracellular ATP, potassium ion (K+) efflux and mitochondrial reactive oxygen species, NLRP3 undergoes oligomerization and recruits pro-caspase-1 via the adapter ASC [2]. The multiprotein complex serves as a signaling platform to trigger the processing of pro-caspase-1 into active caspase-1, which in turn cleaves pro-IL-1β and pro-IL-18 to the mature form and finally leads to the secretion of these proinflammatory cytokines [2]. The process of inflammasome activation is subject to tight regulation, but the underlying mechanism remains elusive.
Emerging evidence indicates that inflammasome activation is regulated by ubiquitination [8, 9]. Ubiquitination corresponds to the conjugation of ubiquitin to the lysine residues of substrate proteins via isopeptide bonds, a process carried out by activating (E1), conjugating (E2) and ligating (E3) enzymes [10]. In addition, deubiquitinating enzymes (DUBs) remove ubiquitin moieties from proteins [10]. NLRP3 is ubiquitinated by Lys-48 (K48)- and Lys-63 (K63)-linked ubiquitin chains, and a series of E3 ligases and DUBs, such as Pellino2 [11], Trim31 [12], BRCC3 [13] and USP7/USP47 [14], have been shown to maintain NLRP3 protein homeostasis by modulating the ubiquitination of NLRP3. ASC functions as the central adapter mediating the formation of various inflammasomes [15, 16], such as NLRP3, AIM2, Pyrin and NLRC4, and has been reported to undergo linear and K63-linked ubiquitination [17,18,19,20,21]. Several E3 ligases have been identified for ASC. The linear ubiquitination assembly complex (LUBAC) mediates the linear ubiquitination of ASC and facilitates NLRP3 inflammasome activation [17]. Peli1 conjugates K63-linked ubiquitin chains to ASC and in turn facilitates ASC/NLRP3 interaction and ASC oligomerization [21]. However, despite being an important regulator for maintaining the ubiquitin balance of target proteins, the DUB of ASC is rarely reported on and needs further elucidation.
Here, we identify ubiquitin-specific peptidase 3 (USP3) as a DUB for ASC that regulates inflammasome activation. USP3 directly interacts with ASC and cleaves its K48-linked polyubiquitin chains in an enzymatic activity-dependent manner, stabilizing the ASC protein by blocking its proteasomal degradation. To the best of our knowledge, this is the first report of the regulation of ASC function by manipulating its K48-linked ubiquitination. Additionally, USP3 deficiency in macrophages or THP-1 cells decreases IL-1β but not TNF-α and IL-6 secretion during inflammasome activation. USP3 overexpression in vivo facilitates inflammasome activation in mouse models of Alum-induced peritonitis, F. novicida infection and flagellin-induced pneumonia. Thus, our findings reveal that USP3 functions as a new regulator of ASC ubiquitination and maintains the physiological role of ASC in mediating inflammasome activation, and we provide new insights into the mechanism by which the ubiquitination of ASC mediates the regulation of inflammasome activation.
Results
Identification of USP3 as a DUB for ASC
To identify the potential deubiquitinating enzymes (DUBs) that regulate ASC deubiquitination, we transfected a total of 22 members of the USP family DUBs into HEK293T cells together with Myc or Flag-tagged ASC and hemagglutinin (HA)-tagged ubiquitin (Ub), followed by immunoprecipitation and immunoblot analysis. This experiment showed that USP3 potently reduced the polyubiquitination of ASC (Supplementary Fig. 1A). To investigate whether USP3 has any effect on inflammasome activation, we transfected a small interfering RNA targeting Usp3 into mouse peritoneal macrophages (PMs) and measured IL-1β secretion triggered by lipopolysaccharide (LPS) and the NLRP3 inflammasome activator ATP. We found that siRNA-mediated knockdown of Usp3 expression in PMs impaired IL-1β secretion (Supplementary Fig. 1B). As a control, knockdown of Brcc3 also reduced the production of IL-1β, which was consistent with a previous report [13] (Supplementary Fig. 1B). Although USP50 has been reported as a DUB for ASC [22], we did not observe attenuation of ASC ubiquitination upon USP50 transfection. Consistently, we found that siRNA-mediated knockdown of Usp50 expression in PMs could not impair IL-1β secretion (Supplementary Fig. 1B). These data collectively indicate that USP3 is a potential DUB of ASC and regulates inflammasome activation.
USP3 directly interacts with ASC
To uncover the molecular mechanism by which USP3 regulates ASC deubiquitination and inflammasome activation, we tested the interaction between ASC and USP3. We transfected Myc-tagged ASC and Flag-tagged USP3 into HEK293T cells, followed by coimmunoprecipitation with an anti-Myc antibody. As shown in Fig. 1A, Myc-ASC pulled down Flag-USP3. Consistently, Flag-USP3 also pulled down Myc-ASC with an anti-Flag antibody (Fig. 1A). The reciprocal immunoprecipitation analysis indicated that USP3 interacts with ASC. Interestingly, we found that USP3 could not pull down NLRP3 and caspase-1, two other components of the NLRP3 inflammasome (Fig. 1B). Furthermore, USP3 and ASC were affinity purified from HEK293T cells transfected with Flag-tagged USP3 and ASC, respectively (Supplementary Fig. 2). In vitro pulldown assays with the recombinant proteins showed that USP3 bound directly with ASC in vitro (Fig. 1C). We then performed confocal microscopy of GFP-ASC and Flag-USP3 in HEK293T, HEK293 and HeLa cells and found that USP3 colocalized with ASC (Fig. 1D). Furthermore, we detected colocalization between endogenous ASC and USP3 in PMs. As shown in Fig. 1E, USP3 colocalized with ASC in the nucleus with PBS or LPS treatment. Upon ATP stimulation, ASCs formed puncta in the periphery of the nucleus, and USP3 still colocalized with the new ASC aggregates. Then, we detected the interaction between USP3 and ASC under physiological conditions. In PMs, the expression of USP3 was increased after LPS plus ATP treatment, whereas USP3 expression remained constant regardless of treatment with inflammatory stimuli in THP-1 cells (Supplementary Fig. 3). We performed coimmunoprecipitation analysis of ASC and USP3 in PMs and THP-1 cells. As shown in Fig. 1F, in PMs, ASC could consistently pull down USP3, and the interaction was enhanced after LPS and ATP treatment, which may be attributed to the increase in USP3 expression. In THP-1 cells, ASC pulled down comparable amounts of USP3 in the presence or absence of LPS plus ATP treatment (Fig. 1G).

