
Abstract
Inflammasomes are multi-protein complexes that regulate the innate immune response by facilitating the release of inflammatory cytokines in response to pathogen exposure or cellular damage. Pro-inflammatory inflammasome signaling is vital to host defense and helps initiate the process of tissue repair following an insult to the host, but can be injurious, when excessive or chronic. As such, inflammasome activity is tightly regulated. Here we discuss one critical mechanism of inflammasome regulation, ubiquitination, that functions as a universal modulator of protein stability and trafficking. Recent studies have provided important insights into the regulation of inflammasome activation by protein ubiquitination. We review the molecular regulation of inflammasome function, specifically, as it relates to ubiquitination, and discuss the implications for the development of therapeutics to specifically target aberrant inflammasome signaling.
Similar content being viewed by others

INTRODUCTION
Inflammation is a non-specific immune response to harmful stimuli. The inflammatory process protects the host by eliminating the noxious agent, be it infectious or not, and by initiating tissue repair by clearing out damaged cells. Recognition of cellular infection by virulent pathogens or cell injury initiates the process of acute inflammation, which is characterized by release of cytokines/chemokines, increased blood flow, vasodilation, extravasation of fluid and cells, and recruitment, influx and activation of immune cells into the involved tissue. Under normal conditions, this process is self-limited, resolving after removal of the activating stimulus. In contrast, excessive or prolonged inflammation can be injurious, leading to simultaneous tissue destruction and repair. As such, regulation of the molecular mediators of the inflammatory process is vital for an appropriate response to host insults. Recently, insights into the molecular regulation of an important mediator of the inflammatory response, the inflammasome, have greatly increased. In this review, we highlight recent discoveries regarding molecular regulation of the inflammasome by ubiquitination and discuss the implications for possible therapeutic interventions targeting the inflammasome to combat injurious inflammatory signaling.
The inflammasome: a key regulator of early immune amplification
Inflammasomes are multimeric protein complexes assembled in the cell to drive the inflammatory process and initiate host defense in response to pathogens and cell injury. Inflammasomes assemble when resident immune cells detect generic molecular motifs from pathogens or cellular injury via their pattern recognition receptors (PRRs). PRRs recognize pathogen-associated molecular patterns (PAMPs) conserved among classes of pathogens or damage-associated molecular patterns (DAMPs) released upon cell injury or death. Inflammasome assembly is initiated by activation of nucleotide-binding oligomerization domain and leucine-rich repeat (LRR) receptors (NOD-like receptors or NLRs) or absent in melanoma 2 (AIM2)-like receptors (ALRs) in response to recognition of a PAMP or DAMP.1, 2 To date, 22 NLRs3 and 4 ALRs4 have been identified in humans. Of those, NLRP1,5 NLRP3,6 NLRC4,7 and AIM2 (refs 8, 9, 10) are known to form inflammasomes, and their role in inflammatory signaling is well established. Other NLRs can form an inflammasome (that is, NLRP7 (ref. 11)) or activate caspase-1 (that is, NLRP2, IFI16 and pyrin12), but are not as well characterized. With few exceptions, NLRs and AIM2 have not been shown to directly interact with their activators, suggesting that other proteins upstream of the inflammasome may function as PRRs that activate NLRs or AIM2, either directly or through an intermediary.2
PAMPs and DAMPs differentially activate the various NLRs and AIM2. The exact composition and regulation of an inflammasome is dependent upon the activated receptor. For example, double-stranded DNA activates AIM2 to assemble the AIM2 inflammasome.8, 9, 10 An adaptor protein ASC (apoptosis-associated speck-like protein containing a caspase activation and recruitment domain) links activated NLRs/AIM2 to the effector protein, caspase-1.13 Activated NLRs/AIM2 self-oligomerize, bridging multiple ternary inflammasome complexes. NLR/AIM2 oligomerization leads to ASC nucleation and polymerization, which subsequently nucleates caspase-1 polymerization resulting in its proximity-induced activation.14 Active caspase-1 facilitates the maturation and release of cytokines, as well as initiating a type of inflammatory cell death known as pyroptosis.1, 12 Although ASC polymerization is self-propagating, the process is greatly enhanced by NLRs/AIM2 oligomerization, suggesting that inflammasome activation is primarily regulated through the sensor proteins.14
Inflammasomes drive inflammation by the maturation and secretion of the potent pro-inflammatory cytokines interleukin (IL)-1β and IL-18.12 The majority of inflammatory cytokines are exclusively regulated by selective transcription and expressed as mature proteins. In contrast, IL-1β and IL-18 are produced as pro-proteins requiring cleavage at the inflammasome before their release and activity. A true multifunctional cytokine, IL-1β, affects nearly every cell type and organ system in the human body.15 Although inflammasome activation and secretion of these cytokines is vital for adequate immune activation in the setting of infection, excessive circulating IL-1β and IL-18 can cause unacceptable toxicity, manifesting as the systemic inflammatory response syndrome and/or acute respiratory distress syndrome and correlating with increased mortality in these settings.16 Furthermore, as inflammasomes also are activated in response to endogenous danger signals, dysregulated inflammasome function and resultant sterile inflammation are associated with autoimmune diseases, known collectively as the cryopyrin-associated periodic syndrome and is thought to contribute to metabolic diseases, including diabetes mellitus and obesity, as well as gout.17 The cytokine products of the inflammasome exhibit a narrow therapeutic window between clinical benefit for host defense or tissue repair and injurious inflammation caused by excessive or inappropriate inflammasome activation.
