
Abstract
The replicative Cdc45-MCM-GINS (CMG) helicase is a large protein complex that functions in the DNA melting and unwinding steps as a component of replisomes during DNA replication in mammalian cells. Although the CMG performs this important role in cell growth, the CMG is not a simple bystander in cell cycle events. Components of the CMG, specifically the MCM precursors, are also involved in maintaining genomic stability by regulating DNA replication fork speeds, facilitating recovery from replicative stresses, and preventing consequential DNA damage. Given these important functions, MCM/CMG complexes are highly regulated by growth factors such as TGF-ß1 and by signaling factors such as Myc, Cyclin E, and the retinoblastoma protein. Mismanagement of MCM/CMG complexes when these signaling mediators are deregulated, and in the absence of the tumor suppressor protein p53, leads to increased genomic instability and is a contributor to tumorigenic transformation and tumor heterogeneity. The goal of this review is to provide insight into the mechanisms and dynamics by which the CMG is regulated during its assembly and activation in mammalian genomes, and how errors in CMG regulation due to oncogenic changes promote tumorigenesis. Finally, and most importantly, we highlight the emerging understanding of the CMG helicase as an exploitable vulnerability and novel target for therapeutic intervention in cancer.
Similar content being viewed by others

Abraham Wald’s “missing bullet hole” genius applies to the CMG helicase
While working for a classified program called the Statistical Research Group in Manhattan during the Second World War, the mathematician Abraham Wald was tasked with determining the best locations on a warplane to increase armor to prevent combat loss from dogfights or ground fire [1]. Placing additional armor on a plane rendered it heavier and less agile, creating a liability in need of careful consideration. Military officers reasoned that the preponderance of bullet holes on the fuselage and wings of aircraft returning to base suggested those were the most vulnerable areas in need of such armor. But the mathematical logic in Wald presented the military with an unorthodox answer: the planes should have their armor increased where the bullet holes were not observed, namely around the engine [1]. His reasoning was that planes with damage to their fuselages and wings returned to base, arguing that those sites of damage, while debilitating, were survivable. However, the lack of planes returning with significant bullet holes against their engine compartments indicated that the engine was a major point of vulnerability that reduced survivability of the planes and flight crews when targeted with gunfire. It was the statistical absence of bullet holes, the “missing bullet holes,” that defined the best location to bring down an aircraft [1].
A type of analogy to Wald’s missing bullet holes and engine vulnerability exists when assessing the roles of the replicative Cdc45-MCM-GINS (CMG) helicase as a driving mechanism underlying cancer development, while also defending the worthiness of the CMG helicase as a druggable target for anti-cancer intervention. Defining the cell cycle and DNA replication as a metaphorical “engine” in propelling a cell forward during proliferation, the presence of mutated or overexpressed oncogenes that drive the cell cycle engine during tumorigenesis is often used to identify potential tumor-specific dependencies. The mutations in such altered genes are analogous to the bullet holes in aircraft assessed by Wald, and are often used to define where one should focus anti-cancer drug development efforts. As will be described in more depth below, CMG helicase activity is central to tumorigenesis and the cell cycle engine, yet human malignancies rarely, if ever, contain mutated CMG components that represent cancer-driving situations [2, 3]. As with Wald’s engine situation, where damage to the engine is not compatible with the survival of aircraft [1], it appears likely that despite elevated mutation rates within tumors, tumor cells cannot survive if they mutate CMG components in ways that reduce or eliminate function. It is this “missing mutation” status of the CMG helicase as a needed survivability factor and lack of cancer-driving CMG mutations, together with mismanagement and alteration of CMG assembly and activation by oncogenic drivers [3,4,5,6,7], that renders the CMG a “never-mutated” tumor-specific vulnerability and justifiable target for anti-cancer drug development.
The CMG helicase: assembly and activation
The replicative helicase is a large enzyme composed of 11 primary subunits, including Cdc45, the tetrameric GINS complex named for its subunits (Go-Ichi-Ni-San, for 5-1-2-3 in Japanese; subunits Sld5, Psf1, Psf2, Psf3), and the MCM2-7 hexameric ATPase core of Mini-Chromosome Maintenance proteins (Fig. 1). Using these subunits as the naming basis (Cdc45-MCM-GINS), the replicative helicase is referred to as the CMG helicase, or simply the CMG [8,9,10,11]. This review will focus on the relevance of the replicative CMG helicase to human cancer by discussing important aspects of CMG assembly and regulation by signaling pathways and how problems with CMG management are involved in cancer development. For an in-depth understanding of the molecular and biochemical details of the initiation or elongation phases of DNA replication, the reader is referred to some elegant research reports and reviews on these topics [9, 10, 12,13,14,15,16,17,18,19,20,21,22,23,24]. In addition, to complement the discussion here, the reader may wish to examine several other reports on the roles of CMG components in cancer for additional insight [25,26,27,28].

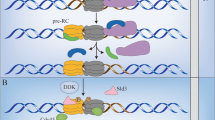
A, B Space-filling and ribbon structures of the human CMG helicase obtained from cryo-electron microscopy (cryo-EM) (PDB accession code 6XTX). C Diagrams showing CMG helicase assembly steps. Double MCM hexamers are loaded first onto DNA, and near the G1/S transition the Cdc45 and GINS subunits are recruited to form double CMGs. Initiation at G1/S leads to establishment of dual CMG helicases on ssDNA, moving in opposite directions within replisome complexes. General locations of DNA polymerases, topoisomerase II, Mcm10, and Ctf4 are shown. Note that during S-phase late-firing DNA replication origins proceed through similar CMG establishment steps. See text for details and references.
The CMG helicase functions during DNA replication to unwind the DNA at replication forks ahead of the DNA polymerases that catalyze the generation of new daughter DNA strands [14,15,16, 18, 19, 29,30,31,32]. The CMG also mediates the initial melting of double-stranded DNA (dsDNA) at replication origins where DNA replication begins in a bidirectional manner [16, 18, 23, 33]. The CMG is the only replicative helicase enzyme that catalyzes these particular melting and unwinding steps during DNA replication, as there are no known enzymes in cells that can replace its function in these processes. The larger multi-protein complex tasked with duplicating the DNA at replication forks during the synthetic S-phase is called a replisome, composed of the CMG helicase, DNA polymerases and primases, topoisomerases, and other associated factors [11, 22, 34,35,36,37]. The CMG helicase is also required for cells to recover DNA replication after encountering fork-stalling stress, or replicative stress, during S-phase [3, 38,39,40,41]. This role of the CMG helicase in recovering from replicative stress is highly relevant to how CMG problems contribute to cancer and requires an understanding of how the CMG helicase is assembled and regulated.
CMG assembly occurs in G1 phase and begins with the recruitment of MCM hexamers to DNA (within chromatin) by the concerted actions of the Origin Recognition Complex (ORC), Cdc6, and Cdt1 (Cdc10-dependent transcript-1) [12, 13, 42,43,44,45,46,47,48,49,50,51,52,53,54]. ORC is analogous in function to Initiator proteins in prokaryotic and viral organisms that bind replicator sequences at DNA replication origins [called ori’s, or autonomous replicating sequences (ARS) in yeast] to facilitate loading of replisome proteins, including the helicase [47, 53, 55,56,57]. However, in higher eukaryotic cells ORC does not have a DNA site-specific binding requirement, instead being influenced in its DNA interactions by general DNA sequence composition (prefers AT-rich) or limited by torsional DNA stress [58,59,60]. Consistent with this, and as discussed more below, mammalian cells do not possess specific DNA replicator sequences at origins [58, 59, 61, 62]. ORC and Cdc6 contain ATPase domains that are involved in the coordination of MCM loading, and MCM loading onto DNA (within chromatin) requires ATP binding and hydrolysis by the MCM subunits [13, 20, 42, 53, 63,64,65,66]. Two MCM2-7 hexamers are loaded by ORC onto DNA in a head-to-head manner encircling the DNA (Fig. 1), with their amino-terminal ends facing each other and the carboxy-terminal ends containing ATPase domains facing outward [12, 13, 20, 23, 24, 42]. The loaded MCM hexamers are referred to as a pre-Replication Complex, or pre-RC [44, 45], and MCM loading is also referred to as licensing DNA for one round of DNA replication [42, 67,68,69,70,71,72].
MCM loading also requires functions of Cdc6 and Cyclin E, and for the latter, there are kinase-dependent (with Cdk2) and kinase-independent roles [45, 48, 66, 73,74,75,76,77,78]. Cyclin E-Cdk2 is recruited to MCM loading sites through interactions with Cdc6 [76, 77, 79], which is also a substrate of Cyclin E-Cdk2 kinase activity [76, 77, 80]. Phosphorylation of Cdc6 prevents its degradation by APC/C-dependent proteolysis [81,82,83]. In the absence of its associated kinase, Cyclin E also interacts with MCM subunits such as Mcm7, and with Cdt1, both events being required for MCM hexamer loading [76,77,78]. These necessary interactions of Cyclin E and its associated kinase during MCM recruitment function upstream or coincident with the functions of Cdt1 in MCM loading [76, 77]. Cyclin E-Cdk2 kinase can phosphorylate Mcm3 and Mcm7 in vitro [84, 85], and phospho-blocking mutation of one targeted site in Mcm3 (T722A) reduces Mcm3 chromatin binding [84], suggesting that phosphorylation of Mcm3 by Cyclin E-Cdk2 is important in MCM assembly. Similarly, a mutation in Mcm7 that blocks phosphorylation of a putative Cyclin E-Cdk2 site (Mcm7-S121A) results in reduced interactions between Mcm7 and Mcm3-Mcm5-Cdc45 complexes [85], suggesting a need for Cyclin E-Cdk2 phosphorylation of this site in pre-RC assembly. However, the Mcm7(S121A) mutant protein appears to be more capable of binding chromatin compared to wildtype Mcm7 [85], suggesting that phosphorylation plays a more complex role in MCM regulation. Indeed, as discussed below, overexpression of Cyclin E causes a genome-wide destabilization of MCM hexamers on chromatin [7]. Xenopus and human Mcm4 protein has been shown to be a substrate of Cdc2 (Cdk1) and Cyclin A-Cdk2 kinases, respectively [86, 87], and phosphorylation by these kinases reduces chromatin association of MCM complexes, part of a process to prevent re-licensing of DNA in late S-phase and G2/M phases [86,87,88]. Although not yet determined, it is possible that deregulation of Cyclin E-Cdk2 (due to Cyclin E overexpression) in mammalian cells might target some of these phosphorylation sites in Mcm4 or other MCM subunits, causing the reduced MCM hexamer chromatin affinity that is observed [7]. Interestingly, for both Mcm3 and Mcm7, overexpression of wildtype proteins causes a block to S-phase entry and checkpoint activation [84, 85], indicating that overexpression of single MCM subunits is not tolerated by mammalian cells. Cyclin E-Cdk2 also phosphorylates Treslin, the homolog of the yeast Sld3 protein, which facilitates interaction with TopBP1 (DNA topoisomerase II binding protein 1; homolog of yeast Dpb11) and promotes recruitment of Cdc45, GINS, and DNA polymerases to chromatin [89,90,91]. Although some of these Cyclin E-Cdk2 mediated events in MCM and CMG assembly are known, a complete mechanistic picture of the contribution of Cyclin E and Cyclin A associated kinases in regulating of MCM/CMG function awaits further investigation.
During the cell cycle, Cyclin E-Cdk2 becomes active in middle-late-G1 phase, which fits nicely with the middle-late-G1 timing of when MCMs are loaded onto chromatin in mammalian cells released from quiescence [92,93,94,95,96,97,98]. Another target of Cyclin E-Cdk2, the Rb tumor suppressor protein, also becomes increasingly phosphorylated coincident with MCM loading, indicating that Cyclin E-Cdk2 (and Cyclin E) dependent MCM loading occurs close to, or at, the Restriction Point of the cell cycle [92, 98, 99]. As will be described below, Rb (and hyperphosphorylated Rb) also plays a role in regulating MCM and CMG function in late-G1 phase. Collectively, these results suggest that, in cells released from quiescence, passage through the Restriction Point (R-Point) parallels MCM loading and licensing of DNA for one round of DNA replication [92]. Interestingly, in cycling mammalian cells (without an intervening quiescent period), MCMs load onto chromatin even earlier, during late mitosis after chromosome separation [100]. The latter indicates that under continuous cycling conditions, the many factors required for MCM loading are present and active prior to or after daughter cells are created.
Cdt1 has no enzymatic domains but plays pivotal roles in the MCM assembly process. Cdt1 contains a carboxy-terminal Mcm6 interacting domain that is required for making contacts with the MCM complex during loading [101,102,103,104]. A small protein called Geminin inhibits Cdt1 function to block MCM assembly, and Geminin achieves this in part by inhibiting the Cdt1-Mcm6 interaction and Cdt1 DNA binding [105,106,107,108,109]. Cdt1 also regulates MCM loading through interactions with chromatin-modifying enzymes, including a histone acetyltransferase (HAT) and histone deacetylase (HDAC) [110, 111]. In G1 phase Cdt1 binds to HBO1 (HAT binding ORC1) to facilitate localized chromatin decondensation and MCM loading onto accessible DNA, which is suppressed by Geminin [110,111,112,113,114]. Once cells enter S-phase and DNA replication begins, Cdt1 interacts with HDAC11 to promote chromatin closure and block further MCM loading to prevent another round of licensing [110]. The interaction between Cdt1 and HDAC11 is enhanced by Geminin binding [110]. Demonstrating its pivotal role in MCM loading and licensing using these mechanisms, overexpression of Cdt1 (during S-phase) can cause cells to re-license their DNA for another round of DNA replication, causing genomic (chromosomal) re-replication within a single cell cycle [106, 110, 115]. In addition, as described more below, Cdt1 overexpression can abrogate growth-inhibitory signals of TGFß1 under certain conditions and force MCM loading when it would normally be blocked [77]. Cdt1 thus appears epistatic to most of the events regulating MCM loading, being able to achieve MCM re-loading as a single deregulated factor. Accordingly, Cdt1 is oncogenic [116, 117], and its role in promoting excessive MCM loading likely contributes to tumorigenesis by creating genomic destabilization [106].
The conversion of a pair of MCM hexamers to a pair of CMG helicases (double CMGs; dCMGs) requires recruitment of Cdc45 and GINS, which occurs near G1/S and at future origins that fire later in S-phase [11, 18, 23, 24, 31, 36] (Fig. 1). Metazoan Cdc45 recruitment requires combined Cdk2 and Cdc7-Dbf4 (DDK) kinase activities, and PP2A phosphatase activity [24, 118,119,120,121,122,123]. GINS recruitment requires Cdc7-Dbf4 and Cdk2 [24, 123]. In yeast, although Cdc45 recruitment to MCMs does not require Cdk2, stable interaction of Cdc45 within CMGs requires GINS and Cdk2 activity [123]. Interestingly, in quiescence-release mammalian cell models CMGs can begin assembly in late-G1 several hours prior to G1/S based on the chromatin association of MCMs and Cdc45 (GINS loading kinetics was not assessed), yet the CMG remains enzymatically inactive until G1/S in terms of processive unwinding of DNA [77, 92, 124]. Yeast cryo-electron microscopic studies have demonstrated that when MCM pairs are converted to two opposing dCMGs prior to, or coincident with, the G1-S transition, changes to CMG-CMG and CMG-DNA interactions produce an ATP-dependent localized destabilization of the DNA within MCM cores of the dCMGs such that a few base pairs are melted and stabilized by each Mcm2 protein [23, 24]. This step likely represents one of the earliest events in the initiation of DNA replication.