USP3 interacts with ASC. A Reciprocal immunoprecipitation analysis of the interaction between USP3 and ASC. HEK293T cells were transfected with the indicated plasmids for 36 h. The cell lysates were subjected to immunoprecipitation with anti-Myc or anti-Flag antibody and then analyzed by immunoblotting. B Coimmunoprecipitation analysis of the interaction between USP3 and the indicated inflammasome components. C Coimmunoprecipitation analysis of the interaction between purified USP3 and ASC in vitro. Recombinant USP3 and ASC proteins were incubated for 60 min, and then the mixture was subjected to immunoprecipitation with anti-ASC antibody followed by immunoblot analysis. D Colocalization of Flag-USP3 (red) with GFP-ASC (green) in HEK293T cells, HEK293 cells, and HeLa cells. Scale bar, 20 μm (top), 10 μm (middle and bottom). E Colocalization between USP3 and ASC in LPS-primed (8 h) PMs treated with ATP (30 min). Scale bars, 10 μm. Coimmunoprecipitation analysis of the interaction between endogenous USP3 and ASC with inflammatory stimulation. PMs in (F) or THP-1 cells in (G) were untreated or primed with LPS for the indicated time points followed by stimulation with ATP for 30 min, and then the cell lysates were subjected to immunoprecipitation with anti-ASC antibody and immunoblot analysis. H Schematic diagram of ASC and USP3 and their truncation mutants. I Coimmunoprecipitation analysis of the interaction between ASC and USP3 truncation mutants. The experimental procedures in (B) and (I) were the same as those in (A). Data are representative of three independent experiments with similar results. Red arrow, IgG; red asterisk, indicated protein. β-Actin was used as a loading control
ASC is composed of a PYD domain and a CARD domain. To determine which domain of ASC is critical for interaction with USP3, we constructed two ASC deletion mutants and performed a coimmunoprecipitation experiment (Fig. 1H). We found that the ASC-CARD domain but not the PYD domain could interact with USP3 (Fig. 1I, left panel). USP3 is composed of a ZnF domain and a UCH domain. We constructed two USP3 deletion mutants containing only ZnF and UCH domains (Fig. 1H). We found that deletion of the USP3 UCH domain ablated the interaction between USP3 and ASC (Fig. 1I, right panel). Collectively, these data prove that USP3 specifically interacts with ASC through the CARD domain of ASC and the UCH domain of USP3.
USP3 abrogates K48-linked ubiquitination of ASC
To investigate the detailed mechanism by which USP3 deubiquitinates ASC, we purified recombinant USP3 and ubiquitin-modified ASC from HEK293T cells transfected with Flag-USP3 or Flag-ASC and HA-Ub and then performed an in vitro deubiquitination assay. As shown in Fig. 2A, purified USP3 potently removed the polyubiquitin chains on ASC, indicating that USP3 directly deubiquitinates ASC. Then, we examined whether the impact of USP3 on ASC ubiquitination relies on its enzymatic activity. We constructed an inactive USP3 mutant, C168S (mutation of cysteine to serine at position 168), which abolishes the deubiquitination activity of USP3 [23]. We cotransfected Myc-ASC, HA-Ub and Flag-tagged wild-type (WT) USP3 or mutant C168S into HEK293T cells. Immunoprecipitation analysis showed that WT-USP3 but not mutant C168S decreased ASC ubiquitination, indicating that the enzymatic activity of USP3 is indispensable for the deubiquitination of ASC (Fig. 2B). To elucidate the types of ASC ubiquitination removed by USP3, we simply transfected HA-Ub mutants K48 and K63, in which all lysine residues except the one at position 48 or 63 are replaced with arginine, together with Myc-ASC with or without Flag-USP3, into HEK293T cells. As shown in Fig. 2C, USP3 substantially reduced the polyubiquitination of ASC in the presence of WT and the K48-Ub mutant but not the K63-Ub mutant. Moreover, USP3 could not abrogate the ubiquitination signal of ASC in cells transfected with the K48R-ubiquitin mutant, which contains a single lysine-to-arginine substitution at position 48. The results suggest that the K48-linked ubiquitin chains should be the major form ubiquitination targeted by USP3 on ASC (Fig. 2D).

USP3 deubiquitinates ASC. A The immunoprecipitation analysis of the ubiquitination of the purified ubiquitin-modified ASC treated with recombinant USP3 protein in deubiquitylation assay buffer in vitro. The recombinant proteins were incubated at 37 °C for 1 h in deubiquitylation assay buffer. Then, the ASC protein was immunoprecipitated and detected by immunoblotting. B Immunoprecipitation analysis of the ubiquitination of ASC in HEK293T cells transfected with Myc-ASC and HA-Ub in the presence of Flag-USP3 or Flag-USP3 mutant C168S. C, D Immunoprecipitation analysis of the ubiquitination of ASC in HEK293T cells transfected with Myc-ASC and HA-Ub or its lysine residue mutants in the presence of Flag-USP3. E Immunoprecipitation analysis of the ubiquitination of ASC and its lysine residue mutants in HEK293T cells transfected with Myc-ASC or its mutants and HA-Ub in the presence of Flag-USP3. F Immunoprecipitation analysis of the ubiquitination of endogenous ASC in PMs. PMs were silenced for Usp3 followed by treatment with LPS for 8 h and stimulation with ATP for 30 min. Then, the cell lysates were immunoprecipitated with anti-ASC antibody and detected by immunoblotting. G Immunoprecipitation analysis of the ubiquitination of endogenous ASC in THP-1 cells. WT or USP3-KO THP-1 cells were treated as in (F), and the cell lysates were immunoprecipitated with anti-ASC antibody and detected by immunoblotting. Data are representative of three independent experiments with similar results. β-Actin was used as a loading control
Given that USP3 interacted with the CARD domain of ASC, we speculate that USP3 may regulate the ubiquitination of the lysine (K) residues in the CARD domain of ASC. We mutated each lysine residue in the ASC-CARD domain to arginine (R). We transfected the WT-ASC-CARD and ASC-CARD mutants into HEK293T cells together with HA-Ub and Flag-USP3. We found that the ASC-CARD mutant K174R was totally resistant to USP3-mediated deubiquitination, indicating that USP3 mainly deubiquitinates ASC-CARD at K174 (Supplementary Fig. 4A). Then, we constructed full-length ASC with the K174R mutation and detected the effect of USP3 on the ubiquitination of this mutant. As shown in Fig. 2E, USP3 could not remove the ubiquitination signal of the ASC-K174R mutant, indicating that USP3 mainly targets the ubiquitin chains on K174 of ASC. In addtion, USP3 could still colocalize and interact with ASC-K174R (Supplementary Fig. 4B, C), indicating that USP3 targets the ubiquitination of K174 on ASC but does not rely on the site for its interaction with ASC. Furthermore, we investigated whether USP3 deubiquitinates endogenous ASC under physiological conditions. We transfected Usp3-siRNA into PMs, primed the PMs with LPS, stimulated them with ATP, and tested the ubiquitination of ASC by immunoprecipitation. As shown in Fig. 2F, ubiquitination of ASC, as well as K48-linked ubiquitination, emerged after treatment with LPS plus ATP (Lane 3), while knockdown of Usp3 potently enhanced the K48-linked ubiquitination signal under this condition (Lane 6). Additionally, we established USP3-knockout (KO) THP-1 cells and examined ASC ubiquitination. DNA sequencing and western blot experiments confirmed the success of USP3 KO in THP-1 cells (Supplementary Fig. 5). WT and USP3-KO THP-1 cells were treated with LPS alone or LPS plus ATP, and then ASC ubiquitination was detected. As shown in Fig. 2G, unlike the trigger of ASC ubiquitination in PMs, only LPS treatment was sufficient to induce ASC ubiquitination in THP-1 cells, and LPS plus ATP cotreatment did not further enhance the ubiquitination signal (Lanes 2 and 3). The level of ASC ubiquitination, especially K48-linked ubiquitination, was obviously increased in USP3-KO THP-1 cells compared with WT cells upon both LPS treatment and LPS plus ATP treatment (Fig. 2G, Lanes 5 and 6). Although the trigger of ASC ubiquitination in PMs and THP-1 cells was different for an unknown reason, the above results consistently indicate that the abrogation of USP3 promotes ASC ubiquitination.
