
Abstract
MCM10 plays a vital role in genome duplication and is crucial for DNA replication initiation, elongation, and termination. It coordinates several proteins to assemble at the fork, form a functional replisome, trigger origin unwinding, and stabilize the replication bubble. MCM10 overexpression is associated with increased aggressiveness in breast, cervical, and several other cancers. Disruption of MCM10 leads to altered replication timing associated with initiation site gains and losses accompanied by genome instability. Knockdown of MCM10 affects the proliferation and migration of cancer cells, manifested by DNA damage and replication fork arrest, and has recently been shown to be associated with clinical conditions like CNKD and RCM. Loss of MCM10 function is associated with impaired telomerase activity, leading to the accumulation of abnormal replication forks and compromised telomere length. MCM10 interacts with histones, aids in nucleosome assembly, binds BRCA2 to maintain genome integrity during DNA damage, prevents lesion skipping, and inhibits PRIMPOL-mediated repriming. It also interacts with the fork reversal enzyme SMARCAL1 and inhibits fork regression. Additionally, MCM10 undergoes several post-translational modifications and contributes to transcriptional silencing by interacting with the SIR proteins. This review explores the mechanism associated with MCM10’s multifaceted role in DNA replication initiation, chromatin organization, transcriptional silencing, replication stress, fork stability, telomere length maintenance, and DNA damage response. Finally, we discuss the role of MCM10 in the early detection of cancer, its prognostic significance, and its potential use in therapeutics for cancer treatment.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
1 Introduction
The genome duplicates in a highly regulated process during DNA replication, which occurs only once in a cell cycle [1, 2]. During the G1 phase, as cells prepare to replicate their DNA, errors in copying the parental DNA strands can lead to mutations as a result of chromosomal breakage, rearrangement, and missegregation of the genetic material. If DNA damage remains unrepaired, it can disrupt the process of DNA replication and trigger the activation of checkpoints [3, 4], which block cellular processes like DNA replication initiation. Consequently, this activates a DNA damage and repair response that provides protective benefits against diseases like cancer [5].
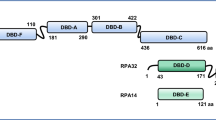
The eukaryotic replicative DNA helicase, responsible for driving the genome duplication process, becomes activated upon the recruitment of several replication factors, including CDC45, the mini chromosome maintenance protein 2–7 (MCM2-7), and GINS. These factors come together to form the CMG complex, which, in the presence of ATP, initiates the process of origin melting [6, 7]. MCMs, being an integral part of the active replisomes in vivo, have been proposed as potential biomarkers in malignancy and dysplasia, highlighting their significance in identifying these conditions [8, 9]. Several hypotheses exist on how dysfunction of replication factors like the MCM proteins can lead to cancer due to chromosomal instability (CIN) and replication stress [10]. One member of the DNA replication-associated MCM family of proteins, MCM10, is involved in the formation of functional CMG complex and helps in the binding of DNA polymerase to replication forks [11, 12]. MCM10 is an evolutionarily conserved DNA-binding protein [11, 13, 14], but its role has remained controversial [15,16,17,18]. It has no sequence homology with the MCM2-7 family members and was first isolated from Saccharomyces cerevisiae mutants that affect the stability of centromeric plasmids [13, 14, 19]. The size of MCM10 proteins differs depending on the organism, from 571 amino acids in yeast to 874 in humans [20, 21] (Table 1). Structurally, it features distinct functional regions, including the N terminal domain (NTD), the C terminal domain (CTD), and the Internal domain (ID). The ID contains two conserved motifs: a PCNA-interacting protein (PIP) box and an oligonucleotide/oligosaccharide-binding (OB) fold consistent with the ability to bind single-stranded DNA (ssDNA) [22]. The PIP box allows deubiquinated MCM10 to interact with PCNA, a critical factor for the processivity of DNA polymerases during replication elongation and repair [23]. It also interacts with MCM2, MCM6, and origin recognition complex subunit 2 (ORC2). The ORC and CDC6 load Cdt1-bound hexameric complexes of MCM2-7 onto the replication origins [24]. Studies from mammalian systems have shown that these interactions are regulated by proteolysis and phosphorylation in a cell cycle-dependent manner [20, 25].
In budding yeast, the MCM double hexamer (DH) activation occurs after the formation of the Cdc45-MCM-GINS (CMG) complex [7]. ATP binding nucleates origin DNA melting with the aid of the CMG complex and maintains replisome stability at initiation [7]. Ctf4/And1 bridges DNA polymerase alpha and the CMG helicase acts as a fork protection complex to ensure stability [26]. Studies from both budding and fission yeast indicate that Cdc45 and GINS can bind the origin DNA to form the CMG without MCM10, but the complex is only functional upon MCM10 binding, which is dependent on Cdc45 and CMG proteins like Sld2. This ultimately leads to the crucial role of MCM10 in unwinding the dsDNA and manifesting in the form of replication initiation [12, 18, 27,28,29,30,31]. However, other studies from Xenopus, yeast, and mammalian cells show that MCM10 is required for the chromatin association of GINS and Cdc45 [15,16,17, 32,33,34]. This difference in opinion on MCM10 chromatin binding and its interdependency with Cdc45 and the GINS during the G1 phase can be explained by the fact that MCM10 recruitment might occur in two affinity modes: one in which loading of CMG happens without MCM10 (low affinity, early G1 phase) and the other in which MCM10 recruitment happens followed by Cdc45 and CMG chromatin binding (high affinity, late G1/S-phase) [35]. Studies from Xenopus have shown that MCM10 cooperates with Cdc45 during elongation and is required for the stable association of Cdc45 with replication forks. Thus, MCM10 is the earliest detectable step in the activation of pre-RCs at the onset of DNA replication before Cdc45 loading and independently of the DBF4-Cdc7 kinase and Cdk2 during the G1/S transition [15]. These cell cycle checkpoint proteins ensure that origins are licensed before they can fire. MCM10 is important for DNA elongation and CMG helicase activity, overcoming replication blocks, and ensuring fork progression and stability as evident from studies in budding yeast [28, 30, 36, 37]. Its deficiency causes replication forks to stall during elongation, indicating that apart from the initiation of DNA synthesis at the origins of DNA replication, it is also required for strand elongation [14]. The stalling is probably due to unfired pre-RCs blocking replication fork progression [38, 39]. Support for the role of MCM10 in elongation also comes from the genetic interactions of MCM10 with subunits of Pol α which primes the replication fork by synthesizing a short RNA primer and initiates short stretches of DNA along the lagging strand. Another protein, DNA2, acts as a quality control check during elongation, ensuring accurate copying of the DNA template by removing mistakes made by the polymerase [40,41,42]. In temperature-sensitive mcm10 yeast mutants, depletion of MCM10 during the S phase results in the degradation of the catalytic subunit of Pol α without affecting Cdc45, indicative of replication arrest [43]. As DNA synthesis continues, the replisome plays a crucial role in maintaining genomic integrity, thereby reducing replicative stress [30, 37, 44]. During replication, MCM10 acts as a crucial checkpoint to ensure DNA unwinds only once per cell cycle and thus prevents a second round of DNA replication [36]. Before S-phase, a double CMG complex forms, partially separating the DNA strands but not fully unwinding them as seen in yeast. MCM10 steps in upon S-phase transition, triggering the complete separation of the CMG complex into two individual units. This separation narrows the channel within the complex, forcing the ejection of the lagging strand template and initiating DNA unwinding. By regulating this process, MCM10 prevents unnecessary re-replication of chromosomes [45].
Studies from Xenopus also support the role of MCM10 in the replication elongation step and the maintenance of genome integrity [30, 46, 47]. MCM10 might have a role in double-strand break (DSB) repair, an important mechanism for restoring damaged DNA, and was found along with the DSB response proteins NBS1 and ATM. Recruitment of Xenopus MCM10 (XMcm10) to origins requires the prior chromatin association of the XMcm2-7 complex, and in response to DSBs, MCM10 was shown to interact with the helicase Dna2 in complex with Ctf4 [46, 47]. MCM10-deficient yeast cells are synthetically lethal with several members of the DSB repair pathway [48]. In mammalian systems, it was shown to contribute to the maintenance of telomeres, the protective caps at the ends of chromosomes, thereby preventing chromosomal instability (CIN) and senescence [49]. MCM2-7 molecules are generally in excess to protect cells from replicative stress and license backup origins of replication [50]. Origins that fire later in the S-phase are prevented from firing early by the activation of the intra-S-phase checkpoint. During cellular damage, MCM10 participates in the DNA damage response, assisting cells in coping with genotoxic stress in yeast and mammalian systems by firing dormant origins [36, 51]. In mammalian cells, this checkpoint inhibits the initiation of DNA replication at these origins after MCM10 loading but before the recruitment of Cdc45 and Ctf4/And1 to the replication fork, indicating checkpoint control of origin firing [52]. Studies from Drosophila have implicated MCM10 in chromatin organization, contributing to the proper packaging of the genetic material [53]. Additional evidence comes from studies in budding yeast where it was shown to be involved in the distribution of silent information regulators (SIRs) like Sir2 on chromatin, emphasizing its role in chromosome organization, and connecting DNA replication with higher-order chromatin structure [54]. Post-translational modifications like acetylation, ubiquitination, and SUMOylation play a pivotal role in fine-tuning MCM10’s activities and interactions with other proteins [25, 55, 56]. MCM10 also plays a role in regulating the phosphorylation of proteins within the DNA replication complex. For example, studies in Fission yeast have shown that MCM10 stimulates Hsk1/Cdc7-mediated phosphorylation of MCM2 and MCM4 [57]. MCM10 is thus a multifaceted protein whose classical functions discovered are mostly linked to DNA initiation function (Fig. 1), and some recently uncovered novel biological functions directly linked to human diseases are discussed in this review (Fig. 2).

Classical functions of MCM10. The classical functions of MCM10 are broadly depicted under DNA replication, DNA damage repair, and post-translational modifications (grouped as white boxes), which are also under cell cycle regulation (represented at the center). MCM10 is required for DNA replication initiation. MCM10 helps in the recruitment and assembly of the CMG complex (comprising Cdc45, MCM2-7 helicase, and GINS) to the origin DNA bound by the ORC complex, Cdc6, and Cdt1. It is crucial in DNA unwinding, helicase activation, and single-stranded binding proteins (SSB). It also helps in loading the polymerase, taking part in elongation and maintaining the replication bubble. MCM10 plays a significant role in DNA damage repair. It monitors fork stability and interacts with MCM7 in fork stalling post-DNA damage. MCM10 interacts with members of the MRN complex (Mre11, Rad50, and Nbs1). It binds the silent information regulators Sir2 and Sir3 and plays a role in chromatin remodeling, which is a prerequisite for DNA damage repair. MCM10 undergoes post-translational modifications. MCM10 undergoes cell cycle-dependent post-translational modifications like ubiquitination, phosphorylation, SUMOylation, and acetylation to perform its multifaceted functions. References are given in brackets

Novel functions of MCM10. The novel functions of MCM10 are broadly grouped under diseases concerning human health, chromatin organization, fork stability, and telomere maintenance (grouped as white boxes). MCM10 is associated with clinical conditions. MCM10 regulates replication timing, and overexpression is linked to the aggressiveness of cancer. Mutants of MCM10 are associated with clinical conditions like restrictive cardiomyopathy (RCM) and classical natural killer cell deficiency (CNKD). MCM10 is required for chromatin organization. It interacts with the histone subunits H3 and H4 and the heterochromatin protein 1 (HP1), showing links with chromatin organization. MCM10 in telomere length maintenance. It interacts with RTEL1 to maintain telomere length and confer telomere stability. MCM10 is essential for fork-related functions. MCM10 exhibits potent strand annealing activity and inhibits fork regression with the help of SMARCAL1. During DNA damage, it inhibits PRIMPOL-mediated repriming and lesion skipping. MCM10 is also involved in replication termination and double hexamer splitting. References are given in brackets
2 MCM10 in DNA replication initiation
During the G1 phase of the yeast cell cycle, a group of proteins comprising the ORC complex and CDC6 loads Cdt1-bound hexameric complexes of MCM2-7 onto the replication origins. This process is called “origin licensing” and signifies that origins are primed to fire during the S-phase [58]. In eukaryotes, replication is initiated from thousands of such licensed replication origins distributed throughout the genome. During DNA replication, origins fire only once during the S-phase, and cells restrict the re-initiation of already fired origins by multiple mechanisms controlled by CDKs [59]. The association of Cdc45 with the MCM 2–7 and the GINS complex is critical for CMG assembly [52] and forms the complex, which creates two replicative helicases positioned in a head-to-head orientation on double-stranded DNA (dsDNA) as seen in budding yeast. MCM10 is essential for the activation of these CMG complexes, enabling it to encircle ssDNA and facilitate bidirectional DNA replication [60]. MCM10 binds to the forked junction and the N-terminal tier of CMG, causing CMG to release its hold on duplex DNA. This places CMG in a steric exclusion mode, encircling ssDNA and freeing the lagging strand from the interior of the MCM2-7 ring. With this configuration, CMG can bypass the impediment on the DNA and continue replication. Additional isomerization reactions involving MCM10 include the cracking open of the N-tier of the MCM2-7 ring to navigate the block and subsequent reclosure, as well as the potential opening of both N- and C-tiers to overcome obstacles without displacement as seen in budding yeast [37, 61].