Once triggered to (further) melt and unwind DNA at G1-S by mechanisms that remain unclear in mammalian cells, CMG pairs pass each other (Fig. 1) and CMG movement within replisomes at replication forks relies on an orchestrated set of ATP hydrolysis steps within the MCM hexameric “core” of the CMG [9, 10, 14, 15, 19]. Neither Cdc45 nor GINS possess enzymatic domains, but their association with the MCM hexamers increases CMG helicase activity upwards of 300-fold, as measured using ATPase and fork-unwinding assays in vitro [10]. The CMG moves along single-stranded DNA (ssDNA) in a 3’-5’ direction (Fig. 1) using ATPase-driven allosteric changes to MCMs as they interact with incoming bases on the ssDNA in the central MCM channel [10, 19, 30, 36, 125]. The ssDNA passing through the CMG central channel is copied by the leading strand polymerase, DNA Polymerase-ε [125]. The lagging strand of DNA is created at the front of the CMG, where the combined efforts of amino-terminal MCM domains and Cdc45 and GINS separate the double-stranded parental DNA [15, 36, 125, 126]. Ctf4 mediates interactions of the CMG with the lagging strand polymerase, initially DNA Polymerase-α-Primase, then transitioning to DNA Polymerase-δ, but Ctf4 also displays some enhancements to CMG processivity on its own in specific in vitro assays [18, 36, 127]. The CMG itself appears to play an important role in managing the distribution of DNA polymerases between leading and lagging strands, as mutations in GINS reduce recruitment of DNA Polymerase-ε to the leading strand and necessitate use of DNA Polymerase-δ instead [128]. Also interacting with CMGs and facilitating CMG and replisome function during DNA replication is a protein called Mcm10, which stabilizes replication forks and manages replicative stresses [24, 129,130,131,132,133,134,135,136]. In some cancers the Mcm10 locus is amplified and Mcm10 protein overexpression is evident, suggesting Mcm10 may play a role in promoting cancer growth through CMG interactions and changes to CMG or replisome function [129]. During CMG unwinding at forks, Cdc45 also recruits histone-modifying enzymes, including Cdk2, to facilitate replisome movement through higher-order chromatin that is modulated by histone-H1-dependent interactions and compaction [137].
An important point to understand from this discussion is that the MCM hexamer pairs that are converted to double CMG helicases and participate in replisomes represent the starting sites of DNA replication in mammalian cells. These MCMs/CMGs are thus the actual origins of DNA replication (analogous to ori’s) even though specific DNA replicator sequences generally do not define such starting sites in mammalian cells.
Reserve MCM “dormant origins”: keys to replication fork management, responses to replicative stress, and genomic stability
A region of a chromosome that is copied bidirectionally from an origin of DNA replication, now defined as the location where a CMG pair becomes activated, is called a replicon. Perhaps paradoxically, mammalian (and other eukaryotic) cells load significantly more MCM hexamers onto chromatin/DNA, on average, for each replicon than are needed to be converted to a single pair of functioning CMG helicases [40, 41, 52, 138]. Many of these extra MCM hexamers that are loaded are derived from nascently-synthesized MCMs prior to the separation of daughter cells during mitosis, and have not functioned in DNA replication prior to their chromatin accumulation in the next G1 and S-phase [139]. These excessive MCM hexamers that are loaded serve at least three purposes. First, MCM hexamers distributed throughout the chromosomes represent the aforementioned licensing step that marks unreplicated DNA and allows (and ensures) DNA replication to occur once, and only once, in each cell cycle. Second, mammalian cells are “smart” in knowing they will encounter issues that lead to replication fork stalling, also called Replicative Stress (RS). These fork-stalling issues could include heterochromatin, topological constraints, transcription interference, DNA mutation-repair events, chemotherapy, or radiation leading to DNA breaks. Importantly, these excessive MCM hexamers provide a means to recover DNA replication after fork-stalling events, leading to their classification as reserve MCMs, or dormant origins [38,39,40,41]. Finally, using mechanisms that remain poorly understood, the excessive MCMs modulate and restrict replisome fork speeds during S-phase to prevent DNA damage and genomic instability [139].
Mammalian cells load 5–10× the number of MCM hexamers than are required to finish an unperturbed S-phase [138]. Estimates derived from quantitative immunoblotting techniques have suggested that for each 100 kb of mammalian DNA, on average, there are ~4–6 MCM hexamers loaded, meaning ~2–3 MCM hexamer pairs (maximal estimates) [138]. Replicons in mammals range in size, from perhaps 20 kb to as high as several hundred kilobases [140, 141]. Thus, a larger 300-kb replicon is predicted to contain as high as ~6–9 MCM hexamer pairs even though only one pair of MCM hexamers is required as an origin within the replicon [138]. Regardless of precise replicon size and MCM loading distribution within replicons, which is likely stochastic in nature in different parts of the genome, it is clear that there is an excess of reserve and required MCM hexamers. Reduction of MCM levels by 80-90% using siRNA-mediated methods, which does not alone hinder cell growth, but depletes the reserve MCMs, results in loss of cell viability and signs of increased DNA damage when RS is induced by fork-stalling drugs [38,39,40,41]. The reserve MCMs are needed to function as CMG helicases to resume DNA replication and recover or complete S-phase, thus maintaining genomic stability after RS and DNA damage [38]. Under unperturbed S-phase conditions or in the absence of RS (i.e., no drugs), reserve MCMs do not act as origins of DNA replication (only licensing and fork speed regulators/suppressors) and are thus dormant origins [38, 40, 41]. Evidence indicates that the ATR and ATM proteins mediate suppression of these dormant origins, regulating origin usage until stalled fork stress or DNA damage is encountered [142,143,144,145,146].
Although reserve MCMs are available to function as CMG helicases under replicative stress conditions, it is accurate to say that we currently do not know whether reserve MCMs are simply hexamers on DNA lacking Cdc45 and GINS, or instead are fully formed CMGs containing Cdc45/GINS that are inactive until an RS event occurs. MCMs are loaded onto chromatin in excess (~4–6 MCM hexamers/100 kb), but the total protein abundance of Cdc45 in mammalian cells (~0.35 molecules total/100 kb, 25–30,000 molecules/cell) is extremely low [138]. In addition, mammalian Cdc45 is rate limiting for CMG formation [138]. At this time, we are not aware of whether GINS levels have been determined. For these reasons, it is more likely that reserve MCM hexamers do not contain Cdc45/GINS due to low stoichiometric levels of such proteins (minimally Cdc45), and MCM-to-CMG conversion occurs only when RS requires activation of reserve helicases. Regardless of the timing of Cdc45/GINS recruitment, going forward, we will refer to reserve helicases as “reserve MCMs” or “reserve CMGs”, as the distinction is not necessarily relevant to further understanding of the role of reserve CMG helicases in cancer.
The extra MCMs loaded onto mammalian chromatin are not just reserves for recovering from RS conditions. Evidence shows that DNA replication fork/replisome speeds are suppressed or modulated by the extra MCMs [139]. Situations that reduce the presence of the extra MCMs do not always hinder the response to RS, but do result in replication fork speeds being increased 20–25% relative to the normal condition in which all extra MCMs are sufficiently loaded prior to S-phase [139]. Although the biochemical mechanisms underlying this ability of reserve MCMs to regulate fork speeds remain unknown at this time, it has been suggested that the extra MCMs may serve as a sort of roadblock, or rate-limiter, for forks established by other MCM/CMGs, sterically limiting replication forks when reserve MCMs are encountered [139]. The steric limitations may also involve time required to disassemble and remove the dormant extra MCMs and unlicense that region of the chromosome [139]. However, other mechanisms involving molecular signaling between MCMs and CMGs (directly or using soluble factors) cannot be ruled out. Loss of control of replication fork rates with reduced MCM availability results in the presence of asymmetrical replisomes and DNA damage, leading to genomic instability [139]. Intriguingly, a reduction of the normal levels of extra MCMs is predicted to create a genome-destabilizing compounding effect: increased fork speeds and DNA damage during S-phase that then requires the presence of reserve MCMs/CMGs to facilitate recovery from the RS and DNA damage induced by the deregulated fork speeds. Application of this concept to tumorigenesis is described below.
A model emerges in which replicons load a large number of MCM hexamers (around the R-point), but only a few are chosen in late-G1 to be converted to CMG helicases that eventually become activated at G1-S to replicate DNA during S-phase (Fig. 2, MCM pairs in orange, CMGs in color). The reserve complement of extra MCMs “waits” for replicative stress to signal their conversion to CMGs for recovery, and also regulates replication fork speed to prevent DNA damage [139]. Intriguingly, this aligns nicely with how DNA replication origins in mammalian cells have been shown to function. Origins are not rare specific DNA sites defined by replicators, but instead are zones of potential replication start sites [61, 62, 147,148,149,150,151,152], or many potential origins where one origin is chosen. Experiments have shown that MCM proteins are indeed loaded into mammalian origin zones in a distributed manner during G1 phase [153], suggesting that ORC loads excessive MCM pairs in a stochastic manner within these zones. Based on the modeling from the above discussion, it appears then that any pair of these loaded MCMs can be converted to a pair of CMG helicases. What specifies a particular MCM pair to be converted to a CMG pair is not known, but it has been shown that parental MCMs that functioned in some manner in the previous S-phase prior to daughter cell creation at mitosis are preferred in the next S-phase for conversion into CMGs [139]. Regardless of the mechanism, the CMG pair activated then defines the origin in that replicon of that cell. The stochastic nature of this process occurring in a population of cells would be seen experimentally as a zone of potential origins, derived from a zone of MCM paired-hexamers, with different MCM pairs randomly chosen in different cells to become CMG helicases within the zone.

Mammalian cells load an excess of MCM pairs (shown in orange as hexamer pairs) onto DNA, with a small number of select MCM pairs being converted into replicative CMG helicases (near G1/S transition). The extra MCMs serve at least three purposes (shown with red text/arrows). The first role is to mark unreplicated DNA, thus licensing DNA for one round of DNA replication. Second, the extra MCMs regulate replication fork speeds of those MCMs that are converted into CMG helicases during S-phase, maintaining a fork rate that prevents DNA damage. Third, following replicative stress (RS), such as fork-stalling or DNA damage, excess reserve MCM pairs can be converted into active CMG helicases for recovery of DNA replication, likely after DNA repair. See text for details and references.
Why would mammalian cells devise such a plastic and non-specific system for initiating DNA replication from random sites/origins, versus from specific replicators that likely require use in every cell cycle? Indeed, although the extra MCMs are clearly involved in licensing, fork speed regulation, and recovery from replicative stresses, mammalian cells could have evolved these genome-stabilizing mechanisms using other factors or methods (other than relying on MCMs), while demanding replication to begin at specific non-redundant sites. One possibility is that this plasticity allows DNA replication to start and complete from any and all regions of the mammalian genome regardless of the transcriptome that is present. Active transcription in a given region can be an opposing force to DNA replication in mammalian cells, with some studies showing that MCM loading and initiation of DNA replication are reduced in actively transcribed genomic regions (i.e., initiation zones have been shown in some cases to be intergenic) [61, 153,154,155]. Perhaps this MCM/CMG plasticity accommodates differences in gene expression during development and differentiation, guaranteeing flexible starting and completion of DNA replication regardless of any limits imposed by transcription or other issues that might interfere with specific replicators in different developing cell types. However, such a model for MCM preferential loading in intergenic regions due to low transcription activity is likely an oversimplification. Active transcription can occur in intergenic regions [156, 157], and transcription itself plays an active role in stimulating DNA replication origin activity or regulating the timing of initiation [158,159,160,161,162]. In fact, differential transcriptional programs in different cell types have been shown to influence origin firing locations, which may also contribute to flexibility of DNA replication initiation (or ensuring initiation) within various transcriptomes [158]. Clearly, more work needs to be done to understand the molecular mechanisms controlling where and how MCMs are ultimately loaded onto DNA in mammalian cells. Regardless of such specific details, the excess MCM/CMG flexibility supplies a needed set of reserve dormant origins that can suffice for completion of DNA replication should unanticipated (or anticipated) problems occur during S-phase, while also managing replication fork speeds. To use the colloquial, the mammalian cell has devised a means to “hit multiple birds with one stone”, the stone being the excessive MCMs necessary for many tasks.
The CMG is a target of TGFß1 growth arrest and the Rb tumor suppressor
CMGs are not passive bystanders in the cell cycle simply waiting to function in a replisome when the time arises. If one defines S-phase as the start of DNA synthesis by polymerases, and late-G1 phase as the time during which double CMGs assemble (and potentially begin localized DNA melting [23]), then G1-S can be defined molecularly as the point in time when CMGs are activated to unwind DNA in a processive manner within replisomes. This would indicate that all stimulatory growth factor signals must ultimately regulate CMG function, which has been illustrated in the above discussion in terms of positive influences of such signals on CMG assembly/activation arising from the cell cycle machinery. Conversely, inhibitory growth factor signals must prevent CMG function, and this is apparent when assessing the mechanisms used by Transforming Growth Factor ß1 (TGFß1) to achieve cell cycle arrest. TGFß1 is a potent growth-suppressive factor for epithelial, endothelial, and many immune cells that inhibits multiple events in the cell cycle to block progression through the G1-S transition [77, 163,164,165]. This growth-suppressive ability of TGFß1 is often lost in human malignancies due to abrogation of the molecular signaling events that mediate TGFß1-induced cell cycle arrest [166, 167]. As such, understanding the mechanisms utilized by TGFß1 to block cell growth can identify molecular targets that are critical in promoting the growth of tumor cells and could thus serve as a focus for drug discovery efforts aimed at blocking tumor progression.
Well-known targets of TGFß1 signals include suppression of Myc and cyclin expression, and inhibition of Cyclin E/Cdk2 and Cyclin D/Cdk4 kinase activities [77, 166,167,168,169,170,171]. TGFß1 also targets proteins that control Cdk function, including suppression of Cdc25A phosphatase and induction of multiple cyclin-dependent kinase inhibitors (CKI) such as p15INK4B, p21Cip1, and p27Kip1 [172,173,174,175,176,177]. Suppression of these kinases prevents phosphorylation of the retinoblastoma protein (Rb), which normally allows cells to progress into S-phase [77, 124, 165, 178, 179]. Important in these events is the role Rb plays in mediating growth arrest by TGFß1, being a collective target of all of these inhibited proteins and enzymes that TGFß1 negatively regulates. However, studies have shown that Rb is not always necessary for TGFß1 to achieve cell cycle arrest, and growth inhibition by TGFß1 can occur in the absence of Myc suppression or in cells lacking the aforementioned CKIs [77, 124, 172, 180,181,182,183]. Added to this, overexpression of Myc alone can override these inhibitory events targeted by TGFß1 and promote G1-S transit, indicating that Myc is epistatic to the negative effects of TGFß1 and positively affects a factor(s) in G1 phase necessary for entry into S-phase [184,185,186]. The mechanisms mediating TGFß1 inhibition of CMG helicase assembly and activation can provide explanations for these findings.
The effects of TGFß1 on the CMG depend on the timing in G1 phase when cells are exposed to TGFß1, and the status of the Rb protein (Fig. 3). In cells expressing Rb, TGFß1 exposure in early G1 blocks MCM expression, along with suppression of Myc expression, and inhibition of Cyclin E/Cdk2 activity later in G1 [77, 124, 165,166,167,168, 170, 171, 178]. The absence of MCM expression will block progress through G1, and lack of Myc and Cyclin E/Cdk2 activity are guarantors of no G1-S transit [77, 124, 165, 168, 170, 178]. However, when Rb-containing cells reach late-G1 and are exposed to TGFß1, these events in early G1 have passed and CMGs have assembled (or are assembling). In late-G1, TGFß1 relies on Rb to bind and inhibit CMGs from becoming active at G1-S, and the more phosphorylated form of Rb can do this [124].

Many of the conventional targets for TGFß1-induced growth arrest, including Myc and Cyclin E-Cdk2 (and Cdk-inhibitors, CKIs), regulate steps involved in MCM/CMG functionality. TGFß1 targets and suppresses Myc, Cyclin E-Cdk2, and MCM expression and abrogates CMG assembly/activation, thereby using a pleiotropic set of inhibitory signals on the CMG to ensure a block to cell cycle progression. TGFß1 also targets MCM hexamer assembly in middle G1, or CMG activation at G1-S, depending on the RB status of the cells (lower right).