USP3 promotes ASC stability
As K48-linked ubiquitination leads to proteasomal degradation [24], we detected whether USP3 regulates the stability of ASC. We transfected Myc-ASC and HA-K48-Ub with increasing amounts of Flag-USP3 into HEK293T cells and detected ASC protein expression by immunoblotting. As shown in Fig. 3A, B, transfection of K48-Ub led to a decrease in ASC protein, and overexpression of Flag-USP3 rescued the ASC protein level in a dose-dependent manner, whereas the USP3 mutant C168S did not have this function. The results indicate that USP3 strengthens ASC stability in an enzymatic activity-dependent manner. Moreover, transfection of the ubiquitin mutant K48R did not lead to a decrease in ASC protein, and Flag-USP3 had a marginal effect under this condition (Fig. 3C). Since we identified K174 as the major site for deubiquitination targeted by USP3 on ASC, we detected the effect of USP3 on the stability of mutant ASC-K174R. The results showed that USP3 had no effect on the expression of this ASC mutant (Fig. 3D). Taken together, the results above indicate that USP3 targets K48-linked ubiquitination on K174 of ASC to modulate the stability of the ASC protein.

USP3 inhibits the proteasomal degradation of ASC. A Immunoblot analysis of ASC expression in HEK293T cells transfected with Myc-ASC and HA-K48-Ub together with increasing amounts of Flag-USP3. B Immunoblot analysis of ASC expression in HEK293T cells transfected with Myc-ASC and HA-K48-Ub in the presence of Flag-USP3 or mutant C168S. C Immunoblot analysis of ASC expression in HEK293T cells transfected with Myc-ASC and HA-K48-Ub or HA-K48R-Ub together with Flag-USP3. D Immunoblot analysis of the expression of ASC and its lysine residue mutant K174R in HEK293T cells transfected with Myc-ASC or its mutant and HA-K48-Ub in the presence of Flag-USP3. E Immunoblot analysis of endogenous ASC expression in WT or USP3-KO THP-1 cells with inflammatory stimulation. The cells were primed with LPS for 12 h and then stimulated with ATP for 30 min. The cell lysates were collected and detected by immunoblot analysis. F, G Immunoblot analysis of the endogenous ASC degradation ratio in WT or USP3-KO THP-1 cells. Cells were primed with LPS for 4 h and then treated with cycloheximide (CHX) for the indicated times. The quantification and statistics of ASC, caspase-1 and NLRP3 protein levels are displayed in (G). H Immunoblot analysis of endogenous ASC degradation in WT or USP3-KO THP-1 cells treated with degradation inhibitors. WT or USP3-KO THP-1 cells were stimulated with LPS for 4 h and then treated with MG132 (10 μM), 3-MA (1 mM) or chloroquine (10 μM) for 6 h, and the cell lysates were collected and analyzed by immunoblotting. Data in (G) are from three independent experiments (n = 3) and are shown as the mean + S.D., and other data are representative of three independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001; two-tailed Student’s t test. β-Actin was used as a loading control. The band density of ASC, caspase-1 and NLRP3 in (A-F, H) was quantified using ImageJ software and normalized to the corresponding β-actin. The protein quantification is displayed above the corresponding band in the western blot figure
Consistent with the above exogenous expression results, knockout of USP3 in THP-1 cells followed by LPS and ATP treatment resulted in the reduction of ASC protein, whereas no effect was observed for NLRP3 and caspase-1 (Fig. 3E). Importantly, USP3 abrogation reduced the amount of ASC only after treatment with inflammatory stimuli (Fig. 3E, Lanes 2 and 4) but showed no effect in the resting state of THP-1 cells (Fig. 3E, Lanes 1 and 3). To further confirm that USP3 regulates endogenous ASC stability, we stimulated THP-1 cells with LPS for 4 h prior to different durations of treatment with the protein synthesis inhibitor cycloheximide (CHX). As shown in Fig. 3F, G, USP3 knockout facilitated the degradation of ASC but had no effect on NLRP3 and caspase-1. Additionally, to explore which pathway mediates USP3-induced degradation of ASC, various inhibitors were employed. We found that the proteasome inhibitor MG132 suppressed USP3-KO-induced ASC degradation, while the autophagy inhibitors 3-methyladenine (3-MA) and chloroquine (CQ) showed no effect (Fig. 3H). Taken together, these results indicate that USP3 promotes ASC stability by inhibiting its proteasome degradation.
USP3 regulates the formation of ASC specks upon inflammasome activation
ASCs form functional prion-like aggregates to propagate immune signaling [25]. The hallmark of NLRP3 inflammasome activation involves the aggregation of ASC and the formation of microscopically visible ASC specks [15]. We performed immunofluorescence staining of ASC specks in PMs primed with LPS and then activated by ATP. The siRNA-mediated Usp3 knockdown led to a significant decrease in ASC specks in PMs compared with that in cells transfected with control siRNA (Fig. 4A, B). Then, we utilized disuccinimidyl suberate (DSS) to cross link ASC aggregates. The results showed that USP3 deficiency reduced the amount of ASC monomer and attenuated the formation of ASC dimers and oligomers in PMs and THP-1 cells primed with LPS and activated by ATP (Fig. 4C, D). These data indicate that USP3 regulates NLRP3 inflammasome activation.