Ubiquitin
Restoration of homeostasis in response to noxious stimuli requires alterations in the abundance and function of cellular proteins. A major mechanism of this ‘proteostasis’ is the reversible post-translational modification, ubiquitination, that functions as a universal modulator of protein trafficking. Ubiquitin linkage occurs by its covalent bonding to a lysine residue within a protein substrate. Containing seven lysine residues itself, ubiquitin can attach to a substrate as a monomer, a linear chain or a branched chain. Ubiquitinated proteins are then directed to other cellular compartments or may be degraded at the proteasome or lysosome, depending on the extent and configuration of the bound ubiquitin.18 For example, ubiquitin chains linked at the lysine 48 (K48) residue often direct substrate proteins to the proteasome for degradation, and K63-linked chains can sort proteins to the lysosome. Recently, a linear mode of ubiquitination was demonstrated by covalent attachment of an incoming ubiquitin to the methionine (M1) residue of another ubiquitin.19 These patterns of ubiquitin linkages to substrates can modulate the destiny and function of proteins. Substrate ubiquitination occurs by activity of three distinct enzymes. First, the ubiquitin protein is added to a ubiquitin activating enzyme (E1) and then transferred to a ubiquitin-conjugating enzyme (E2); finally, a ubiquitin (E3) ligase covalently attaches ubiquitin to the target protein. E3 ligases, of which over 700 exist, recognize substrates specifically through recognition of a substrate motif, often consisting of another post-translational modification, such as phosphorylation or acetylation.20 There exist two major E3 ligase families, the HECT and Cullin-RING ligases, both of which contain conserved E2 association domains, but display significant variation in their substrate binding domain structures. Most of the E3 ligases characterized are multi-subunit complexes, such as the Skp1-Cullin-F-box (SCF) E3 ligases, comprising the ubiquitin-loaded E2 conjugation enzyme (Cullin), a linker protein (SKP1) and substrate recognition module termed an F-box protein (FBX), to mediate ubiquitination and degradation. Opposing the activity of the E3 ligases, the de-ubiquitinase enzymes (DUBs) remove ubiquitin chains.21
The dynamic balance between E3 ligases and DUBs regulates the abundance and activity of critical cellular proteins to mediate immune responses, among other cellular processes. Dysregulation of this balance is known to have a role in various disease processes.22 As one example of proteostatic inflammatory signaling, the interplay between two E3 ligase FBX O3 and FBXL2, results in an increase in the pro-inflammatory tumor necrosis factor (TNF) receptor-associated factors (TRAFs) in response to bacterial endotoxin. In this model, FBXL2 constitutively ubiquitinates TRAFs for degradation. Under inflammatory stimulation, FBXO3 increases in abundance and ubiquitinates FBXL2 for degradation with resultant accumulation of TRAFs.23 As another example, the quintessential pro-inflammatory transcription factor NF-κB (nuclear factor of κ-light polypeptide gene enhancer in B cells) induces expression of many important immune mediators including cytokines, chemokines, adhesion molecules, matrix metalloproteases and leukocyte growth factors.24 Before this activity, inhibitor of κB (IκB) binds to NF-κB, thereby preventing its nuclear translocation.25 With stimulation, IκB is phosphorylated, allowing for recognition and ubiquitination by the E3 ligase component FBW1. Ub-IκB is degraded at the proteasome, liberating NF-κB to translocate to the nucleus.26 This review will discuss how the inflammasome is similarly regulated by ubiquitination through the activity of both E3 ligases and DUBs.
UBIQUITIN IN REGULATION OF INFLAMMASOME ACTIVITY
NLRP3: general overview
In contrast to most innate immune receptors,27 NLRP3 (NALP3, CIAS1, cryopyrin and PYPAF1) is activated by a wide variety of stimuli including bacterial RNA, bacterial RNA/DNA hybrids, proteins from Gram-positive and Gram-negative bacteria, RNA and DNA viruses, fungi, protozoa, monosodium urate crystals, calcium pyrophosphate dihydrate crystals, alum, ATP and nigericin.2, 28 These ligands have not been shown to interact directly with NLRP3. With such a diversity of activating stimuli, it is more likely that NLRP3 is activated downstream of multiple PRRs converging on a common mechanism.2 One example is sensing of dsRNA by DHX33. With dsRNA exposure, an E3 ligase, TRIM33, ubiquitinates DHX33. Ub-DHX33 then binds NLRP3 with resultant NLRP3 activation.29 Activated NLRP3 forms a complex with ASC and caspase-1, thereby forming the classically described inflammasome. In macrophages and dendritic cells, two signals are required for NLRP3 inflammasome activation. The first is mediated by TLRs, NOD2, TNFR1 or TNFR2. The second signal is provided by a pore-forming toxin or by activation of purinergic receptors. Monocytes require only the first signal.30
Despite considerable study, there is no consensus on the mechanism of NLRP3 inflammasome priming and activation. The two-step model of inflammasome activation is well established, but mechanistic studies remain challenging as they can be difficult to separate priming and activation experimentally. The activated inflammasome is easily identifiable by cytokine release or by polymerization of ASC and caspase-1, all being measurable readouts. In contrast, it is more difficult to identify the ‘primed’ inflammasome, best defined as an inflammasome licensed for activation by a second signal. Study has focused on identifying those events and signaling pathways that are required for priming and subsequent activation, and results have been conflicting. Multiple reports agree that signaling through transcriptionally active receptors such as TLRs, NOD2, TNFR1 and TNFR2 is necessary for NLRP3 activation.31, 32, 33 However, it remains unclear whether their transcriptional activity is important or whether some other mediator is driving the process.
