
Abstract
Background
The efficacy of FOLFIRI plus an antiangiogenesis biologic agent as 2nd line therapy for metastatic colorectal adenocarcinoma is limited. TAS-102 is a novel oral antimetabolite with a distinct mechanism of action from fluoropyrimidines. We evaluated the antitumour efficacy of TAS-102, irinotecan and bevacizumab in patients with pre-treated, advanced colorectal adenocarcinoma in a multicenter, phase II, single-arm study.
Methods
Patients with advanced colorectal adenocarcinoma who had progressed after oxaliplatin and fluoropyrimidine and were eligible for treatment with bevacizumab were treated with irinotecan, bevacizumab, and TAS-102 in 28-day cycles. The primary endpoint was progression-free survival (PFS).
Results
We enrolled 35 evaluable patients. The study was positive. The median PFS was 7.9 (90% CI 6.2–11.8) months (vs. 6 months in historical control, p = 0.018). The median overall survival was 16.5 (90% CI 9.8–17.5) months. Sixty-seven per cent of patients experienced grade 3 or higher treatment-related adverse events. The most common toxicities were hematological (neutropenia) and gastrointestinal (diarrhoea, nausea, and vomiting).
Conclusions
Irinotecan, TAS-102 and bevacizumab is an active 2nd line therapy for patients with metastatic colorectal adenocarcinoma. Neutropenia is common and can affect dose density/intensity mandating use of G-CSF. A randomized study versus standard-of-care therapy is warranted.
Clinical trial registration
ClinicalTrials.gov NCT04109924.
Similar content being viewed by others


Background
Colorectal adenocarcinoma (CRC) remains a major cause of cancer-related death in the United States [1, 2]. Despite improvements in systemic therapy, a deeper understanding of the molecular underpinnings of this disease, and personalized treatment options, the expected median overall survival (OS) for patients with metastatic disease after progression on 1st line therapy is only 11–13 months [3,4,5]. The combination of bevacizumab, irinotecan, and fluorouracil (5-FU) given as continuous infusion (FOLFIRI) remains one of the most used 2nd line treatments [6, 7]. Combinations of FOLFIRI and different antiangiogenic biologic (anti-VEGF: bevacizumab, aflibercept, ramucirumab) agents have produced a median progression-free survival (PFS) in the range of 6 months in the 2nd line setting [3,4,5]. While such regimens are clearly active, further gains are desperately needed.
TAS-102 consists of trifluorothymidine, a thymidine-based nucleoside analog, and the thymidine phosphorylase inhibitor tipiracil. Following uptake into cancer cells, trifluorothymidine is phosphorylated and incorporated into DNA, inhibits cell proliferation, and increases cell death [8]. Tipiracil increases trifluorothymidine exposure by inhibiting its metabolism by thymidine phosphorylase [9]. TAS-102 has a distinct mechanism of action from 5-FU and in preclinical models can overcome 5-FU resistance [10]. TAS-102 has demonstrated antitumour activity against CRC in vivo [10]. DNA incorporation is thought to be the major mechanism of action of TAS-102 resulting in G2 arrest in p53-proficient and -deficient cells [11, 12] and double strand DNA breaks [12, 13]; the base excision repair (BER) pathway and glycosylation responses also differ from that seen with 5-FU-mediated DNA damage [14]. In the phase III RECOURSE trial, TAS-102 was superior to placebo as treatment for patients with advanced, refractory CRC who had received prior oxaliplatin- and irinotecan-fluoropyrimidine treatment [15]. Subsequently, in the phase III SUNLIGHT study, the combination of TAS-102 and bevacizumab was found to be superior to TAS-102 alone in the same patient population [16].
The TASCO-1 study evaluated bevacizumab with TAS-102 or capecitabine in treatment-naïve CRC patients unfit for intensive chemotherapy [17]. The observed median PFS and OS in the TAS-102/bevacizumab arm was higher suggesting that TAS-102 may confer greater benefit than 5-FU. Additionally, there is additive effect when TAS-102 is combined with irinotecan in 5-FU sensitive and resistant cell lines [18]. Interestingly, induction of apoptosis is most efficient in vitro when the irinotecan metabolite SN-38 is administered 24 h prior to TAS-102 [19]. In a phase I study, TAS-102 in combination with irinotecan (180 mg/m2 every 14 days) and bevacizumab (5 mg/kg every 14 days) was safely dosed at 25 mg/m2 twice daily, days 1–5 every 14 days [20]. Notably, in the expansion cohort of this study, where 88% of the patients had received prior irinotecan, 3 (13%) had partial responses (PR), and two in previously irinotecan-exposed patients. The median PFS was 7.9 months, comparing favorably to FOLFIRI plus an anti-VEGF agent in the 2nd line setting [3,4,5].