Although CMG assembly can occur in the absence of MCM10 in budding yeast, DNA replication origin firing and the recruitment of replication protein A (RPA) to replication origins are compromised [18, 27, 29], implying that MCM10’s unwinding function occurs downstream of helicase assembly. MCM10 appears to have two potential mechanisms for activating CMG helicase. Firstly, it actively assists in converting the replicative helicase from a double to a single CMG complex stimulated by the activity of DDK [18], an observation also supported in mammalian cells [62, 63]. Secondly, MCM10 stabilizes ssDNA due to its stronger affinity for unwound DNA and is thus crucial for fork stability [34, 48, 64]. In addition, it maintains the replication bubble by initially binding to ssDNA, and later it is displaced by RPA, which protects longer stretches of ssDNA and has a much higher affinity for ssDNA than MCM10 [22, 65]. In another study, it has been proven biochemically that MCM10 is also capable of stripping the single-strand binding protein RPA and reannealing complementary DNA strands, a process that is important for fork reversal and DNA unwinding [66]. In budding yeast mutants having a defective MCM10 DNA binding domain, there is a significant decrease in RPA association at specific origin sequences, associated with a severe reduction in viability [34]. MCM10 stimulates MCM2 phosphorylation in vitro and allows CMG to melt long stretches of dsDNA, aiding in origin melting [60]. The presence of an intact MCM10 coiled-coil interaction surface at the NTD along with Ctf4 facilitates the oligomerization required for helicase assembly and DNA polymerase recruitment to MCM2-7 [42, 67, 68].
MCM10 helps in bridging the crucial components of origin activation like the mammalian RECQ4 and MCM2-7 helicases, which makes the MCM-RECQ4 complex essential for origin activation and influences normal cell cycle progression by ensuring robust origin firing [62, 63, 69]. In budding yeast, the temperature-sensitive dna43-1 (originally isolated mutant allele of MCM10) fails to replicate DNA at the restrictive temperature of 37 °C and causes cells to arrest [13]. Furthermore, the mcm10-1 allele in yeast also affects the frequency of DNA replication initiation and causes DNA replication forks to stall during elongation at origins that have not initiated [14]. Evidence from the following studies in fission yeast involving cdc23 (mutant allele of MCM10) indicates that MCM10 is an essential gene for proliferation as fungal spores failed to germinate displaying a cell cycle arrest phenotype [70]. Cdc23 also interacts with the CENP-B homolog Abp1, indicating its role in the origin firing of centromeric repeats [71]. Interestingly, insights from two-hybrid studies revealed that Rad4, a DNA repair protein particularly involved in nucleotide excision repair (NER), interacts with MCM10, and deletion mapping studies indicated that the C-terminus of MCM10 is important for repair. The rad4-116 allele was unable to associate MCM10 with the replication origin at a restrictive temperature, leading to replication arrest; hence, MCM10 plays an important role in replication restart post-DNA damage repair [72]. The fact that MCM10 is also essential during the elongation process was shown by a hydroxyurea block experiment, in which cells that retain chromatin-bound Cdc45 are incapable of completing DNA replication in the absence of Cdc23 [16]. Thus, MCM10 is required in multiple aspects of the DNA replication process (Fig. 1, Table 1).
In a recent study using budding yeast, the function of MCM10 in the transition of CMG mode-switching between ssDNA and dsDNA was examined. MCM10 preserves the CMG complex on DNA and significantly enhances the CMG transition by facilitating its loading onto ssDNA [61]. Without MCM10, most CMGs dissociated from DNA after the collapse of the fork junction, leading to a significant decrease in mode-switching probability. This indicates that it plays a crucial role in stabilizing the CMG complex by enhancing CMG’s affinity for DNA [37]. Truncated versions of Mcm10, specifically Mcm10-N terminal and Mcm10-C terminal deletions, demonstrated different abilities to support CMG mode switching, correlating with cellular viability. Interestingly, the C-terminal domain, a unique feature of MCM10 in higher eukaryotes, is lacking in yeast [73]. This highlights the importance of specific domains of MCM10 in regulating CMG function among diverse groups of organisms. MCM10 has the unique ability to bind to the DH helicase even before the recruitment of firing factors, potentially facilitating the immediate activation of the CMG complex as a result of strong affinity for ssDNA [74]. Although the exact order and timing of firing factor release remain unknown, MCM10 is implicated in the elongation phase of DNA replication, indicating that it may remain bound to the active helicase during this stage. Additionally, the crossing of CMG complexes in an “N-terminus first” manner ensures the complete unwinding of the origin DNA and aids in coordinating the assembly of the two leading strand replisomes. This coordination is crucial for efficient and accurate DNA replication as observed in both the yeast and mammalian systems [34, 74].
MCM10 has been implicated in multiple functions during replication initiation, and two major mechanisms have been proposed to explain its actions in budding yeast like the regulation of CMG activity and recruitment of other proteins to replication forks (Fig. 1). Firstly, MCM10 interacts with the CMG complex and has been proposed to stabilize the helicase, promote DNA unwinding, and coordinate replication fork progression [37, 61, 74]. Secondly, MCM10 has been implicated in recruiting other proteins to the replication forks. This is shown by the ability of MCM10 to interact with and recruit factors such as DNA polymerases, helicases, PCNA, and DNA repair proteins to the replication machinery. MCM10 also exhibits genetic interactions with several DNA replication elongation factors, including Cdt1 [40, 75]. These interactions facilitate the coordination of various enzymatic activities at the replication fork and ensure proper DNA replication and repair processes [23], observations which are also supported by studies in Xenopus [73, 76]. By combining these two mechanisms, MCM10 emerges as a critical player in regulating DNA replication initiation and fork progression.
3 MCM10 in the regulation of replication timing
Earlier studies from fission yeast and mammalian cells have shown that DNA replication is heterogeneous across the S-phase and exhibits a stochastic mode of origin firing [77,78,79]. Aberrant MCM10 function has been linked to replication timing alterations, highlighting its indispensable role in establishing and maintaining the order of DNA replication. MCM10’s involvement in DNA unwinding and its interaction with other replication proteins could indirectly influence origin selection. By affecting the efficiency or timing of origin firing, MCM10 contributes towards determining which origins are activated first during S-phase. Since certain origins fire early, others fire late during the S-phase. Our previous study with budding yeast showed that DNA origin firing efficiency depends on the number of MCM hexamers loaded onto the origins of DNA replication. Further, we showed that mutations in the MCM binding domain severely reduced MCM association with the origin DNA and altered the replication timing profile [80]. This observation is supported by recent studies on the replication timing of 184 cell lines from 3 cell types, which uncovered mutations in 2 genes that consistently resulted in altered replication timing [81]. The first gene identified was RIF1 (Rap-interacting factor 1), a known replication timing regulator, while the second gene was MCM10. Mutation of MCM10 is associated with replication delay and initiation site gains and losses, clearly indicating its role in the regulation of replication timing in mammalian cells [81]. Thus, MCM complex depletion affects replication origin activity and chromosomal translocation and disrupts global replication timing [81,82,83] (Fig. 2). Notably, similar replication timing aberrations were not observed in cells mutated for other central components of the DNA replication initiation machinery, such as GINS4 and RECQL4. The replication timing phenotype of mcm10 cells therefore appears to exhibit a high level of specificity, in line with the diverse roles of MCM10 in stabilizing the CMG helicase complex and the replisome [81]. A study on breast cancer cell lines showed an increase in cell proliferation following MCM10 overexpression which hints at a potential link between MCM10 and replication timing. While the exact mechanism remains elusive, several possibilities emerge. Overexpressed MCM10 could accelerate S-phase [5] leading to faster cell division. Alternatively, it might increase the number of replication origins initiated, thereby influencing replication fork distribution. Furthermore, the accelerated replication process induced by MCM10 overexpression could potentially generate replication stress, prompting cellular responses that might include alterations in replication timing. These indirect effects on epigenetic modifications cannot be ruled out, as these factors are known to influence replication timing [5]. In summary, while MCM10’s primary function is in DNA unwinding during S-phase, emerging evidence suggests its broader involvement in replication timing and potentially in origin selection.
4 MCM10 in chromatin organization and transcriptional silencing
MCM protein’s role in chromatin organization is evidenced by studies involving Drosophila, mice, zebrafish, yeast, and Xenopus. Members of the MCM family promote stem cell differentiation through their ability to bind to nucleosome components, including the core histones H3 and H4, and this direct interaction of the MCM protein with chromatin is facilitated by the parental histone transfer to daughter chromosomes during DNA replication as evidenced from studies in mice [84]. Loss of MCM10 was recently shown to affect hematopoietic stem cell (HSC) emergence in zebrafish embryos signifying the importance of MCM10 in HSC development [85]. In Arabidopsis, MCM10 (AtMCM10) was shown to directly bind to histone H3-H4 and promote nucleosome assembly [86]. As in Xenopus, where it is not required for bulk DNA replication [44], it is also not essential for plant growth and development [86]. However, the loss of both AtMCM10 and the histone chaperone CAF-1 is lethal and hence absolutely needed for replication-coupled nucleosome assembly [86]. Thus, MCM10 plays a major role in chromatin organization, as its depletion has been shown to result in chromatin condensation and loss of DNA content in Drosophila [87]. One such study that points towards a role in chromatin organization reported that MCM10 and Sirtuin 1 levels affect origin activation and replication fork velocity. Sirtuin 1 regulates MCM10 acetylation, and its concentration might modulate MCM10 levels. Furthermore, depletion of Sirtuin 1 by siRNA resulted in an increased level of MCM10 and chromatin binding activity in mammalian cells [88] indicating that Sirtuin-dependent acetylation might modulate MCM10 binding (loading and unloading) to the chromatin. It was observed that budding yeast cells carrying the mcm10 mutation exhibit a loss of chromosome integrity when attempting DNA replication at non-permissive temperatures. Interestingly, the double mutant containing both the mcm10-1 and cdc47-1 (mcm7-1 allele) restores the interaction between MCM10 and MCM7, thereby correcting the defects observed in each single mutant, such as stalling of replication forks at origins [21]. The ability of budding yeast MCM10 to bind to the DNA sliding clamp indicates that it enhances protein–protein interactions during the pre-replication complex (pre-RC) and its transition to the elongation stage [89]. During the G1 phase, MCM10 exhibits weak loading onto the DHs, and as the cell enters the S phase, the connection between MCM10 and DHs strengthens, coinciding with the increased recruitment of CDC45 [35, 90]. In vitro studies using yeast extract have shown that MCM10 directly interacts with Cdc45 and recruits it to the MCM2-7 complex [34] an observation also supported by mammalian studies [91]. Budding yeast MCM10 binds to the N-terminal domains of MCM2, MCM4, and MCM6 within the replication machinery, and it is essential for the proper regulation of helicase activity. In vivo DH-splitting assays have demonstrated that the interaction between MCM10 and DH is vital for helicase separation, during initiation, which in turn correlates with relatively slow S-phase progression in the absence of MCM10 [90].
In Xenopus, MCM10 interacts with MCM2-7 through its C-terminal domain (CTD), which is necessary to recruit and activate the helicase complex. Homologous regions of the MCM10 protein from other organisms are clustered in the CTD, and NTD regions, suggesting the presence of structured domains connected by the highly conserved IDs [73]. This modular architecture of MCM10, with distinct domains for dimerization and interactions with DNA and DNA polymerase α-primase, likely enables efficient coordination of MCM10’s biochemical activities within the replisome [73]. Functionally, the CTD, similar to the NTD, facilitates interactions with DNA and Pol-α, and their combined utilization enhances the binding affinity of MCM10 to DNA [73, 91]. In budding yeast, the zinc finger motif is required for self-interaction to form homo complex assembly, which might provide the structural basis for interaction with the MCM2-7 complex [92]. Interestingly, yeast homologs lack the major C-terminal region, which indicates that the NTD and ID are responsible for the essential functions of MCM10 in lower organisms [93]. Additionally, MCM10 was shown to play a role in nuclear localization, though the exact mechanism remains unclear [94]. In budding yeast, the short CTD of MCM10 interacts with MCM2 and MCM6, but this region is not conserved in higher eukaryotes [35, 90]. MCM10 CTD in Drosophila is known to interact with the heterochromatin protein 1a (HP1a) and is involved in chromatin condensation and precisely controlled cellular processes, like cell cycle regulation and differentiation. Knockdown of either HP1 or MCM10 activates the G1 checkpoint [53] and has been shown to result in semi-lethality in Drosophila [95].