An earlier model of NLRP3 inflammasome priming suggested that the process is transcriptionally regulated. Activation of NF-κB through TLRs or NOD2 increases the transcription of NLRP3 and pro-IL-1β. A critical mass of NLRP3 is necessary before inflammasome activation, and upregulation of NLRP3 is sufficient for priming-independent activation by a single signal.31 Indeed, upregulation of NLRP3 expression does seem to lower the threshold for activation as well as increase the amplitude of caspase-1 cleavage.32 Hours after TLR stimulation, pro-IL-1β levels peak, providing substrate for the enzymatic activity of the inflammasome.34 It is important to separate IL-1β release from inflammasome activation, as the two events are not necessarily related temporally, owing to the delay in availability of pro-IL-1β. In contrast, pro-IL-18 is constitutively expressed and available for inflammasome processing. Release of IL-18 can occur within minutes of appropriate stimulation,34 illustrating that inflammasome activation may occur well before any pro-IL-1β is available for maturation. In lieu of being necessary for priming,35 transcriptional upregulation of NLRP3 and pro-IL-1β may serve to amplify inflammasome activity.
NLRP3: ubiquitination
Numerous studies have demonstrated that NLRP3 inflammasome priming is independent of new protein synthesis; these studies suggest an important role for ubiquitin in NLRP3 inflammasome regulation. Inhibition of transcription or translation fails to prevent inflammasome priming and activation.32, 33, 34 Also, release of IL-18 within minutes of stimulation, faster than the observed increases in NLRP3 protein abundance, supports post-translational modification as the activating mechanism.34 Treatment of macrophages with bacterial endotoxin increases the ubiquitination of the NLRP3 inflammasome complex, shown to be modified by K48, K63, linear and unanchored polyubiquitin.36 Chemical inhibition of the proteasome, a well-known destination for many ubiquitinated substrates, inhibits the release of both IL-1β37 and IL-18.34 However, the ubiquitination and protein abundance of NLRP3 protein is unaffected by inhibition of the proteasome or autophagy.38 The effect on IL-1β release is most likely due to decreased synthesis of the pro-protein, as proteasomal inhibition is known to abrogate NF-κB-dependent transcription. Although the relationship between IL-18 and the proteasome is less clear, it may also be mediated by IκBα, a known inhibitor of NF-κB, thus suggesting a role for ubiquitination in the regulation of the NLRP3 inflammasome. Additional work suggests a role for ubiquitin in the regulation of inflammasomes because treatment of cells with inhibitors of DUB enzymes decreases caspase activation and IL-1β secretion in response to ligands for the NLRP3 (refs 32, 39) and AIM2 inflammasomes.39
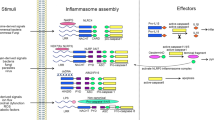
Ubiquitination of the NLRP3 receptor critically regulates its protein stability and inflammasome function (Figure 1). Although the NLRP3 inflammasome complex shows increased ubiquitination when stimulated, multiple reports agree that the NLRP3 protein itself is deubiquitinated in response to pro-inflammatory stimuli,32, 40 providing a mechanism for the increased NLRP3 protein abundance and perhaps licensing NLRP3 for further post-translational modifications involved in inflammasome activation. Studies demonstrate that NLRP3 is both K48- and K63 ubiquitinated,38 suggesting that regulation may be more complex. Recently, NLRP3 was shown to be constitutively ubiquitinated by the SCF complex subunit F-box L2 (FBXL2) for degradation at the proteasome. Exposure to bacterial endotoxin decreases the interaction between FBXL2 and NLRP3, thus decreasing its ubiquitination and preserving the substrate for inflammasome activity.40 In a separate study, the neurotransmitter dopamine functions as an anti-inflammatory stimulus and activates the E3 ligase, membrane-associated ring finger (C3HC4) 7 (MARCH7) to ubiquitinate NLRP3, targeting it for degradation by autophagy.41 Yet another study demonstrates that the DUB enzyme BRCA1/BRCA2-containing complex, subunit 3 (BRCC3), is required for NLRP3 inflammasome activity.38 Enteropathogenic and enterohemorrhagic Escherichia coli secrete a protein NleA that inhibits deubiquitination of NLRP3 to limit inflammasome activation.42 The strategy used by enteropathogenic and enterohemorrhagic Escherichia coli to hijack the ubiquitin machinery for immune evasion highlights the importance of this deubiquitination event to NLRP3 inflammasome function. These studies detail the multiple ubiquitin modifying enzymes involved in regulation of NLRP3 stability and protein abundance. Although different signals may activate different E3 ligases, ubiquitination may serve as a final common pathway for NLRP3 activation and subsequent inflammasome priming and activity.