We hypothesized that the combination of TAS-102, irinotecan, and bevacizumab is more active than the commonly used FOLFIRI plus bevacizumab in patients with advanced CRC who have progressed on 1st line therapy with a fluoropyrimidine (5-FU or capecitabine) and oxaliplatin.
Materials and methods
Design
TABAsCO was an open-label, single-arm, multicenter, phase II study of TAS-102, irinotecan, and bevacizumab in patients with advanced (metastatic or unresectable) CRC who had received 1st line treatment with or had disease recurrence within 12 months of adjuvant therapy with fluoropyrimidine and oxaliplatin. The study was conducted according to the Declaration of Helsinki principles and approved by the Institutional Review Boards (IRB) from all participating institutions (Roswell Park Comprehensive Cancer Center, Rutgers Cancer Institute of New Jersey, Moffitt Cancer Center, and Fox Chase Comprehensive Cancer Center). All patients provided informed consent prior to study participation. The study was registered at ClinicalTrials.gov (NCT04109924).
Objectives
The primary objective was to evaluate the antineoplastic efficacy of TAS-102 plus irinotecan and bevacizumab in participants with pre-treated, advanced (metastatic or unresectable) CRC. The secondary objective was to evaluate the adverse event (AE) profile of this regimen. The evaluation of prognostic and predictive biomarkers was a post hoc exploratory objective.
Eligibility
We enrolled patients age 18 years or older with advanced CRC who had received prior treatment with fluoropyrimidine plus oxaliplatin in the metastatic or unresectable setting or had disease recurrence within 12 months of adjuvant therapy with a fluoropyrimidine plus oxaliplatin. Patients had to have an ECOG performance status (PS) of 0 or 1, measurable disease per RECIST 1.1 criteria [21], and adequate bone marrow, liver, and kidney function. Patients had to be eligible for treatment with bevacizumab (i.e., had controlled blood pressure at baseline; no recent surgery, arterial or venous thrombotic or embolic events within 3 months of study initiation, or recent grade 3 or higher haemorrhage; and no significant proteinuria). Patients who had prior treatment with TAS-102 or irinotecan, other anti-cancer therapy within 2 weeks of the planned first dose of study medication, and/or history of abnormal glucuronidation of bilirubin were excluded from this study. Patients with unstable angina, symptomatic congestive heart failure or cardiac arrhythmia requiring anti-arrhythmic therapy, untreated brain metastases, unresolved grade 1 or higher toxicities from prior therapy, excluding alopecia and grade 2 neuropathy, were similarly not allowed to participate.
Study treatment and procedures
Patients were treated with irinotecan 180 mg/m2 IV and bevacizumab 5 mg/kg IV days 1 and 15 of each 28-day treatment cycle. TAS-102 25 mg/m2 was administered twice daily, orally, on days 2–6 and 16–20 of each cycle. The dosing rationale is based on preclinical agent sequencing [19] and clinical safety data [20]. Patients could be dosed only when the absolute neutrophil count (ANC) and platelet count were ≥ 1,500/ mm3 and ≥ 75,000 /mm3 respectively. Dose adjustments were allowed (Supplementary Table 1). After the enrollment of the first 8 patients, we noted increased rates of grade 2 neutropenia precluding dosing every 2 weeks. We then implemented amendment 4 where use of non-pegylated granulocyte-colony stimulating factor (G-CSF) was suggested after the 1st episode of neutropenia grade 2 or higher lasting ≥ 7 days (initiated 24 h after the last TAS-102 dose and for up to 5 days).
The disease status was evaluated with CT or MRI at baseline and every 8 weeks after the initiation of study treatment. Patients could remain on study until disease progression, unacceptable toxicity, or discontinuation of both TAS-102 and irinotecan for toxicities.
Study Endpoints
The primary endpoint of the study was PFS. Secondary endpoints included OS, overall response rate (ORR), and the rate of AEs. The final analysis was to be conducted 12 months after enrollment of the final patient or once all patients experienced a progression event, whichever occurs earlier.
Statistical Considerations
PFS was treated as bivariate time-to-event data and defined as the time from treatment until disease progression, death, or last follow-up. Historically, the FOLFIRI plus bevacizumab combination can achieve a median PFS of approximately 6 months [3,4,5]. We tested the following hypothesis using a one-sided, one-sample log-rank test:
Ho:M50 = 6 versus HA:M50 > 6, where M50 is the true median PFS for patients treated with TAS-102 + irinotecan + bevacizumab.
If the true median PFS of the experimental treatment is 9 months and the PFS times are exponentially distributed, a sample size of n = 36 subjects would achieve 80.5% power (at α = 0.1) to detect such an effect (hazard ratio ≈ 0.67) compared to historical control. To account for potential deviations from assumptions and potential dropouts, a total of n = 42 patients could be accrued.