USP3 promotes ASC speck formation and oligomerization. A Immunostaining of endogenous ASC specks in PMs. PMs were primed with LPS for 8 h and then stimulated with ATP for 30 min. Scale bar, 50 μm. B The ASC specks in (A) were counted and presented as the percentage of total cell numbers. C PMs silenced for Usp3 were primed with LPS for 8 h and then stimulated with ATP for 30 min. The cell lysates were cross-linked with DSS, and ASC oligomerization was detected by immunoblot analysis with anti-ASC antibody. D WT or USP3-KO THP-1 cells were primed with LPS for 8 h and then stimulated with ATP for 30 min. The cell lysates were treated as in (C). Data in (B) are from three independent experiments (n = 3) and are shown as the mean + S.D., and other data are representative of three independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001; two-tailed Student’s t test. β-Actin was used as a loading control
USP3 promotes inflammasome activation
To determine the function of USP3 in regulating inflammasome activation, we tested the effect of USP3 deficiency on the inflammasome-mediated secretion of inflammatory cytokines, caspase-1 activation, and cell death. We employed a widely used two-signal model of inflammasome activation in PMs, including priming cells with LPS followed by stimulation with activators of the NLRP3 inflammasome (ATP, nigericin (Nig), and Alum). As shown in Fig. 5A, immunoblot analysis showed that Usp3 knockdown reduced LPS- and ATP-triggered caspase-1 cleavage (P20) and IL-1β secretion (P17), whereas the expression of pro-caspase-1 and pro-IL-1β was not changed. In addition, LPS-induced NF-κB signaling and MAPK signaling, including the phosphorylation of IKK-β, IκB-α, p65, ERK, JNK and p38, were comparable between control-siRNA transfected PMs and Usp3-siRNA transfected PMs (Fig. 5B). The results indicate that USP3 deficiency inhibits NLRP3 inflammasome activation but does not affect the LPS-triggered transcriptional response. To further corroborate the function of USP3 in modulating inflammasome activation, we detected the expression and secretion of inflammatory cytokines by qRT‒PCR and ELISA. The PMs were primed with LPS and then activated with ATP or Nig. We observed that knockdown of Usp3 in PMs significantly reduced IL-1β secretion, but the secretion of TNF-α and IL-6 was not influenced (Fig. 5C). The mRNA levels of Il1b, Tnf and Il6 were not changed by Usp3 knockdown (Supplementary Fig. 6A). In addition, pyroptotic cell death triggered by inflammasome activation was also detected. The PMs were primed with LPS and activated with the NLRP3 inflammasome activator ATP, Nig or Alum, and then lactate dehydrogenase (LDH) release was detected. LPS and NLRP3 activator treatment triggered cell death, while knockdown of Usp3 significantly blocked this phenomenon (Fig. 5D). To examine the function of USP3 in other inflammasomes, we utilized double-stranded DNA (poly (dA: dT)), a trigger of the AIM2 (absent in melanoma 2) inflammasome; flagellin, a trigger of the NLRC4 (NLR family CARD domain containing 4) inflammasome; and C3 toxin, a trigger of the Pyrin inflammasome. As shown in Fig. 5E, siRNA-mediated knockdown of Usp3 decreased the IL-1β secretion triggered by all inflammasome activators, while the secretion of TNF-α and IL-6 was comparable to that in control-siRNA treated cells. The mRNA levels of inflammatory cytokines were not changed by the various stimuli (Supplementary Fig. 6B). Furthermore, we confirmed the above results in USP3-KO THP-1 cells. WT and USP3-KO THP-1 cells were treated with LPS and stimulated with ATP, and then IL-1β, IL-6 and TNF-α secretion was measured. USP3 knockout significantly reduced IL-1β secretion but had no influence on IL-6 and TNF-α secretion (Fig. 5F). Then, we reconstituted USP3 or USP3-C168S in USP3-KO THP-1 cells. We treated these THP-1 cells with LPS and ATP and detected IL-1β production. We found that reconstitution of USP3, but not USP3-C168S, in USP3-KO THP-1 cells rescued ASC expression and IL-1β production (Fig. 5G, H). The results prove that USP3 stabilizes ASC and promotes inflammasome activation in a manner dependent on its DUB catalytic activity with an on-target effect. Since MG132 could block the degradation of ASC caused by USP3 KO (Fig. 3H), we tested whether MG132 could rescue the inflammasome inhibition caused by USP3 deficiency. We found that the addition of MG132 could rescue the effect of LPS and ATP treatment on IL-1β production in a dose-dependent manner in USP3-KO THP-1 cells (Supplementary Fig. 6C). To further elucidate the mechanistic link by which USP3 regulates ASC ubiquitination and inflammasome activation, we reconstituted WT ASC and ASC-K172R mutant (corresponding to K174 of human ASC) in RAW264.7 macrophages, which have been reported to be ASC-deficient cell lines [26]. The mutation increased ASC expression and IL-1β production upon inflammatory stimulation (Fig. 5I, J). We knocked down Usp3 in RAW264.7 macrophages, treated the cells with LPS and ATP, and then detected ASC ubiquitination and IL-1β production (Fig. 5I, J). The results showed that knockdown of Usp3 increased ASC ubiquitination and simultaneously reduced the amount of ASC protein. Moreover, knockdown of Usp3 did not exhibit an obvious effect on the ubiquitination or protein amount of ASC-K172R. Additionally, knockdown of Usp3 reduced IL-1β production after ASC transfection but showed no effect after ASC-K172R transfection. Our results prove that USP3 targets K172 (human K174) for ASC ubiquitination and inflammasome activation. Together, these results indicate that USP3 promotes inflammasome activation.

USP3 promotes NLRP3 inflammasome activation. A Immunoblot analysis of the supernatants (SN) or cell lysates (CL) of PMs with Usp3 silenced, primed with LPS for the indicated times, and stimulated with ATP for 30 min. B Immunoblot analysis of NF-κB and MAPK signaling in cell lysates of PMs with Usp3 silenced, followed by stimulation with LPS for the indicated times. C ELISA analysis of IL-1β, TNF-α and IL-6 secretion from PMs with Usp3 silenced, followed by treatment with LPS plus ATP (30 min) or Nig (45 min). D LDH release from PMs with Usp3 silencing, primed with LPS for 8 h, and stimulated with ATP (30 min), Nig (45 min) or Alum (6 h). Bars represent the mean percentage of LDH release relative to the total cells lysed. E ELISA analysis of IL-1β, TNF-α and IL-6 secretion from PMs with Usp3 silenced, primed with LPS for 8 h, and stimulated with ATP (30 min), Nig (45 min), Alum (6 h), poly (dA: dT) (1 h), flagellin (1 h) or C3 toxin (6 h). F ELISA analysis of IL-1β, TNF-α and IL-6 secretion from WT or USP3-KO THP-1 cells primed with LPS for the indicated times, followed by stimulation with ATP (30 min). G Immunoblot analysis of the protein levels of USP3 and ASC in WT, USP3-KO THP-1 cells and USP3-KO THP-1 cells reconstituted with USP3 or USP3-C168S. Cells were primed with LPS for 8 h, followed by stimulation with ATP for 30 min. H ELISA analysis of IL-1β secretion from WT, USP3-KO THP-1 cells and USP3-KO THP-1 cells reconstituted with USP3 or USP3-C168S. Cells were treated as in (G). I Immunoprecipitation analysis of the ubiquitination of ASC in RAW264.7 cells. RAW264.7 cells with Usp3 silenced were transfected with Flag-ASC or its lysine residue mutant K172R, treated with LPS for 8 h and stimulated with ATP for 30 min. Then, the cell lysates were immunoprecipitated with anti-ASC antibody and detected by immunoblotting. J ELISA analysis of IL-1β secretion from RAW264.7 cells. RAW264.7 cells were silenced for Usp3, transfected with Flag-ASC or its lysine residue mutant K172R, treated with LPS for 8 h and stimulated with ATP for 30 min. Data in (C–F, H, J) are from three independent experiments (n = 3) and are shown as the mean + S.D., and other data are representative of three independent experiments with similar results. *p < 0.05, **p < 0.01, ***p < 0.001; two-tailed Student’s t test. β-Actin was used as a loading control. The band density of ASC in (G, I) was quantified using ImageJ software and normalized to corresponding β-actin. The protein quantification is displayed above the corresponding band in the western blot figure
USP3 overexpression facilitates NLRP3, AIM2 and NLRC4 inflammasome activation in vivo