Curiously, work performed by our group in two related studies has shown that in certain mammalian cells lacking Rb (mouse keratinocytes, MK) these TGFß1-induced mechanisms controlling the CMG are upended. Myc, Cyclin E/Cdk2 kinase, and MCM expression are no longer inhibited by TGFß1 exposure in early G1 when Rb is missing, and in late-G1, without Rb, TGFß1 cannot inhibit any CMGs that do form [77, 124]. Nonetheless, despite these severely debilitating effects on TGFß1’s ability to block cell growth in the absence of Rb, TGFß1 continues to inhibit the cell cycle, albeit only when added to cells that are in early G1 [77, 124]. While future work by other groups may expand upon these findings with additional insight, one mechanism identified involves an ability of TGFß1 to inhibit MCM hexamer assembly/loading, derived at least in part from the suppression of a Cyclin E-Mcm7 interaction and reduced association of active Cyclin E/Cdk2 kinase with chromatin [77]. Overexpression of Cdt1 can override these particular TGFß1 arrest mechanisms in Rb-lacking MK cells, restoring MCM assembly and G1-S transit [77]. This would suggest that another oncogenic role for Cdt1 may include abrogation of growth-inhibitory TGFß1 signals in cells lacking Rb.
The mechanisms by which Rb binds and inhibits the CMG provide answers to some questions regarding how Rb regulates G1-S transit and plays an important role in tumorigenesis. The combined work from our group and others has found that Rb uses a bi-partite mechanism to block the start of DNA replication: inhibition of the CMG and abrogation of Ctf4 and DNA polymerase interactions with the CMG/replisome [32, 187, 188]. These functions of Rb derive from specific exons located in the amino-terminal half of Rb (RbN). A domain in RbN comprised of exon5/6 (called the Projection) interferes with Ctf4/Polα recruitment to replisomes/CMGs, while exon7 (Ex7) inhibits the CMG [187, 189]. These exons are often lost in familial inherited retinoblastomas that are lower penetrance diseases, compared to higher penetrance retinoblastomas derived from mutations or loss of the carboxy-terminus of Rb where the E2F proteins are known to interact [187, 189,190,191,192]. Importantly, both of these RbN exon domains are additive and independent in their ability to control DNA replication, but are only individually lost in a particular inherited cancer lineage [187, 189, 190]. Thus, partial penetrance can be explained by an inherited Rb allele with reduced inhibitory functions toward either the CMG or Ctf4/Polα, but not both, depending on the specific exon that is lost from the RbN domain [187]. Notably, there are other proteins that interact with RbN [189, 193], so the mechanisms mediating partial penetrance are likely more complex than simply claiming the CMG and Ctf4/Polα as explanations.
RbN has been shown to provide a necessary function for inhibiting G1-S transit, independent of the carboxy-terminus of Rb [187, 194, 195]. The control over Ctf4/ Polα and the CMG using specific exon domains of RbN discussed above provides a molecular understanding [32, 187]. RbN binds to the CMG through direct interactions with Mcm7, which are diminished when Ex7 is missing from RbN [32, 124, 187, 188]. Full-length Rb can also bind to the CMG, and this is independent of phosphorylation status of the carboxy-terminal region of Rb [124, 187, 188]. As such, the CMG remains a direct target of Rb in late-G1, after the Restriction Point when Rb becomes phosphorylated [98, 124, 187, 196]. This explains why Rb can mediate TGFß1 inhibitory signals toward the CMG in late-G1, and why RbN is required to mediate control over G1-S transit [124, 187, 194, 195]. Importantly, it is not known how Rb/RbN biochemically controls the CMG via Mcm7 interactions [187]. Does Rb inhibit ATP hydrolysis or fork-unwinding activities of the CMG? Or does Rb interfere with an unknown partner for the CMG? Future studies may uncover answers to these questions. Besides regulating G1-S transit, Rb is also important for mediating ongoing DNA replication arrest in cells exposed to ionizing radiation or fork-stalling drugs [197,198,199]. The absence of Rb leads to errant and excessive DNA replication, creating a form of genomic instability called hyperploidy [197]. Though not formally shown, a plausible target of Rb in suppressing DNA replication and hyperploidy under such conditions may be the CMG helicase.
Reserve MCM/CMGs resemble tumor suppressors in experimental models
CMG helicases are quite logically involved in promoting cell cycle progression and DNA replication, responding to positive growth factor pathways and the cell cycle, and being targeted for inhibition by negative growth factors such as TGFß1. At first glance, this could suggest the possibility that CMGs (and MCMs) might be found in some human malignancies to function as growth drivers, analogous to oncogenic growth drivers. However, there is an absence of genetic evidence in human cancers showing that components of the CMG (or the CMG helicase on the whole) are oncogenic drivers of tumorigenesis, mutated or amplified with gain-of-function outcomes as occurs with known oncogenic drivers (see discussion below). On the contrary, in certain experimental models MCM/CMGs actually display qualities of tumor suppressors, and this almost certainly derives from mismanagement of the extra reserve MCM/CMG complement in cells. Although there are currently no genetic demonstrations that MCM/CMGs are, in fact, tumor suppressors in any particular human malignancy, genetic studies in mice suggest that intact wildtype MCM proteins function to suppress tumor initiation [200,201,202,203].
In one study, mice carrying a single engineered mutation in the Mcm4 gene referred to as the Mcm4Chaos3 allele, but no other genetic changes in oncogenes or tumor suppressor loci, are tumor prone [203]. Tumors that arise include primarily breast adenocarcinomas, but also some lymphomas or histiocytic sarcomas [203]. Why this limited set of tumors arises when Mcm4 is mutated, which could affect many cell types in the animals, is a curiosity that remains unexplained. Fibroblasts from Mcm4Chaos3 mice have increased DNA damage, stalled replication forks, and activated fork recovery events even though such cells were not subjected to replicative stress from outside influences such as fork-stalling drugs [202, 203]. The unperturbed S-phases in Mcm4Chaos3 fibroblasts are stressed for DNA replication processes, and biochemically this is derived from the presence of weakened MCM hexamers [202]. The mutant CMGs created from these Mcm4Chaos3 MCM hexamers are enzymatically unhindered when tested in vitro in assays examining fork-unwinding ability [202]. However, the mutation in Mcm4 results in an apparently destabilized Mcm4 protein and an associated reduction in Mcm7 protein level [203]. This leads to a lowered chromatin binding capability by the entire complement of MCM hexamers, yielding functionally weakened MCM hexamers, including the extra reserve MCMs [202]. Based on our understanding of the roles the extra reserve MCMs play in cells, weakened MCM reserves render such cells less capable of responding to any normal fork-stalling events that occur during DNA replication, but likely also contribute to changes in replication fork speeds that yield faster, lower fidelity replisome movement [139]. Together, these events lead to DNA damage and increased replicative stress, destabilizing the genome [139]. Intriguingly, the RS induced in these unperturbed fibroblasts from the MCM4Chaos3 mice appears to be accepted by the cells, being low enough to evade cell cycle checkpoint arrest of cells [202]. This ongoing RS causes problems for chromosome segregation and allows acquisition of genomic deficiencies, likely driving the tumorigenesis that is seen [202]. These results indicate that failure to developmentally maintain a proper, healthy complement of MCMs, particularly MCM reserves, leads to DNA stress over time that can promote genomic instability and evolutionarily drive tumorigenesis.
A similar situation exists in mice with reduced expression of Mcm2 protein. Using genetic modifications to the Mcm2 locus, investigators found that reducing Mcm2 expression to approximately one-third of normal levels, which would diminish the extra reserve MCMs (and did co-reduce Mcm7 protein), also results in early onset cancer development [200, 201]. Tumors that appear include B-cell and T-cell lymphomas, thymomas, liver cancers, and lung cancer, and polyps were present at times in the intestine and colon [200, 201]. While there is some overlap of lymphoma development between these Mcm2-deficient mice and the MCM4Chaos3 mice, it is again curious that only a limited number of tumor types arise. The authors noted that the genetic background of the Mcm2-deficient mice influences tumor outcomes [200, 201], suggesting that tumor development due to MCM mutations involves additional unknown genetic conditions, perhaps in certain cell types, to promote specific tumor formation in these mouse models. Under conditions of replicative stress, fibroblasts from mice with diminished Mcm2 display low levels of replication origin usage [200]. Under normal conditions, slight elevation in DNA damage response indicators was present but the cells seemed accepting of it [200], similar to the Mcm4Chaos3 situation [202]. Although not shown experimentally, the small increase in damage could be due to reduced origin usage in the context of lowered Mcm2, leading to incomplete DNA replication. When mated to mice lacking p53 expression, a significant reduction in viable offspring is seen, since the presence of low levels of Mcm2 concurrent with an absence of p53 reduces viability of cells during embryogenesis [200]. However, in the small number of mice that are born a more rapid onset of tumorigenesis is seen. One reason for this appears to be an increase in genomic damage at the cellular level in mice with both reduced Mcm2 and p53 loss compared to mice with only Mcm2 deficiencies [200]. Reduced MCM hexamers produce an environment that renders cells less capable of controlling replication fork speeds and recovering from resultant DNA damage [139]. Loss of p53 likely intensifies the genomic instability that ensues, removing an important tumor-suppressive DNA damage sensor, thereby leading to a synthetically lethal condition in many embryonic cells or allowing increased DNA damage to remain in cells that survive [200].
The results from these mice studies demonstrate that small changes in MCM reserves (dormant origins, and extra MCMs that regulate fork speeds) lead to increased genomic instability and tumorigenic outcomes. Importantly, mutations or reduced functioning in MCMs resemble that of p53 or Rb mutations/loss in tumorigenesis, where genome stability is reduced in the tumor evolutionary process due to loss of tumor suppressor function. Thus, cells must maintain proper reserve MCM functionality, which is tumor-suppressive, while mismanagement of reserve MCMs is a tumor-driving situation.
Oncogene overexpression mismanages CMGs and creates replicative stress
Certain oncogenes have been found to mismanage MCM/CMG assembly and/or activation, suggesting a mechanism by which they can drive tumorigenesis though debilitation of the reserve complement of MCM/CMGs [3]. Although Myc is conventionally thought of as a transcriptional regulator, Myc also has an important non-transcriptional role in regulating activity of CMG helicases [4,5,6, 204]. Myc regulates the assembly of CMGs by promoting the recruitment of Cdc45 and GINS to MCM hexamers [5, 6, 204]. Myc achieves this in part by recruiting two histone acetyltransferases, GCN5 and Tip60, to Myc-bound chromatin sites, thereby leading to decondensation of higher-order chromatin and creation of access for Cdc45 and GINS to bind to resident MCM hexamers [204]. A specific domain of Myc, Myc-Box II, is required for this stimulation of CMG assembly and activation [204]. Myc promotion of CMG function also involves regulation of necessary kinase activity [5]. Intriguingly, there are hints that Myc may stimulate CMG activity in a more direct manner, since Myc is found in complexes with Cdc45 when CMGs are stimulated and Myc can interact with the Mcm7 protein [204, 205]. However, a direct role for Myc in CMG binding and stimulation awaits further investigation. These mechanisms underlying how Myc regulates CMG assembly, together with the many other roles for Myc in promoting transcription and other cell cycle events, collectively explain why Myc can override TGFß1 growth arrest [184] (Fig. 3) and promote S-phase entry. Intriguingly, Myc cannot override TGFß1 if Myc is overexpressed specifically in late-G1 [184], perhaps because Myc will have no MCM hexamers or CMG components to stimulate if TGFß1 has blocked MCM/CMG presence earlier in G1 [77].
Overexpression of Myc leads to over-activation of CMG helicases (Fig. 4) [3, 5, 6, 204]. These over-activated CMGs decrease inter-origin distances and are necessarily derived from the reserve pool of dormant reserve MCMs being converted to CMGs [5]. Such effects of Myc overexpression on the CMGs are a problem for cells, as excessive Myc is known to induce replicative stress in an acute manner, far too quickly to be explained by changes in transcription profiles such as metabolic gene stimulation [5, 6, 206]. Myc overexpression produces acute onset of genomic instability, fork rate slowing, fork asymmetry, and DNA damage [3, 5, 6, 206], with such outcomes aligning with observations seen when extra MCM reserves are reduced [139]. The DNA damage induced by Myc strictly requires CMG over-activation, and experimental approaches that promote excessive Cdc45 or GINS recruitment to MCMs can phenocopy these effects of Myc in creating acute DNA damage responses [5]. Thus, one mechanism by which overexpressed Myc can drive tumorigenesis is through excessive CMG stimulation and consequent mismanagement of the reserve complement of extra MCM/CMGs, which creates DNA damage. Another mechanism derives from the Myc-induced increase in replication fork density and resultant structural problems within certain segments of the genome [5]. Myc-induced fork density changes and reduction of MCM reserves likely compound this situation to reduce genome stability. Ironically, deregulation of the CMGs (and MCM reserves) by Myc may create the initial replicative stress in terms of altered DNA replication fidelity and fork stress, and to recover from such RS cells need the full complement of reserve MCM/CMGs that Myc has perturbed. Thus, elevated Myc would appear to cause and exacerbate genomic instability. This ability of Myc, a cancer-driver overexpressed in the majority of human malignancies, to create MCM reserve deficiencies as a likely secondary driver for cancer development is similar in concept to reduced MCM functionality driving cancer in the Mcm mutant mouse models.

Oncoproteins such as Cyclin E or Myc, when overexpressed, cause problems in the dynamics of MCM loading and usage. Cyclin E elevation suppresses MCM loading (black squares), while Myc elevation leads to excessive stimulation of CMG helicases (black arrows). Altering MCM/CMG levels adversely affects replication fork speeds and origin usage, and reduces MCM/CMG reserves. These oncogene-driven effects on the extra reserve MCM/CMGs generate replicative stress (RS) and DNA damage, and create a CMG vulnerability wherein tumor cells have difficulty responding to the RS, or to additional fork-stalling or fork-destabilizing stresses.
Overexpression of Cyclin E also causes mismanagement of MCM reserves [3, 7]. Elevated Cyclin E is associated with acute onset of genomic instability, and promotes abnormal origin firing, collapsed replication forks, and double-strand DNA breaks [207,208,209], again aligning with that seen when the extra reserve MCMs are reduced [139]. Cyclin E deregulation also appears to contribute to chromosomal rearrangements and genome duplication that are present in cancers [207, 209]. At the mechanistic level, these events likely derive in part from an ability of Cyclin E overexpression to cause a reduction in the number of MCM hexamers that are loaded onto DNA (Fig. 4) [7]. This necessarily depletes the reserve pool of MCM/CMGs that are needed for responding to RS during S-phase and limiting DNA replication fork speeds to prevent DNA damage [139]. As discussed above, Cyclin E-Cdk2 plays complex roles in MCM assembly, appearing to promote MCM subunit chromatin binding, but limit MCM hexamer assembly when deregulated [7, 84, 85]. Deregulated Cyclin E also causes premature entry into S-phase, which may contribute to the MCM deficiency by promoting S-phase entry before enough MCMs have loaded [209]. Thus, similar to the situation for Myc, elevated Cyclin E creates RS due to MCM reduction and other mechanisms that is then difficult to recover from due to a reduction of dormant reserve MCM origins.
Notably, Myc or Cyclin E may not affect all MCM (reserve) hexamers in all parts of the genome evenly. Depending on the timing and degree of Myc or Cyclin E overexpression, or their physical locations of DNA interactions, certain regions of the genome may be more susceptible to altered MCM/CMG management by these oncogenes. In addition, the transcriptome of a particular cell may influence chromatin states and locations where oncogenes more adversely affect MCM/CMG loading levels. It is currently unclear whether mammalian MCMs are loaded onto chromatin in an evenly-distributed manner or asymmetrically throughout the mammalian genome, nor if certain chromosomal regions contain more or fewer local MCMs relative to other domains. However, in yeast, it has been shown that certain regions of the genome have enriched levels of dormant MCMs loaded onto chromatin [210]. As such, one could speculate that if regions of the mammalian genome exist with a lower density of local MCM reserves, then oncogene mismanagement of MCMs in such regions could yield more RS and DNA damage relative to regions that have higher MCM densities. In sum, oncoproteins such as Myc or Cyclin E may produce tumor heterogeneity at the genomic level as a result of stochastic MCM mismanagement and DNA damage that drives evolutionary selection of tumor cells with certain growth advantages.