NLRP3 protein stability is regulated by the activity of multiple E3 ligases and DUBs. NLRP3 comprises three domains: an LRR domain, an NBD and a pyrin domain. E3 ligases, SCF-FBXL2 and MARCH7, ubiquitinate the LRR domain of NLRP3, which provides a signal for its degradation. SCF-FBXL2 constitutively ubiquitinates NLRP3 for degradation; this process is decreased in the presence of bacterial endotoxin. MARCH7 is stimulated by DA to increase ubiquitination of NLRP3. As oligomerization of activated NLRP3 enhances inflammasome formation, lowering NLRP3 protein abundance has an overall anti-inflammatory effect. A DUB, BRCC3, deubiquitinates the LRR domain of NLRP3, which is necessary for NLRP3 activation. Ubiquitination can serve as an activating modification in NLRP3. An unknown Cullin ring ligase ubiquitinates the NBD of NLRP3 before its activation and function in the inflammasome complex. DA, dopamine; DUBs, de-ubiquitinase enzymes; LRR, leucine-rich repeat; NBD, nucleotide-binding domain; NLR, NOD-like receptor.
As ubiquitination of NLRP3 is complex, inflammasome activation may involve multiple ubiquitination events. NLRP3 is modified by both K48- and K63 polyubiquitin, and covalent attachment sites have been identified in both the domains of LRR and nucleotide-binding domain (NBD). Deubiquitination of NLRP3 at certain lysine residues may preserve the protein to perpetuate inflammasome signaling. However, for other inflammasome components, such as NLRC4 and ASC, ubiquitination provides a non-degradative signal necessary for activation. The NLRP3 inflammasome may similarly require a non-degradative ubiquitination signal for activation. A ubiquitin modifying enzyme, A20 or TNFAIP3 (TNF alpha-induced protein 3), is a negative regulator of NLRP3 protein, pro-IL-1β protein and IL-1β secretion.43, 44 A20 deficiency leads to an increase in IL-1β complex ubiquitination, exaggerated IL-1β secretion as well as NLRP3 inflammasome activation in response to only a single signal in macrophages, which usually require two signals for activation.36 These findings suggest that A20 regulates a ubiquitin signal within the NLRP3 inflammasome complex required for activation. Notably, the direct effects on NLRP3 protein are likely mediated through A20 interactions with NF-κB.44 In macrophages, both NLRP3 and NLRC4 inflammasome activity is attenuated by a Cullin ring ubiquitin ligase inhibitor termed glomulin (GLMN). Shigella species target GLMN by translocation of an E3 ligase (invasion plasmid antigen H7.8) into host cells; IpaH7.8 ubiquitinates GLMN, resulting in its degradation at the proteasome and relieving its inhibition of an unidentified Cullin ring ligase.45 This study provides indirect evidence that activity of a Cullin ring ligase may be important for NLRP3 activation, and proposes that ubiquitination provides a non-degradative, activating stimulus for the NLRP3 inflammasome.
Other NLRs
In comparison with the NLRP3 inflammasome, study of other NLRs and their inflammasomes is limited and less is known about their regulation. As NLRs share common domains, such as the LRR and NBD, some common themes in their mechanisms of action and regulation would be expected. Ubiquitination has been suggested to regulate NLRC4 inflammasome activation and caspase-8-mediated cell death. NLRC4 ubiquitination is not only required for inflammasome activation but it also serves as a signal for degradation at the proteasome.46 Further study is needed to address regulation of other NLRs, which may provide mechanistic insight or uncover common pathways of inflammasome activity.
ASC
Many NLRs/ALRs including human NLRP1,5 NLRP3,6 NLRP7,11 NLRC4 (ref. 7) and AIM2 (refs 8, 9, 10) can assemble an inflammasome. In addition, NLRP1 and NLRC4 inflammasomes are capable of cytokine maturation and pyroptosis with or independent of ASC.2 For inflammasomes dependent on ASC, such as NLRP3 or AIM2, ASC polymerization enhances the nucleation and polymerization of caspase-1.14 Regulation of ASC governs the activity of the ASC-dependent inflammasomes and likely impacts the NLRP1 and NLRC4 inflammasomes, which can assemble with ASC without true dependence for function. Similar to NLRP3, differential ubiquitination of ASC regulates its activity and stability (Figure 2).

Differential ubiquitination of ASC determines inflammasome activation or ASC degradation. The NLRP3, NLRP7 and AIM2 inflammasomes are dependent on ASC for inflammasome formation and function. In addition, NLRP1 and NLRC4 form inflammasomes with ASC, but can function independently of ASC. Containing a PYRIN and CARD domain, ASC is differentially ubiquitinated by either K63 ubiquitin chain or linear ubiquitin. The linear ubiquitin chain is covalently attached through the activity of LUBAC and is specifically dependent on HOIL-1L, a component of LUBAC. Linear ubiquitination provides an activating signal, and HOIL-1L activity is necessary for inflammasome activation. K63-linked ubiquitination provides both a degradative and an activating signal. The E3 ligase TRAF3 directly ubiquitinates ASC at Lys174; this modification is required for inflammasome function while concurrently tagging ASC for recruitment to the autophagosome. AIM2, absent in melanoma 2; ASC, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain; LUBAC, linear ubiquitin chain assembly complex; NLR, NOD-like receptor; TRAF, tumor necrosis factor receptor-associated factor.