Chromatin silencing plays a crucial role in regulating the transcript levels of cells as evidenced from studies in budding yeast. Transcriptional silencing occurs at the silent mating type loci HML, HMR, and the yeast telomeres. MCM10 plays a role in the maintenance but not in the establishment of the silent loci at HML in the budding yeast Saccharomyces cerevisiae. Silencing of the expression of genes coding Uracil (URA3) and Adenine (ADE2) reporter genes inserted into telomeric and HMR loci is significantly reduced in two recessive alleles of MCM10 [96], indicating a defect in the maintenance or inheritance of telomeric silencing. MCM10 has been found to interact with members of the silent information regulators (SIR), Sir2 and Sir3, confirming that it plays a functional role at the cryptic mating-type loci in yeast, and these interactions with the SIR proteins promote transcriptional silencing [97]. MCM10 is necessary for the coupling of replication and the silencing machinery via the short C-terminal domain, and studies from yeast have indicated that the silencing function of MCM10 is separate from its replication function [54].
5 MCM10 in genome amplification and telomere maintenance
Gene amplification is a common phenomenon in cancers, characterized by an increase in the copy number of a specific region of a chromosome arm which leads to the overexpression of genes [98]. In several cancers, amplification of the MCM10 gene is observed [56, 99,100,101,102], which is responsible for the overexpression phenotype and might also alter the CMG or replisome function [103]. Elevated levels of MCM10 can disrupt normal DNA replication processes, leading to accelerated DNA synthesis and cell proliferation [104]. Additionally, increased MCM10 expression can cause aberrant replication fork dynamics, promoting genomic instability [56], which in turn can lead to uncontrolled cell proliferation and tumorigenesis [105].
Recent studies from mammalian cells indicate the critical role of MCM10 in maintaining telomere length. Chronic MCM deficiency is associated with defects in telomere maintenance leading to telomere erosion, which is directly linked to aging [49]. In HCT116 (human colorectal carcinoma cell line) MCM10 mutants, shorter telomeres were observed compared to the wild-type samples. MCM10 mutants utilize the repair protein MUS81 to process stalled replication forks and improve cell survival [49]. MCM10 also interacts with the regulator of telomere length 1 (RTEL1), an essential DNA helicase involved in mammalian telomere replication, and promotes the progression of stalled forks, especially during termination [49, 76]. Recent reports point towards a link between compromised replisome machinery and inherited cardiomyopathy. Patients with bi-allelic MCM10 variants suffered from the clinical condition restrictive cardiomyopathy (RCM) [49]. Biallelic mutations of MCM10 were associated with a significant reduction in proliferation rates caused by a decrease in active replication forks, eventually leading to increased cell death irrespective of the cell type [49].
6 MCM10 in DNA replication stress, fork stability, and damage response
DNA replication stress involves the stalling or slowing of replication due to various mechanisms interfering with the normal replication process that leads to fork collapse and DNA strand breaks as evidenced by the studies in mammalian cells [51, 106]. Lesions occurring during DNA replication have been shown to generate mutations, copy number changes, and chromosomal rearrangements associated with cancer development [107]. The MCM complex is important in influencing the ability to replicate genetic material, respond to DNA damage, and interact with elements of DNA repair pathways in mammalian cells [108]. It is degraded through the Cul4-DDB1-VprBP ubiquitin ligase complex as a consequence of UV stress-induced proteolysis [109]; however, in another study, endogenous MCM10 of Xenopus was found to be stable in egg extracts, even in the presence of active DNA damage signaling [110]. This might be because the degradation complex is active later in the developmental phase. In reaction to replication stress induced by UV irradiation, budding yeast MCM10 was shown to interact with the Mec3 subunit of the 9–1-1 clamp, probably having a role in DNA repair or replication fork restart [89]. In response to replication stress like UV, many checkpoint and repair pathways are activated to maintain genomic stability [111], and mcm10 mutants are synthetically lethal in combination with the DNA replication checkpoint mutant rad53-1 [40]. Recent experimentation in budding yeast has shown that Rad53 phosphorylation of MCM10 leads to reduced fork rate under certain conditions, indicating that MCM10 functionality is under the control of the cell cycle checkpoint [112]. Inactivation of MCM10 in these cells during the S phase leads to loss of chromosome integrity, and the absence of S-phase checkpoint function leads to catastrophic mitosis. These findings indicate that mcm10 cells rely on a checkpoint mechanism to preserve genetic stability [40].
A key telomere-associated protein, the Regulator of Telomere Elongation Helicase 1 (RTEL1), and MCM10 play a crucial role in promoting the progression of stalled forks during termination as a result of stress in Xenopus [76]. In budding yeast, CMG ubiquitination is blocked in the absence of MCM10, a signal required for replication termination and CMG disassembly [55]. Further studies from mammalian cells have shown that MCM10 associated with self-ubiquitination initiates degradation before the onset of mitosis to prevent aberrant initiation of DNA replication [113] (Fig. 1). Recently, it has been shown that pathogenic mutations associated with DNA replication genes can lead to classical natural killer cell deficiency (CNKD), pointing to a potential link between DNA replication and NK cell development [106, 114]. MCM4, GINS, and MCM10 are affected in individuals with CNKD, suggesting that the disease is caused by defects in origin licensing or firing [115]. Specific bi-allelic variations in MCM10 lead to the premature differentiation arrest of distinct cardiac and immune cell lineages, contributing to the clinical phenotype seen in CNKD patients [49]. Also, this NK cell deficiency has been linked to compound heterozygous variants of MCM10. A recent study identified patients with significant decreases in functional MCM10 due to these variants. MCM10 haploinsufficiency in induced pluripotent stem cell (iPSC) lines demonstrated impaired clonogenic survival and increased genomic instability, including micronuclei formation and telomere erosion. These defects were correlated with reduced yields of hematopoietic stem cells (HSCs) and impaired NK cell differentiation [116]. The human germline MCM10 variants were also observed in restrictive cardiomyopathy (RCM) associated with thymic and splenic hypoplasia, emphasizing the clinical significance of DNA replication-associated proteins in stress [49] (Fig. 2).
Genomic instability has been demonstrated to increase mammalian cellular heterogeneity, resulting in aggressive tumor behavior [117]. BRCA2, a tumor suppressor protein crucial for maintaining genome integrity through its interaction with MCM10, inhibits lesion skipping and primase polymerase-mediated repriming to control fork progression post-DNA damage [118] in mammalian cells. MCM10 recruits BRCA2 upon stalling DNA polymerase epsilon at DNA lesions (stalled forks) and inhibits PRIMPOL-mediated repriming, thereby restraining fork progression and preventing ssDNA gap formation (Fig. 2). Loss of the association between MCM10 and BRCA2 leads to unrestrained fork progression after DNA damage, and depletion of MCM10 leads to reduced DNA synthesis and slower replication fork progression, as determined by BrdU incorporation and DNA combing [118]. Previous studies using mammalian cells demonstrated an increased expression of MCM10 during the G1/S transition and a decreased expression level of MCM10 in the G2/M phase of the cell cycle, which indicates that MCM10 is depleted post-genome duplication [113, 119]. Reduction of MCM10 in S-phase results in prolonged DNA synthesis as there is inhibition of new origin firing [75]. MCM10 is required for replication fork elongation and replisome stability, and it interacts with the members of the MRN complex, the S-phase checkpoint kinases ATM and ATR to avoid replication catastrophe in the event of DNA damage in Xenopus [120, 121]. MCM10 was also found to be essential for the peri-implantation development of the mouse embryo since its absence leads to defective cellular proliferation, which underlies developmental failure [122]. MCM10 knockdown also activates the G2 checkpoint (comprising Chk1 and Cdc25). This arrest of the cell cycle and prolonged depletion lead to DNA damage followed by cell death [121]. Additionally, MCM10 deficiency reduces the activity of the DNA polymerase subunit p180, leading to a delay in S-phase entry [120]. Loss of both Mcm10 and p180 in HeLa cells was shown to inhibit S phase entry and accumulation of cells in late S/G2 due to DNA damage, triggering apoptosis in a subset of cells. Interestingly, similar studies in Drosophila embryonic cells did not result in a similar arrest, suggesting differences in cellular responses between organisms [120]. On the other hand, overexpression of MCM10 induces enhanced proliferation by activating the replicative complexes, resulting in decreased DNA replication accuracy and an increase in single-strand DNA accumulation. Excess ssDNA activates the DNA damage response (DDR) pathway, recruiting several proteins that drive uncontrolled cell cycle progression in mammalian cells [123, 124]. To counter replication stress, the SUMOylated MCM10 variant rs2274110, located at the 15th exon of MCM10, is found to be overexpressed in oesophageal squamous cell carcinoma (OSCC) [56]. Aberrant overexpression of MCM10 facilitates cancer cells’ proliferation and metastatic abilities by inducing DNA over-replication and genomic instability [56]. In budding yeast loss of MCM10 can indeed result in replisome instability and DNA damage, which require DSB repair. In cases where the DNA damage response checkpoints are compromised or unable to efficiently repair, the breaks can persist and contribute to a severe form of fork collapse, leading to irreversible replication and fork failure [125]. MCM10 inhibits fork regression by binding the fork reversal enzyme SMARCAL1 to the fork junction or helps to reanneal unwound nascent strands to their parental template [126]. Interestingly, in MCM10 deficient fission yeast cells, the pre-initiation complex is formed, but DNA unwinding does not occur, which is required for replication fork assembly. These findings imply that MCM10 self-interaction might directly influence DNA replication [127].
7 MCM10 in disease, cancer diagnostics, and therapeutics
One of the early events during cancer development in mammalian cells is the accumulation of genomic instability among healthy cells, and MCM10 plays a crucial role in its progression [117]. It has been reported that MCM10 controls prostate cancer’s progression by regulating several biological processes, including cell death and apoptosis [128]. MCM10 controls the cell cycle by regulating the expression of genes that are essential for DNA replication and cell division. It enhances tumorigenic characteristics in non-tumorigenic mammary cells through increased proliferation and a shortened cell cycle [129]. In such cells, mesenchymal markers are affected, including the up-regulation of Vimentin, the transcription factors Snail and Twist2, and the down-regulation of the tumor suppressor protein E-cadherin, which are part of the complex process of cancer progression and metastasis. Elevated MCM10 expression in normal epithelial cells leads to increased mRNA levels of the transcription factors Snail and Twist2, along with elevated phospho-AKT(ser473) and phospho-Gsk-3beta levels, which indicates MCM10’s role in cancer development [5]. This is supported by the fact that phosphorylation of AKT, GSK3β, and Snail promotes tumorigenesis, particularly in squamous cell carcinoma and breast cancer [130, 131]. Additionally, increased accumulation of ssDNA has been correlated with overexpression of MCM10, as evidenced by enhanced expression of ATR and CHK1 proteins which are the downstream targets of ssDNA [5]. Recently, it was shown that increased c-Myc activity is a major factor of DNA replication stress in breast cancer stem cells and that MCM10 is a c-Myc-dependent firing factor [51]. c-Myc is induced by the expression of the E2F1 and E2F2 genes and can indirectly promote MCM expression [132]. Similarly, MYCN (N-MYC), another MYC family of proto-oncogenes, was shown to be involved in the regulation of the MCM10 protein [132] possibly via the E2F/pRB pathway, which induces the expression of MCM10 in the human colorectal carcinoma cell line HCT116 [133, 134]. In neuroblastoma, a strong correlation was observed between MCM10 and N-MYC expression with accompanying promoter binding [132]. All these observations point towards the role of these protooncogenes in regulating MCM10 expression. The Wnt/β-catenin signaling pathway is critical in cell proliferation and differentiation and contributes to the progression of cancer. Wnt signaling controls the G1-S phase transition, which leads to genome duplication and mitosis. Increased levels of β-catenin could potentially stimulate the transformation of normal cells into a cancerous state by inducing the expression of the Cyclin D1 gene (CCND1), leading to unregulated cell cycle progression [135]. Interestingly, among the MCM10 knockout group, substantial downregulation was observed in cyclin D1 levels responsible for the G1/S transition, which indicates that reduced expression of this cyclin might leads to a cell cycle arrest phenotype. Analyzing the expression levels of both MCM10 and CCND1 could serve as a valuable prognostic indicator for early-stage lung cancer [136].