To investigate the physiological role of USP3 in regulating inflammasome activation in vivo, we overexpressed USP3 in mice through tail vein injection of a plasmid encoding Flag-tagged mouse Usp3 (Flag-mUsp3) with in vivo transfection reagent. The expression of USP3 was confirmed in various organs (Fig. 6A). We extracted PMs from USP3-overexpressing mice, treated the cells with LPS plus Alum, and detected IL-1β production in vitro. The results showed that USP3 was successfully overexpressed in these cells and that IL-1β production was increased (Supplementary Fig. 7A). Then, we employed a model of peritonitis induced by the NLRP3 inflammasome activator Alum, a widely used adjuvant that promotes leukocyte recruitment via the induction of NLRP3 inflammasome-dependent IL-1β secretion [27, 28]. As shown in Fig. 6B, the overexpression of USP3 enhanced the secretion of IL-1β in vivo, while the production of TNF-α and IL-6 was not affected. In addition, more cleaved caspase-1 (P20) and IL-1β (P17) in lavage fluid were observed after USP3 overexpression (Fig. 6C). Alum induced a drastic increase in the number of peritoneal exudate cells (PECs) [29]. We found that the numbers of Alum-induced PECs were increased in USP3-overexpressing mice (Fig. 6D). Analysis of the PEC populations revealed that Alum treatment increased the numbers of Ly6G neutrophils and Ly6C inflammatory monocytes in WT mice, and overexpression of USP3 further enhanced their population (Fig. 6D, Supplementary Fig. 7B). Then, we generated AIM2- and NLRC4-dependent mouse models to explore the effect of USP3 on the regulation of the AIM2 and NLRC4 inflammasomes in vivo. The AIM2 inflammasome has been reported to participate in host defense against F. novicida infection [30]. We overexpressed USP3 as described in a previous method and then subcutaneously infected mice with F. novicida (1.5 × 105 CFU per mouse) and detected the bacterial burdens and IL-1β production. As shown in Fig. 6E, the bacterial burdens in the livers and spleens of USP3-overexpressing mice were significantly lower than those in control mice. Meanwhile, IL-1β production in the sera of USP3-overexpressing mice was enhanced (Fig. 6F). The results indicate that overexpression of USP3 can facilitate AIM2 inflammasome activation and host defense against F. novicida infection. Then, we used a mouse model of flagellin-induced pneumonia to evaluate the role of USP3 in NLRC4 inflammasome activation in vivo. We overexpressed USP3 and intranasally instilled the mice with in vivo transfection reagent and flagellin to induce pneumonia. We found that IL-1β secretion in bronchoalveolar lavage fluid (BALF) was increased in flagellin-challenged USP3-overexpressing mice compared to flagellin-challenged control mice (Fig. 6G). In addition, we tested bronchoalveolar lavage cells (BALCs) and flagellin-induced recruitment of inflammatory cells by flow cytometry (Fig. 6H and Supplementary Fig. 7C). The total number of BALCs recruited upon flagellin challenge was increased in USP3-overexpressing mice (Fig. 6H). Furthermore, the flagellin-induced recruitment of neutrophils and Ly6C+ monocytes was also strongly increased in USP3-overexpressing mice (Fig. 6H and Supplementary Fig. 7C). Collectively, the above results indicate that USP3 modulates NLRP3, AIM2 and NLRC4 inflammasome activation in vivo.

USP3 overexpression facilitates NLRP3, AIM2 and NLRC4 inflammasome activation in vivo. A Immunoblot analysis of the protein level of USP3 in various organs of WT- or USP3-overexpressing mice after i.p. Alum injection for 12 h. B ELISA analysis of IL-1β, TNF-α and IL-6 secretion from the peritoneal lavage fluid of WT- or USP3-overexpressing mice after i.p. Alum injection for 12 h. (mice: n = 5 for each group). C Immunoblot analysis of supernatants (SN) or cell lysates (CL) of lavage fluid from WT- or USP3-overexpressing mice after i.p. Alum injection for 12 h. (mice: n = 3 for each group). D Absolute numbers of PECs, percentages of neutrophils and Ly6C+ monocytes from WT- or USP3-overexpressing mice after i.p. Alum injection for 12 h. (mice: n = 5 for each group). E WT- or USP3-overexpressing mice were infected subcutaneously with 1.5 × 105 CFU F. novicida, and the bacterial burden in the spleen and liver was measured on Day 2 after infection. (mice: n = 5 for each group). F ELISA analysis of IL-1β in sera from WT- or USP3-overexpressing mice infected subcutaneously with 1.5 × 105 CFU F. novicida. G ELISA analysis of IL-1β in the BALF from WT- or USP3-overexpressing mice 12 h after intranasal instillation of isotonic saline or flagellin. H Absolute numbers of BALCs, percentages of neutrophils and Ly6C+ monocytes from WT- or USP3-overexpressing mice after isotonic saline or flagellin intranasal instillation for 12 h. Data are shown as the mean + S.D. in (B, F, G). Similar results were obtained in three independent experiments. *p < 0.05, **p < 0.01, ***p < 0.001; two-tailed Student’s t test
Discussion
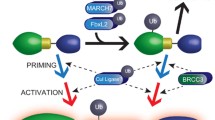
Inflammasomes are crucial for defense against pathogenic microorganisms, and dysregulation of inflammasome activation is closely associated with various human diseases. Therefore, it is of great importance to strictly control inflammasome activation to maintain immune homeostasis and prevent detrimental effects. In this study, we identified USP3 as a direct DUB for ASC. USP3 removes the K48-linked ubiquitination of ASC in an enzymatic activity-dependent manner and strengthens ASC stability by blocking its proteasomal degradation. Moreover, USP3 positively regulates inflammasome activation in response to several inflammasome stimuli, and this function was confirmed in mouse models of Alum-induced peritonitis, F. novicida infection and flagellin-induced pneumonia in vivo. Our work reveals USP3 as a novel regulator of ASC ubiquitination and inflammasome activation (Fig. 7).

Working model for USP3 promoting inflammasome activation
Ubiquitination functions as an essential regulator of inflammasome activation [8]. To date, most studies have concentrated on the ubiquitination of the ligand-sensor protein NLRP3, and a series of E3s and DUBs have been identified [8]. The major way ubiquitination controls NLRP3 function is by regulating its proteasomal or autophagy degradation [8]. In contrast to the detailed description of the ubiquitination of NLRP3, the function and mechanism by which ubiquitination regulates ASC, the central adapter for most inflammasomes, is still incompletely understood. ASC has been proposed as a target for both K63-linked and linear ubiquitination, involving the mechanism of autophagy degradation and oligomerization [8, 17,18,19,20,21]. However, the function and regulation of K48-linked ubiquitination of ASC have not yet been investigated. Here, we found that the K48-linked ubiquitination of ASC substantially increased during inflammasome activation (Fig. 2F, G). USP3 removed the K48-linked but not K63-linked ubiquitination of ASC (Fig. 2C). Additionally, USP3 could not remove the ubiquitin chains on ASC formed by transfection of the K48R mutant plasmid (Fig. 2D). The K48R mutant plasmid contains a single lysine-to-arginine substitution at position 48 and an HA-tag at its end, so this plasmid can mediate the formation of any other types of ubiquitination except K48-linked and linear ubiquitination. Our results cannot exclude the possibility that USP3 may also affect the linear ubiquitination of ASC. However, given that the linear ubiquitination of ASC does not affect ASC degradation [17], we conclude that USP3 mainly targets K48-linked ubiquitin chains on ASC to regulate ASC stability. Elimination of the K48-linked ubiquitin chains by USP3 blocks the proteasomal degradation of ASC and strengthens its stability. USP3 deficiency not only induces the degradation of ASC monomer, but also reduces the amounts of ASC dimer and oligomer, which are crucial for inflammasome assembly. ASC functions as the central adapter for several inflammasomes, such as NLRP3, AIM2, Pyrin and NLRC4 [31]. The function of USP3 in regulating ASC K48-linked ubiquitination and stability also affects various inflammasomes in vitro and in vivo (Figs. 5E and 6). To the best of our knowledge, this is the first study to elucidate the function of the K48-linked ubiquitination of ASC and establish USP3 as a direct regulator of this type of ASC ubiquitination and inflammasome activation.