Related to the above oncogene-induced effects, MCM mismanagement and genomic instability can also be induced by whole genome duplication events, independent of oncogene activation [211]. Cells that are induced to become tetraploid display elevated DNA damage during DNA replication in the first S-phase following a genome duplication [211]. Regions of the tetraploid genomes are under-replicated, while other regions are over-replicated. At the mechanistic level this appears to be due to a diminished level of necessary DNA replication factors, including MCM and Cdc45 components [211]. Intriguingly, a logical interpretation of these results is that MCM/CMG reserves are mismanaged in the tetraploid cells, being reduced stoichiometrically relative to the increased DNA content. Once MCM/Cdc45 levels (and other factors) were increased to accommodate the extra genomic material, the cells displayed less genomic instability [211]. Such findings are consistent with a need to tightly regulate the levels of MCM/CMG complexes in a cell to maintain genomic stability.
Human cancers and the “missing mutations” in MCM/CMGs
Given that mismanagement of reserve MCM/CMGs can (experimentally) promote tumorigenesis, and that oncogenic pathways can cause such MCM/CMG mismanagement and DNA damage, one might predict that at the genetic or protein levels the CMG itself or some of its subunits will be mutated, overexpressed, or under-expressed in human cancers, with such CMG genetic alterations resulting in cancer-driving conditions or loss of tumor-suppressive function, similar to that observed for well-established cancer-drivers or tumor suppressors (e.g., Myc, Ras, p53, or Rb genes). However, to date, genetic evidence is lacking that can demonstrate CMG components are oncogenic cancer-drivers or tumor suppressors in human cancers.
When examining many human tumor tissue samples by immunohistochemical (IHC) methods one typically sees elevated expression of all CMG subunits analyzed, often referred to as “overexpression” of CMG subunits that might suggest a cancer-driving or cancer-promoting situation. For a thorough review and collation of such tumor analysis findings the reader is referred to a comprehensive discussion [212]. Analysis of elevated CMG subunit expression in human tumor tissue has also been used as a novel biomarker to identify malignant and pre-malignant tissue and as a predictor of survival, with higher MCM, Cdc45, or GINS levels, for example, often associated with worse outcomes [25, 26, 213,214,215]. However, elevated tumor tissue expression of CMG components in histopathological samples does not provide conclusive evidence that, on a per cell basis, the CMG subunits (all 11) are actually overexpressed stoichiometrically relative to the number of CMG subunits expressed in normal (non-tumor) proliferating cells. There is evidence that in some established cancer cell lines several of the CMG components may be overexpressed, sometimes based on mRNA levels [25, 27, 28, 212]. However, it has also been shown in a quantitative study that proliferating tumor lines and non-tumor proliferating cells contain roughly equal protein numbers of the CMG subunits [138]. Quite clearly, elevated IHC staining for CMG subunits indicates that CMGs are more visible in proliferating tumor tissue relative to that seen in non-tumor, differentiated, and non-proliferating (or lower-proliferating) neighboring tissue. However, elevated IHC staining observations will be evident for many proteins involved in cell cycle progression because tumor tissue is in a proliferative state. Importantly, higher IHC staining or elevated expression in a tumor cell line does not demonstrate that such “overexpressed” proteins are drivers of the cancers.
Information from large-scale genomic sequencing efforts on human tumor samples indicates that the CMG enzyme is a “never-mutated” protein complex in tumor cells, from the perspective of failing to find demonstrable CMG cancer-driving or inactivating mutations. A search of more than 68,000 human tumor samples analyzed in 205 studies in the publicly available database cBioPortal (www.cbioportal.org; accessed October 23, 2022) [216, 217] finds that, while the loci coding for the 11 CMG subunits are mutated or amplified to a small degree in some cancers (~1–3% for each CMG subunit in this analysis), a refined search for each of the CMG subunit loci finds that known cancer-driving or inactivating alterations or hot-spot mutation sites are not evident (Fig. 5A, B). This is in contrast to known cancer-driving mutations/amplifications in Myc, Ras, or other oncogenic loci that are highly evident across human cancers, as are loss-of-function or inactivating mutations in the loci for tumor suppressors such as p53 and Rb. Human cancers have elevated DNA damage and mutation rates, which predicts that even the 11 CMG subunits will be subject to stochastic genetic changes in at least some human cancers, consistent with the data in the cBioPortal collection. However, using Mcm7 coding region mutations/alterations as an example (Fig. 5C), the mutation profile residing in human cancers for Mcm7 is indicative of randomly distributed passive mutations throughout the coding region, and lacking potentially interesting hot-spot driver alterations. The cBioPortal dataset shows that other CMG subunits have similar mutation profiles to Mcm7, and not unlike that also seen for ß-Actin, which is arguably not a cancer-driver.

A Search results from the publicly available cBioPortal database of genetic analyses on 68,088 tumor samples from 64,959 patients in 205 independent studies (performed October 23, 2022) for mutations, amplifications, deletions or other alterations in the 11 CMG helicase components. The small percentages that each CMG subunit are altered across all human tumor samples analyzed are indicated on the left (asterisks), and grouped by alteration type. CMG components are altered in 4949 (7%) of all tumor samples. Study origins used for tumor analyses are indicated at the top by color coding (see cBioPortal site for publicly accessible information and details of study origins). GINS1-3 are Psf1-3, respectively; GINS4 is Sld5. B Refined cBioPortal search removing mutations, variants, and copy number changes (n = 7280) of unknown significance of the same 68,088 human tumor sample cohort based on sites-option set to filter based on current understanding of such genetic changes. C Mutation profile for Mcm7 obtained from cBioPortal search of 68,088 human tumor samples (majority are missense mutations). Mcm7 N-terminal and ATPase domains with homology to other MCM subunits are shown in green and red, respectively. The number of times a given change in Mcm7 is found in sampled human cancers is indicated by the Y-axis. Note that unlike that seen for many oncogenes or tumor suppressors, hot-spot mutation sites that might indicate cancer-promoting changes are not evident for Mcm7, and the profile is instead that of randomly distributed passive mutations across human cancer samples.
Importantly, while current research information suggests that there are no CMG mutations in human cancers known to be capable of driving tumorigenesis, future studies might uncover such a situation (e.g., an Mcm cancer-driving mutant) in at least some human malignancies. In addition, it is possible that deregulation of individual CMG subunits might affect cell growth for reasons independent of CMG enzyme control itself. For example, elevated Mcm7 expression has been seen in prostate cancers, and can promote aggressive characteristics of prostatic tumors [218]. Although unclear, one mechanism underlying this could be derived from the ability of Mcm7 to bind Rb and interfere with Rb signaling pathways [32, 124, 187, 188]. However, it has also been found that overexpression of Mcm3, Mcm7, or Cdc45 as single subunits is not easily tolerated by mammalian cells and can elicit a checkpoint response with failure to enter S-phase [84, 85, 138, 219], suggesting that the stoichiometry of the 11 CMG subunits is important and necessary for cell cycle progression and cell viability. Although CMG (and MCM) deficiencies can cause tumorigenesis, as shown in genetic-based animal studies, and CMGs/MCMs can be mismanaged by oncogenes to create replicative stress that promotes tumor formation, the paradox currently exists that the CMG helicase is simply not mutated in human cancers in a cancer-driving or function-inactivating manner similar to other known oncogenic drivers or tumor-suppressors. To use Abraham Wald’s analogy to the missing bullet holes [1], the CMG is “missing mutations”.
Why would this paradox exist for the CMG? The tumors analyzed by scientists are the final products of an evolutionary selection process for survival. It is quite possible that early in the tumorigenic conversion process CMG mutations might occur, or even later in cancer development due to elevated mutation rates in cancers. Importantly, any such mutations in CMG subunits would affect the entire complement of CMG helicases (and MCMs) throughout the genome. However, this may not be compatible with survival if other necessary oncogenic or tumor-suppressive genetic changes occur in the same tumor cell. For example, most human tumors display genetic changes to p53, leading to its loss or diminished function. However, loss of p53 function in the presence of reduced reserve MCMs, which promotes tumorigenesis in the small number of mice that are born, also creates a synthetically lethal situation that significantly reduces viability due to increased genomic DNA damage [200]. When a p53 mutant tumor cell also acquires Myc overexpression, for example, to drive changes in CMG reserves (and other Myc-driven changes to transcription), then survival of the tumor cell may be seriously compromised if the entire complement of MCM/CMG helicases is also mutated or altered in function in any further way. In this manner, the CMG will be “missing mutations” due to a loss of CMG-mutated cells from the final evolutionarily determined tumor profile, similar to how Wald’s aircraft with missing bullet holes fails to arrive at the airbase due to their loss in battle [1].
Targeting CMGs: a specific liability for tumor cells
The ability of oncogenes such as Myc or Cyclin E to drive MCM/CMG mismanagement, while considered a “strength” or selection advantage that promotes genomic instability, heterogeneity, and tumorigenesis from the overall tumor’s perspective, actually creates an exploitable vulnerability for individual tumor cell survival. Normal, non-tumor (host) cells that are proliferating have no (or fewer) mutations/alterations in Myc, Cyclin E, p53, or Rb, all of which are involved in MCM/CMG reserve mismanagement when deregulated in tumor cells. Therefore, non-tumor cells contain a full complement of MCM/CMGs, including reserves, and have the capacity to properly respond to replicative stresses that they encounter, and maintain proper replications fork speeds [139]. However, tumor cells that have acquired MCM/CMG reserve deficiencies due to oncogenic changes will likely have a reduced capacity to respond to replicative stresses and manage fork speeds. Such tumors would have a selective disadvantage relative to normal cells in responding to chemotherapy drugs that stall replication forks [38]. But more importantly, compared to non-tumor cells, tumor cells with reduced MCM/CMG reserve capacity would be predicted to be sensitive to CMG inhibitors (CMGi) that might deregulate replication fork speeds, create further replicative stress, and simultaneously inhibit any remaining MCM/CMG reserves from aiding in recovery from CMGi-induced stress. Thus, whereas normal cells have a “buffer” of reserve MCM/CMG capacity, tumor cells with MCM-mismanaging changes lack such a buffer, or their buffer is diminished, and are predicted to be selectively vulnerable to further CMG inhibition by a pharmacologic CMGi drug or other RS-inducing insults, or both (Fig. 6).

Deficiencies in MCM/CMG management in tumor cells create a selectiveness weakness in cancers that can be taken advantage of in the clinic with drugs designed to target MCM/CMG complexes. Oncogene-induced reduced functionality of MCM/CMG complexes, together with synthetically lethal conditions when p53 is lost and MCMs are co-reduced, renders the MCM/CMG complexes a unique vulnerability. The “missing mutation” status of MCM and CMG complexes also defines the helicase as a needed survivability factor in cancers.
The genetic interactions between p53 loss and reduced MCM/CMG reserves present another argument for tumor cells having an inherent and selective vulnerability to CMG inhibition. Loss of p53 function in a background of reduced MCM levels leads to appreciable embryonic lethality [200], indicating that a synthetic lethality is created, or nearly so, such that viability of cells is severely diminished. Replacing MCM loss/reduction with a CMG inhibitor would potentially phenocopy this loss of viability in cells lacking proper p53 function. Again, this would be a tumor-specific situation, likely exacerbated by gains in Myc or Cyclin E expression that further debilitate MCM/CMG reserves. As such, p53 mutations/loss alone are a likely predictor of sensitivity to pharmacological CMG inhibition (Fig. 6).
Illustrating that tumor cells are indeed selectively sensitive to CMG inhibition are results showing that pancreatic ductal adenocarcinoma (PDAC) and colorectal cancer (CRC) cells are reduced in viability when RS is applied to them under conditions where MCM levels are reduced by siRNA-mediated genetic methods [38]. In these experiments, the reserve pool of MCMs was reduced, which did not alone affect tumor or normal cell growth. However, both tumor types were sensitized to RS-inducing standard-of-care chemotherapy drugs when MCM reserves were diminished [38]. The PDAC cells in particular were shown to be selectively reduced in proliferative capacity relative to non-tumor human skin cells, the latter of which would have a buffer of MCM reserves [38]. PDAC are often driven by mutations in K-Ras, which is known to have downstream positive effects on Myc and Cyclin E protein expression levels [220,221,222,223,224,225], and CRC are associated with changes to Myc and Ras [226, 227], consistent with such tumor cells likely mismanaging the MCM/CMG reserves. These studies indicate that a therapeutic window exists in these and likely other tumor cells for sensitivity to pharmacologic inhibition of the CMG.
Drugging the CMG with CMG inhibitors (CMGi)
In the anti-cancer arsenal, there are many efficacious drugs targeting enzymes directly or indirectly that comprise the replisome and associated factors necessary for DNA replication. Topoisomerases are targeted with doxorubicin, etoposide, and the camptothecan family of drugs, while DNA polymerases are indirectly targeted by interfering with nucleotide pools (e.g., methotrexate inhibiting DHFR, gemcitabine inhibiting ribonucleotide reductase). Clearly, the human CMG (and MCM complex) also has merits as a DNA replication target for anti-cancer approaches, and it is a druggable enzyme (see also discussion below on therapeutic considerations). An attractive and tractable means of developing CMGi against the CMG could focus on identifying compounds that inhibit one or more ATP clefts of the MCM core of the CMG.
The binding and hydrolysis of ATP by kinases typically occurs within a pocket generated by a single polypeptide chain. The ATP catalytic clefts of the CMG helicase are distinct from this design. ATP binding and hydrolysis occurs within catalytic clefts formed between each pair of MCM dimers that make up the MCM hexamer [10, 19, 30, 228, 229]. This mode of ATP binding and hydrolysis by the CMG is similar to that found in other eukaryotic and viral hexameric helicases such as SV40 large-T antigen [230]. In each cleft that is created by adjacent MCM subunits, one MCM subunit (the cis subunit [230]) contributes the P-loop (canonical sequence GXXGXG) with phosphate-interacting “GKT/S” motif, while the other subunit (the trans subunit [230]) contributes a necessary arginine finger motif [10, 19, 42, 228, 229]. Using the ATP cleft of the Mcm3-7 pair as an example, the Mcm3 trans subunit provides the catalytic arginine residue, while the Mcm7 cis subunit provides the ATP binding P-loop with phosphate-interacting lysine residue (in Mcm7 sequence is MGDPGVAKSQ; K is amino acid 387). The six distinct ATP hydrolytic clefts of the MCM hexameric core of the CMG work in a non-symmetrical and combinatorial manner to hydrolyze ATP and alter MCM subunit structures to achieve CMG movement during DNA unwinding, and are required for MCM hexamer loading during G1 phase [9, 10, 19, 20, 42]. Thus, CMGi targeting one or more ATP clefts will likely inhibit MCM assembly and CMG function and would represent a new class of anti-cancer compounds.
An alternative approach to developing CMGi could focus on the identification of compounds that mimic and interfere with the binding site interactions between Cdc45 and MCM subunits, or between the GINS complex and the MCM hexamers (or between GINS subunits). Although more difficult to develop than ATPase inhibitors, such peptidomimetic chemical compounds would likely have a different mode of action for cell growth inhibition relative to compounds that inhibit ATPase activity of the MCM hexamers. Blocking Cdc45 binding, for example, would inhibit CMG assembly and possibly cause dissociation of Cdc45 from active CMG helicases during S-phase, thereby inhibiting only CMG activity. MCM assembly onto DNA occurs in G1 and requires ATP binding and hydrolysis [20, 42], but does not require Cdc45 or GINS association. As such, drugs that interfere with Cdc45 binding to the CMG would not affect MCM binding to DNA, sparing MCM hexamers and particularly reserve MCMs, from drug effects. In contrast, drugs that target MCM ATPase function would affect MCM assembly, reserve MCM levels, CMG assembly, and CMG activity. Due to the wider set of events that would be suppressed by MCM ATPase inhibition, it is possible that such an approach could elicit more toxic side effects in the clinic, particularly when considering systemic off-tumor effects on non-tumor host cells. However, it is also possible that MCM ATPase targeting, versus inhibiting Cdc45 binding and CMGs alone, might offer clinical anti-cancer advantages due to the ability to target and reduce the assembly of reserve MCMs in tumor cells that already contain weakened MCM reserves due to oncogenic signals.