Recent studies have shown MCM10 to be overexpressed in various cancer tissues, including the ovarian, breast, pancreas, prostate, bone, brain, and lung [101, 102, 128, 132, 137, 138] (Fig. 3). Overexpression of MCM10 can up-regulate the DNA damage response during the early stages of cancer, and recent studies have investigated the link between MCM10 expression and the aggressiveness of breast and cervical cancer [5, 139]. In addition, elevated MCM10 levels were found to correspond to increased genomic amplification in cancer cells [25, 140, 141]. Breast cancer patients expressing a lower level of MCM10 had significantly longer survival than those patients whose tumors expressed a higher level of MCM10 [5], indicating that MCM10 levels are linked to tumor progression. All the above findings imply that MCM10 expression correlates with cancer progression and defines the aggressiveness of cancer. Recently, insulin-like growth factors (IGFs) have been shown to play significant roles in mammalian growth, development, aging, and disease processes [142]. Current research indicates that MCM10 is regulated by IGF-1, and dysregulated IGF signaling can contribute to malignant transformations leading to the advancement of tumors [128, 142, 143]. MCM10 expression can also be influenced by microRNA like miR-146-5p, known for its involvement in the regulation of cancer gene expression such as CXCL8 and UHRF1. miR-146-5p may target specific mRNA molecules that modulate the levels of proteins, including those related to DNA replication [144]. In addition, it also sheds light on the possible mechanisms of action and regulation of different pathways that lead to tumorigenesis. In liver cancer, MCM10 is known to be one of the genes related to clonal expression and shows poor prognosis [145,146,147], and patients with lung adenocarcinoma were shown to have considerably higher MCM2, MCM4, and MCM10 mRNA expression [148]. In the neuroblastoma cell line IMR32, the overexpression of DAX1 (an orphan nuclear receptor that is induced by the EWS/FLI1 oncoprotein, which is highly expressed in Ewing’s tumor), leads to a substantial increase in the expression of MCM10, which indicates that certain oncoproteins can directly influence MCM10 levels [140]. Increased MCM10 immunoexpression was found to be substantially correlated with an advanced primary tumor, nodal status, and vascular invasion in urothelial carcinoma [141]. Also, it is highly expressed in osteosarcoma samples compared to their normal counterparts [149]. Similarly, a recent study revealed a significant upregulation of MCM10 in prostate cancer (PCa) with a strong correlation between elevated MCM10 expression and advanced clinical stages [128]. In glioma cells, MCM10 mRNA was quantitated using qRT-PCR and microarray methods and validated with increasing expression in grade II, III, and IV gliomas [150].

Schematic representation of MCM10 overexpression in different types of cancer. MCM10 overexpression is a promising biomarker for cancer diagnosis, prognosis, and is a potential therapeutic target. Elevated MCM10 levels in specific cancer types could aid in identifying high-risk patients, facilitating early detection, and guiding personalized treatment approaches. References are given in brackets
MCM10 knockdown drastically reduced colony formation and cell proliferation in prostate cancer cell lines and induced cell apoptosis [128]. Another study found that the knockdown of MCM10 in glioma cells showed disrupted cell proliferation, invasion, and migration, which indicates that MCM10 is involved in the regulation of glioblastoma multiforme [143]. In addition, a recent study identified high-frequency rare variants (c.849G > T) in MCM10 among Han Chinese women affected with Polycystic ovarian syndrome (PCOS) [151] that have been linked to the progression of the disease. MCM10 expression has been shown as a prognostic biomarker for patients with uterine corpus endometrial carcinoma and glioma [152, 153]. MCM genes may activate the ERK pathway, and it is thought that the ERK-1 and ERK-2 pathways may be related to the pathogenesis of PCOS [154]. The increased demand in cancer cells suggests that compounds inhibiting MCM10 might be important therapeutic agents to control cancer. MCM10 analogs like suramin (an antiparasitic agent) have recently been reported as anti-cancer treatments [155]. Therefore, MCM10 is a potential target with prognostic, diagnostic, and therapeutic significance [101, 139, 145, 152].
8 Discussion
MCM10 has long remained an enigmatic molecule, and recent research points towards its multifaceted nature, which includes novel and classical functions (Figs. 1 and 2). This evolutionary conserved protein physically interacts with members of the MCM2-7 helicase complex and with other DNA replication-associated proteins (Table 1). It is involved in DNA replication initiation, elongation, termination, and the maintenance of genome stability, as evident from the studies conducted in both vertebrates and non-vertebrates [15, 21, 30, 76, 87, 156]. MCM10 is not a stable component of the replisome, unlike DNA polymerase α or MCM2-7 core helicase as evidenced in yeast [27, 31]. It binds to chromatin through interactions with MCM2-7, followed by the loading of CDC45 and DNA polymerase α [42] as seen in human/Xenopus, and this ultimately leads to the initiation of genome duplication [75]. Overexpression of MCM10 is linked to the accumulation of DNA damage, genomic instability, and accelerated tumorigenesis [5, 139]. In addition, it has been shown that MCM10-deficient mammalian cells are incompetent in correcting replication-associated DNA damage, suggesting that MCM10 might have a role in repair [49]. The impact of mutations in MCM10 and their effects on the expression of reporter gene at the telomeres and HM loci of budding yeast revealed its role in maintaining heterochromatin (involving acetylation and deacetylation of the SIR proteins) and influencing transcriptional silencing [54].
Apart from its crucial role during DNA replication, other functions like chromatin organization, telomere maintenance, and replication timing might be second-order effects of the DNA replication process since all these functions (Figs. 1 and 2) are directly related to the genome duplication process, starting from DNA unwinding, replicating the genetic material, and repackaging of the newly synthesized DNA into a higher order chromatin structure. MCM10 also undergoes several cell cycle-linked post-translational modifications, like ubiquitination [55], SUMOylation [56], phosphorylation [25], and acetylation [88] as shown in studies from yeast and mammalian cells (Fig. 1). During periods of replicative stress, MCM10 regulates DNA fork speed and ensures timely completion by firing backup (dormant) replication origins so that the genome is duplicated on time, thus helping in recovery from DNA damage during the period of replicative stress [51, 66, 103, 157]. In mammalian cells, a small amount of MCM10 is sufficient for DNA replication, as shown by the fact that MCM10 ablation does not significantly affect cell proliferation in normal cells. However, this is not the case in cancer cells, where MCM10 appears to be a limiting factor and hence MCM10 levels dictate the extensiveness of origin firing and define the aggressiveness of cancer [5, 45, 51]. We speculate an increased number of MCM10 molecules can trigger a greater number of licensed origins with higher MCMs loaded (affinity), and this changes the replication dynamics between a normal and cancer cell. Recent observations in mammalian cells have demonstrated how deletion of MCM10 disrupts the replication timing program and plays an important role similar to the telomere-binding protein RIF1, which also modulates replication timing [81]. Telomerase activity is constrained in mcm10 mutants, resulting in the accumulation of abnormal replication fork structures and shortened telomeres [49]. MCM10 interacts with the tumor suppressor protein BRCA2 during DNA damage and recruits it to stabilize the fork [118]. In addition, it prevents lesion skipping and inhibits PRIMPOL-mediated repriming [118]. In budding yeast, MCM10 has been shown to interact with the fork reversal enzyme SMARCAL1 and inhibit fork regression, providing an overall protective function [126]. In such situations, either MCM10 nonspecifically binds the single-stranded region of a forked DNA that is stalled at the lesion on the leading strand or MCM10 prevents fork reversal by direct strand annealing activity [126]. All these findings from both yeast and mammalian studies support the hypothesis that loss of MCM10 leads to replication fork arrest, fork instability, DNA damage, and cell death.
The significance of MCM10 in cancer extends beyond its DNA replication-associated functions (discussed above), where it plays a role in firing backup origins. It differs from members of the MCM2-7 core helicase complex, which is not a limiting factor for cancer cell proliferation. Indeed, an environment with more licensed origins in cancer cells [158] creates a perfect opportunity to fire (aided by the dynamic nature of MCM10 to activate origins of DNA replication). It is tempting to speculate that increased MCM10 molecules can be attributed as a result of genomic amplification in cancer cells. We think that this feature probably controls the aggressiveness of cancers seen in breast, cervical, and ovarian, which is also defined by increased collisions between transcription and the replication machinery [51, 139]. Recently, it was shown that increased replication origin firing is linked to enhanced chromosomal missegregation and instability (CIN) in cancer cells [159]. We hypothesize that random origin firing by MCM10 can be a crucial factor behind increased microtubule dynamics in mitosis, leading to whole chromosome missegregation and CIN seen in cancer cells. We believe that MCM10, though involved throughout the replication process (initiation, elongation, and termination), is not an integral part of the replisome but can be envisioned acting in a loading/unloading mechanism where MCM10 transiently interacts with the DNA to accomplish its function. This hypothesis, which is also supported by studies from yeasts [12, 27, 28], needs further confirmation. Increased levels of MCM10 as a result of gene amplification might be regulating the number of active origins (which are crucial for the determination of the cancerous stage of the cervical cells) [139]. Overexpression of MCM10 leads to shortened cell cycle, particularly the S-phase which indicates a high rate of DNA replication. MCM10 binding to licensed origins can lead to multiple origin firing, and this can be tested for accumulation of ssDNA. An increased measure of RPA2 among the breast cancer biopsy samples compared to normal indicated a higher possibility of ssDNA accumulation in the presence of increased MCM10 molecules [5]. Interestingly, epigenetic and non-mutational regulatory mechanisms control MCM10 expression. This insight opens avenues for exploring MCM10 as a diagnostic and therapeutic target across various malignancies [160]. In addition, MCM10’s role in uncontrolled cellular proliferation makes it a prime target for drug design [161].
Since MCM10 acetylation is regulated by the mammalian histone deacetylase SIRT1 [88], its regulation points to the loading and unloading nature of MCM10 chromatin binding. Given the fact that MCM10 is upregulated in several cancers, it is a good candidate for early detection since its expression has been found to correlate with the tumor stages, which warrants the need to develop therapeutics like the antiparasitic drug Suramin [155] and also small molecule inhibitors designed against the posttranslational modifications of MCM10 (Fig. 1). MCM10 expression was shown to correlate with the stages of breast cancer and Gliomas [5, 150], and our ongoing studies with MCM10 and clinical cases of cervical cancer indicate a link between MCM10 expression and tumor progression (unpublished observation). Such studies can translate into kits for the early detection of cancer based on MCM10 expression levels. As we learn more about MCM10, we gain insights into its clinical manifestations in diseases like CNKD and RCM [49, 106, 115, 116] and get to know about the rare variants and mutations in MCM10 that have been identified in various diseases, highlighting its potential as a prognostic biomarker. This again urges the scientific community to investigate more into pathogenic mutations related to dysregulated replication initiation factors. MCM10 is thus a multifaceted molecule of clinical importance that should be explored further for the benefit of human health.
Data availability
No datasets were generated or analysed during the current study.
Abbreviations
- ATP:
-
Adenosine triphosphate
- BRCA2:
-
Breast cancer gene 2
- BrdU:
-
Bromodeoxyuridines
- CAF-1:
-
Chromatin assembly factor 1
- CCND1:
-
Cyclin D1
- CDC:
-
Cell division cycle
- CDK:
-
Cyclin-dependent kinases
- Cdt1:
-
Chromatin licensing and DNA replication factor 1
- Chk1:
-
Checkpoint kinase 1
- CIN:
-
Chromosomal instability
- CMG:
-
Cdc45-MCM2-7-GINS
- CNKD:
-
Classical natural killer cell deficiency
- CTD:
-
C-terminal domain
- Ctf4:
-
Chromosome transmission fidelity 4
- DH:
-
Double hexamers
- DDK:
-
DBF4-dependent kinase
- DDR:
-
DNA damage response
- DSB:
-
Double-strand breaks
- dsDNA:
-
Double-stranded DNA
- ERK:
-
Extracellular signal-regulated kinase
- EMT:
-
Epithelial-mesenchymal transition
- GINS:
-
Go-ichi-ni-san (5–1-2–3 in Japanese, Sld5, and Psf1,2,3)
- HSC:
-
Hematopoietic stem cell
- HP1:
-
Heterochromatin protein 1
- ID:
-
Internal domain
- IGF:
-
Insulin-like growth factor
- MCM:
-
Minichromosome maintenance protein
- MYCN(N-MYC):
-
One of the Myc family protooncogene
- NTD:
-
N-terminal domain
- NER:
-
Nucleotide excision repair
- NK cells:
-
Natural killer cells
- ORC:
-
Origin recognition complex
- OSCC:
-
Oesophageal squamous cell carcinoma
- PCa :
-
Prostate cancer
- PCOS:
-
Polycystic ovarian syndrome
- PIP:
-
PCNA interacting protein
- pre-RC:
-
Pre-replication complex
- PRIMPOL:
-
Primase and DNA-directed polymerase
- RCM:
-
Restrictive cardiomyopathy
- RECQ:
-
DNA helicase encoded by the RECQL4 gene
- RTEL1:
-
Regulator of telomere length 1
- RPA:
-
Replication protein A
- RIF1:
-
Rap-interacting factor 1
- ssDNA:
-
Single-stranded DNA
- SIR:
-
Silent information regulators
- SIRT1:
-
Sirtuin 1
- SMARCAL1:
-
SW1/SNF-related, matrix-associated, actin-dependent regulator of chromatin, subfamily A Like 1
References
Sclafani, R. A., & Holzen, T. M. (2007). Cell cycle regulation of DNA replication. Annual Review of Genetics, 41, 237–280. https://doi.org/10.1146/annurev.genet.41.110306.130308
DePamphilis, M. L. (2016). Genome duplication: The heartbeat of developing organisms. Current Topics in Developmental Biology, 116, 201–229. https://doi.org/10.1016/bs.ctdb.2015.10.002
Gaillard, H., García-Muse, T., & Aguilera, A. (2015). Replication stress and cancer. Nature Reviews Cancer, 15, 276–289. https://doi.org/10.1038/nrc3916
Waterman, D. P., Haber, J. E., & Smolka, M. B. (2020). Checkpoint responses to DNA double-strand breaks. Annual Review of Biochemistry, 89, 103–133. https://doi.org/10.1146/annurev-biochem-011520-104722
Mughal, M. J., Chan, K. I., Mahadevappa, R., Wong, S. W., Wai, K. C., & Kwok, H. F. (2022). Over-Activation of Minichromosome Maintenance Protein 10 Promotes Genomic Instability in Early Stages of Breast Cancer. International Journal of Biological Sciences, 18, 3827–3844. https://doi.org/10.7150/ijbs.69344
Bell, S. P. (2017). Rethinking origin licensing. eLife, 6, e24052. https://doi.org/10.7554/eLife.24052
Lewis, J. S., Gross, M. H., Sousa, J., Henrikus, S. S., Greiwe, J. F., Nans, A., Diffley, J. F. X., & Costa, A. (2022). Mechanism of replication origin melting nucleated by CMG helicase assembly. Nature, 606, 1007–1014. https://doi.org/10.1038/s41586-022-04829-4
Polasek-Sedlackova, H., Miller, T. C. R., Krejci, J., Rask, M.-B., & Lukas, J. (2022). Solving the MCM paradox by visualizing the scaffold of CMG helicase at active replisomes. Nature Communications, 13, 6090. https://doi.org/10.1038/s41467-022-33887-5
Wang, Y., Chen, H., Zhang, J., Cheng, A. S. L., Yu, J., To, K. F., & Kang, W. (2020). MCM family in gastrointestinal cancer and other malignancies: From functional characterization to clinical implication. Biochimica et Biophysica Acta - Reviews on Cancer, 1874, 188415. https://doi.org/10.1016/j.bbcan.2020.188415
Briu, L.-M., Maric, C., & Cadoret, J.-C. (2021). Replication Stress, Genomic Instability, and Replication Timing: A Complex Relationship. International Journal of Molecular Sciences, 22, 4764. https://doi.org/10.3390/ijms22094764
Warren, E. M., Huang, H., Fanning, E., Chazin, W. J., & Eichman, B. F. (2009). Physical Interactions between Mcm10, DNA, and DNA Polymerase α *. Journal of Biological Chemistry, 284, 24662–24672. https://doi.org/10.1074/jbc.M109.020438
Kanke, M., Kodama, Y., Takahashi, T. S., Nakagawa, T., & Masukata, H. (2012). Mcm10 plays an essential role in origin DNA unwinding after loading of the CMG components. EMBO Journal, 31, 2182–2194. https://doi.org/10.1038/emboj.2012.68
Solomon, N. A., Wright, M. B., Chang, S., Buckley, A. M., Dumas, L. B., & Gaber, R. F. (1992). Genetic and molecular analysis of DNA43 and DNA52: Two new cell-cycle genes in Saccharomyces cerevisiae. Yeast Chichester Engl., 8, 273–289. https://doi.org/10.1002/yea.320080405
Merchant, A. M., Kawasaki, Y., Chen, Y., Lei, M., & Tye, B. K. (1997). A lesion in the DNA replication initiation factor Mcm10 induces pausing of elongation forks through chromosomal replication origins in Saccharomyces cerevisiae. Molecular and Cellular Biology, 17, 3261–3271.