Linear ubiquitination of ASC by the linear ubiquitin chain assembly complex (LUBAC) has been described to provide an activating signal for certain ASC-dependent inflammasomes. The multi-protein LUBAC consists of three proteins, HOIL-1, HOIP and SHARPIN. Both HOIL-1 (ref. 47) and SHARPIN48 are necessary for NF-κB activation following stimulation of TLRs. In addition to regulating the transcriptional activity of TLRs, HOIL-1 (ref. 49) and SHARPIN50 participate in inflammasome activation. Deficiency of these LUBAC components results in decreased activation of caspase-1 and IL-1β secretion. LUBAC interacts directly with the inflammasome through ASC, catalyzing the linear ubiquitination of ASC, but not NLRP3 or AIM2, through the E3 ligase activity of HOIL-1 and HOIP.49 SHARPIN inhibits inflammasome activation without effects on NLRP3 transcription, suggesting that it likely mediates inflammasome activity through ubiquitination.50 However, a direct inflammasome substrate for SHARPIN is yet to be identified.
K63 ubiquitination can provide both a degradative and an activating signal for ASC, and the ASC-dependent inflammasomes. In macrophages, autophagosome induction occurs concurrently with and in response to the same stimuli as activation of the NLRP3 and AIM2 inflammasomes. Increased activation of autophagy results in decreased inflammasome activity, and blockade of autophagy increases inflammasome signaling. ASC displays increased K63-linked ubiquitination and autophagosome recruitment with activation by bacterial endotoxin, an NLRP3 stimulus, or double-stranded DNA, an AIM2 inflammasome stimulus.51 In response to viral RNAs or vesicular stomatitis virus infection, the mitochondrial antiviral signaling protein engages ASC and TRAF3, an E3 ligase. Through TRAF3 activity, ASC is K63 poly-ubiquitinated at Lys174. K63-linked ubiquitination of ASC is necessary for inflammasome activation and IL-1β release,52 while also tagging ASC for autophagosome recruitment.51 Activation of the inflammasome and autophagy by the same signal provides a possible mechanism by which inflammasome function is self-limiting, highlighting the importance of regulating its potent pro-inflammatory potential.
IL-1β
Unlike the constitutively expressed pro-IL-18, transcriptional upregulation of pro-IL-1β is necessary before its maturation and release. Levels of pro-IL-1β increase over the course of hours following TLR ligation. In the absence of an inflammasome activating signal (the second signal), pro-IL-1β is ubiquitinated and degraded at the proteasome with resultant decrease in its abundance. Secreted mature IL-1β is not ubiquitinated, suggesting that deubiquitination occurs just before or during processing by the inflammasome.53 One high-risk strain of human papillomavirus (HPV16) exploits this biology to evade the innate immune response. HPV16 harboring the E6 oncogene encodes the ubiquitin ligase E6-AP, which increases the ubiquitination and proteasomal degradation of pro-IL-1β.54 This exemplifies a possible therapeutic strategy to reduce IL-1β signaling by targeting ubiquitin.
TARGETING UBIQUITINATION TO INHIBIT THE INFLAMMASOME
The functional inflammasome is a potent mediator of inflammatory signaling, making it an attractive therapeutic target. Ongoing efforts to develop an anti-inflammasome therapeutic have resulted in numerous publications proposing a wide variety of compounds.55 A number of these inhibitors are repurposed medications, such as glyburide,56, 57 nucleoside reverse transcriptase inhibitors,58 parthenolide59 and Bay 11-7082.59 Other compounds, such as β-hydroxybutyrate,60 type 1 interferon61 and MCC950,62 have been shown to inhibit inflammasome activation, but their mechanism of action remains unclear.
Despite great advancements in our understanding of inflammasome biology, rationally designed, specific inhibitors of the inflammasome are lacking. Multiple studies have established E3 ligases and DUBs as viable targets for novel anti-inflammatory proteins and small molecules. This review has highlighted a number of E3 ligases and a DUB that critically regulate the protein constituents of the inflammasome, and inflammasome activation relies on adequate protein abundance of NLRs and ASC. Destabilizing inflammasome constituents by targeting ubiquitination is therefore a rational approach in inhibiting the inflammasome. One study has validated this approach. The small-molecule inhibitor BC-1215 was developed to inhibit the pro-inflammatory E3 ligase subunit, FBXO3, which ubiquitinates and degrades another E3 ligase component, FBXL2. NLRP3 is ubiquitinated by FBXL2 and degraded at the proteasome. Treatment with BC-1215 inhibits inflammasome activity by decreasing NLRP3 abundance.40 Analogs of BC-1215 also inhibit FBXO3 and have been shown to inhibit IL-1β secretion and tissue damage in sepsis mouse models including cecal puncture and ligation, bacterial-induced pneumonia and H1N1 infection.23 Although BC-1215 also inhibits TRAFs and is not specific to the inflammasome, these studies serves as a proof of concept for therapeutic targeting of the inflammasome through inhibition of the ubiquitin machinery.