The PFS and OS were summarized using the Kaplan–Meier method. The ORR was treated as a dichotomous variable and summarized using frequencies and relative frequencies. Using Jeffrey’s prior method, a 90% confidence interval (CI) about the true ORR was obtained for each treatment group.
There was no formal power calculation for the exploratory analyses including gene expression in the de-identified tumour tissue protocols.
Results
Baseline characteristics
We enrolled 48 patients from 1/8/2020 to 8/3/2022 within the 4 participating institutions. Of those, 42 were eligible and received at least one dose of the study treatment (Fig. 1). The baseline characteristics are presented in Table 1. All patients had prior treatment with fluoropyrimidine and oxaliplatin, 62% of the patients had prior treatment with bevacizumab; 2 patients had received prior treatment with cetuximab or panitumumab as part of a 1st line regimen. Two patients had received prior treatment with immune checkpoint inhibitors in combination with chemotherapy as 1st line treatment. Of these patients, one (evaluable) had a microsatellite-stable (MS-S)/mismatch repair (MMR) proficient tumour and was treated with nivolumab while the second (non-evaluable, MS and MMR statuses were not reported) was treated with pembrolizumab.

Patient enrollment, allocation and follow-up.
Efficacy
Thirty-eight patients had at least one disease assessment, and 35 patients were evaluable for the primary endpoint. At the time of data cutoff (3/13/2023), 94% of all evaluable patients were off treatment. The most common reason for treatment discontinuation (51%) was progressive disease. The median follow-up among all evaluable patients was 14.1 (90% CI 0.9–17.2) months. The study was positive for the primary endpoint. The median PFS in the 35 evaluable patients was 7.9 (90% CI 6.2–11.8) months (p = 0.018, Fig. 2a). The 12-month PFS rate was 35% (90% CI 18–52%). With a median follow-up of 14.5 (90% CI 3.1–26) months, the median OS was 16.5 (90% CI 9.8–17.5) months (Fig. 2b). Additionally, when patients with MS-high (MS-H)/MMR deficient or BRAF mutant tumour were excluded from the analysis, the results remained statistically significant. In patients with MS-S/MMR proficient and BRAF wild-type tumours the median PFS was 7.5 (90% CI 5.3–9.0, p = 0.094) months while the median OS was 16.5 (90% CI 9.8–17.5) months. The PFS and OS in all patients who received at least one dose of the study treatment were comparable to the evaluable population results (Supplementary Figure 1).

PFS a and OS b in all evaluable patients. C censored, E events, T total.
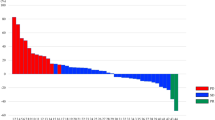
The ORR within the evaluable patient population was 14.3% (90% CI: 6.8–26.0%) with one patient attaining a complete response (CR, Fig. 3). The disease control rate (CR, partial response [PR] and stable disease [SD]) was 74.3% (90% CI: 61–84.8%).

Waterfall Plot, best % change in target tumour burden.
Safety
All patients experienced at least one treatment-related AE (TRAE, Table 2). Twenty-eight patients (67%) experienced a grade 3 or higher TRAE and 4 (9%) experienced a grade 4 or higher TRAE. The most common toxicities were haematological (neutropenia) and gastrointestinal (diarrhoea, nausea, and vomiting) as expected based on the individual toxicity profiles of irinotecan and TAS-102. The incidence of grade 3 or higher hypertension was 5%. Fifty-seven per cent of all treated patients required treatment with G-CSF. While neutropenia was common, there was only one episode of febrile neutropenia (grade 3). A total of 12 (29%) patients experienced a treatment-related SAE (Supplementary Table 2). There was one death on a study from treatment-related colonic perforation. Four patients stopped treatment due to an AE. Twenty-seven and 17 patients had at least one dose adjustment or treatment delay for an AE, respectively.
Exploratory Analyses
There was no difference in OS or PFS in patients with KRAS (KRASmt) or NRAS (NRASmt) mutant vs. wild-type tumours (Supplementary Figure 2A/B). There was a trend for improved OS and PFS in patients with left- vs. right-sided primary tumours and the absence of liver metastases (Supplementary Table 3); neither reached statistical significance. Contrary, patients without prior bevacizumab treatment had significantly higher median OS (16.5 months) and PFS (13.7 months) compared to patients with prior bevacizumab treatment (14.2 and 6.4 months respectively, Supplementary Table 3). Similarly, patients who received the last dose of oxaliplatin >6 months from study entry had superior outcomes compared to patients who were treated with oxaliplatin dose within 6 months from study entry (median PFS 17 vs. 6.7 months and median OS 25.3 vs. 13.7 months, Supplementary Table 3).