Therapeutic hypotheses and considerations for using CMGi as anti-cancer agents
Several arguments have been made in this review justifying the use of pharmacologic inhibitors of the CMG as anti-cancer agents in the clinic. First, tumors carrying mutations or elevated expression in Myc or Cyclin E (and likely Ras) are predicted to have mismanaged the CMG helicases by reducing reserves. This leads to increased RS, a diminished response to RS, and faster (lower fidelity) replication fork speeds [139]. Second, tumors with p53 (TP53) mutations or loss of function are predicted to contain an environment that is sensitive to CMG inhibition, producing synthetically lethal conditions. Third, the CMG is absolutely required for G1-S transit and tumor cell survival, and this particularly applies to any remaining levels of MCM/CMG reserves after oncogenic mismanagement. Indeed, the fact that the CMG is “missing mutations” (activating, or loss of function) in human cancers is a tumor-evolutionary indicator that at least a nominal level of CMG function is necessary for tumor survival. Notably, these arguments and oncogene-induced CMG weaknesses are specific for tumor cells relative to normal host cells, suggesting tumor cells will be selectively sensitive to CMGi, ideal for a chemotherapeutic target (Fig. 6).
A question that can be raised is whether future CMGi will have advantages to current chemotherapeutic treatments in the clinic. Many standard-of-care chemotherapy drugs directly or indirectly target enzymes involved in DNA replication, such as topoisomerases, ribonucleotide reductase, dihydrofolate reductase, or DNA polymerases (doxorubicin, etoposide, gemcitabine, methotrexate), or create DNA damage (platinum drugs, alkylating agents). Tumor cells are generally more sensitive to these compounds due to increased proliferation, heightened ongoing DNA damage, or deficiencies in DNA repair relative to normal host cells. However, most of these chemotherapy drugs have elevated toxicities in patients, with narrow therapeutic windows. One argument for this is likely that normal host cells also require these enzyme functions, particularly cells that are proliferating, and as a general rule these chemotherapy enzyme targets are not themselves known to be functionally weakened in tumor cells (nor in normal cells). The CMG helicase is likely also a viable chemotherapy target for these same reasons, being necessary for DNA replication and responses to DNA damage, and it is also true that CMGi could elicit some off-tumor toxicities due to similar roles for the CMG in normal host cells. As such, the development of future CMGi would provide an alternative anti-cancer chemotherapy agent, with different pharmacologic properties, that could be similarly useful against select cancers.
However, there are also unique advantages to using CMGi in place of chemotherapy (or with chemotherapy as a sensitizer) due to inherent weaknesses and mismanagement of the MCM/CMG complexes created by oncogenic changes (e.g., Myc or Cyclin E overexpression) and other tumor-specific vulnerabilities discussed above. In tumor cells, mismanagement problems render the MCM/CMG complexes, as drug targets, mechanistically distinct weaknesses compared to current chemotherapy enzyme targets. We propose that targeting the MCM/CMG complexes with future CMGi/MCMi offers the potential for an innovative tumor-selective molecular targeting approach with unique advantages over chemotherapy. Indeed, certain tumors with overexpressed Myc or Cyclin E might be particularly sensitive to CMGi versus standard chemotherapy, and inherent weaknesses in the MCM/CMG complex might provide a wider therapeutic window for CMGi/MCMi use against other cancers. In addition, as described above, developing inhibitors that target the CMG specifically (CMGi) or the MCM ATPase domains (MCMi) could provide different clinical outcomes in terms of off-tumor toxicities.
As an example for an innovative use of CMGi in the clinic, osteosarcoma (OS) is an aggressive bone tumor in adolescents and young adults that has been examined by a large-scale genomic sequencing effort called the TARGET-OS Project [231]. Although no single mutated driver of OS was identified in these analyses, the vast majority of tumors demonstrate cell cycle dysregulation with subgroups of OS with gains in Myc or Cyclin E (CCNE1), loss of Rb, or gain of Cdk4 (which regulates Rb). In addition, the vast majority of OS have completely lost p53 protein expression, or TP53 is mutated [232]. Although each of these genetic alterations in OS affect proteins that are not themselves druggable, the collective nature of these genetic changes would imply that the CMG helicase is a potential vulnerability across many OS subtypes. As such, new clinical approaches to OS treatment, or for other tumor situations where a known driver does not exist, might consider incorporating CMGi as a pharmacologic intervention in patients stratified for known CMG-modifying genetic changes such as amplified Myc, CCNE1, and/or TP53 loss.
Related to the above discussion on chemotherapy versus targeting the CMG, CMGi/MCMi might be useful in enhancing the effectiveness of existing chemotherapy drugs that induce replicative stresses requiring proper function of the CMG in recovery efforts. The combination of CMGi plus chemotherapy could potentially allow a reduced level of chemotherapy drug to be administered to alleviate off-tumor toxicities, while the CMGi would inhibit the CMG enzyme required for recovering from chemotherapy insults. A clinical consideration for use of CMGi in this manner is the timing or scheduling of CMGi administration in combination with standard-of-care chemotherapeutic regimens. If a CMG inhibitor blocks MCM loading in G1, then it is possible that pre-treatment with a CMGi may reduce the effectiveness of fork-stalling chemotherapy if treated cells fail to enter S-phase. Conversely, it may be more advantageous to treat with a CMGi after such chemotherapy exposure, where the chemotherapy induces replicative stress in tumors that then critically require (remaining) CMG reserves to be functional.
It is also important to consider the duration of exposure to CMGi, as the mouse studies would suggest that long-term CMGi treatment might create reduced genomic stability and DNA damage even in normal cells, leading to unwanted secondary tumor development [200,201,202,203]. Clearly, this is also true of conventional chemotherapeutic drugs that damage, crosslink, or alkylate/modify DNA. However, tumor development in mice required conditions in which MCM levels were constantly reduced during the entire developmental timeframe of embryogenesis to adulthood [200, 201, 203], which is arguably not a situation that would be encountered in the clinic when using CMGi.
The CMG could be targeted clinically using indirect methods, with inhibitors to proteins that regulate CMG assembly and/or activation. For example, Cdc7 kinase inhibitors are being tested in the clinic, and Cdc7 is required for CMG activation [233,234,235,236,237,238,239,240,241,242]. However, kinase inhibitors sometimes have activity against other kinases, or the targeted kinases regulate other important cell cycle events aside from CMG management, making interpretation of their clinical effects difficult to assign specifically to CMG inhibition [3]. Other future druggable targets that regulate the CMG include ORC and Cdc6, but specific inhibitors/drugs against these enzymes do not currently exist.
Finally, an emerging clinical argument for using CMGi may derive from the unique roles of the CMG helicase in DNA replication and recovering from DNA damage or fork-stalling conditions [243]. Many solid tumors contain deficiencies in DNA repair, either as an early genetic driving condition (e.g., inherited Brca1/2 mutations), or acquired during the tumorigenic process [244, 245]. These deficiencies can be in homologous repair (HR), non-homologous end-joining repair (NHEJ), base-excision repair (BER), or nucleotide-excision repair (NER). It is possible that signatures in tumors indicative of DNA repair deficiencies may predict increased sensitivity to CMGi, as the co-existence of multiple DNA repair/recovery deficits may create a synthetically lethal situation specific to certain tumors. Emerging characterization signatures for chromosomal instability may be biomarkers to help guide patient selection for future trials investigating this possibility [246, 247]. Some of the genes to consider in this regard include mutations in Brca1/2, Fanconi anemia (FANC) genes, ATM, PalB2, or NBS. Similarly, combination of CMGi with existing DNA repair inhibitors such as PARP inhibitors or ATR inhibitors may show promise in the clinic for certain malignancies.
Closing thoughts
Although we have focused on a select few cancer-promoting gene products as being MCM/CMG helicase (mis)regulators, namely Myc, Cyclin E, p53, and Rb, there are undoubtedly other genes and protein products that will be found to have tumor-driving influences on the CMG and genome stability. Similarly, we cannot rule out that Mcm, Cdc45, or GINS mutants may be discovered in certain human malignancies that might slightly alter CMG function but remain compatible with survival. There are also other cancer-relevant CMG issues awaiting further investigation that we have not discussed here. For example, Mcm3 interacts with Keap1, a regulator of Nrf2 and responses to oxidative stresses [248, 249]. What this interaction means functionally for the CMG has not been unraveled. MCMs also interact with cohesins in regulating chromosome topology [250, 251], and MCMs are required for loading of cohesins [252,253,254], which link daughter chromatids after DNA replication and may influence condensation in mitosis [255,256,257]. There are also associations of MCM and ORC subunits with centrosomes, which play a role in DNA replication and mitosis [258, 259]. Mechanisms by which changes in MCM/CMG reserves affect cohesion, centrosomes, and mitosis may predict chemosensitizing synthetic effects for CMGi and mitotic drugs such as taxanes.
Our goal here was to dissect out some of the known cancer-driving influences on MCM/CMG assembly and activation dynamics, to illustrate that while the CMG may obviously appear to be passively directed by many cell cycle processes and signals, once properly assembled and regulated, the CMGs and MCMs also perform active roles in the fidelity of DNA replication, recovery from stresses, and regulation of fork speeds, all of which collectively maintains genomic stability. Abraham Wald’s mathematical and theoretical genius clearly applies to the CMG helicase and offers explanations for why the CMG is “missing mutations” in cancer. Human cancers are telling us that the CMG is, like Wald’s airplane engine, critical for tumor cell survival. Weakened by oncogenic changes, wildtype CMG becomes a tumor-specific weakness that should be targeted with drugs to selectively inhibit cancer growth.
References
Ellenberg J. How not to be wrong: the power of mathematical thinking. The Penguin Press: New York; 2014.
Reed DR, Metts J, Pressley M, Fridley BL, Hayashi M, Isakoff MS, et al. An evolutionary framework for treating pediatric sarcomas. Cancer. 2020;126:2577–87.
Reed DR, Alexandrow MG. Myc and the replicative CMG helicase: the creation and destruction of cancer: Myc over-activation of CMG helicases drives tumorigenesis and creates a vulnerability in CMGs for therapeutic intervention. Bioessays. 2020;42:e1900218.
Dominguez-Sola D, Gautier J. MYC and the control of DNA replication. Cold Spring Harb Perspect Med. 2014;4:a014423.
Srinivasan SV, Dominguez-Sola D, Wang LC, Hyrien O, Gautier J. Cdc45 is a critical effector of myc-dependent DNA replication stress. Cell Rep. 2013;3:1629–39.
Dominguez-Sola D, Ying CY, Grandori C, Ruggiero L, Chen B, Li M, et al. Non-transcriptional control of DNA replication by c-Myc. Nature. 2007;448:445–51.
Ekholm-Reed S, Mendez J, Tedesco D, Zetterberg A, Stillman B, Reed SI. Deregulation of cyclin E in human cells interferes with prereplication complex assembly. J Cell Biol. 2004;165:789–800.
Aparicio T, Ibarra A, Mendez J. Cdc45-MCM-GINS, a new power player for DNA replication. Cell Div. 2006;1:18.
Costa A, Ilves I, Tamberg N, Petojevic T, Nogales E, Botchan MR, et al. The structural basis for MCM2-7 helicase activation by GINS and Cdc45. Nat Struct Mol Biol. 2011;18:471–7.
Ilves I, Petojevic T, Pesavento JJ, Botchan MR. Activation of the MCM2-7 helicase by association with Cdc45 and GINS proteins. Mol Cell. 2010;37:247–58.
Gambus A, Jones RC, Sanchez-Diaz A, Kanemaki M, van Deursen F, Edmondson RD, et al. GINS maintains association of Cdc45 with MCM in replisome progression complexes at eukaryotic DNA replication forks. Nat Cell Biol. 2006;8:358–66.
Miller TCR, Locke J, Greiwe JF, Diffley JFX, Costa A. Mechanism of head-to-head MCM double-hexamer formation revealed by cryo-EM. Nature. 2019;575:704–10.
Remus D, Beuron F, Tolun G, Griffith JD, Morris EP, Diffley JF. Concerted loading of Mcm2-7 double hexamers around DNA during DNA replication origin licensing. Cell. 2009;139:719–30.
Yao NY, Zhang D, Yurieva O, O’Donnell ME. CMG helicase can use ATPgammaS to unwind DNA: implications for the rate-limiting step in the reaction mechanism. Proc Natl Acad Sci USA. 2022;119:e2119580119.
Yuan Z, Georgescu R, Bai L, Zhang D, Li H, O’Donnell ME. DNA unwinding mechanism of a eukaryotic replicative CMG helicase. Nat Commun. 2020;11:688.
Yao NY, O’Donnell ME. Getting ready for DNA duplication. Elife. 2019;8:e51291.
O’Donnell ME, Li H. The ring-shaped hexameric helicases that function at DNA replication forks. Nat Struct Mol Biol. 2018;25:122–30.
Li H, O’Donnell ME. The eukaryotic CMG helicase at the replication fork: emerging architecture reveals an unexpected mechanism. Bioessays. 2018;40.
Eickhoff P, Kose HB, Martino F, Petojevic T, Abid Ali F, Locke J, et al. Molecular basis for ATP-hydrolysis-driven DNA translocation by the CMG helicase of the eukaryotic replisome. Cell Rep. 2019;28:2673–88.e2678.
Kang S, Warner MD, Bell SP. Multiple functions for Mcm2-7 ATPase motifs during replication initiation. Mol Cell. 2014;55:655–65.
Liu L, Zhang Y, Zhang J, Wang JH, Cao Q, Li Z, et al. Characterization of the dimeric CMG/pre-initiation complex and its transition into DNA replication forks. Cell Mol Life Sci. 2020;77:3041–58.
Bell SP. DNA replication. Terminating the replisome. Science. 2014;346:418–9.
Lewis JS, Gross MH, Sousa J, Henrikus SS, Greiwe JF, Nans A, et al. Mechanism of replication origin melting nucleated by CMG helicase assembly. Nature. 2022;606:1007–14.
Costa A, Diffley JFX. The initiation of eukaryotic DNA replication. Annu Rev Biochem. 2022;91:107–31.
Usman M, Okla MK, Asif HM, AbdElgayed G, Muccee F, Ghazanfar S, et al. A pan-cancer analysis of GINS complex subunit 4 to identify its potential role as a biomarker in multiple human cancers. Am J Cancer Res. 2022;12:986–1008.
He Z, Wang X, Yang Z, Jiang Y, Li L, Wang X, et al. Expression and prognosis of CDC45 in cervical cancer based on the GEO database. PeerJ. 2021;9:e12114.
Zhang K, Zhou J, Wu T, Tian Q, Liu T, Wang W, et al. Combined analysis of expression, prognosis and immune infiltration of GINS family genes in human sarcoma. Aging (Albany NY). 2022;14:5895–907.
Yu S, Wang G, Shi Y, Xu H, Zheng Y, Chen Y. MCMs in cancer: prognostic potential and mechanisms. Anal Cell Pathol (Amst). 2020;2020:3750294.
Rzechorzek NJ, Hardwick SW, Jatikusumo VA, Chirgadze DY, Pellegrini L. CryoEM structures of human CMG-ATPgammaS-DNA and CMG-AND-1 complexes. Nucleic Acids Res. 2020;48:6980–95.
Bochman ML, Schwacha A. The Mcm2-7 complex has in vitro helicase activity. Mol Cell. 2008;31:287–93.
Pacek M, Tutter AV, Kubota Y, Takisawa H, Walter JC. Localization of MCM2-7, Cdc45, and GINS to the site of DNA unwinding during eukaryotic DNA replication. Mol Cell. 2006;21:581–7.
Pacek M, Walter JC. A requirement for MCM7 and Cdc45 in chromosome unwinding during eukaryotic DNA replication. EMBO J. 2004;23:3667–76.