CONCLUSIONS
The pro-inflammatory cytokines IL-1β and IL-18 are important amplifiers of the inflammatory process and contribute to activation of both the innate and adaptive immune response. These cytokines are both multifunctional and potent, which likewise makes them potentially dangerous. Inflammasomes facilitate maturation of pro-IL-1β and pro-IL-18, acting as an additional barrier for release. Mechanisms of NLR and inflammasome activation remain controversial, and many more discoveries await us to provide clarity. Nonetheless, mounting evidence suggests that post-translational modification by ubiquitin has important roles in the activation and regulation of the inflammasomes. Pathogens target the ubiquitin machinery to promote their survival and propagation in the host. Chemical inhibition of both E3 ligases and DUBs modulate inflammasome activity and cytokine secretion, validating ubiquitin enzymes as viable therapeutic targets to modify inflammasome signaling. Efforts to develop inflammasome-specific therapeutics are ongoing, yet it is difficult to prognosticate whether ubiquitin-targeting of the inflammasome will prove to be a fruitful strategy in combating injurious inflammatory signaling. Regardless, further study of ubiquitin and the inflammasome will help us to elucidate the complex biology of this vital orchestrator of inflammation.
References
Schroder K, Tschopp J . The inflammasomes. Cell 2010; 140: 821–832.
Man SM, Kanneganti TD . Regulation of inflammasome activation. Immunol Rev 2015; 265: 6–21.
Harton JA, Linhoff MW, Zhang J, Ting JP . Cutting edge: CATERPILLER: a large family of mammalian genes containing CARD, pyrin, nucleotide-binding, and leucine-rich repeat domains. J Immunol 2002; 169: 4088–4093.
Cridland JA, Curley EZ, Wykes MN, Schroder K, Sweet MJ, Roberts TL et al. The mammalian PYHIN gene family: phylogeny, evolution and expression. BMC Evol Biol 2012; 12: 140.
Martinon F, Burns K, Tschopp J . The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell 2002; 10: 417–426.
Mariathasan S, Weiss DS, Newton K, McBride J, O'Rourke K, Roose-Girma M et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006; 440: 228–232.
Poyet JL, Srinivasula SM, Tnani M, Razmara M, Fernandes-Alnemri T, Alnemri ES . Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J Biol Chem 2001; 276: 28309–28313.
Fernandes-Alnemri T, Yu JW, Datta P, Wu J, Alnemri ES . AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009; 458: 509–513.
Hornung V, Ablasser A, Charrel-Dennis M, Bauernfeind F, Horvath G, Caffrey DR et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 2009; 458: 514–518.
Burckstummer T, Baumann C, Blüml S, Dixit E, Dürnberger G, Jahn H et al. An orthogonal proteomic-genomic screen identifies AIM2 as a cytoplasmic DNA sensor for the inflammasome. Nat Immunol 2009; 10: 266–272.
Khare S, Dorfleutner A, Bryan NB, Yun C, Radian AD, de Almeida L et al. An NLRP7-containing inflammasome mediates recognition of microbial lipopeptides in human macrophages. Immunity 2012; 36: 464–476.
Lamkanfi M, Dixit VM . Mechanisms and functions of inflammasomes. Cell 2014; 157: 1013–1022.
Fernandes-Alnemri T, Wu J, Yu JW, Datta P, Miller B, Jankowski W et al. The pyroptosome: a supramolecular assembly of ASC dimers mediating inflammatory cell death via caspase-1 activation. Cell Death Differ 2007; 14: 1590–1604.
Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell 2014; 156: 1193–1206.
Dinarello CA . Biologic basis for interleukin-1 in disease. Blood 1996; 87: 2095–2147.
Meduri GU, Headley S, Kohler G, Stentz F, Tolley E, Umberger R et al. Persistent elevation of inflammatory cytokines predicts a poor outcome in ARDS. Plasma IL-1 beta and IL-6 levels are consistent and efficient predictors of outcome over time. Chest 1995; 107: 1062–1073.
Hoffman HM, Wanderer AA . Inflammasome and IL-1beta-mediated disorders. Curr Allergy Asthma Rep 2010; 10: 229–235.
Ciechanover A . Proteolysis: from the lysosome to ubiquitin and the proteasome. Nat Rev Mol Cell Biol 2005; 6: 79–87.
Sasaki K, Iwai K . Roles of linear ubiquitinylation, a crucial regulator of NF-kappaB and cell death, in the immune system. Immunol Rev 2015; 266: 175–189.
Glickman MH, Ciechanover A . The ubiquitin-proteasome proteolytic pathway: destruction for the sake of construction. Physiol Rev 2002; 82: 373–428.
Komander D, Clague MJ, Urbe S . Breaking the chains: structure and function of the deubiquitinases. Nat Rev Mol Cell Biol 2009; 10: 550–563.
Weathington NM, Sznajder JI, Mallampalli RK . The emerging role of the ubiquitin proteasome in pulmonary biology and disease. Am J Respir Crit Care Med 2013; 188: 530–537.