In this study, we found that the K48-linked ubiquitination of ASC, which leads to proteasomal degradation, is substantially induced by LPS and inflammasome activator treatment. However, the expression of ASC remains relatively stable during the process of inflammasome activation. This phenomenon suggests that there may be other important mechanisms, such as transcriptional regulation or autophagic degradation, that cooperate with K48-linked ubiquitination to dynamically maintain the homeostasis of ASC during the inflammatory response. Although USP3 binds consistently with ASC, it stabilizes ASC only when ASC is substantially modified by K48-linked ubiquitination, which is triggered by transfection of K48-Ub plasmid or inflammatory stimulation. Disturbance of the K48-linked ubiquitination of ASC by USP3 knockdown leads to a drastic reduction in ASC levels during inflammasome activation, suggesting that the ubiquitin balance is essential for ASC stability and cannot be rescued by other regulatory mechanisms. Given the above, we propose a model to illustrate how USP3 regulates inflammasome activation (Fig. 7). In resting cells (unactivated phase), USP3 consistently binds with ASC but shows no effect on ASC stability. Upon treatment with inflammatory stimuli (activated phase), ASC is loaded with K48-linked ubiquitin chains catalyzed by unknown E3s, which results in the degradation of ASC. Simultaneously, USP3 cleaves the ubiquitin chains to prevent proteasomal degradation, dynamically maintains the homeostasis of ASC and ensures the physiological function of ASC in mediating inflammasome activation.
USP3 has been reported as a regulator of several cellular targets, such as checkpoint kinase 1 [23], RIG-I [32], p53 [33], histones [34] and Aurora A kinase [35]. However, the role of USP3 in modulating inflammasome activation has not been previously investigated. Here, we identify for the first time that USP3 targets ASC to regulate inflammasome activation in vitro and in vivo. USP3 binds with and regulates the stability of ASC but not NLRP3 or caspase-1 (Figs. 1B and 3E). Additionally, USP3 modulates ubiquitination and IL-1β production with the reconstitution of WT ASC but not the ASC-K172R mutant in ASC-deficient RAW264.7 cells (Fig. 5I, J). The results indicate that ASC is the specific substrate for USP3 in the inflammasome and that USP3 specifically targets the ubiquitination of ASC to regulate inflammasome activation. These findings provide a new perspective for understanding the function of USP3 in cells and in vivo.
In summary, we report that USP3 functions as a direct DUB for ASC to remove its K48-linked ubiquitination. USP3 strengthens ASC stability and maintains the physiological function of ASC in mediating inflammasome activation. Our work identifies USP3 as a novel regulator of ASC ubiquitination and inflammasome activation. These findings expand the current understanding of the molecular mechanism controlling inflammasome activation.
Methods
Plasmid constructs and small interfering RNA
The coding region of human USP3 was amplified by standard PCR and cloned into pReceiver-Lv242. The coding regions of human NLRP3, CASP1 and PYCARD were amplified by standard PCR and cloned into pCMV-N-Myc. The coding region of mouse Usp3 was amplified and cloned into pEnCMV-FLAG-SV40-Neo. A truncated form of human USP3 was amplified and cloned into p3×FLAG-CMV-10, and a truncated form of human PYCARD was amplified and cloned into pcDNA3.1. pLVX-IRES-Puro-hUSP3 was generated by subcloning the hUSP3-coding sequence into the pLVX-IRES-Puro vector. Point mutations were generated using the KOD-Plus-Mutagenesis kit (Toyobo). Plasmids encoding HA-ubiquitin or ubiquitin mutants have been described previously [36].
The small interfering RNA sequence was as follows: mouse Usp3: 5′- GGACAGCAAUGUGUCUUUATT -3′. Scramble small interfering RNA (siRNA) was used as a negative control in all RNA interference experiments. These siRNAs were obtained from GenePharma. For the transient transfection of siRNA into PMs, Lipofectamine RNAi-MAX (Invitrogen) reagent was used according to the manufacturer’s manual.
Generation of USP3-KO THP-1 cells through CRISPR‒Cas9
THP-1 cells with USP3 knockout were generated by a lenti-CRISPR/Cas9-v2 system, and the sequence of the target USP3 guide RNA was as follows: 3′-GTCCGGAGCAATGCAGACGC-5′. The isolated single clonal knockout cells, which survived after puromycin killing, were confirmed by immunoblot analysis. THP-1 cells were differentiated to a macrophage-like state by incubation with 100 ng/mL PMA (Sigma Aldrich, P1585) overnight before treatment with inflammatory stimuli.
Mice and cells
WT mice were purchased from Charles River Laboratories. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, with the approval of the Scientific Investigation Board of School of Basic Medical Science, Shandong University. Human HEK293T, HEK293, HeLa, RAW264.7 and THP-1 cells were purchased from American Type Culture Collection. THP-1 cells were cultured in RPMI-1640 supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. The rest of the cells were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin. PMs were harvested from mice 3 days after starch injection. PMs were cultured in DMEM supplemented with 10% FBS, 100 U/mL penicillin, and 100 μg/mL streptomycin.
Reagents and antibodies
Antibodies were purchased as follows: anti-USP3 (12490-1-AP) was from Proteintech; anti-ASC (67824 and 13833), anti-K48-Ub (8081), anti-IL-1β (12242) and p17 (85658), anti-IκB-α (4812), anti-p-IκB-α (2859), anti-p-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) (4377), anti-p-p38 MAPK (Thr180/Tyr182) (9215), anti-IKKβ (8943), p-IKKα (Ser176)/IKKβ (Ser177) (2078), anti-p-MAPK/JNK (9252), anti-p-RELA/p65 (3033), anti-p-MAPK/p38 (4511), anti-p-MAPK/ERK (4695), anti-MAPK/JNK (9252), anti-RELA/p65 (8242), anti-MAPK/p38 (8690), anti-MAPK1/ERK2-MAPK3/ERK1 (4370), anti-K63-Ub (12930) were from Cell Signaling Technology; mouse anti-c-Myc (TA150121-1) was from OriGene; rabbit anti-c-Myc (A190-105A) was from Bethl Laboratories; mouse anti-Flag (M20008) was from Abmart; rabbit anti-HA was from Rockland Immunochemicals; mouse anti-Ub (P4D1) (sc-8017) and Protein A/G PLUS-Agarose were from Santa Cruz Biotechnology; and anti-caspase-1 (AG-20B-0042-C100) and anti-NLRP3 (AG-20B-0014-C100) were from AdipoGen. Alexa Fluor 568 (A11004) goat anti-mouse and Alexa Fluor 488 (A11034) goat anti-rabbit secondary antibodies were purchased from Thermo Fisher Scientific. Horseradish peroxidase-conjugated secondary antibodies and antibodies against β-actin were purchased from Beijing Zhongshan Jin-qiao Biotechnology. LPS, ATP, CHX, 3-MA, MG132, and chloroquine were purchased from Sigma‒Aldrich and were used at final concentrations of 200 ng/mL, 2 mM, 100 μg/mL, 1 mM, 10 μM, and 10 μM, respectively. ImjectTM Alum Adjuvant was obtained from Thermo Fisher Scientific and was used at a final concentration of 200 μg/mL. Nigericin, flagellin and poly (dA: dT) were purchased from Invitrogen and were used at final concentrations of 10 μM, 200 ng/mL, and 200 ng/mL, respectively. Poly (dA: dT) and flagellin were transfected into PMs. C3 toxin was purchased from Cytoskeleton and used at a final concentration of 1 μg/mL. Pierce BS3 (Thermo Scientific) was purchased from Thermo Scientific and used at a final concentration of 3 mM. PMA was purchased from Sigma‒Aldrich and used at a final concentration of 100 ng/mL. Flag-peptide was purchased from Sigma‒Aldrich.