Furthermore, we evaluated the effect of mutations other than RAS in OS and PFS in patients with clinical multigene molecular testing data (Supplementary Table 4). Patients with TP53 mutant (p53mt) tumours had significantly inferior PFS compared to patients with wild-type (p53wt) tumours (median 7.5 vs. 13 months; p = 0.084); there was no statistically significant difference in OS (Supplementary Figure 2C/D). Additionally, patients with APC wild-type (APCwt) tumours had superior PFS compared to patients with APC mutant (APCmt) tumours (Supplementary Figure 2E/F, median 13.7 vs. 6.8 months; p = 0.065).
The presence of mutations in homologous recombination (HR) repair pathway and or DNA damage repair (DDR) has been associated with improved outcomes with FOLFIRI in patients with metastatic pancreatic cancer [22]. Four patients in the study had tumours with BRCA1/2 mutation. In our dataset, we did not identify an effect of HR and or DDR mutations in OS or PFS (Supplementary Figure 3).
We also evaluated the significance of expression of genes involved in different types of DNA repair, trifluorothymidine metabolism, cell cycle control, and replication in treatment outcomes. We identified 12 consecutive patients treated on the study at Roswell Park with available archival tumour tissue. Under a separate de-identified IRB-approved Roswell Park protocol (BDR 155422), we extracted RNA from these tumours for whole transcriptome sequencing (see Supplementary Methods). Additionally, we identified 21 consecutive patients treated with TAS-102 monotherapy with available archival tumour samples for similar analyses to be used as controls (IRB-approved Roswell Park protocol BDR 151721). All patients participating in this translational study provided universal informed consent for use of tumour specimens for research. Genes of interest were grouped in the following categories: HR, BER, MMR, nucleotide excision repair (NER), proofreading polymerases, non-homologous end joining (NHEJ), DNA damage response, cell cycle checkpoints, replication, and trifluorothymidine/nucleotide metabolism. There was a trend for improved PFS and OS in patients with tumours having decreased expression of genes involved in NER, trifluorothymidine/nucleotide metabolism, and proofreading polymerases. There was a trend for improved PFS with low expression of BER and MMR genes, cell cycle checkpoints and replication-related genes (Supplementary Figure 4, Supplementary Table 5). In the control samples, while low expression of NER repair genes was similarly associated with improved outcomes, low expression of TK1 and higher expression of TP was associated with worse OS (Supplementary Table 5).
Discussion
Historically, the median PFS and OS with FOLFIRI plus an anti-VEGF agent as 2nd line therapy in patients with advanced CRC ranges from 5.7 to 6.9 and 11.2 to 13.5 months, respectively [3,4,5]. In this phase II study, we show that the combination of irinotecan, TAS-102 and bevacizumab is an active 2nd line regimen for patients with pre-treated, advanced CRC. The observed median PFS and OS of 7.9 and 16.5 months, respectively, were higher compared to historical controls while the ORR of 14.3% was within the range observed in the landmark studies (ranging between 5 and 19.8%) [3,4,5]. The main toxicities were hematological and gastrointestinal as expected. While the incidence of gastrointestinal toxicity appears similar to FOLFIRI plus anti-VEGF agent, the incidence of grade 3 or worse neutropenia is lower compared to FOLFIRI plus ramucirumab or aflibercept and higher compared to FOLFIRI plus bevacizumab [3,4,5]. Importantly, the incidence of grade 2 neutropenia in our study was 31% necessitating use of G-CSF support to maintain dose density in 57% of all treated patients.
TAS-102 was recently combined with liposomal irinotecan in a phase II study in patients with pre-treated CRC (n = 22 patients) [23]. Contrary to our study, 2 or more prior therapies were allowed (median 1, range 1–4 prior therapies); 3 patients had prior irinotecan. Compared to our study, TAS-102 was dosed at a higher dose (35 mg/m2) starting on day 1 (vs. day 2) for 5 days while liposomal irinotecan was dosed at 60 mg/m2 on day 1. Additionally, approximately half of the patients in this study received bevacizumab. The observed ORR (15%) was almost identical to the ORR observed in our study while the median PFS and OS were 9.7 and 10.1 months, respectively. The incidence of grade 3 or higher neutropenia was similar compared to our study; the rate of grade 3 or worse diarrhoea was lower. A randomized phase III study is required to determine the effectiveness of irinotecan (conventional or liposomal), TAS-102 and bevacizumab as 2nd line therapy in patients with advanced CRC over FOLFIRI plus bevacizumab.