Botchan M, Berger J. DNA replication: making two forks from one prereplication complex. Mol Cell. 2010;40:860–1.
Deegan TD, Mukherjee PP, Fujisawa R, Polo Rivera C, Labib K. CMG helicase disassembly is controlled by replication fork DNA, replisome components and a ubiquitin threshold. Elife. 2020;9:e60371.
Yeeles JTP, Janska A, Early A, Diffley JFX. How the eukaryotic replisome achieves rapid and efficient DNA replication. Mol Cell. 2017;65:105–16.
Georgescu R, Yuan Z, Bai L, de Luna Almeida Santos R, Sun J, Zhang D, et al. Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proc Natl Acad Sci USA. 2017;114:E697–E706.
Gambus A, van Deursen F, Polychronopoulos D, Foltman M, Jones RC, Edmondson RD, et al. A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase alpha within the eukaryotic replisome. EMBO J. 2009;28:2992–3004.
Bryant VL, Elias RM, McCarthy SM, Yeatman TJ, Alexandrow MG. Suppression of reserve MCM complexes chemosensitizes to gemcitabine and 5-fluorouracil. Mol Cancer Res. 2015;13:1296–305.
Ibarra A, Schwob E, Mendez J. Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proc Natl Acad Sci USA. 2008;105:8956–61.
Ge XQ, Jackson DA, Blow JJ. Dormant origins licensed by excess Mcm2-7 are required for human cells to survive replicative stress. Genes Dev. 2007;21:3331–41.
Woodward AM, Gohler T, Luciani MG, Oehlmann M, Ge X, Gartner A, et al. Excess Mcm2-7 license dormant origins of replication that can be used under conditions of replicative stress. J Cell Biol. 2006;173:673–83.
Coster G, Frigola J, Beuron F, Morris EP, Diffley JF. Origin licensing requires ATP binding and hydrolysis by the MCM replicative helicase. Mol Cell. 2014;55:666–77.
Tanaka S, Diffley JF. Interdependent nuclear accumulation of budding yeast Cdt1 and Mcm2-7 during G1 phase. Nat Cell Biol. 2002;4:198–207.
Labib K, Kearsey SE, Diffley JF. MCM2-7 proteins are essential components of prereplicative complexes that accumulate cooperatively in the nucleus during G1-phase and are required to establish, but not maintain, the S-phase checkpoint. Mol Biol Cell. 2001;12:3658–67.
Donovan S, Harwood J, Drury LS, Diffley JFX. Cdc6p-dependent loading of Mcm proteins onto pre-replicative chromatin in budding yeast. Proc Natl Acad Sci USA. 1997;94:5611–6.
Austin RJ, Orr-Weaver TL, Bell SP. Drosophila ORC specifically binds to ACE3, an origin of DNA replication control element. Genes Dev. 1999;13:2639–49.
Bell SP, Mitchell J, Leber J, Kobayashi R, Stillman B. The multidomain structure of Orc1p reveals similarity to regulators of DNA replication and transcriptional silencing. Cell. 1995;83:563–8.
Santocanale C, Diffley JFX. ORC- and Cdc6-dependent complexes at active and inactive chromosomal replication origins in Saccharomyces cerevisiae. EMBO J. 1996;15:6671–9.
Liang C, Weinreich M, Stillman B. ORC and Cdc6p interact and determine the frequency of initiation of DNA replication in the genome. Cell. 1995;81:667–76.
Madine MA, Swietlik M, Pelizon C, Romanowski P, Mills AD, Laskey RA. The roles of the MCM, ORC, and Cdc6 proteins in determining the replication competence of chromatin in quiescent cells. J Struct Biol. 2000;129:198–210.
Frigola J, Remus D, Mehanna A, Diffley JF. ATPase-dependent quality control of DNA replication origin licensing. Nature. 2013;495:339–43.
Edwards MC, Tutter AV, Cvetic C, Gilbert CH, Prokhorova TA, Walter JC. MCM2-7 complexes bind chromatin in a distributed pattern surrounding the origin recognition complex in Xenopus egg extracts. J Biol Chem. 2002;277:33049–57.
Bell SP, Stillman B. ATP-dependent recognition of eukaryotic origins of DNA replication by a multiprotein complex. Nature. 1992;357:128–34.
Fernandez-Cid A, Riera A, Tognetti S, Herrera MC, Samel S, Evrin C, et al. An ORC/Cdc6/MCM2-7 complex is formed in a multistep reaction to serve as a platform for MCM double-hexamer assembly. Mol Cell. 2013;50:577–88.
Gavin KA, Hidaka M, Stillman B. Conserved initiator proteins in eukaryotes. Science. 1995;270:1667–71.
Rowley A, Cocker JH, Harwood J, Diffley JFX. Initiation complex assembly at budding yeast replication origins begins with the recognition of a bipartite sequence by limiting amounts of the initiator, ORC. EMBO J. 1995;14:2631–41.
Chesnokov I, Gossen M, Remus D, Botchan M. Assembly of functionally active Drosophila origin recognition complex from recombinant proteins. Genes Dev. 1999;13:1289–96.
Remus D, Beall EL, Botchan MR. DNA topology, not DNA sequence, is a critical determinant for Drosophila ORC-DNA binding. EMBO J. 2004;23:897–907.
Cvetic C, Walter JC. Eukaryotic origins of DNA replication: could you please be more specific? Semin Cell Dev Biol. 2005;16:343–53.
Vashee S, Cvetic C, Lu W, Simancek P, Kelly TJ, Walter JC. Sequence-independent DNA binding and replication initiation by the human origin recognition complex. Genes Dev. 2003;17:1894–908.
Hamlin JL, Mesner LD, Dijkwel PA. A winding road to origin discovery. Chromosome Res. 2010;18:45–61.
Hamlin JL, Mesner LD, Lar O, Torres R, Chodaparambil SV, Wang L. A revisionist replicon model for higher eukaryotic genomes. J Cell Biochem. 2008;105:321–9.
Klemm RD, Austin RJ, Bell SP. Coordinate binding of ATP and origin DNA regulates the ATPase activity of the origin recognition complex. Cell. 1997;88:493–502.
Lee DG, Makhov AM, Klemm RD, Griffith JD, Bell SP. Regulation of origin recognition complex conformation and ATPase activity: differential effects of single-stranded and double-stranded DNA binding. EMBO J. 2000;19:4774–82.
Weinreich M, Liang C, Stillman B. The Cdc6p nucleotide-binding motif is required for loading mcm proteins onto chromatin. Proc Natl Acad Sci USA. 1999;96:441–6.
Perkins G, Diffley JFX. Nucleotide-dependent prereplicative complex assembly by Cdc6p, a homolog of eukaryotic and prokaryotic clamp-loaders. Mol Cell. 1998;2:23–32.
Blow JJ, Hodgson B. Replication licensing-defining the proliferative state? Trends Cell Biol. 2002;12:72–78.
Tada S, Blow JJ. The replication licensing system. Biol Chem. 1998;379:941–9.
Thommes P, Kubota Y, Takisawa H, Blow JJ. The RLF-M component of the replication licensing system forms complexes containing all six MCM/P1 polypeptides. EMBO J. 1997;16:3312–9.
Chong JP, Blow JJ. DNA replication licensing factor. Prog Cell Cycle Res. 1996;2:83–90.
Chong JPJ, Mahbubani HM, Khoo C-Y, Blow JJ. Purification of an MCM-containing complex as a component of the DNA replication licensing system. Nature. 1995;375:418–21.
Cvetic CA, Walter JC. Getting a grip on licensing: mechanism of stable Mcm2-7 loading onto replication origins. Mol Cell. 2006;21:143–4.
Cocker JH, Piatti S, Santocanale C, Nasmyth K, Diffley JFX. An essential role for the Cdc6 protein in forming the pre-replicative complexes of budding yeast. Nature. 1996;379:180–2.
Cook JG, Park CH, Burke TW, Leone G, DeGregori J, Engel A, et al. Analysis of Cdc6 function in the assembly of mammalian prereplication complexes. Proc Natl Acad Sci USA. 2002;99:1347–52.
Yan Z, DeGregori J, Shohet R, Leone G, Stillman B, Nevins JR, et al. Cdc6 is regulated by E2F and is essential for DNA replication in mammalian cells. Proc Natl Acad Sci USA. 1998;95:3603–8.
Furstenthal L, Kaiser BK, Swanson C, Jackson PK. Cyclin E uses Cdc6 as a chromatin-associated receptor required for DNA replication. J Cell Biol. 2001;152:1267–78.
Nepon-Sixt BS, Alexandrow MG. TGFbeta1 cell cycle arrest is mediated by inhibition of MCM assembly in Rb-deficient conditions. Mol Cancer Res. 2019;17:277–88.
Geng Y, Lee YM, Welcker M, Swanger J, Zagozdzon A, Winer JD, et al. Kinase-independent function of cyclin E. Mol Cell. 2007;25:127–39.
Saha P, Chen J, Thome KC, Lawlis SJ, Hou ZH, Hendricks M, et al. Human CDC6/Cdc18 associates with Orc1 and cyclin-cdk and is selectively eliminated from the nucleus at the onset of S phase. Mol Cell Biol. 1998;18:2758–67.
Alexandrow MG, Hamlin JL. Cdc6 chromatin affinity is unaffected by serine-54 phosphorylation, S-phase progression, and overexpression of cyclin A. Mol Cell Biol. 2004;24:1614–27.
Lau E, Zhu C, Abraham RT, Jiang W. The functional role of Cdc6 in S-G2/M in mammalian cells. EMBO Rep. 2006;7:425–30.
Walter D, Hoffmann S, Komseli ES, Rappsilber J, Gorgoulis V, Sorensen CS. SCF(Cyclin F)-dependent degradation of CDC6 suppresses DNA re-replication. Nat Commun. 2016;7:10530.
Mailand N, Diffley JF. CDKs promote DNA replication origin licensing in human cells by protecting Cdc6 from APC/C-dependent proteolysis. Cell. 2005;122:915–26.
Li J, Deng M, Wei Q, Liu T, Tong X, Ye X. Phosphorylation of MCM3 protein by cyclin E/cyclin-dependent kinase 2 (Cdk2) regulates its function in cell cycle. J Biol Chem. 2011;286:39776–85.
Wei Q, Li J, Liu T, Tong X, Ye X. Phosphorylation of minichromosome maintenance protein 7 (MCM7) by cyclin/cyclin-dependent kinase affects its function in cell cycle regulation. J Biol Chem. 2013;288:19715–25.
Hendrickson M, Madine M, Dalton S, Gautier J. Phosphorylation of MCM4 by cdc2 protein kinase inhibits the activity of the minichromosome maintenance complex. Proc Natl Acad Sci USA. 1996;93:12223–8.
Moritani M, Ishimi Y. Inhibition of DNA binding of MCM2-7 complex by phosphorylation with cyclin-dependent kinases. J Biochem. 2013;154:363–72.
Findeisen M, El-Denary M, Kapitza T, Graf R, Strausfeld U. Cyclin A-dependent kinase activity affects chromatin binding of ORC, Cdc6, and MCM in egg extracts of Xenopus laevis. Eur J Biochem. 1999;264:415–26.
Boos D, Sanchez-Pulido L, Rappas M, Pearl LH, Oliver AW, Ponting CP, et al. Regulation of DNA replication through Sld3-Dpb11 interaction is conserved from yeast to humans. Curr Biol. 2011;21:1152–7.
Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Treslin collaborates with TopBP1 in triggering the initiation of DNA replication. Cell. 2010;140:349–59.
Kumagai A, Shevchenko A, Shevchenko A, Dunphy WG. Direct regulation of Treslin by cyclin-dependent kinase is essential for the onset of DNA replication. J Cell Biol. 2011;193:995–1007.
Mukherjee P, Cao TV, Winter SL, Alexandrow MG. Mammalian MCM loading in late-G(1) coincides with Rb hyperphosphorylation and the transition to post-transcriptional control of progression into S-phase. PLoS One. 2009;4:e5462.
Ohtsubo M, Theodoras AM, Schumacher J, Roberts JM, Pagano M. Human cyclin E, a nuclear protein essential for the G1-to-S phase transition. Mol Cell Biol. 1995;15:2612–24.
Dou Q-P, Levin AH, Zhao S, Pardee AB. Cyclin E and cyclin A as candidates for the restriction point protein. Cancer Res. 1993;53:1493–7.
Koff A, Giordano A, Desai D, Yamashita K, Harper JW, Elledge S, et al. Formation and activation of a cyclin E-cdk2 complex during the G1 phase of the human cell cycle. Science. 1992;257:1689–94.
Koff A, Cross F, Fisher A, Schumacher J, Leguellec K, Philippe M, et al. Human cyclin E, a new cyclin that interacts with two members of the CDC2 gene family. Cell. 1991;66:1217–28.
Dulic V, Lees E, Reed SI. Association of human cyclin E with a periodic G1-S phase protein kinase. Science. 1992;257:1958–61.
Lundberg AS, Weinberg RA. Functional inactivation of the retinoblastoma protein requires sequential modification by at least two distinct cyclin-cdk complexes. Mol Cell Biol. 1998;18:753–61.
Keenan SM, Lents NH, Baldassare JJ. Expression of cyclin E renders cyclin D-CDK4 dispensable for inactivation of the retinoblastoma tumor suppressor protein, activation of E2F, and G1-S phase progression. J Biol Chem. 2004;279:5387–96.
Mendez J, Stillman B. Chromatin association of human origin recognition complex, cdc6, and minichromosome maintenance proteins during the cell cycle: assembly of prereplication complexes in late mitosis. Mol Cell Biol. 2000;20:8602–12.
Zhang J, Yu L, Wu X, Zou L, Sou KK, Wei Z, et al. The interacting domains of hCdt1 and hMcm6 involved in the chromatin loading of the MCM complex in human cells. Cell Cycle. 2010;9:4848–57.
Wei Z, Liu C, Wu X, Xu N, Zhou B, Liang C, et al. Characterization and structure determination of the Cdt1 binding domain of human minichromosome maintenance (Mcm) 6. The. J Biol Chem. 2010;285:12469–73.
Liu C, Wu R, Zhou B, Wang J, Wei Z, Tye BK, et al. Structural insights into the Cdt1-mediated MCM2-7 chromatin loading. Nucleic Acids Res. 2012;40:3208–17.
Yuan Z, Riera A, Bai L, Sun J, Nandi S, Spanos C, et al. Structural basis of Mcm2-7 replicative helicase loading by ORC-Cdc6 and Cdt1. Nat Struct Mol Biol. 2017;24:316–24.
Yanagi K, Mizuno T, You Z, Hanaoka F. Mouse geminin inhibits not only Cdt1-MCM6 interactions but also a novel intrinsic Cdt1 DNA binding activity. J Biol Chem. 2002;277:40871–80.
Saxena S, Dutta A. Geminin-Cdt1 balance is critical for genetic stability. Mutat Res. 2005;569:111–21.
Hodgson B, Li A, Tada S, Blow JJ. Geminin becomes activated as an inhibitor of Cdt1/RLF-B following nuclear import. Curr Biol. 2002;12:678–83.
Madine M, Laskey R. Geminin bans replication licence. Nat Cell Biol. 2001;3:E49–50.
Wohlschlegel JA, Dwyer BT, Dhar SK, Cvetic C, Walter JC, Dutta A. Inhibition of eukaryotic DNA replication by geminin binding to Cdt1. Science. 2000;290:2309–12.
Wong PG, Glozak MA, Cao TV, Vaziri C, Seto E, Alexandrow M. Chromatin unfolding by Cdt1 regulates MCM loading via opposing functions of HBO1 and HDAC11-geminin. Cell Cycle. 2010;9:4351–63.
Miotto B, Struhl K. HBO1 histone acetylase is a coactivator of the replication licensing factor Cdt1. Genes Dev. 2008;22:2633–8.
Iizuka M, Matsui T, Takisawa H, Smith MM. Regulation of replication licensing by acetyltransferase Hbo1. Mol Cell Biol. 2006;26:1098–108.