Wohlschlegel, J. A., Dhar, S. K., Prokhorova, T. A., Dutta, A., & Walter, J. C. (2002). Xenopus Mcm10 binds to origins of DNA replication after Mcm2-7 and stimulates origin binding of Cdc45. Molecular Cell, 9, 233–240. https://doi.org/10.1016/s1097-2765(02)00456-2
Gregan, J., Lindner, K., Brimage, L., Franklin, R., Namdar, M., Hart, E. A., Aves, S. J., & Kearsey, S. E. (2003). Fission yeast Cdc23/Mcm10 functions after pre-replicative complex formation to promote Cdc45 chromatin binding. Molecular Biology of the Cell, 14, 3876–3887. https://doi.org/10.1091/mbc.e03-02-0090
Sawyer, S. L., Cheng, I. H., Chai, W., & Tye, B. K. (2004). Mcm10 and Cdc45 cooperate in origin activation in Saccharomyces cerevisiae. Journal of Molecular Biology, 340, 195–202. https://doi.org/10.1016/j.jmb.2004.04.066
Heller, R. C., Kang, S., Lam, W. M., Chen, S., Chan, C. S., & Bell, S. P. (2011). Eukaryotic origin-dependent DNA replication in vitro reveals sequential action of DDK and S-CDK kinases. Cell, 146, 80–91. https://doi.org/10.1016/j.cell.2011.06.012
Maine, G. T., Sinha, P., & Tye, B.-K. (1984). Mutants of s. cerevisiae defective in the maintenance of minichromosomes. Genetics, 106, 365–385. https://doi.org/10.1093/genetics/106.3.365
Izumi, M., Yanagi, K., Mizuno, T., Yokoi, M., Kawasaki, Y., Moon, K.-Y., Hurwitz, J., Yatagai, F., & Hanaoka, F. (2000). The human homolog of Saccharomyces cerevisiae Mcm10 interacts with replication factors and dissociates from nuclease-resistant nuclear structures in G2 phase. Nucleic Acids Research, 28, 4769–4777.
Homesley, L., Lei, M., Kawasaki, Y., Sawyer, S., Christensen, T., & Tye, B. K. (2000). Mcm10 and the MCM2–7 complex interact to initiate DNA synthesis and to release replication factors from origins. Genes & Development, 14, 913–926.
Fien, K., Cho, Y.-S., Lee, J.-K., Raychaudhuri, S., Tappin, I., & Hurwitz, J. (2004). Primer utilization by DNA polymerase alpha-primase is influenced by its interaction with Mcm10p. Journal of Biological Chemistry, 279, 16144–16153. https://doi.org/10.1074/jbc.M400142200
Das-Bradoo, S., Ricke, R. M., & Bielinsky, A.-K. (2006). Interaction between PCNA and Diubiquitinated Mcm10 Is Essential for Cell Growth in Budding Yeast. Molecular and Cellular Biology, 26, 4806–4817. https://doi.org/10.1128/MCB.02062-05
Fernández-Cid, A., Riera, A., Tognetti, S., Herrera, M. C., Samel, S., Evrin, C., Winkler, C., Gardenal, E., Uhle, S., & Speck, C. (2013). An ORC/Cdc6/MCM2-7 complex is formed in a Multistep reaction to serve as a platform for MCM double-hexamer assembly. Molecular Cell, 50, 577–588. https://doi.org/10.1016/j.molcel.2013.03.026
Izumi, M., Yatagai, F., & Hanaoka, F. (2001). Cell cycle-dependent proteolysis and phosphorylation of human Mcm10. Journal of Biological Chemistry, 276, 48526–48531. https://doi.org/10.1074/jbc.M107190200
Simon, A. C., Zhou, J. C., Perera, R. L., van Deursen, F., Evrin, C., Ivanova, M. E., Kilkenny, M. L., Renault, L., Kjaer, S., Matak-Vinković, D., Labib, K., Costa, A., & Pellegrini, L. (2014). A Ctf4 trimer couples the CMG helicase to DNA polymerase α in the eukaryotic replisome. Nature, 510, 293–297. https://doi.org/10.1038/nature13234
van Deursen, F., Sengupta, S., De Piccoli, G., Sanchez-Diaz, A., & Labib, K. (2012). Mcm10 associates with the loaded DNA helicase at replication origins and defines a novel step in its activation. EMBO Journal, 31, 2195–2206. https://doi.org/10.1038/emboj.2012.69
Watase, G., Takisawa, H., & Kanemaki, M. T. (2012). Mcm10 plays a role in functioning of the eukaryotic replicative DNA helicase, Cdc45-Mcm-GINS. Curr. Biol. CB, 22, 343–349. https://doi.org/10.1016/j.cub.2012.01.023
Yeeles, J. T. P., Deegan, T. D., Janska, A., Early, A., & Diffley, J. F. X. (2015). Regulated eukaryotic DNA replication origin firing with purified proteins. Nature, 519, 431–435. https://doi.org/10.1038/nature14285
Lõoke, M., Maloney, M. F., & Bell, S. P. (2017). Mcm10 regulates DNA replication elongation by stimulating the CMG replicative helicase. Genes & Development, 31, 291–305. https://doi.org/10.1101/gad.291336.116
Gambus, A., van Deursen, F., Polychronopoulos, D., Foltman, M., Jones, R. C., Edmondson, R. D., Calzada, A., & Labib, K. (2009). A key role for Ctf4 in coupling the MCM2-7 helicase to DNA polymerase α within the eukaryotic replisome. EMBO Journal, 28, 2992–3004. https://doi.org/10.1038/emboj.2009.226
Im, J.-S., Ki, S.-H., Farina, A., Jung, D.-S., Hurwitz, J., & Lee, J.-K. (2009). Assembly of the Cdc45-Mcm2-7-GINS complex in human cells requires the Ctf4/And-1, RecQL4, and Mcm10 proteins. Proceedings of the National Academy of Sciences USA, 106, 15628–15632. https://doi.org/10.1073/pnas.0908039106
Karnani, N., & Dutta, A. (2011). The effect of the intra-S-phase checkpoint on origins of replication in human cells. Genes & Development, 25, 621–633. https://doi.org/10.1101/gad.2029711
Perez-Arnaiz, P., & Kaplan, D. L. (2016). An Mcm10 Mutant Defective in ssDNA Binding Shows Defects in DNA Replication Initiation. Journal of Molecular Biology, 428, 4608–4625. https://doi.org/10.1016/j.jmb.2016.10.014
Douglas, M. E., & Diffley, J. F. X. (2016). Recruitment of Mcm10 to Sites of Replication Initiation Requires Direct Binding to the Minichromosome Maintenance (MCM) Complex *. Journal of Biological Chemistry, 291, 5879–5888. https://doi.org/10.1074/jbc.M115.707802
Baxley, R. M., & Bielinsky, A.-K. (2017). Mcm10: A Dynamic Scaffold at Eukaryotic Replication Forks. Genes, 8, 73. https://doi.org/10.3390/genes8020073
Langston, L. D., Mayle, R., Schauer, G. D., Yurieva, O., Zhang, D., Yao, N. Y., Georgescu, R. E., & O’Donnell, M. E. (2017). Mcm10 promotes rapid isomerization of CMG-DNA for replisome bypass of lagging strand DNA blocks. eLife, 6, e29118. https://doi.org/10.7554/eLife.29118
Deegan, T. D., Baxter, J., Ortiz Bazán, M. Á., Yeeles, J. T. P., & Labib, K. P. M. (2019). Pif1-family helicases support fork convergence during DNA replication termination in eukaryotes. Molecular Cell, 74, 231-244.e9. https://doi.org/10.1016/j.molcel.2019.01.040
Hill, J., Eickhoff, P., Drury, L. S., Costa, A., & Diffley, J. F. X. (2020). The eukaryotic replisome requires an additional helicase to disarm dormant replication origins. bioRxiv, 17, 301366. https://doi.org/10.1101/2020.09.17.301366
Kawasaki, Y., Hiraga, S., & Sugino, A. (2000). Interactions between Mcm10p and other replication factors are required for proper initiation and elongation of chromosomal DNA replication in Saccharomyces cerevisiae. Genes to Cells, 5, 975–989. https://doi.org/10.1046/j.1365-2443.2000.00387.x
Liu, Q., Choe, W., & Campbell, J. L. (2000). Identification of the Xenopus laevis homolog of Saccharomyces cerevisiae DNA2 and its role in DNA replication. Journal of Biological Chemistry, 275, 1615–1624. https://doi.org/10.1074/jbc.275.3.1615
Zhu, W., Ukomadu, C., Jha, S., Senga, T., Dhar, S. K., Wohlschlegel, J. A., Nutt, L. K., Kornbluth, S., & Dutta, A. (2007). Mcm10 and And-1/CTF4 recruit DNA polymerase alpha to chromatin for initiation of DNA replication. Genes & Development, 21, 2288–2299. https://doi.org/10.1101/gad.1585607
Ricke, R. M., & Bielinsky, A.-K. (2004). Mcm10 regulates the stability and chromatin association of DNA polymerase-alpha. Molecular Cell, 16, 173–185. https://doi.org/10.1016/j.molcel.2004.09.017
Chadha, G. S., Gambus, A., Gillespie, P. J., & Blow, J. J. (2016). Xenopus Mcm10 is a CDK-substrate required for replication fork stability. Cell Cycle Georget. Tex, 15, 2183–2195. https://doi.org/10.1080/15384101.2016.1199305
Henrikus, S. S., Gross, M. H., Willhoft, O., Pühringer, T., Lewis, J. S., McClure, A. W., Greiwe, J. F., Palm, G., Nans, A., Diffley, J. F. X., & Costa, A. (2024). Unwinding of a eukaryotic origin of replication visualized by cryo-EM. Nature Structural & Molecular Biology, 17, 1–12. https://doi.org/10.1038/s41594-024-01280-z
Wawrousek, K. E., Fortini, B. K., Polaczek, P., Chen, L., Liu, Q., Dunphy, W. G., & Campbell, J. L. (2010). Xenopus DNA2 is a helicase/nuclease that is found in complexes with replication proteins And-1/Ctf4 and Mcm10 and DSB response proteins Nbs1 and ATM. Cell Cycle Georget. Tex, 9, 1156–1166. https://doi.org/10.4161/cc.9.6.11049
Thu, Y. M., & Bielinsky, A.-K. (2013). Enigmatic roles of Mcm10 in DNA replication. Trends in Biochemical Sciences, 38, 184–194. https://doi.org/10.1016/j.tibs.2012.12.003
Lee, C., Liachko, I., Bouten, R., Kelman, Z., & Tye, B. K. (2010). Alternative mechanisms for coordinating polymerase alpha and MCM helicase. Molecular and Cellular Biology, 30, 423–435. https://doi.org/10.1128/MCB.01240-09
Baxley, R. M., Leung, W., Schmit, M. M., Matson, J. P., Yin, L., Oram, M. K., Wang, L., Taylor, J., Hedberg, J., Rogers, C. B., Harvey, A. J., Basu, D., Taylor, J. C., Pagnamenta, A. T., Dreau, H., Craft, J., Ormondroyd, E., Watkins, H., Hendrickson, E. A., … Bielinsky, A.-K. (2021). Bi-allelic MCM10 variants associated with immune dysfunction and cardiomyopathy cause telomere shortening. Nature Communications, 12, 1626. https://doi.org/10.1038/s41467-021-21878-x
Ibarra, A., Schwob, E., & Méndez, J. (2008). Excess MCM proteins protect human cells from replicative stress by licensing backup origins of replication. Proceedings of the National Academy of Sciences of the United States of America, 105, 8956–8961. https://doi.org/10.1073/pnas.0803978105
Murayama, T., Takeuchi, Y., Yamawaki, K., Natsume, T., Li, M., Marcela, R.-C.N., Nishimura, T., Kogure, Y., Nakata, A., Tominaga, K., Sasahara, A., Yano, M., Ishikawa, S., Ohta, T., Ikeda, K., Horie-Inoue, K., Inoue, S., Seki, M., Suzuki, Y., … Gotoh, N. (2021). MCM10 compensates for Myc-induced DNA replication stress in breast cancer stem-like cells. Cancer Science, 112, 1209–1224. https://doi.org/10.1111/cas.14776
Georgescu, R., Yuan, Z., Bai, L., de Luna Almeida, R., Santos, J., Sun, D., Zhang, O., Yurieva, H., & O’Donnell, M. E. (2017). Structure of eukaryotic CMG helicase at a replication fork and implications to replisome architecture and origin initiation. Proceedings of the National Academy of Sciences of the United States of America, 114, E697–E706. https://doi.org/10.1073/pnas.1620500114