TAS-102 alone [15] and in combination with bevacizumab [16] has improved outcomes in patients with oxaliplatin-, irinotecan- and fluoropyrimidine-refractory CRC. To this day, there is no predictive molecular biomarker to guide treatment decisions. In vitro studies reveal that KRASG12 mutant CRC are more resistant to trifluorothymidine causing limited DNA damage [24]. ln a secondary biomarker analysis from the RECOURSE study, KRASG12 mutations were associated with worse OS with TAS-102 monotherapy while KRASG13 mutations with improved OS [24]. On the contrary, in a pooled analysis from 3 randomized controlled studies testing TAS-102 vs. placebo (including RECOURSE), no difference in OS was observed based on the presence of KRASG12 or KRASG13 mutations after adjustment for confounding factors [25]. Similarly, the OS benefit of TAS-102 plus bevacizumab in the SUNLIGHT study appears to be independent of the KRASG12 mutation status [26]. The small sample size of our study precludes codon-specific analyses. With this limitation, we did not identify a difference in outcomes based on the KRAS and NRAS status of the tumour. Additionally, retrospective data indicate that the clinical benefit for TAS-102 as a single agent in p53mt CRC is extremely limited [27]. Similarly, in our study, patients with p53mt had inferior outcomes compared to patients with p53wt tumours.
We have previously reported that p53mt tumours have high expression of BER, MMR and replication-related genes [12, 28]. In CRC, low expression of genes involved in the HR is associated with improved outcomes with DNA-damaging agents such as oxaliplatin and irinotecan [29]. Additionally, highly proliferative CRC are more sensitive to DNA-damaging agents [30]. Trifluorothymidine enters cells via the nucleoside transporters hCNT and hENT [31, 32]. It is then phosphorylated and converted to triphosphate by thymidine kinase-1 (TK1) [14, 33]. Subsequently, DNA polymerase incorporates trifluorothymidine to DNA; the DNA lesion is then repaired with BER and single-strand DNA (ssDNA) break repair mechanisms [14, 28, 33]. TK1 expression is essential for the antitumour effect of trifluorothymidine in preclinical models [34, 35]. Patients with high TK1 tumour expression have significantly improved clinical outcomes with TAS-102 monotherapy [36]. Thymidine phosphorylase (TP), the target of tipiracil, controls the bioavailability of trifluorothymidine; TP is produced and excreted by the proximal renal tubule through regulation by organic cation transporter 2 (OCT2) and toxin extrusion 1 (MATE1) [37]. Notably, germline polymorphisms in hENT1, OCT2, and MATE1 are linked to clinical outcomes for TAS-102 monotherapy [38]. Further, alterations in DDR and replication-related genes may also affect clinical outcomes in CRC patients receiving TAS-102 [39].
In a small number of patients from our study with available archival tumour tissue, we observed that there is a trend for improved outcomes in patients whose tumours had low expression of trifluorothymidine/nucleotide metabolism genes while patients with p53wt tumours had better PFS on study treatment. Contrary to previous studies [29, 30], low expression of HR and high expression replication-related genes in our small translational experiment was not associated with improved PFS with DNA damaging agents. Additionally, low expression of genes implicated in alternative DNA repair pathways (NER, BER, MMR) had a non-significant association with improved outcomes. It is plausible that prior exposure to and disease progression while on oxaliplatin may affect the significance of HR deficiency and high proliferation as predictive biomarkers, especially when these are assessed in specimens collected from treatment-naïve patients. Furthermore, tumours with low expression of cell cycle checkpoints, TK1/TP and replication-related genes may behave more indolently in general thus explaining improved PFS while on the study treatment. Further evaluation of DNA repair pathways as integrated biomarkers in future larger studies is warranted.
Our study has several limitations. First, the historical control was based on phase 3 clinical trials that evaluated FOLFIRI plus an anti-VEGF agent as 2nd line therapy. In the ML18147 and RAISE studies, where all patients had received prior bevacizumab, the median PFS was identical (5.7 months) and lower than the one observed in the aflibercept study (6.9 months), where only 30% of the patients had received prior bevacizumab [3,4,5]. In our clinical trial, approximately 40% of the patients had not previously received bevacizumab. These patients had inferior outcomes compared to patients who had not previously received bevacizumab. These results must be interpreted with caution as the study was not powered to detect differences between bevacizumab-pre-treated vs. not patients and the CI are wide and overlap. In support, subgroup analysis of the larger (N = 1226) FOLFIRI plus aflibercept study did not identify differences in PFS or OS between bevacizumab-pre-treated vs. not patients [4]. Finally, we performed multiple and not prespecified exploratory analyses in this small cohort without accounting for false positive results. The results of these analyses are hypothesis-generating and not yet appropriate as biomarkers for treatment decisions including eligibility for future clinical trials. Genetic biomarkers such as the TP53 and APC status of the tumour should be integrated and further evaluated in future, larger clinical trials.
In conclusion, the combination of irinotecan, TAS-102 and bevacizumab is active in pre-treated, metastatic CRC with improved outcomes compared to historical control (FOLFIRI plus anti-VEGF agent). While toxicity is manageable, G-CSF support is required to maintain dose density/intensity. Further exploration of this regimen in randomized studies is warranted.