Iizuka M, Stillman B. Histone acetyltransferase HBO1 interacts with the ORC1 subunit of the human initiator protein. J Biol Chem. 1999;274:23027–34.
Miotto B, Struhl K. HBO1 histone acetylase activity is essential for DNA replication licensing and inhibited by Geminin. Mol Cell. 2010;37:57–66.
Vaziri C, Saxena S, Jeon Y, Lee C, Murata K, Machida Y, et al. A p53-dependent checkpoint pathway prevents rereplication. Mol Cell. 2003;11:997–1008.
Arentson E, Faloon P, Seo J, Moon E, Studts JM, Fremont DH, et al. Oncogenic potential of the DNA replication licensing protein CDT1. Oncogene. 2002;21:1150–8.
Seo J, Chung YS, Sharma GG, Moon E, Burack WR, Pandita TK, et al. Cdt1 transgenic mice develop lymphoblastic lymphoma in the absence of p53. Oncogene. 2005;24:8176–86.
Walter JC. Evidence for sequential action of cdc7 and cdk2 protein kinases during initiation of DNA replication in Xenopus egg extracts. J Biol Chem. 2000;275:39773–8.
Lin XH, Walter J, Scheidtmann K, Ohst K, Newport J, Walter G. Protein phosphatase 2A is required for the initiation of chromosomal DNA replication. Proc Natl Acad Sci USA. 1998;95:14693–8.
Ballabeni A, Zamponi R, Caprara G, Melixetian M, Bossi S, Masiero L, et al. Human CDT1 associates with CDC7 and recruits CDC45 to chromatin during S phase. J Biol Chem. 2009;284:3028–36.
Chou DM, Petersen P, Walter JC, Walter G. Protein phosphatase 2A regulates binding of Cdc45 to the prereplication complex. J Biol Chem. 2002;277:40520–7.
Greiwe JF, Miller TCR, Locke J, Martino F, Howell S, Schreiber A, et al. Structural mechanism for the selective phosphorylation of DNA-loaded MCM double hexamers by the Dbf4-dependent kinase. Nat Struct Mol Biol. 2022;29:10–20.
Yeeles JT, Deegan TD, Janska A, Early A, Diffley JF. Regulated eukaryotic DNA replication origin firing with purified proteins. Nature. 2015;519:431–5.
Mukherjee P, Winter SL, Alexandrow MG. Cell cycle arrest by transforming growth factor beta1 near G1/S is mediated by acute abrogation of prereplication complex activation involving an Rb-MCM interaction. Mol Cell Biol. 2010;30:845–56.
Kang YH, Galal WC, Farina A, Tappin I, Hurwitz J. Properties of the human Cdc45/Mcm2-7/GINS helicase complex and its action with DNA polymerase epsilon in rolling circle DNA synthesis. Proc Natl Acad Sci USA. 2012;109:6042–7.
Baretic D, Jenkyn-Bedford M, Aria V, Cannone G, Skehel M, Yeeles JTP. Cryo-EM structure of the fork protection complex bound to CMG at a replication fork. Mol Cell. 2020;78:926–40.e913.
Kang YH, Farina A, Bermudez VP, Tappin I, Du F, Galal WC, et al. Interaction between human Ctf4 and the Cdc45/Mcm2-7/GINS (CMG) replicative helicase. Proc Natl Acad Sci USA. 2013;110:19760–5.
Dmowski M, Jedrychowska M, Makiela-Dzbenska K, Denkiewicz-Kruk M, Sharma S, Chabes A, et al. Increased contribution of DNA polymerase delta to the leading strand replication in yeast with an impaired CMG helicase complex. DNA Repair (Amst). 2022;110:103272.
Baxley RM, Bielinsky AK. Mcm10: a dynamic scaffold at eukaryotic replication forks. Genes (Basel). 2017;8:73.
Mayle R, Langston L, Molloy KR, Zhang D, Chait BT, O’Donnell ME. Mcm10 has potent strand-annealing activity and limits translocase-mediated fork regression. Proc Natl Acad Sci USA. 2019;116:798–803.
Langston LD, Mayle R, Schauer GD, Yurieva O, Zhang D, Yao NY, et al. Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. Elife. 2017;6:e29118.
Ricke RM, Bielinsky AK. Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Mol Cell. 2004;16:173–85.
Izumi M, Yanagi K, Mizuno T, Yokoi M, Kawasaki Y, Moon KY, et al. The human homolog of Saccharomyces cerevisiae Mcm10 interacts with replication factors and dissociates from nuclease-resistant nuclear structures in G(2) phase. Nucleic Acids Res. 2000;28:4769–77.
Homesley L, Lei M, Kawasaki Y, Sawyer S, Christensen T, Tye BK. Mcm10 and the MCM2-7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes Dev. 2000;14:913–26.
Wohlschlegel JA, Dhar SK, Prokhorova TA, Dutta A, Walter JC. Xenopus Mcm10 binds to origins of DNA replication after Mcm2-7 and stimulates origin binding of Cdc45. Mol Cell. 2002;9:233–40.
Douglas ME, Diffley JF. Recruitment of Mcm10 to sites of replication initiation requires direct binding to the minichromosome maintenance (MCM) complex. J Biol Chem. 2016;291:5879–88.
Alexandrow MG, Hamlin JL. Chromatin decondensation in S-phase involves recruitment of Cdk2 by Cdc45 and histone H1 phosphorylation. J Cell Biol. 2005;168:875–86.
Wong PG, Winter SL, Zaika E, Cao TV, Oguz U, Koomen JM, et al. Cdc45 limits replicon usage from a low density of preRCs in mammalian cells. PLoS One. 2011;6:e17533.
Sedlackova H, Rask MB, Gupta R, Choudhary C, Somyajit K, Lukas J. Equilibrium between nascent and parental MCM proteins protects replicating genomes. Nature. 2020;587:297–302.
Huberman JA. Prokaryotic and eukaryotic replicons. Cell. 1995;82:535–42.
Berezney R, Dubey DD, Huberman JA. Heterogeneity of eukaryotic replicons, replicon clusters, and replication foci. Chromosoma. 2000;108:471–84.
Luciani MG, Oehlmann M, Blow JJ. Characterization of a novel ATR-dependent, Chk1-independent, intra-S-phase checkpoint that suppresses initiation of replication in Xenopus. J Cell Sci. 2004;117:6019–30.
Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell. 2003;11:203–13.
Shechter D, Gautier J. ATM and ATR check in on origins: a dynamic model for origin selection and activation. Cell Cycle. 2005;4:235–8.
Shechter D, Costanzo V, Gautier J. ATR and ATM regulate the timing of DNA replication origin firing. Nat Cell Biol. 2004;6:648–55.
Shechter D, Costanzo V, Gautier J. Regulation of DNA replication by ATR: signaling in response to DNA intermediates. DNA Repair (Amst). 2004;3:901–8.
Dijkwel PA, Wang S, Hamlin JL. Initiation sites are distributed at frequent intervals in the Chinese hamster dihydrofolate reductase origin of replication but are used with very different efficiencies. Mol Cell Biol. 2002;22:3053–65.
Dijkwel PA, Hamlin JL. The Chinese hamster dihydrofolate reductase origin consists of multiple potential nascent-strand start sites. Mol Cell Biol. 1995;15:3023–31.
Dijkwel PA, Vaughn JP, Hamlin JL. Replication initiation sites are distributed widely in the amplified CHO dihydrofolate reductase domain. Nucleic Acids Res. 1994;22:4989–96.
Hamlin JL, Mosca PJ, Dijkwel PA, Lin H-B. Initiation of replication at a mammalian chromosomal origin. Cold Spring Harb Symp Quant Biol. 1993;58:467–74.
Vaughn JP, Dijkwel PA, Hamlin JL. Replication initiates in a broad zone in the amplified CHO dihydrofolate reductase domain. Cell. 1990;61:1075–87.
Mesner LD, Valsakumar V, Karnani N, Dutta A, Hamlin JL, Bekiranov S. Bubble-chip analysis of human origin distributions demonstrates on a genomic scale significant clustering into zones and significant association with transcription. Genome Res. 2011;21:377–89.
Alexandrow MG, Ritzi M, Pemov A, Hamlin JL. A potential role for mini-chromosome maintenance (MCM) proteins in initiation at the dihydrofolate reductase replication origin. J Biol Chem. 2002;277:2702–8.
Saha S, Shan Y, Mesner LD, Hamlin JL. The promoter of the Chinese hamster ovary dihydrofolate reductase gene regulates the activity of the local origin and helps define its boundaries. Genes Dev. 2004;18:397–410.
Mesner LD, Hamlin JL. Specific signals at the 3’ end of the DHFR gene define one boundary of the downstream origin of replication. Genes Dev. 2005;19:1053–66.
Tippens ND, Liang J, Leung AK, Wierbowski SD, Ozer A, Booth JG, et al. Transcription imparts architecture, function and logic to enhancer units. Nat Genet. 2020;52:1067–75.
Henriques T, Scruggs BS, Inouye MO, Muse GW, Williams LH, Burkholder AB, et al. Widespread transcriptional pausing and elongation control at enhancers. Genes Dev. 2018;32:26–41.
Comoglio F, Schlumpf T, Schmid V, Rohs R, Beisel C, Paro R. High-resolution profiling of Drosophila replication start sites reveals a DNA shape and chromatin signature of metazoan origins. Cell Rep. 2015;11:821–34.
MacAlpine DM, Rodriguez HK, Bell SP. Coordination of replication and transcription along a Drosophila chromosome. Genes Dev. 2004;18:3094–105.
Karnani N, Taylor CM, Malhotra A, Dutta A. Genomic study of replication initiation in human chromosomes reveals the influence of transcription regulation and chromatin structure on origin selection. Mol Biol Cell. 2010;21:393–404.
Dellino GI, Cittaro D, Piccioni R, Luzi L, Banfi S, Segalla S, et al. Genome-wide mapping of human DNA-replication origins: levels of transcription at ORC1 sites regulate origin selection and replication timing. Genome Res. 2013;23:1–11.
Chen YH, Keegan S, Kahli M, Tonzi P, Fenyo D, Huang TT, et al. Transcription shapes DNA replication initiation and termination in human cells. Nat Struct Mol Biol. 2019;26:67–77.
Moses HL, Yang EY, Pietenpol JA. Regulation of epithelial proliferation by TGF-beta. Ciba Found Symp. 1991;157:66–74.
Bierie B, Moses HL. TGF-beta and cancer. Cytokine Growth Factor Rev. 2006;17:29–40.
Murphy CS, Pietenpol JA, Munger K, Howley PM, Moses HL. c-myc and pRB: Role in TGFb1 inhibition of keratinocyte proliferation. Cold Spring Harb Symp Quant Biol. 1991;56:129–35.
Massague J. TGFbeta in cancer. Cell. 2008;134:215–30.
Massague J, Blain SW, Lo RS. TGFbeta signaling in growth control, cancer, and heritable disorders. Cell. 2000;103:295–309.
Koff A, Ohtsuki M, Polyak K, Roberts JM, Massagué J. Negative regulation of G1 in mammalian cells: Inhibition of cyclin E-dependent kinase by TGF-b. Science. 1993;260:536–9.
Ewen ME, Sluss HK, Whitehouse LL, Livingston DM. TGFb inhibition of Cdk4 synthesis is linked to cell cycle arrest. Cell. 1993;74:1009–20.
Pietenpol JA, Holt JT, Stein RW, Moses HL. Transforming growth factor b-1 suppression of c-myc gene transcription: role in inhibition of keratinocyte proliferation. Proc Natl Acad Sci USA. 1990;87:3758–62.
Massague J, Chen YG. Controlling TGF-beta signaling. Genes Dev. 2000;14:627–44.
Iavarone A, Massague J. Repression of the Cdk activator Cdc25A and cell cycle arrest by cytokine TGF-b in cells lacking the Cdk inhibitor p15. Nature. 1997;387:417–22.
Reynisdottir I, Polyak K, Iavarone A, Massague J. Kip/Cip and ink4 cdk inhibitors cooperate to induce cell cycle arrest in response to TGFb. Genes Dev. 1995;9:1831–45.
Polyak K, Lee M-H, Erdjument-Bromage H, Koff A, Roberts JM, Tempst P, et al. Cloning of p27Kip1, a cyclin-dependent kinase inhibitor and a potential mediator of extracellular antimitogenic signals. Cell. 1994;78:59–66.
Polyak K, Kato J, Solomon MJ, Sherr CJ, Massague J, Roberts JM, et al. p27Kip1, A cyclin-Cdk inhibitor, links transforming growth factor-b and contact inhibition to cell cycle arrest. Genes Dev. 1994;8:9–22.
Seoane J, Pouponnot C, Staller P, Schader M, Eilers M, Massague J. TGFbeta influences Myc, Miz-1 and Smad to control the CDK inhibitor p15INK4b. Nat Cell Biol. 2001;3:400–8.
Alexandrow MG, Moses HL. Kips off to Myc: implications for TGF beta signaling. J Cell Biochem. 1997;66:427–32.
Pietenpol JA, Stein RW, Moran E, Yaciuk P, Schlegel R, Lyons RM, et al. TGF-b1 inhibition of c-myc transcription and growth in keratinocytes is abrogated by viral transforming proteins with pRB binding domains. Cell. 1990;61:777–85.
Laiho M, DeCaprio JA, Ludlow JW, Livingston DM, Massague J. Growth inhibition by TGF-beta linked to suppression of retinoblastoma protein phosphorylation. Cell. 1990;62:175–85.
Wolfraim LA, Walz TM, James Z, Fernandez T, Letterio JJ. p21Cip1 and p27Kip1 act in synergy to alter the sensitivity of naive T cells to TGF-beta-mediated G1 arrest through modulation of IL-2 responsiveness. J Immunol. 2004;173:3093–102.
Nakayama K, Ishida N, Shirane M, Inomata A, Inoue T, Shishido N, et al. Mice lacking p27Kip1 display increased body size, multiple organ hyperplasia, retinal dysplasia, and pituitary tumors. Cell. 1996;85:707–20.
Voss M, Wolff B, Savitskaia N, Ungefroren H, Deppert W, Schmiegel W, et al. TGFbeta-induced growth inhibition involves cell cycle inhibitor p21 and pRb independent from p15 expression. Int J Oncol. 1999;14:93–101.
Baldwin RL, Korc M. Growth inhibition of human pancreatic carcinoma cells by transforming growth factor beta-1. Growth Factors. 1993;8:23–34.
Alexandrow MG, Kawabata M, Aakre M, Moses HL. Overexpression of the c-Myc oncoprotein blocks the growth-inhibitory response but is required for the mitogenic effects of transforming growth factor beta 1. Proc Natl Acad Sci USA. 1995;92:3239–43.
Claassen GF, Hann SR. A role for transcriptional repression of p21CIP1 by c-Myc in overcoming transforming growth factor beta -induced cell-cycle arrest. Proc Natl Acad Sci USA. 2000;97:9498–503.
Sun P, Dong P, Dai K, Hannon GJ, Beach D. p53-independent role of MDM2 in TGF-b1 resistance. Science. 1998;282:2270–2.
Borysov SI, Nepon-Sixt BS, Alexandrow MG. The N-terminus of the Rb protein inhibits DNA replication via a bipartite mechanism disrupted in partially penetrant retinoblastomas. Mol Cell Biol. 2016;36:832–45.
Sterner JM, Dew-Knight S, Musahl C, Kornbluth S, Horowitz JM. Negative regulation of DNA replication by the retinoblastoma protein is mediated by its association with MCM7. Mol Cell Biol. 1998;18:2748–57.
Hassler M, Singh S, Yue WW, Luczynski M, Lakbir R, Sanchez-Sanchez F, et al. Crystal structure of the retinoblastoma protein N domain provides insight into tumor suppression, ligand interaction, and holoprotein architecture. Mol Cell. 2007;28:371–85.
Riley DJ, Liu CY, Lee WH. Mutations of N-terminal regions render the retinoblastoma protein insufficient for functions in development and tumor suppression. Mol Cell Biol. 1997;17:7342–52.