Vo, N., Anh Suong, D. N., Yoshino, N., Yoshida, H., Cotterill, S., & Yamaguchi, M. (2017). Novel roles of HP1a and Mcm10 in DNA replication, genome maintenance and photoreceptor cell differentiation. Nucleic Acids Research, 45, 1233–1254. https://doi.org/10.1093/nar/gkw1174
Liachko, I., & Tye, B. K. (2009). Mcm10 mediates the interaction between DNA replication and silencing machineries. Genetics, 181, 379–391. https://doi.org/10.1534/genetics.108.099101
Deegan, T. D., Mukherjee, P. P., Fujisawa, R., Polo Rivera, C., & Labib, K. (2020). CMG helicase disassembly is controlled by replication fork DNA, replisome components and a ubiquitin threshold. eLife, 9, e60371. https://doi.org/10.7554/eLife.60371
Tian, J., Lu, Z., Niu, S., Zhang, S., Ying, P., Wang, L., Zhang, M., Cai, Y., Dong, T., Zhu, Y., Zhong, R., Wang, Z., Chang, J., & Miao, X. (2021). Aberrant MCM10 SUMOylation induces genomic instability mediated by a genetic variant associated with survival of esophageal squamous cell carcinoma. Clinical and Translational Medicine, 11, e485. https://doi.org/10.1002/ctm2.485
Lee, J.-K., Seo, Y.-S., & Hurwitz, J. (2003). The Cdc23 (Mcm10) protein is required for the phosphorylation of minichromosome maintenance complex by the Dfp1-Hsk1 kinase. Proceedings of the National Academy of Sciences USA, 100, 2334–2339. https://doi.org/10.1073/pnas.0237384100
Evrin, C., Clarke, P., Zech, J., Lurz, R., Sun, J., Uhle, S., Li, H., Stillman, B., & Speck, C. (2009). A double-hexameric MCM2-7 complex is loaded onto origin DNA during licensing of eukaryotic DNA replication. Proceedings of the National Academy of Sciences USA, 106, 20240–20245. https://doi.org/10.1073/pnas.0911500106
Nguyen, V. Q., Co, C., & Li, J. J. (2001). Cyclin-dependent kinases prevent DNA re-replication through multiple mechanisms. Nature, 411, 1068–1073. https://doi.org/10.1038/35082600
Langston, L. D., & O’Donnell, M. E. (2019). An explanation for origin unwinding in eukaryotes. eLife, 8, e46515. https://doi.org/10.7554/eLife.46515
Wasserman, M. R., Schauer, G. D., O’Donnell, M. E., & Liu, S. (2019). Replication Fork Activation Is Enabled by a Single-Stranded DNA Gate in CMG Helicase. Cell, 178, 600-611.e16. https://doi.org/10.1016/j.cell.2019.06.032
Xu, X., Rochette, P. J., Feyissa, E. A., Su, T. V., & Liu, Y. (2009). MCM10 mediates RECQ4 association with MCM2-7 helicase complex during DNA replication. EMBO Journal, 28, 3005–3014. https://doi.org/10.1038/emboj.2009.235
Kliszczak, M., Sedlackova, H., Pitchai, G. P., Streicher, W. W., Krejci, L., & Hickson, I. D. (2015). Interaction of RECQ4 and MCM10 is important for efficient DNA replication origin firing in human cells. Oncotarget, 6, 40464–40479. https://doi.org/10.18632/oncotarget.6342
Xiang, S., Luo, X., Welch, D., Reed, D. R., & Alexandrow, M. G. (2023). Identification of selective ATP-competitive CMG helicase inhibitors for cancer intervention that disrupt CMG-replisome function. Research Square, 11, 355–3182731. https://doi.org/10.21203/rs.3.rs-3182731/v1
Bruck, I., & Kaplan, D. (2009). Dbf4-Cdc7 Phosphorylation of Mcm2 Is Required for Cell Growth. Journal of Biological Chemistry, 284, 28823–28831. https://doi.org/10.1074/jbc.M109.039123
Brosh, R. M., & Trakselis, M. A. (2019). Fine-tuning of the replisome: Mcm10 regulates fork progression and regression. Cell Cycle, 18, 1047–1055. https://doi.org/10.1080/15384101.2019.1609833
Ricke, R. M., & Bielinsky, A.-K. (2006). A Conserved Hsp10-like Domain in Mcm10 Is Required to Stabilize the Catalytic Subunit of DNA Polymerase-α in Budding Yeast *. Journal of Biological Chemistry, 281, 18414–18425. https://doi.org/10.1074/jbc.M513551200
Perez-Arnaiz, P., Bruck, I., Colbert, M. K., & Kaplan, D. L. (2017). An intact Mcm10 coiled-coil interaction surface is important for origin melting, helicase assembly and the recruitment of Pol-α to Mcm2-7. Nucleic Acids Research, 45, 7261–7275. https://doi.org/10.1093/nar/gkx438
Im, J.-S., Park, S.-Y., Cho, W.-H., Bae, S.-H., Hurwitz, J., & Lee, J.-K. (2015). RecQL4 is required for the association of Mcm10 and Ctf4 with replication origins in human cells. Cell Cycle, 14, 1001–1009. https://doi.org/10.1080/15384101.2015.1007001
Aves, S. J., Tongue, N., Foster, A. J., & Hart, E. A. (1998). The essential schizosaccharomyces pombe cdc23 DNA replication gene shares structural and functional homology with the Saccharomyces cerevisiae DNA43 (MCM10) gene. Current Genetics, 34, 164–171. https://doi.org/10.1007/s002940050382
Locovei, A. M., Spiga, M.-G., Tanaka, K., Murakami, Y., & D’Urso, G. (2006). The CENP-B homolog, Abp1, interacts with the initiation protein Cdc23 (MCM10) and is required for efficient DNA replication in fission yeast. Cell Division, 1, 27. https://doi.org/10.1186/1747-1028-1-27
Taylor, M., Moore, K., Murray, J., Aves, S. J., & Price, C. (2011). Mcm10 interacts with Rad4/Cut5(TopBP1) and its association with origins of DNA replication is dependent on Rad4/Cut5(TopBP1). DNA Repair, 10, 1154–1163. https://doi.org/10.1016/j.dnarep.2011.09.001
Robertson, P. D., Warren, E. M., Zhang, H., Friedman, D. B., Lary, J. W., Cole, J. L., Tutter, A. V., Walter, J. C., Fanning, E., & Eichman, B. F. (2008). Domain architecture and biochemical characterization of vertebrate Mcm10. Journal of Biological Chemistry, 283, 3338–3348. https://doi.org/10.1074/jbc.M706267200
Douglas, M. E., Ali, F. A., Costa, A., & Diffley, J. F. X. (2018). The mechanism of eukaryotic CMG helicase activation. Nature, 555, 265–268. https://doi.org/10.1038/nature25787
Izumi, M., Mizuno, T., Yanagi, K.-I., Sugimura, K., Okumura, K., Imamoto, N., Abe, T., & Hanaoka, F. (2017). The Mcm2-7-interacting domain of human mini-chromosome maintenance 10 (Mcm10) protein is important for stable chromatin association and origin firing. Journal of Biological Chemistry, 292, 13008–13021. https://doi.org/10.1074/jbc.M117.779371
Campos, L. V., Van Ravenstein, S. X., Vontalge, E. J., Greer, B. H., Heintzman, D. R., Kavlashvili, T., McDonald, W. H., Rose, K. L., Eichman, B. F., & Dewar, J. M. (2023). RTEL1 and MCM10 overcome topological stress during vertebrate replication termination. Cell Reports, 42, 112109. https://doi.org/10.1016/j.celrep.2023.112109
Rhind, N. (2006). DNA replication timing: Random thoughts about origin firing. Nature Cell Biology, 8, 1313–1316. https://doi.org/10.1038/ncb1206-1313
Wang, W., Klein, K. N., Proesmans, K., Yang, H., Marchal, C., Zhu, X., Borrman, T., Hastie, A., Weng, Z., Bechhoefer, J., Chen, C.-L., Gilbert, D. M., & Rhind, N. (2021). Genome-wide mapping of human DNA replication by optical replication mapping supports a stochastic model of eukaryotic replication. Molecular Cell, 81, 2975-2988.e6. https://doi.org/10.1016/j.molcel.2021.05.024
Windhager, J., Paine, A., Nathanailidou, P., Tasiudi, E., Martínez, M. R., Lygeros, J., Lygerou, Z., & Rapsomaniki, M. A. (2019). A stochastic hybrid model of DNA replication incorporating 3D protein mobility dynamics. BioRxiv, 583187. https://doi.org/10.1101/583187
Das, S. P., Borrman, T., Liu, V. W. T., Yang, S.C.-H., Bechhoefer, J., & Rhind, N. (2015). Replication timing is regulated by the number of MCMs loaded at origins. Genome Research, 25, 1886–1892. https://doi.org/10.1101/gr.195305.115
Caballero, M., Ge, T., Rebelo, A. R., Seo, S., Kim, S., Brooks, K., Zuccaro, M., Kanagaraj, R., Vershkov, D., Kim, D., Smogorzewska, A., Smolka, M., Benvenisty, N., West, S. C., Egli, D., Mace, E. M., & Koren, A. (2022). Comprehensive analysis of DNA replication timing across 184 cell lines suggests a role for MCM10 in replication timing regulation. Human Molecular Genetics, 31, 2899–2917. https://doi.org/10.1093/hmg/ddac082
Peycheva, M., Neumann, T., Malzl, D., Nazarova, M., Schoeberl, U. E., & Pavri, R. (2022). DNA replication timing directly regulates the frequency of oncogenic chromosomal translocations. Science, 377, eabj5502. https://doi.org/10.1126/science.abj5502
Orr, S., Gaymes, T., Ladon, D., Chronis, C., Czepulkowski, B., Wang, R., Mufti, G., Marcotte, E., & Thomas, N. (2010). Reducing MCM levels in human primary T cells during the G0→G1 transition causes genomic instability during the first cell cycle. Oncogene, 29, 3803–3814. https://doi.org/10.1038/onc.2010.138
Xu, X., Hua, X., Brown, K., Ren, X., & Zhang, Z. (2022). Mcm2 promotes stem cell differentiation via its ability to bind H3–H4. ELife, 11, e80917. https://doi.org/10.7554/eLife.80917
Cacialli, P., Dogan, S., Linnerz, T., Pasche, C., & Bertrand, J. Y. (2023). Minichromosome maintenance protein 10 (mcm10) regulates hematopoietic stem cell emergence in the zebrafish embryo. Stem Cell Rep., 18, 1534–1546. https://doi.org/10.1016/j.stemcr.2023.05.022
Zhao, X., Wang, J., Jin, D., Cheng, J., Chen, H., Li, Z., Wang, Y., Lou, H., Zhu, J.-K., Du, X., & Gong, Z. (2023). AtMCM10 promotes DNA replication-coupled nucleosome assembly in Arabidopsis. Journal of Integrative Plant Biology, 65, 203–222. https://doi.org/10.1111/jipb.13438
Christensen, T. W., & Tye, B. K. (2003). Drosophila Mcm10 interacts with members of the prereplication complex and is required for proper chromosome condensation. Molecular Biology of the Cell, 14, 2206–2215. https://doi.org/10.1091/mbc.e02-11-0706
Fatoba, S. T., Tognetti, S., Berto, M., Leo, E., Mulvey, C. M., Godovac-Zimmermann, J., Pommier, Y., & Okorokov, A. L. (2013). Human SIRT1 regulates DNA binding and stability of the Mcm10 DNA replication factor via deacetylation. Nucleic Acids Research, 41, 4065–4079. https://doi.org/10.1093/nar/gkt131
Alver, R. C., Zhang, T., Josephrajan, A., Fultz, B. L., Hendrix, C. J., Das-Bradoo, S., & Bielinsky, A.-K. (2014). The N-terminus of Mcm10 is important for interaction with the 9–1-1 clamp and in resistance to DNA damage. Nucleic Acids Research, 42, 8389–8404. https://doi.org/10.1093/nar/gku479
Quan, Y., Xia, Y., Liu, L., Cui, J., Li, Z., Cao, Q., Chen, X. S., Campbell, J. L., & Lou, H. (2015). Cell-cycle-regulated interaction between Mcm10 and double hexameric Mcm2-7 is required for helicase splitting and activation during S phase. Cell Reports, 13, 2576–2586. https://doi.org/10.1016/j.celrep.2015.11.018