RNA purification and qRT‒PCR
Total RNA was extracted from cells by using the RNA Fast 200 Kit according to the manufacturer’s instructions (Fastagen, Shanghai, China). RNA (500 ng) was reverse transcribed using reverse transcriptase (Takara Bio). The reverse-transcription products of each sample were amplified by the CFX-Connect Detection System (Bio-Rad Laboratories) using SYBR Green PCR Master Mix (Roche Diagnostics) according to the manufacturer’s instructions. Data were quantitatively normalized to the expression of β-actin in each sample. The primer sequences are shown in Supplementary Table S1.
Enzyme-linked immunosorbent assay (ELISA)
The concentrations of TNF-α, IL-6, and IL-1β were measured by using ELISA kits (Dakewe Biotech, Shenzhen, China) according to the manufacturer’s instructions.
Coimmunoprecipitation assay
For the coimmunoprecipitation (Co-IP) assay, cells were collected after transfection for 36 h and lysed in lysis buffer (1% [v/v] NP-40, 50 mM Tris-HCl [pH 7.4], 50 mM EDTA, 0.15 M NaCl) with a protease inhibitor mixture (Sigma‒Aldrich). After centrifugation for 15 min at 15000 × g at 4 °C, the supernatants were collected and incubated with the indicated antibodies followed by the addition of protein A/G beads (Santa Cruz). After incubation overnight at 4 °C, the beads were washed five times with lysis buffer. Immunoprecipitates were eluted by boiling with 2 × SDS loading buffer.
Immunoblot analysis
For immunoblot analysis, cells or tissues were lysed with lysis buffer supplemented with protease inhibitor mixture. Total cell lysates or immunoprecipitates were separated by SDS‒PAGE, transferred onto a nitrocellulose membrane (Millipore), blocked with 5% (w/v) bovine serum albumin (BSA), probed with the indicated antibodies, and visualized by ECL western blotting detection reagent. Images were taken with SageCapture and processed with Adobe Photoshop and Adobe Illustrator.
In vitro deubiquitination assay
HEK293T cells were transfected with HA-Ub and Flag-ASC plasmids. After transfection for 36 h, the cells were lysed with lysis buffer. Anti-Flag M2-conjugated beads (Sigma‒Aldrich) were applied to immunoprecipitate Flag-ASC, in which ASC was ubiquitinated. Ubiquitinated ASC was then eluted with the Flag peptide (Sigma‒Aldrich). Recombinant Flag-USP3 was purified in the same way as ASC. Then, ubiquitinated Flag-ASC was incubated with recombinant Flag-USP3 in deubiquitylation assay buffer (50 mM Tris-HCL [pH 7.5], 5 mM DTT, and 50 mM NaCl) at 37 °C for 1 h, followed by immunoprecipitation and immunoblot analysis.
Confocal microscopy
HeLa, HEK293 or HEK293T cells were grown on 12-well slides and transfected with the indicated plasmids. PMs were grown on 12-well slides and stimulated with LPS plus ATP treatment. The cells were then fixed in 4% paraformaldehyde, permeabilized with 0.2% Triton X-100, and blocked with PBS containing 1% BSA. The fixation, permeabilization, and blocking buffers were all purchased from Beyotime Biotechnology. The cells were then reacted with the indicated primary antibodies at 4 °C overnight, rinsed, and reacted with the corresponding secondary antibodies (Invitrogen). Nuclei were counterstained with DAPI (Abcam). Images were taken with a Zeiss LSM780 confocal microscope and processed with Zeiss Zen software.
hUSP3 overexpression lentivirus
The lentivirus was produced by transient transfection of the pLVX-IRES-Puro- hUSP3 construct or control vector into HEK293T cells using Lipofectamine 2000 (Thermo Fisher Scientific) with psPAX2 and pMD2.G.
In vivo transfection
Flag-mUSP3 was cloned into pEnCMV and injected into the tail veins of mice (40 μg/mouse) in the presence of in vivo DNA transfection reagent (Entranster-in vivo; Engreen).
In vivo Alum-induced peritonitis
WT- or USP3-overexpressing mice (6-7 weeks old) were injected intraperitoneally with Alum (700 μg) for 12 h. The peritoneal lavage fluid was collected and concentrated for ELISA analysis, and the PECs were analyzed by flow cytometry as described previously [12].
F. novicida infection of mice
The F. novicida strain U112 was provided by Dr. Xiaopeng Qi (Shandong University). WT- or USP3-overexpressing mice (8-10 weeks old) were infected subcutaneously with F. novicida U112 (1.5 × 105 CFU per mouse) and were euthanized on Day 2 after infection. Then, the livers and spleens were harvested to determine the bacterial burden. The sera were collected for ELISA analysis.
In vivo flagellin-induced pneumonia
WT- or USP3-overexpressing mice (males, 8 weeks old) were intranasally instilled with a mixture of flagellin (1 μg per mouse) and in vivo transfection reagent (Entranster-in vivo; Engreen) in a total volume of 50 μL for 12 h to induce pneumonia or with isotonic saline for 12 h as a negative control. BALF was obtained by lavaging the lung with 3 mL PBS and concentrated for ELISA analysis. The BALCs were collected and analyzed by flow cytometry.
Flow cytometry
Suspensions of PECs and BALCs were prepared and stained with antibodies against surface markers. The markers 7AAD (BioLegend, 559925), LY6G (BioLegend, 127605) and ITGAM/CD11b (BioLegend, 17-0012-81) were used for analysis of the recruitment of neutrophils. 7AAD, LY6C (BioLegend, 128007) and ITGAM were used for the analysis of monocytes. Data were collected by an LSRFortessa flow cytometer (BD Biosciences) and analyzed with FlowJo software.
Statistical analysis and immunoblot quantification
All the data were obtained from at least three independent experiments with comparable results. The data are presented as the mean + S.D. Statistical analysis was conducted using the two-tailed Student’s t test, and statistical significance is indicated as follows: *p < 0.05, **p < 0.01, ***p < 0.001. The density of the immunoblot bands was obtained using ImageJ (Rawak Software, Inc.) in the linear range.
References
Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20.
Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19:477–89.
Schroder K, Tschopp J. The inflammasomes. Cell. 2010;140:821–32.
Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87.