Otterson GA, Chen W, Coxon AB, Khleif SN, Kaye FJ. Incomplete penetrance of familial retinoblastoma linked to germ-line mutations that result in partial loss of RB function. Proc Natl Acad Sci USA. 1997;94:12036–40.
Lefevre SH, Chauveinc L, Stoppa-Lyonnet D, Michon J, Lumbroso L, Berthet P, et al. A T to C mutation in the polypyrimidine tract of the exon 9 splicing site of the RB1 gene responsible for low penetrance hereditary retinoblastoma. J Med Genet. 2002;39:E21.
Sterner JM, Murata Y, Kim HG, Kennett SB, Templeton DJ, Horowitz JM. Detection of a novel cell cycle-regulated kinase activity that associates with the amino terminus of the retinoblastoma protein in G2/M phases. J Biol Chem. 1995;270:9281–8.
Goodrich DW. How the other half lives, the amino-terminal domain of the retinoblastoma tumor suppressor protein. J Cell Physiol. 2003;197:169–80.
Barrientes S, Cooke C, Goodrich DW. Glutamic acid mutagenesis of retinoblastoma protein phosphorylation sites has diverse effects on function. Oncogene. 2000;19:562–70.
Wu JR, Gilbert DM. The replication origin decision point is a mitogen-independent, 2-aminopurine-sensitive, G1-phase event that precedes restriction point control. Mol Cell Biol. 1997;17:4312–21.
Avni D, Yang H, Martelli F, Hofmann F, ElShamy WM, Ganesan S, et al. Active localization of the retinoblastoma protein in chromatin and its response to S phase DNA damage. Mol Cell. 2003;12:735–46.
Stengel KR, Dean JL, Seeley SL, Mayhew CN, Knudsen ES. RB status governs differential sensitivity to cytotoxic and molecularly-targeted therapeutic agents. Cell Cycle. 2008;7:1095–103.
Knudsen KE, Booth D, Naderi S, Sever-Chroneos Z, Fribourg AF, Hunton IC, et al. RB-dependent S-phase response to DNA damage. Mol Cell Biol. 2000;20:7751–63.
Kunnev D, Rusiniak ME, Kudla A, Freeland A, Cady GK, Pruitt SC. DNA damage response and tumorigenesis in Mcm2-deficient mice. Oncogene. 2010;29:3630–8.
Pruitt SC, Bailey KJ, Freeland A. Reduced Mcm2 expression results in severe stem/progenitor cell deficiency and cancer. Stem Cells. 2007;25:3121–32.
Kawabata T, Luebben SW, Yamaguchi S, Ilves I, Matise I, Buske T, et al. Stalled fork rescue via dormant replication origins in unchallenged S phase promotes proper chromosome segregation and tumor suppression. Mol Cell. 2011;41:543–53.
Shima N, Alcaraz A, Liachko I, Buske TR, Andrews CA, Munroe RJ, et al. A viable allele of Mcm4 causes chromosome instability and mammary adenocarcinomas in mice. Nat Genet. 2007;39:93–98.
Nepon-Sixt BS, Bryant VL, Alexandrow MG. Myc-driven chromatin accessibility regulates Cdc45 assembly into CMG helicases. Commun Biol. 2019;2:110.
Koch HB, Zhang R, Verdoodt B, Bailey A, Zhang CD, Yates JR III, et al. Large-scale identification of c-MYC-associated proteins using a combined TAP/MudPIT approach. Cell Cycle. 2007;6:205–17.
Maya-Mendoza A, Ostrakova J, Kosar M, Hall A, Duskova P, Mistrik M, et al. Myc and Ras oncogenes engage different energy metabolism programs and evoke distinct patterns of oxidative and DNA replication stress. Mol Oncol. 2015;9:601–16.
Costantino L, Sotiriou SK, Rantala JK, Magin S, Mladenov E, Helleday T, et al. Break-induced replication repair of damaged forks induces genomic duplications in human cells. Science. 2014;343:88–91.
Teixeira LK, Reed SI. Cyclin E deregulation and genomic instability. Adv Exp Med Biol. 2017;1042:527–47.
Macheret M, Halazonetis TD. Intragenic origins due to short G1 phases underlie oncogene-induced DNA replication stress. Nature. 2018;555:112–6.
Das SP, Borrman T, Liu VW, Yang SC, Bechhoefer J, Rhind N. Replication timing is regulated by the number of MCMs loaded at origins. Genome Res. 2015;25:1886–92.
Gemble S, Wardenaar R, Keuper K, Srivastava N, Nano M, Mace AS, et al. Genetic instability from a single S phase after whole-genome duplication. Nature. 2022;604:146–51.
Seo YS, Kang YH. The human replicative helicase, the CMG complex, as a target for anti-cancer therapy. Front Mol Biosci. 2018;5:26.
Lu HP, Du XF, Li JD, Huang SN, He RQ, Wu HY, et al. Expression of cell division cycle protein 45 in tissue microarrays and the CDC45 gene by bioinformatics analysis in human hepatocellular carcinoma and patient outcomes. Med Sci Monit. 2021;27:e928800.
Tan DF, Huberman JA, Hyland A, Loewen GM, Brooks JS, Beck AF, et al. MCM2-a promising marker for premalignant lesions of the lung: a cohort study. BMC Cancer. 2001;1:6.
Ramnath N, Hernandez FJ, Tan DF, Huberman JA, Natarajan N, Beck AF, et al. MCM2 is an independent predictor of survival in patients with non-small-cell lung cancer. J Clin Oncol. 2001;19:4259–66.
Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2012;2:401–4.
Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal. 2013;6:pl1.
Ren B, Yu G, Tseng GC, Cieply K, Gavel T, Nelson J, et al. MCM7 amplification and overexpression are associated with prostate cancer progression. Oncogene. 2006;25:1090–8.
Zhang R, Liu Z, Zhang G. CDC45 modulates MCM7 expression and inhibits cell proliferation by suppressing the PI3K/AKT pathway in acute myeloid leukemia. Am J Transl Res. 2021;13:10218–32.
Minella AC, Welcker M, Clurman BE. Ras activity regulates cyclin E degradation by the Fbw7 pathway. Proc Natl Acad Sci USA. 2005;102:9649–54.
Kapeli K, Hurlin PJ. Differential regulation of N-Myc and c-Myc synthesis, degradation, and transcriptional activity by the Ras/mitogen-activated protein kinase pathway. J Biol Chem. 2011;286:38498–508.
Tsai WB, Aiba I, Long Y, Lin HK, Feun L, Savaraj N, et al. Activation of Ras/PI3K/ERK pathway induces c-Myc stabilization to upregulate argininosuccinate synthetase, leading to arginine deiminase resistance in melanoma cells. Cancer Res. 2012;72:2622–33.
Leone G, DeGregori J, Sears R, Jakoi L, Nevins JR. Myc and Ras collaborate in inducing accumulation of active cyclin E/Cdk2 and E2F. Nature. 1997;387:422–6.
Sears R, Leone G, DeGregori J, Nevins JR. Ras enhances Myc protein stability. Mol Cell. 1999;3:169–79.
Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev. 2000;14:2501–14.
Kruszewski W, Kowara R, Rzepko R, Warezak C, Zielinski J, Gryglewski G, et al. K-RAS point mutation, and amplification of C-MYC and C-ERBB2 in colon adenocarcinoma. Folia Histochem Cytobiol. 2004;42:173–9.
Field JK, Spandidos DA. The role of ras and myc oncogenes in human solid tumours and their relevance in diagnosis and prognosis (review). Anticancer Res. 1990;10:1–22.
Bochman ML, Bell SP, Schwacha A. Subunit organization of Mcm2-7 and the unequal role of active sites in ATP hydrolysis and viability. Mol Cell Biol. 2008;28:5865–73.
Schwacha A, Bell SP. Interactions between two catalytically distinct MCM subgroups are essential for coordinated ATP hydrolysis and DNA replication. Mol Cell. 2001;8:1093–104.
Wang D, Alvarez-Cabrera AL, Chen XS. Study of SV40 large T antigen nucleotide specificity for DNA unwinding. Virol J. 2017;14:79.
Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, et al. Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature. 2018;555:371–6.
Chen X, Bahrami A, Pappo A, Easton J, Dalton J, Hedlund E, et al. Recurrent somatic structural variations contribute to tumorigenesis in pediatric osteosarcoma. Cell Rep. 2014;7:104–12.
OR E, Dhami SPS, Baev DV, Ortutay C, Halpin-McCormick A, Morrell R, et al. Repression of Mcl-1 expression by the CDC7/CDK9 inhibitor PHA-767491 overcomes bone marrow stroma-mediated drug resistance in AML. Sci Rep. 2018;8:15752.
Gad SA, Ali HEA, Gaballa R, Abdelsalam RM, Zerfaoui M, Ali HI, et al. Targeting CDC7 sensitizes resistance melanoma cells to BRAF(V600E)-specific inhibitor by blocking the CDC7/MCM2-7 pathway. Sci Rep. 2019;9:14197.
Iwai K, Nambu T, Dairiki R, Ohori M, Yu J, Burke K, et al. Molecular mechanism and potential target indication of TAK-931, a novel CDC7-selective inhibitor. Sci Adv. 2019;5:eaav3660.
Cheng AN, Lo YK, Lin YS, Tang TK, Hsu CH, Hsu JT, et al. Identification of novel Cdc7 kinase inhibitors as anti-cancer agents that target the interaction with Dbf4 by the fragment complementation and drug repositioning approach. EBioMedicine. 2018;36:241–51.
McLaughlin RP, He J, van der Noord VE, Redel J, Foekens JA, Martens JWM, et al. A kinase inhibitor screen identifies a dual cdc7/CDK9 inhibitor to sensitise triple-negative breast cancer to EGFR-targeted therapy. Breast Cancer Res. 2019;21:77.
Menichincheri M, Albanese C, Alli C, Ballinari D, Bargiotti A, Caldarelli M, et al. Cdc7 kinase inhibitors: 5-heteroaryl-3-carboxamido-2-aryl pyrroles as potential antitumor agents. 1. Lead finding. J Med Chem. 2010;53:7296–315.
Masai H, Arai K. Cdc7 kinase complex: a key regulator in the initiation of DNA replication. J Cell Physiol. 2002;190:287–96.
Bousset K, Diffley JFX. The Cdc7 protein kinase is required for origin firing during S phase. Genes Dev. 1998;12:480–90.
Pasero P, Duncker BP, Schwob E, Gasser SM. A role for the Cdc7 kinase regulatory subunit Dbf4p in the formation of initiation-competent origins of replication. Genes Dev. 1999;13:2159–76.
Labib K. How do Cdc7 and cyclin-dependent kinases trigger the initiation of chromosome replication in eukaryotic cells? Genes Dev (Rev). 2010;24:1208–19.
Long DT, Joukov V, Budzowska M, Walter JC. BRCA1 promotes unloading of the CMG helicase from a stalled DNA replication fork. Mol Cell. 2014;56:174–85.
Merajver SD, Pham TM, Caduff RF, Chen M, Poy EL, Cooney KA, et al. Somatic mutations in the BRCA1 gene in sporadic ovarian tumours. Nat Genet. 1995;9:439–43.
Mori T, Aoki T, Matsubara T, Iida F, Du X, Nishihira T, et al. Frequent loss of heterozygosity in the region including BRCA1 on chromosome 17q in squamous cell carcinomas of the esophagus. Cancer Res. 1994;54:1638–40.
Steele CD, Abbasi A, Islam SMA, Bowes AL, Khandekar A, Haase K, et al. Signatures of copy number alterations in human cancer. Nature. 2022;606:984–91.
Drews RM, Hernando B, Tarabichi M, Haase K, Lesluyes T, Smith PS, et al. A pan-cancer compendium of chromosomal instability. Nature. 2022;606:976–83.
Tamberg N, Tahk S, Koit S, Kristjuhan K, Kasvandik S, Kristjuhan A, et al. Keap1-MCM3 interaction is a potential coordinator of molecular machineries of antioxidant response and genomic DNA replication in metazoa. Sci Rep. 2018;8:12136.
Mulvaney KM, Matson JP, Siesser PF, Tamir TY, Goldfarb D, Jacobs TM, et al. Identification and characterization of MCM3 as a Kelch-like ECH-associated Protein 1 (KEAP1) substrate. J Biol Chem. 2016;291:23719–33.
Emerson DJ, Zhao PA, Cook AL, Barnett RJ, Klein KN, Saulebekova D, et al. Cohesin-mediated loop anchors confine the locations of human replication origins. Nature. 2022;606:812–9.
Dequeker BJH, Scherr MJ, Brandao HB, Gassler J, Powell S, Gaspar I, et al. MCM complexes are barriers that restrict cohesin-mediated loop extrusion. Nature. 2022;606:197–203.
Ivanov MP, Ladurner R, Poser I, Beveridge R, Rampler E, Hudecz O, et al. The replicative helicase MCM recruits cohesin acetyltransferase ESCO2 to mediate centromeric sister chromatid cohesion. EMBO J. 2018;37:e97150.
Takahashi TS, Basu A, Bermudez V, Hurwitz J, Walter JC. Cdc7-Drf1 kinase links chromosome cohesion to the initiation of DNA replication in Xenopus egg extracts. Genes Dev. 2008;22:1894–905.
Takahashi TS, Yiu P, Chou MF, Gygi S, Walter JC. Recruitment of Xenopus Scc2 and cohesin to chromatin requires the pre-replication complex. Nat Cell Biol. 2004;6:991–6.
Bloom MS, Koshland D, Guacci V. Cohesin function in cohesion, condensation, and DNA repair is regulated by Wpl1p via a common mechanism in Saccharomyces cerevisiae. Genetics. 2018;208:111–24.
Heidinger-Pauli JM, Mert O, Davenport C, Guacci V, Koshland D. Systematic reduction of cohesin differentially affects chromosome segregation, condensation, and DNA repair. Curr Biol. 2010;20:957–63.
Guacci V, Koshland D, Strunnikov A. A direct link between sister chromatid cohesion and chromosome condensation revealed through the analysis of MCD1 in S. cerevisiae. Cell. 1997;91:47–57.
Knockleby J, Lee H. Same partners, different dance: involvement of DNA replication proteins in centrosome regulation. Cell Cycle. 2010;9:4487–91.
Prasanth SG, Prasanth KV, Siddiqui K, Spector DL, Stillman B. Human Orc2 localizes to centrosomes, centromeres and heterochromatin during chromosome inheritance. EMBO J. 2004;23:2651–63.
Funding
This work was supported by the National Pediatric Cancer Foundation (nationalpcf.org; DRR) and research support to MGA from the National Institutes of Health (R01 GM140140-01) and the Adolescent and Young Adult Program at the Moffitt Cancer Center and Research Institute.
Author information
Authors and Affiliations
Contributions
Composed and edited manuscript: SX. Designed figures, composed, and edited manuscript: DRR and MGA.
Corresponding authors
Ethics declarations
Competing interests
The authors declare no competing interests.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Xiang, S., Reed, D.R. & Alexandrow, M.G. The CMG helicase and cancer: a tumor “engine” and weakness with missing mutations. Oncogene 42, 473–490 (2023). https://doi.org/10.1038/s41388-022-02572-8
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41388-022-02572-8
- Springer Nature Limited
This article is cited by
-
Combined therapy of dabrafenib and an anti-HER2 antibody–drug conjugate for advanced BRAF-mutant melanoma
Cellular & Molecular Biology Letters (2024)
-
GINS2 regulates temozolomide chemosensitivity via the EGR1/ECT2 axis in gliomas
Cell Death & Disease (2024)
-
Unwinding Helicase MCM Functionality for Diagnosis and Therapeutics of Replication Abnormalities Associated with Cancer: A Review
Molecular Diagnosis & Therapy (2024)
-
Multifaceted role of the DNA replication protein MCM10 in maintaining genome stability and its implication in human diseases
Cancer and Metastasis Reviews (2024)
-
Modeling phenotypic heterogeneity towards evolutionarily inspired osteosarcoma therapy
Scientific Reports (2023)