Data availability
De-identified clinical data will be available upon request to the corresponding author. The raw RNA sequencing data can be accessed from https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE275628.
References
Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33.
Siegel RL, Wagle NS, Cercek A, Smith RA, Jemal A. Colorectal cancer statistics, 2023. CA Cancer J Clin. 2023;73:233–54.
Bennouna J, Sastre J, Arnold D, Osterlund P, Greil R, Van Cutsem E, et al. Continuation of bevacizumab after first progression in metastatic colorectal cancer (ML18147): a randomised phase 3 trial. Lancet Oncol. 2013;14:29–37.
Van Cutsem E, Tabernero J, Lakomy R, Prenen H, Prausova J, Macarulla T, et al. Addition of aflibercept to fluorouracil, leucovorin, and irinotecan improves survival in a phase III randomized trial in patients with metastatic colorectal cancer previously treated with an oxaliplatin-based regimen. J Clin Oncol. 2012;30:3499–506.
Tabernero J, Yoshino T, Cohn AL, Obermannova R, Bodoky G, Garcia-Carbonero R, et al. Ramucirumab versus placebo in combination with second-line FOLFIRI in patients with metastatic colorectal carcinoma that progressed during or after first-line therapy with bevacizumab, oxaliplatin, and a fluoropyrimidine (RAISE): a randomised, double-blind, multicentre, phase 3 study. Lancet Oncol. 2015;16:499–508.
Bikov KA, Mullins CD, Hung A, Seal B, Onukwugha E, Hanna N. Patterns of biologics use across treatment lines in elderly (age >65) medicare patients with metastatic colon cancer. oncologist. 2016;21:676–83.
Abrams TA, Meyer G, Schrag D, Meyerhardt JA, Moloney J, Fuchs CS. Chemotherapy usage patterns in a US-wide cohort of patients with metastatic colorectal cancer. J Natl Cancer Inst. 2014;106:djt371.
Tanaka N, Sakamoto K, Okabe H, Fujioka A, Yamamura K, Nakagawa F, et al. Repeated oral dosing of TAS-102 confers high trifluridine incorporation into DNA and sustained antitumor activity in mouse models. Oncol Rep. 2014;32:2319–26.
Fukushima M, Suzuki N, Emura T, Yano S, Kazuno H, Tada Y, et al. Structure and activity of specific inhibitors of thymidine phosphorylase to potentiate the function of antitumor 2’-deoxyribonucleosides. Biochem Pharm. 2000;59:1227–36.
Emura T, Murakami Y, Nakagawa F, Fukushima M, Kitazato K. A novel antimetabolite, TAS-102 retains its effect on FU-related resistant cancer cells. Int J Mol Med. 2004;13:545–9.
Matsuoka K, Iimori M, Niimi S, Tsukihara H, Watanabe S, Kiyonari S, et al. Trifluridine induces p53-dependent sustained G2 phase arrest with its massive misincorporation into DNA and few DNA strand breaks. Mol Cancer Ther. 2015;14:1004–13.
Alruwaili MM, Zonneville J, Naranjo MN, Serio H, Melendy T, Straubinger RM, et al. A synergistic two-drug therapy specifically targets a DNA repair dysregulation that occurs in p53-deficient colorectal and pancreatic cancers. Cell Rep Med. 2024:101434.
Suzuki N, Nakagawa F, Nukatsuka M, Fukushima M. Trifluorothymidine exhibits potent antitumor activity via the induction of DNA double-strand breaks. Exp Ther Med. 2011;2:393–7.
Emura T, Suzuki N, Yamaguchi M, Ohshimo H, Fukushima M. A novel combination antimetabolite, TAS-102, exhibits antitumor activity in FU-resistant human cancer cells through a mechanism involving FTD incorporation in DNA. Int J Oncol. 2004;25:571–8.
Mayer RJ, Van Cutsem E, Falcone A, Yoshino T, Garcia-Carbonero R, Mizunuma N, et al. Randomized trial of TAS-102 for refractory metastatic colorectal cancer. N. Engl J Med. 2015;372:1909–19.
Prager GW, Taieb J, Fakih M, Ciardiello F, Van Cutsem E, Elez E, et al. Trifluridine-tipiracil and bevacizumab in refractory metastatic colorectal cancer. N. Engl J Med. 2023;388:1657–67.
Van Cutsem E, Danielewicz I, Saunders MP, Pfeiffer P, Argiles G, Borg C, et al. Trifluridine/tipiracil plus bevacizumab in patients with untreated metastatic colorectal cancer ineligible for intensive therapy: the randomized TASCO1 study. Ann Oncol. 2020;31:1160–8.
Nukatsuka M, Nakagawa F, Saito H, Sakata M, Uchida J, Takechi T. Efficacy of combination chemotherapy using a novel oral chemotherapeutic agent, TAS-102, with irinotecan hydrochloride on human colorectal and gastric cancer xenografts. Anticancer Res. 2015;35:1437–45.