Milner MT, Maddugoda M, Gotz J, Burgener SS, Schroder K. The NLRP3 inflammasome triggers sterile neuroinflammation and Alzheimer’s disease. Curr Opin Immunol. 2021;68:116–24.
Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35.
Ge Q, Chen X, Zhao Y, Mu H, Zhang J. Modulatory mechanisms of NLRP3: Potential roles in inflammasome activation. Life Sci. 2021;267:118918.
Lopez-Castejon G. Control of the inflammasome by the ubiquitin system. Febs j. 2020;287:11–26.
Shim DW, Lee KH. Posttranslational Regulation of the NLR Family Pyrin Domain-Containing 3 Inflammasome. Front Immunol. 2018;9:1054.
Popovic D, Vucic D, Dikic I. Ubiquitination in disease pathogenesis and treatment. Nat Med. 2014;20:1242–53.
Humphries F, Bergin R, Jackson R, Delagic N, Wang B, Yang S, et al. The E3 ubiquitin ligase Pellino2 mediates priming of the NLRP3 inflammasome. Nat Commun. 2018;9:1560.
Song H, Liu B, Huai W, Yu Z, Wang W, Zhao J, et al. The E3 ubiquitin ligase TRIM31 attenuates NLRP3 inflammasome activation by promoting proteasomal degradation of NLRP3. Nat Commun. 2016;7:13727.
Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J. Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell. 2013;49:331–8.
Palazon-Riquelme P, Worboys JD, Green J, Valera A, Martin-Sanchez F, Pellegrini C, et al. USP7 and USP47 deubiquitinases regulate NLRP3 inflammasome activation. EMBO Rep. 2018;19:e44766.
Hoss F, Rodriguez-Alcazar JF, Latz E. Assembly and regulation of ASC specks. Cell Mol Life Sci. 2017;74:1211–29.
Dick MS, Sborgi L, Ruhl S, Hiller S, Broz P. ASC filament formation serves as a signal amplification mechanism for inflammasomes. Nat Commun. 2016;7:11929.
Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q, et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med. 2014;211:1333–47.
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA, et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol. 2012;13:255–63.
Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y, et al. MAVS Promotes Inflammasome Activation by Targeting ASC for K63-Linked Ubiquitination via the E3 Ligase TRAF3. J Immunol. 2015;194:4880–90.
Siu KL, Yuen KS, Castaño-Rodriguez C, Ye ZW, Yeung ML, Fung SY, et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. Faseb j. 2019;33:8865–77.
Zhang L, Ko CJ, Li Y, Jie Z, Zhu L, Zhou X, et al. Peli1 facilitates NLRP3 inflammasome activation by mediating ASC ubiquitination. Cell Rep. 2021;37:109904.
Lee JY, Seo D, You J, Chung S, Park JS, Lee JH, et al. The deubiquitinating enzyme, ubiquitin-specific peptidase 50, regulates inflammasome activation by targeting the ASC adaptor protein. FEBS Lett. 2017;591:479–90.
Cheng YC, Shieh SY. Deubiquitinating enzyme USP3 controls CHK1 chromatin association and activation. Proc Natl Acad Sci USA. 2018;115:5546–51.
Grice GL, Nathan JA. The recognition of ubiquitinated proteins by the proteasome. Cell Mol Life Sci. 2016;73:3497–506.
Cai X, Xu H, Chen ZJ. Prion-Like Polymerization in Immunity and Inflammation. Cold Spring Harb Perspect Biol. 2017;9:a023580.
Pelegrin P, Barroso-Gutierrez C, Surprenant A. P2X7 receptor differentially couples to distinct release pathways for IL-1beta in mouse macrophage. J Immunol. 2008;180:7147–57.
Eisenbarth SC, Colegio OR, O’Connor W, Sutterwala FS, Flavell RA. Crucial role for the Nalp3 inflammasome in the immunostimulatory properties of aluminium adjuvants. Nature. 2008;453:1122–6.
Sokolovska A, Hem SL, HogenEsch H. Activation of dendritic cells and induction of CD4(+) T cell differentiation by aluminum-containing adjuvants. Vaccine. 2007;25:4575–85.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Forster I, et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity. 2011;34:213–23.
Guo Y, Li L, Xu T, Guo X, Wang C, Li Y, et al. HUWE1 mediates inflammasome activation and promotes host defense against bacterial infection. J Clin Investig. 2020;130:6301–16.
Xue Y, Enosi Tuipulotu D, Tan WH, Kay C, Man SM. Emerging Activators and Regulators of Inflammasomes and Pyroptosis. Trends Immunol. 2019;40:1035–52.
Cui J, Song Y, Li Y, Zhu Q, Tan P, Qin Y, et al. USP3 inhibits type I interferon signaling by deubiquitinating RIG-I-like receptors. Cell Res. 2014;24:400–16.
Fu S, Shao S, Wang L, Liu H, Hou H, Wang Y, et al. USP3 stabilizes p53 protein through its deubiquitinase activity. Biochem Biophys Res Commun. 2017;492:178–83.
Nicassio F, Corrado N, Vissers JH, Areces LB, Bergink S, Marteijn JA, et al. Human USP3 is a chromatin modifier required for S phase progression and genome stability. Curr Biol. 2007;17:1972–7.
Shi K, Zhang JZ, Yang L, Li NN, Yue Y, Du XH, et al. Protein deubiquitylase USP3 stabilizes Aurora A to promote proliferation and metastasis of esophageal squamous cell carcinoma. BMC Cancer. 2021;21:1196.
Liu B, Zhang M, Chu H, Zhang H, Wu H, Song G, et al. The ubiquitin E3 ligase TRIM31 promotes aggregation and activation of the signaling adaptor MAVS through Lys63-linked polyubiquitination. Nat Immunol. 2017;18:214–24.
Acknowledgements
This work was supported by grants from the National Key Research and Development Program (2021YFC2300603), the National Natural Science Foundation of China (32000633, 31730026, 81930039), the Natural Science Foundation of Shandong Province (ZR2020QH136), the China Postdoctoral Science Foundation (2020M682187), and the Postdoctoral Innovation Project of Shandong Province (202002012). We thank Dr. Xiaopeng Qi (Shandong University) for the F. novicida strain U112.
Author information
Authors and Affiliations
Contributions
CG conceived the research; CG and FL designed the experiments; WZ and FL performed the research; LZ, YZ, BL, CM, WZ, and SL, provided discussions; CG and FL analyzed the data; CG and FL wrote the paper.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Supplementary information
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Zhuang, W., Zhang, L., Zheng, Y. et al. USP3 deubiquitinates and stabilizes the adapter protein ASC to regulate inflammasome activation. Cell Mol Immunol 19, 1141–1152 (2022). https://doi.org/10.1038/s41423-022-00917-7
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41423-022-00917-7
- Springer Nature Limited
Keywords
This article is cited by
-
MiR-223 enhances lipophagy by suppressing CTSB in microglia following lysolecithin-induced demyelination in mice
Lipids in Health and Disease (2024)
-
Ubiquitin specific peptidase 3: an emerging deubiquitinase that regulates physiology and diseases
Cell Death Discovery (2024)
-
Elucidating Microglial Heterogeneity and Functions in Alzheimer’s Disease Using Single-cell Analysis and Convolutional Neural Network Disease Model Construction
Scientific Reports (2024)