Di Perna, R., Aria, V., De Falco, M., Sannino, V., Okorokov, A. L., Pisani, F. M., & De Felice, M. (2013). The physical interaction of Mcm10 with Cdc45 modulates their DNA-binding properties. The Biochemical Journal, 454, 333–343. https://doi.org/10.1042/BJ20130059
Cook, C. R., Kung, G., Peterson, F. C., Volkman, B. F., & Lei, M. (2003). A Novel Zinc Finger Is Required for Mcm10 Homocomplex Assembly *. Journal of Biological Chemistry, 278, 36051–36058. https://doi.org/10.1074/jbc.M306049200
Du, W., Stauffer, M. E., & Eichman, B. F. (2012). Structural Biology of Replication Initiation Factor Mcm10 (pp. 197–216). In S. MacNeill (Ed.), Eukaryot. Replisome Guide Protein Struct. Funct., Springer Netherlands, Dordrecht. https://doi.org/10.1007/978-94-007-4572-8_11
Burich, R., & Lei, M. (2003). Two bipartite NLSs mediate constitutive nuclear localization of Mcm10. Current Genetics, 44, 195–201. https://doi.org/10.1007/s00294-003-0443-y
Apger, J., Reubens, M., Henderson, L., Gouge, C. A., Ilic, N., Zhou, H. H., & Christensen, T. W. (2010). Multiple functions for drosophila Mcm10 suggested through analysis of two Mcm10 mutant alleles. Genetics, 185, 1151–1165. https://doi.org/10.1534/genetics.110.117234
Liachko, I., & Tye, B. K. (2005). Mcm10 is required for the maintenance of transcriptional silencing in Saccharomyces cerevisiae. Genetics, 171, 503–515. https://doi.org/10.1534/genetics.105.042333
Douglas, N. L., Dozier, S. K., & Donato, J. J. (2005). Dual Roles for Mcm10 in DNA Replication Initiation and Silencing at the Mating-type Loci. Molecular Biology Reports, 32, 197–204. https://doi.org/10.1007/s11033-005-2312-x
Albertson, D. G. (2006). Gene amplification in cancer. Trends in Genetics, 22, 447–455. https://doi.org/10.1016/j.tig.2006.06.007
Thu, Y. M., & Bielinsky, A.-K. (2014). MCM10: one tool for all - integrity, maintenance and damage control. Seminars in Cell and Developmental Biology, 30, 121–130. https://doi.org/10.1016/j.semcdb.2014.03.017
Chen, D., Zhong, N., Guo, Z., Ji, Q., Dong, Z., Zheng, J., Ma, Y., Zhang, J., He, Y., & Song, T. (2023). MCM10, a potential diagnostic, immunological, and prognostic biomarker in pan-cancer. Science and Reports, 13, 17701. https://doi.org/10.1038/s41598-023-44946-2
Shao, M., Yang, S., & Dong, S. (2021). High expression of MCM10 is predictive of poor outcomes in lung adenocarcinoma. PeerJ, 9, e10560. https://doi.org/10.7717/peerj.10560
Wu, Z., Wang, Y., Li, J., Wang, H., Tuo, X., & Zheng, J. (2022). MCM10 is a prognostic biomarker and correlated with immune checkpoints in ovarian cancer. Frontiers in Genetics, 13, 864578. https://www.frontiersin.org/articles/10.3389/fgene.2022.864578 (Accessed September 2, 2022).
Xiang, S., Reed, D. R., & Alexandrow, M. G. (2023). The CMG helicase and cancer: A tumor “engine” and weakness with missing mutations. Oncogene, 42, 473–490. https://doi.org/10.1038/s41388-022-02572-8
Baxley, R. M., Thu, Y. M., & Bielinsky, A. K. (2016). The role of Mcm10 in replication initiation (pp. 319–341). In D.L. Kaplan (Ed.), Initiat. DNA Replication Eukaryotes, Springer International Publishing, Cham. https://doi.org/10.1007/978-3-319-24696-3_16
Shen, Z. (2011). Genomic instability and cancer: An introduction. Journal of Molecular Cell Biology, 3, 1–3. https://doi.org/10.1093/jmcb/mjq057
Mace, E. M., Paust, S., Conte, M. I., Baxley, R. M., Schmit, M. M., Patil, S. L., Guilz, N. C., Mukherjee, M., Pezzi, A. E., Chmielowiec, J., Tatineni, S., Chinn, I. K., Akdemir, Z. C., Jhangiani, S. N., Muzny, D. M., Stray-Pedersen, A., Bradley, R. E., Moody, M., Connor, P. P., … Orange, J. S. (2020). Human NK cell deficiency as a result of biallelic mutations in MCM10. The Journal of Clinical Investigation, 130, 5272–5286. https://doi.org/10.1172/JCI134966
Zeman, M. K., & Cimprich, K. A. (2014). Causes and consequences of replication stress. Nature Cell Biology, 16, 2–9. https://doi.org/10.1038/ncb2897
Drissi, R., Chauvin, A., McKenna, A., Lévesque, D., Blais-Brochu, S., Jean, D., & Boisvert, F.-M. (2018). Destabilization of the MiniChromosome Maintenance (MCM) complex modulates the cellular response to DNA double strand breaks. Cell Cycle Georget Tex, 17, 2593–2609. https://doi.org/10.1080/15384101.2018.1553336
Kaur, M., Khan, M. D. M., Kar, A., Sharma, A., & Saxena, S. (2012). CRL4–DDB1–VPRBP ubiquitin ligase mediates the stress triggered proteolysis of Mcm10. Nucleic Acids Research, 40, 7332–7346. https://doi.org/10.1093/nar/gks366
Jevitt, A. M., Rankin, B. D., Chen, J., & Rankin, S. (2023). The cohesin modifier ESCO2 is stable during DNA replication. Chromosome Research, 31, 6. https://doi.org/10.1007/s10577-023-09711-1
Lopes, M., Foiani, M., & Sogo, J. M. (2006). Multiple Mechanisms Control Chromosome Integrity after Replication Fork Uncoupling and Restart at Irreparable UV Lesions. Molecular Cell, 21, 15–27. https://doi.org/10.1016/j.molcel.2005.11.015
McClure, A. W., & Diffley, J. F. (2021). Rad53 checkpoint kinase regulation of DNA replication fork rate via Mrc1 phosphorylation. eLife, 10, e69726. https://doi.org/10.7554/eLife.69726
Kaur, M., Sharma, A., Khan, M., Kar, A., & Saxena, S. (2010). Mcm10 proteolysis initiates before the onset of M-phase. BMC Cell Biology, 11, 84. https://doi.org/10.1186/1471-2121-11-84
Guilz, N. C. Ahn, Y. O., Fatima, H., Pedroza, L. A., Seo, S., Soni, R. K., Wang, N., Egli, D., & Mace, E. M. (2024). Replication stress in activated human NK cells induces sensitivity to apoptosis. Replication Stress in Activated Human NK Cells Induces Sensitivity to Apoptosis. Journal of Immunology (Baltimore, Md. : 1950), 213(1), 40–51. https://doi.org/10.4049/jimmunol.2300843
Schmit, M., & Bielinsky, A.-K. (2021). Congenital Diseases of DNA Replication: Clinical Phenotypes and Molecular Mechanisms. International Journal of Molecular Sciences, 22, E911. https://doi.org/10.3390/ijms22020911
Schmit, M. M., Baxley, R. M., Wang, L., Hinderlie, P., Kaufman, M., Simon, E., Raju, A., Miller, J. S., & Bielinsky, A.-K. (2024). A critical threshold of MCM10 is required to maintain genome stability during differentiation of induced pluripotent stem cells into natural killer cells. Open Biology, 14, 230407. https://doi.org/10.1098/rsob.230407
Kalimutho, M., Nones, K., Srihari, S., Duijf, P. H. G., Waddell, N., & Khanna, K. K. (2019). Patterns of Genomic Instability in Breast Cancer. Trends in Pharmacological Sciences, 40, 198–211. https://doi.org/10.1016/j.tips.2019.01.005
Kang, Z., Fu, P., Alcivar, A. L., Fu, H., Redon, C., Foo, T. K., Zuo, Y., Ye, C., Baxley, R., Madireddy, A., Buisson, R., Bielinsky, A.-K., Zou, L., Shen, Z., Aladjem, M. I., & Xia, B. (2021). BRCA2 associates with MCM10 to suppress PRIMPOL-mediated repriming and single-stranded gap formation after DNA damage. Nature Communications, 12, 5966. https://doi.org/10.1038/s41467-021-26227-6
Das, M., Prasad, S. B., Yadav, S. S., Govardhan, H. B., Pandey, L. K., Singh, S., Pradhan, S., & Narayan, G. (2013). Over Expression of Minichromosome Maintenance Genes is Clinically Correlated to Cervical Carcinogenesis. PLoS ONE, 8, e69607. https://doi.org/10.1371/journal.pone.0069607
Chattopadhyay, S., & Bielinsky, A.-K. (2007). Human Mcm10 Regulates the Catalytic Subunit of DNA Polymerase-α and Prevents DNA Damage during Replication. Molecular Biology of the Cell, 18, 4085–4095. https://doi.org/10.1091/mbc.E06-12-1148
Park, J. H., Bang, S. W., Kim, S. H., & Hwang, D. S. (2008). Knockdown of human MCM10 activates G2 checkpoint pathway. Biochemical and Biophysical Research Communications, 365, 490–495. https://doi.org/10.1016/j.bbrc.2007.11.004
Lim, H. J., Jeon, Y., Jeon, C. H., Kim, J. H., & Lee, H. (1813). Targeted disruption of Mcm10 causes defective embryonic cell proliferation and early embryo lethality. Biochimica et Biophysica Acta, 2011, 1777–1783. https://doi.org/10.1016/j.bbamcr.2011.05.012
Pilié, P. G., Tang, C., Mills, G. B., & Yap, T. A. (2019). State-of-the-art strategies for targeting the DNA damage response in cancer. Nature Reviews. Clinical Oncology, 16, 81–104. https://doi.org/10.1038/s41571-018-0114-z
Zou, L., & Elledge, S. J. (2003). Sensing DNA Damage Through ATRIP Recognition of RPA-ssDNA Complexes. Science, 300, 1542–1548. https://doi.org/10.1126/science.1083430
Cobb, J. A., Schleker, T., Rojas, V., Bjergbaek, L., Tercero, J. A., & Gasser, S. M. (2005). Replisome instability, fork collapse, and gross chromosomal rearrangements arise synergistically from Mec1 kinase and RecQ helicase mutations. Genes & Development, 19, 3055–3069. https://doi.org/10.1101/gad.361805
Mayle, R., Langston, L., Molloy, K. R., Zhang, D., Chait, B. T., & O’Donnell, M. E. (2019). Mcm10 has potent strand-annealing activity and limits translocase-mediated fork regression. Proceedings of the National Academy of Sciences of the United States of America, 116, 798–803. https://doi.org/10.1073/pnas.1819107116
Tanaka, S., & Ogawa, S. (2022). Dimerization of Firing Factors for Replication Origin Activation in Eukaryotes: A Crucial Process for Simultaneous Assembly of Bidirectional Replication Forks? Biology, 11, 928. https://doi.org/10.3390/biology11060928
Cui, F., Hu, J., Ning, S., Tan, J., & Tang, H. (2018). Overexpression of MCM10 promotes cell proliferation and predicts poor prognosis in prostate cancer. The Prostate, 78, 1299–1310. https://doi.org/10.1002/pros.23703
Chee, C. W., Mohd Hashim, N., Abdullah, I., & Nor Rashid, N. (2024). RNA sequencing and bioinformatics analysis reveals the downregulation of DNA replication genes by morindone in colorectal cancer cells. Biotechnology and Applied Biochemistry, 196, 3216–3233. https://doi.org/10.1007/s12010-023-04690-9
Sun, M., Guo, X., Qian, X., Wang, H., Yang, C., Brinkman, K. L., Serrano-Gonzalez, M., Jope, R. S., Zhou, B., Engler, D. A., Zhan, M., Wong, S. T. C., Fu, L., & Xu, B. (2012). Activation of the ATM-Snail pathway promotes breast cancer metastasis. Journal of Molecular Cell Biology, 4, 304–315. https://doi.org/10.1093/jmcb/mjs048
Grille, S. J., Bellacosa, A., Upson, J., Klein-Szanto, A. J., van Roy, F., Lee-Kwon, W., Donowitz, M., Tsichlis, P. N., & Larue, L. (2003). The protein kinase Akt induces epithelial mesenchymal transition and promotes enhanced motility and invasiveness of squamous cell carcinoma lines. Cancer Research, 63, 2172–2178.