Matsuoka K, Takechi T. Combined efficacy and mechanism of trifluridine and SN-38 in a 5-FU-resistant human colorectal cancer cell lines. Am J Cancer Res. 2017;7:2577–86.
Varghese AM, Cardin DB, Hersch J, Benson AB, Hochster HS, Makris L, et al. Phase I study of trifluridine/tipiracil plus irinotecan and bevacizumab in advanced gastrointestinal tumors. Clin Cancer Res. 2020;26:1555–62.
Eisenhauer EA, Therasse P, Bogaerts J, Schwartz LH, Sargent D, Ford R, et al. New response evaluation criteria in solid tumours: revised RECIST guideline (version 1.1). Eur J Cancer. 2009;45:228–47.
Chiorean EG, Guthrie KA, Philip PA, Swisher EM, Jalikis F, Pishvaian MJ, et al. Randomized phase II study of PARP inhibitor ABT-888 (Veliparib) with modified FOLFIRI versus FOLFIRI as second-line treatment of metastatic pancreatic cancer: SWOG S1513. Clin Cancer Res. 2021;27:6314–22.
Alese OB, Gbolahan OB, Diab M, Botrus G, Coleman K, McCook-Veal A, et al. OniLon: Phase II trial of trifluridine/tipiracil (TAS-102) and nanoliposomal irinotecan (nal-IRI) in advanced colorectal cancer. J Clin Oncol. 2023;41:3580.
van de Haar J, Ma X, Ooft SN, van der Helm PW, Hoes LR, Mainardi S, et al. Codon-specific KRAS mutations predict survival benefit of trifluridine/tipiracil in metastatic colorectal cancer. Nat Med. 2023;29:605–14.
Yoshino T, Van Cutsem E, Li J, Shen L, Kim TW, Sriuranpong V, et al. Effect of KRAS codon 12 or 13 mutations on survival with trifluridine/tipiracil in pretreated metastatic colorectal cancer: a meta-analysis. ESMO Open. 2022;7:100511.
Tabernero J, Taieb J, Fakih M, Prager GW, Van Cutsem E, Ciardiello F, et al. Impact of KRAS(G12) mutations on survival with trifluridine/tipiracil plus bevacizumab in patients with refractory metastatic colorectal cancer: post hoc analysis of the phase III SUNLIGHT trial. ESMO Open. 2024;9:102945.
Giampieri R, Zizzi A, Bittoni A, Pecci F, Giglio E, Giulia M, et al. P-27 Retrospective observational analysis of p53 mutational status as a prognostic factor in TAS-102 treated metastatic colorectal cancer patients. Ann Oncol. 2020;31:S98.
Zonneville J, Wang M, Alruwaili MM, Smith B, Melnick M, Eng KH, et al. Selective therapeutic strategy for p53-deficient cancer by targeting dysregulation in DNA repair. Commun Biol. 2021;4:862.
Walden D, Deshmukh S, Batalini F, Zheng-Lin B, Wu S, Xiu J, et al. Chemotherapeutic sensitivity in colorectal cancer expressing low RNA of wild type homologous recombination genes. J Clin Oncol. 2023;41:3531.
Spurr LF, Martinez CA, Katipally RR, Iyer SC, Pugh SA, Bridgewater JA, et al. A proliferative subtype of colorectal liver metastases exhibits hypersensitivity to cytotoxic chemotherapy. NPJ Precis Oncol. 2022;6:72.
Takahashi K, Yoshisue K, Chiba M, Nakanishi T, Tamai I. Involvement of concentrative nucleoside transporter 1 in intestinal absorption of trifluridine using human small intestinal epithelial cells. J Pharm Sci. 2015;104:3146–53.
Takahashi K, Yoshisue K, Chiba M, Nakanishi T, Tamai I. Contribution of equilibrative nucleoside transporter(s) to intestinal basolateral and apical transports of anticancer trifluridine. Biopharm Drug Dispos. 2018;39:38–46.
Sakamoto K, Yokogawa T, Ueno H, Oguchi K, Kazuno H, Ishida K, et al. Crucial roles of thymidine kinase 1 and deoxyUTPase in incorporating the antineoplastic nucleosides trifluridine and 2’-deoxy-5-fluorouridine into DNA. Int J Oncol. 2015;46:2327–34.
Kataoka Y, Iimori M, Niimi S, Tsukihara H, Wakasa T, Saeki H, et al. Cytotoxicity of trifluridine correlates with the thymidine kinase 1 expression level. Sci Rep. 2019;9:7964.