Chen BB, Coon TA, Glasser JR, McVerry BJ, Zhao J, Zhao Y et al. A combinatorial F box protein directed pathway controls TRAF adaptor stability to regulate inflammation. Nat Immunol 2013; 14: 470–479.
Gilmore TD . Introduction to NF-kappaB: players, pathways, perspectives. Oncogene 2006; 25: 6680–6684.
Jacobs MD, Harrison SC . Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998; 95: 749–758.
Tanaka K, Kawakami T, Tateishi K, Yashiroda H, Chiba T . Control of IkappaBalpha proteolysis by the ubiquitin-proteasome pathway. Biochimie 2001; 83: 351–356.
Akira S, Uematsu S, Takeuchi O . Pathogen recognition and innate immunity. Cell 2006; 124: 783–801.
Bryant C, Fitzgerald KA . Molecular mechanisms involved in inflammasome activation. Trends Cell Biol 2009; 19: 455–464.
Weng L, Mitoma H, Trichot C, Bao M, Liu Y, Zhang Z et al. The E3 ubiquitin ligase tripartite motif 33 is essential for cytosolic RNA-induced NLRP3 inflammasome activation. J Immunol 2014; 193: 3676–3682.
Netea MG, Nold-Petry CA, Nold MF, Joosten LA, Opitz B, van der Meer JH et al. Differential requirement for the activation of the inflammasome for processing and release of IL-1beta in monocytes and macrophages. Blood 2009; 113: 2324–2335.
Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol 2009; 183: 787–791.
Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES . Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem 2012; 287: 36617–36622.
Fernandes-Alnemri T, Kang S, Anderson C, Sagara J, Fitzgerald KA, Alnemri ES . Cutting edge: TLR signaling licenses IRAK1 for rapid activation of the NLRP3 inflammasome. J Immunol 2013; 191: 3995–3999.
Ghonime MG, Shamaa OR, Das S, Eldomany RA, Fernandes-Alnemri T, Alnemri ES et al. Inflammasome priming by lipopolysaccharide is dependent upon ERK signaling and proteasome function. J Immunol 2014; 192: 3881–3888.
Schroder K, Sagulenko V, Zamoshnikova A, Richards AA, Cridland JA, Irvine KM et al. Acute lipopolysaccharide priming boosts inflammasome activation independently of inflammasome sensor induction. Immunobiology 2012; 217: 1325–1329.
Duong BH, Onizawa M, Oses-Prieto JA, Advincula R, Burlingame A, Malynn BA et al. A20 restricts ubiquitination of pro-interleukin-1beta protein complexes and suppresses NLRP3 inflammasome activity. Immunity 2015; 42: 55–67.
Ortiz-Lazareno PC, Hernandez-Flores G, Dominguez-Rodriguez JR, Lerma-Diaz JM, Jave-Suarez LF, Aguilar-Lemarroy A et al. MG132 proteasome inhibitor modulates proinflammatory cytokines production and expression of their receptors in U937 cells: involvement of nuclear factor-kappaB and activator protein-1. Immunology 2008; 124: 534–541.
Py BF, Kim MS, Vakifahmetoglu-Norberg H, Yuan J . Deubiquitination of NLRP3 by BRCC3 critically regulates inflammasome activity. Mol Cell 2013; 49: 331–338.
Lopez-Castejon G, Luheshi NM, Compan V, High S, Whitehead RC, Flitsch S et al. Deubiquitinases regulate the activity of caspase-1 and interleukin-1beta secretion via assembly of the inflammasome. J Biol Chem 2013; 288: 2721–2733.
Han S, Lear TB, Jerome JA, Rajbhandari S, Snavely CA, Gulick DL et al. Lipopolysaccharide primes the NALP3 inflammasome by inhibiting its ubiquitination and degradation mediated by the SCFFBXL2 E3 ligase. J Biol Chem 2015; 290: 18124–18133.
Yan Y, Jiang W, Liu L, Wang X, Ding C, Tian Z et al. Dopamine controls systemic inflammation through inhibition of NLRP3 inflammasome. Cell 2015; 160: 62–73.
Yen H, Sugimoto N, Tobe T . Enteropathogenic Escherichia coli uses NleA to inhibit NLRP3 inflammasome activation. PLoS Pathog 2015; 11: e1005121.
Li M, Shi X, Qian T, Li J, Tian Z, Ni B et al. A20 overexpression alleviates pristine-induced lupus nephritis by inhibiting the NF-kappaB and NLRP3 inflammasome activation in macrophages of mice. Int J Clin Exp Med 2015; 8: 17430–17440.
Vande Walle L, Van Opdenbosch N, Jacques P, Fossoul A, Verheugen E, Vogel P et al. Negative regulation of the NLRP3 inflammasome by A20 protects against arthritis. Nature 2014; 512: 69–73.
Suzuki S, Mimuro H, Kim M, Ogawa M, Ashida H, Toyotome T et al. Shigella IpaH7.8 E3 ubiquitin ligase targets glomulin and activates inflammasomes to demolish macrophages. Proc Natl Acad Sci USA 2014; 111: E4254–E4263.