Koppen, A., Ait-Aissa, R., Koster, J., van Sluis, P. G., Ora, I., Caron, H. N., Volckmann, R., Versteeg, R., & Valentijn, L. J. (2007). Direct regulation of the minichromosome maintenance complex by MYCN in neuroblastoma. European Journal of Cancer, 1990(43), 2413–2422. https://doi.org/10.1016/j.ejca.2007.07.024
Schuldiner, O., & Benvenisty, N. (2001). A DNA microarray screen for genes involved in c-MYC and N-MYC oncogenesis in human tumors. Oncogene, 20, 4984–4994. https://doi.org/10.1038/sj.onc.1204459
Yoshida, K., & Inoue, I. (2004). Expression of MCM10 and TopBP1 is regulated by cell proliferation and UV irradiation via the E2F transcription factor. Oncogene, 23, 6250–6260. https://doi.org/10.1038/sj.onc.1207829
Shtutman, M., Zhurinsky, J., Simcha, I., Albanese, C., D’Amico, M., Pestell, R., & Ben-Ze’ev, A. (1999). The cyclin D1 gene is a target of the beta-catenin/LEF-1 pathway. Proceedings of the National Academy of Sciences of the United States of America, 96, 5522–5527. https://doi.org/10.1073/pnas.96.10.5522
Wang, M., Xie, S., Yuan, W., Xie, T., Jamal, M., Huang, J., Yin, Q., Song, H., & Zhang, Q. (2019). Minichromosome maintenance protein 10 as a marker for proliferation and prognosis in lung cancer. International Journal of Oncology, 55, 1349–1360. https://doi.org/10.3892/ijo.2019.4899
Mahadevappa, R., Neves, H., Yuen, S. M., Jameel, M., Bai, Y., Yuen, H.-F., Zhang, S.-D., Zhu, Y., Lin, Y., & Kwok, H. F. (2018). DNA Replication Licensing Protein MCM10 Promotes Tumor Progression and Is a Novel Prognostic Biomarker and Potential Therapeutic Target in Breast Cancer. Cancers, 10, 282. https://doi.org/10.3390/cancers10090282
Peng, Y.-P., Zhu, Y., Yin, L.-D., Zhang, J.-J., Guo, S., Fu, Y., Miao, Y., & Wei, J.-S. (2016). The Expression and Prognostic Roles of MCMs in Pancreatic Cancer. PLoS ONE, 11, e0164150. https://doi.org/10.1371/journal.pone.0164150
Ahmed, S. M. Q., Laha, S., Das, R., Ifthikar, M. A., & Das, S. P. (2023). MCM10 expression is linked to cervical cancer aggressiveness. Frontiers in Molecular Medicine, 3, 1009903. https://www.frontiersin.org/articles/10.3389/fmmed.2023.1009903 (Accessed March 7, 2023).
García-Aragoncillo, E., Carrillo, J., Lalli, E., Agra, N., Gómez-López, G., Pestaña, A., & Alonso, J. (2008). DAX1, a direct target of EWS/FLI1 oncoprotein, is a principal regulator of cell-cycle progression in Ewing’s tumor cells. Oncogene, 27, 6034–6043. https://doi.org/10.1038/onc.2008.203
Li, W. M., Huang, C. N., Ke, H. L., Li, C. C., Wei, Y. C., Yeh, H. C., Chang, L. L., Huang, C. H., Liang, P. I., Yeh, B. W., Chan, T. C., Li, C. F., & Wu, W. J. (2016). MCM10 overexpression implicates adverse prognosis in urothelial carcinoma. Oncotarget, 7, 77777–77792. https://doi.org/10.18632/oncotarget.12795.
Hua, H., Kong, Q., Yin, J., Zhang, J., & Jiang, Y. (2020). Insulin-like growth factor receptor signaling in tumorigenesis and drug resistance: A challenge for cancer therapy. Journal of Hematology and Oncology, 13, 64. https://doi.org/10.1186/s13045-020-00904-3
Kang, P., Han, Z., Liao, Z., Zhang, H., Jia, W., & Tian, Y. (2020). Knockdown of MCM10 gene impairs glioblastoma cell proliferation, migration and invasion and the implications for the regulation of tumorigenesis. Journal of Molecular Neuroscience, 70, 759–768. https://doi.org/10.1007/s12031-020-01486-y
Wotschofsky, Z., Gummlich, L., Liep, J., Stephan, C., Kilic, E., Jung, K., Billaud, J.-N., & Meyer, H.-A. (2016). Integrated microRNA and mRNA signature associated with the transition from the locally confined to the metastasized clear cell renal cell carcinoma exemplified by miR-146-5p. PLoS ONE, 11, e0148746. https://doi.org/10.1371/journal.pone.0148746
Chen, Y.-R., Li, Y.-T., Wang, M.-Q., & Zhu, S.-L. (2021). Prognostic significance and function of MCM10 in human hepatocellular carcinoma. Future Oncology, 17, 4457–4470. https://doi.org/10.2217/fon-2021-0225
Zhang, W., Huang, F., Tang, X., & Ran, L. (2022). The clonal expression genes associated with poor prognosis of liver cancer. Frontiers in Genetics, 13, 808273. https://doi.org/10.3389/fgene.2022.808273
Wan, W., Shen, Y., & Li, Q. (2020). MCM10 acts as a potential prognostic biomarker and promotes cell proliferation in hepatocellular carcinoma: Integrated bioinformatics analysis and experimental validation. Cancer Management and Research, 12, 9609–9619. https://doi.org/10.2147/CMAR.S267493
Li, S., Jiang, Z., Li, Y., & Xu, Y. (2019). Prognostic significance of minichromosome maintenance mRNA expression in human lung adenocarcinoma. Oncology Reports, 42, 2279–2292. https://doi.org/10.3892/or.2019.7330
Zhou, J., Wang, M., Zhou, Z., Wang, W., Duan, J., & Wu, G. (2021). Expression and Prognostic Value of MCM Family Genes in Osteosarcoma. Frontiers in Molecular Biosciences, 8, 668402. https://doi.org/10.3389/fmolb.2021.668402
Hua, C., Zhao, G., Li, Y., & Bie, L. (2014). Minichromosome Maintenance (MCM) Family as potential diagnostic and prognostic tumor markers for human gliomas. BMC Cancer, 14, 526. https://doi.org/10.1186/1471-2407-14-526
Zhou, J., Liu, F., Tian, L., Yang, M., Tan, J., Liu, X., Li, P., Chen, J., Chen, G., Xu, L., Peng, L., Wu, Q., & Zou, Y. (2023). Mutational analysis of minichromosome maintenance complex component (MCM) family genes in Chinese Han women with polycystic ovarian syndrome. Gynecological Endocrinology Official Journal of International Society of Gynecological Endocrinology, 39, 2206912. https://doi.org/10.1080/09513590.2023.2206912
Chen, J., Wu, S., Wang, J., Han, C., Zhao, L., He, K., Jia, Y., & Cui, M. (2023). MCM10: An effective treatment target and a prognostic biomarker in patients with uterine corpus endometrial carcinoma. Journal of Cellular and Molecular Medicine, 27, 1708–1724. https://doi.org/10.1111/jcmm.17772
Tian, Q.-S., Zhang, Q., & Huang, W. (2023). MCM10 as a novel prognostic biomarker and its relevance to immune infiltration in gliomas. Technology and Health Care: Official Journal of European Society For Emergency Medicine, 31, 1301–1317. https://doi.org/10.3233/THC-220576
Guney, G., Taşkın, M. I., Sener, N., Tolu, E., Dodurga, Y., Elmas, L., Cetin, O., & Sarigul, C. (2022). The role of ERK-1 and ERK-2 gene polymorphisms in PCOS pathogenesis. Reproductive Biology and Endocrinology, 20, 95. https://doi.org/10.1186/s12958-022-00967-6
Paulson, C. N., John, K., Baxley, R. M., Kurniawan, F., Orellana, K., Francis, R., Sobeck, A., Eichman, B. F., Chazin, W. J., Aihara, H., Georg, G. I., Hawkinson, J. E., & Bielinsky, A.-K. (2019). The anti-parasitic agent suramin and several of its analogues are inhibitors of the DNA binding protein Mcm10. Open Biology, 9, 190117. https://doi.org/10.1098/rsob.190117
Araki, Y., Kawasaki, Y., Sasanuma, H., Tye, B. K., & Sugino, A. (2003). Budding yeast mcm10/dna43 mutant requires a novel repair pathway for viability. Genes to Cells, 8, 465–480. https://doi.org/10.1046/j.1365-2443.2003.00648.x
Ge, X. Q., Jackson, D. A., & Blow, J. J. (2007). Dormant origins licensed by excess Mcm2–7 are required for human cells to survive replicative stress. Genes & Development, 21, 3331–3341. https://doi.org/10.1101/gad.457807
Valenzuela, M. S., Hu, L., Lueders, J., Walker, R., & Meltzer, P. S. (2012). Broader utilization of origins of DNA replication in cancer cell lines along a 78 kb region of human chromosome 2q34. Journal of Cellular Biochemistry, 113, 132–140. https://doi.org/10.1002/jcb.23336
Böhly, N., Schmidt, A.-K., Zhang, X., Slusarenko, B. O., Hennecke, M., Kschischo, M., & Bastians, H. (2022). Increased replication origin firing links replication stress to whole chromosomal instability in human cancer. Cell Reports, 41, 111836. https://doi.org/10.1016/j.celrep.2022.111836
Abdel-Maksoud, M. A., Iqbal, K., Gull, S., Kumar, S. K., Mastoor, M., Almutairi, S. M., Almanaa, T. N., Alfuraydi, A. A., Mubarak, A., Kotob, M. H., Jamil, M., & Zaky, M. Y. (2023). Immune modulation and prognostic significance of MCM10 in pan-cancer: A comprehensive analysis. Am. J. Transl. Res., 15, 6451–6463.
Alghamdi, Y. S., Mashraqi, M. M., Alsalmi, O., Alharthi, A. A., & Gharib, A. F. (2024). Multitargeted Docking, DFT-Based Optimisation, Pharmacokinetics, and MD Simulation Reveal 6-Oxidopamine HBr as a Multitargeted Inhibitor of Cervical Cancer Proteins. Current Medicinal Chemistry. https://doi.org/10.2174/0109298673289824240529063416
Acknowledgements
The authors would like to thank Yenepoya Research Centre for providing laboratory facilities.
Funding
The authors would like to acknowledge funding from the Indian Council of Medical Research (ICMR), New Delhi, India, for funding our research program. SPD and SMQA are supported by the ICMR grant 8/1/17/2019-RMC. JS is supported by a junior research fellowship from Yenepoya (Deemed to be University).
Author information
Authors and Affiliations
Contributions
SPD conceptualised, drafted, reviewed and approved the manuscript. SMQA drafted and contributed to the artwork. SL contributed towards providing critical suggestions and reviewing the manuscript. JS contributed towards the manuscript revision and artwork.
Corresponding author
Ethics declarations
Ethical approval
Not applicable.
Competing interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Ahmed, S.M.Q., Sasikumar, J., Laha, S. et al. Multifaceted role of the DNA replication protein MCM10 in maintaining genome stability and its implication in human diseases. Cancer Metastasis Rev (2024). https://doi.org/10.1007/s10555-024-10209-3
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s10555-024-10209-3