Edahiro K, Iimori M, Kobunai T, Morikawa-Ichinose T, Miura D, Kataoka Y, et al. Thymidine kinase 1 loss confers trifluridine resistance without affecting 5-fluorouracil metabolism and cytotoxicity. Mol Cancer Res. 2018;16:1483–90.
Yoshino T, Yamazaki K, Shinozaki E, Komatsu Y, Nishina T, Baba H, et al. Relationship between thymidine kinase 1 expression and trifluridine/tipiracil therapy in refractory metastatic colorectal cancer: a pooled analysis of 2 randomized clinical trials. Clin Colorectal Cancer. 2018;17:e719–e32.
Yoshisue K, Takahashi K, Okayama T, Yamashita F, Chiba M, editors. Investigation of transporters that play an important role in urinary secretion of thymidine phosphorylase inhibitor combined with a novel anti-cancer agent of TAS-102. DRUG METABOLISM REVIEWS; 2015: TAYLOR & FRANCIS LTD 4 PARK SQUARE, MILTON PARK, ABINGDON OX14 4RN, OXON.
Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Dadduzio V, et al. Potential role of polymorphisms in the transporter genes ENT1 and MATE1/OCT2 in predicting TAS-102 efficacy and toxicity in patients with refractory metastatic colorectal cancer. Eur J Cancer. 2017;86:197–206.
Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Murgioni S, et al. Genetic variants of DNA repair-related genes predict efficacy of TAS-102 in patients with refractory metastatic colorectal cancer. Ann Oncol. 2017;28:1015–22.
Acknowledgements
TAS-102 was provided for free by Taiho Oncology. This work was partially supported by the National Cancer Institute (NCI) grants R37CA282430 to Dr. Christos Fountzilas and P30CA016056 involving the use of Roswell Park Comprehensive Cancer Center’s Biostatistics and Bioinformatics Shared Resource, Genomics Shared Resource, and the Pathology Network Shared Resource. The initial results of this study were presented in the American Society of Clinical Oncology 2023 Annual Meeting (Abstract 3590, Poster 290).
Funding
This study was funded by the National Comprehensive Cancer Network Oncology Research Program through a grant provided by Taiho Oncology. The funder had no role in the design or interpretation of study results.
Author information
Authors and Affiliations
Contributions
Patrick Boland - conceptualization, formal analysis, funding acquisition, data curation, methodology, project administration, and writing (review and editing), Sarbajit Mukherjee - data curation, and writing (review and editing), Iman Imanirad - project administration, data curation, and writing (review and editing), Namrata Vijayvergia project administration, data curation, and writing (review and editing), Seth Cohen - data curation, and writing (review and editing), Medhavi Gupta - methodology, and writing (review and editing), Renuka Iyer - data curation, and writing (review and editing), Andrei Bakin - formal analysis, and writing (review and editing), Jianxin Wang - formal analysis, data curation, methodology, and writing (review and editing), Sarah Chatley - data curation, and writing (review and editing), Beth Cahill - data curation, and writing (review and editing), Deepak Vadehra - data curation, and writing (review and editing), Kristopher Attwood - formal analysis, data curation, methodology, and writing (review and editing), Howard Hochster - data curation, and writing (review and editing), Christos Fountzilas - formal analysis, funding acquisition, data curation, methodology, project administration, and writing (original draft, review and editing).
Corresponding author
Ethics declarations
Competing interests
Dr. Christos Fountzilas has research support for this clinical trial from the National Comprehensive Cancer Network Oncology Research Program (paid to the institute). He has research support from the National Comprehensive Cancer Network Foundation, Taiho Oncology, Pfizer Inc, and Merck Sharp & Dohme Corp (paid to the institute) unrelated to this study. Dr. Sarbajit Mukherjee has received research funding from Ipsen Biopharmaceuticals (paid to the institute) unrelated to this study. Dr. Iman Imanirad has received advisory board compensation from Eisai Co, Ltd, unrelated to this study.
Ethics approval and consent to participate
The study was conducted according to the Declaration of Helsinki principles and approved by the Institutional Review Boards (IRB) from all participating institutions (Roswell Park Comprehensive Cancer Center, Rutgers Cancer Institute of New Jersey, Moffitt Cancer Center, and Fox Chase Comprehensive Cancer Center). All patients provided informed consent prior to study participation. All patients providing tumor samples for whole transcriptome sequencing (de-identified Roswell Park IRB-approved protocols BDR 155422 and BDR 151721) had provided universal informed consent for use of tumor samples in research.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Boland, P.M., Mukherjee, S., Imanirad, I. et al. TAS-102, Irinotecan, and bevacizumab in pre-treated metastatic colorectal cancer (TABAsCO), a phase II clinical trial. Br J Cancer (2024). https://doi.org/10.1038/s41416-024-02845-x
Received:
Revised:
Accepted:
Published:
DOI: https://doi.org/10.1038/s41416-024-02845-x
- Springer Nature Limited