Kumar Y, Radha V, Swarup G . Interaction with Sug1 enables Ipaf ubiquitination leading to caspase 8 activation and cell death. Biochem J 2010; 427: 91–104.
Tokunaga F, Iwai K . [Involvement of LUBAC-mediated linear polyubiquitination of NEMO in NF-kappaB activation]. Tanpakushitsu Kakusan Koso 2009; 54: 635–642.
Tokunaga F, Nakagawa T, Nakahara M, Saeki Y, Taniguchi M, Sakata S et al. SHARPIN is a component of the NF-kappaB-activating linear ubiquitin chain assembly complex. Nature 2011; 471: 633–636.
Rodgers MA, Bowman JW, Fujita H, Orazio N, Shi M, Liang Q et al. The linear ubiquitin assembly complex (LUBAC) is essential for NLRP3 inflammasome activation. J Exp Med 2014; 211: 1333–1347.
Gurung P, Lamkanfi M, Kanneganti TD . Cutting edge: SHARPIN is required for optimal NLRP3 inflammasome activation. J Immunol 2015; 194: 2064–2067.
Shi CS, Shenderov K, Huang NN, Kabat J, Abu-Asab M, Fitzgerald KA et al. Activation of autophagy by inflammatory signals limits IL-1beta production by targeting ubiquitinated inflammasomes for destruction. Nat Immunol 2012; 13: 255–263.
Guan K, Wei C, Zheng Z, Song T, Wu F, Zhang Y et al. MAVS promotes inflammasome activation by targeting ASC for K63-linked ubiquitination via the E3 ligase TRAF3. J Immunol 2015; 194: 4880–4890.
Ainscough JS, Frank Gerberick G, Zahedi-Nejad M, Lopez-Castejon G, Brough D, Kimber I et al. Dendritic cell IL-1alpha and IL-1beta are polyubiquitinated and degraded by the proteasome. J Biol Chem 2014; 289: 35582–35592.
Niebler M, Qian X, Höfler D, Kogosov V, Kaewprag J, Kaufmann AM et al. Post-translational control of IL-1beta via the human papillomavirus type 16 E6 oncoprotein: a novel mechanism of innate immune escape mediated by the E3-ubiquitin ligase E6-AP and p53. PLoS Pathog 2013; 9: e1003536.
Guo H, Callaway JB, Ting JP . Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med 2015; 21: 677–687.
Lamkanfi M, Mueller JL, Vitari AC, Misaghi S, Fedorova A, Deshayes K et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 2009; 187: 61–70.
Masters SL, Dunne A, Subramanian SL, Hull RL, Tannahill GM, Sharp FA et al. Activation of the NLRP3 inflammasome by islet amyloid polypeptide provides a mechanism for enhanced IL-1beta in type 2 diabetes. Nat Immunol 2010; 11: 897–904.
Fowler BJ, Gelfand BD, Kim Y, Kerur N, Tarallo V, Hirano Y et al. Nucleoside reverse transcriptase inhibitors possess intrinsic anti-inflammatory activity. Science 2014; 346: 1000–1003.
Juliana C, Fernandes-Alnemri T, Wu J, Datta P, Solorzano L, Yu JW et al. Anti-inflammatory compounds parthenolide and Bay 11-7082 are direct inhibitors of the inflammasome. J Biol Chem 2010; 285: 9792–9802.
Youm YH, Nguyen KY, Grant RW, Goldberg EL, Bodogai M, Kim D et al. The ketone metabolite beta-hydroxybutyrate blocks NLRP3 inflammasome-mediated inflammatory disease. Nat Med 2015; 21: 263–269.
Guarda G, Braun M, Staehli F, Tardivel A, Mattmann C, Förster I et al. Type I interferon inhibits interleukin-1 production and inflammasome activation. Immunity 2011; 34: 213–223.
Coll RC, Robertson AA, Chae JJ, Higgins SC, Muñoz-Planillo R, Inserra MC et al. A small-molecule inhibitor of the NLRP3 inflammasome for the treatment of inflammatory diseases. Nat Med 2015; 21: 248–255.
Acknowledgements
This work was supported by the Flight Attendant Medical Research Institute.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Competing interests
The authors declare no conflict of interest.
Rights and permissions
About this article

Cite this article
Bednash, J., Mallampalli, R. Regulation of inflammasomes by ubiquitination. Cell Mol Immunol 13, 722–728 (2016). https://doi.org/10.1038/cmi.2016.15
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/cmi.2016.15
- Springer Nature Limited
Keywords
This article is cited by
-
Deubiquitinase USP16 induces gouty arthritis via Drp1-dependent mitochondrial fission and NLRP3 inflammasome activation
Arthritis Research & Therapy (2023)
-
E3 ubiquitin ligase SYVN1 is a key positive regulator for GSDMD-mediated pyroptosis
Cell Death & Disease (2022)
-
Inflammatory, regulatory, and autophagy co-expression modules and hub genes underlie the peripheral immune response to human intracerebral hemorrhage
Journal of Neuroinflammation (2019)
-
Important roles of C-terminal residues in degradation of capsid protein of classical swine fever virus
Virology Journal (2019)
-
Extracellular ADP facilitates monocyte recruitment in bacterial infection via ERK signaling
Cellular & Molecular Immunology (2018)