
Abstract
As a result of preterm birth, immature kidneys are exposed to interventions in the NICU that promote survival, but are nephrotoxic. Furthermore, the duration of renal development may be truncated in these vulnerable neonates. Immaturity and nephrotoxic exposures predispose preterm newborns to acute kidney injury (AKI), particularly in the low birth weight and extremely preterm gestational age groups. Several studies have associated preterm birth as a risk factor for future chronic kidney disease (CKD). However, only a few publications have investigated the impact of neonatal AKI on CKD development. Here, we will review the evidence linking preterm birth and AKI in the NICU to CKD and highlight the knowledge gaps and opportunities for future research. For neonatal intensive care studies, we propose the inclusion of AKI as an important short-term morbidity outcome and CKD findings such as a reduced glomerular filtration rate in the assessment of long-term outcomes.
Similar content being viewed by others

Introduction
Preterm birth is common: 10.5% of infants are born at <37 weeks gestation worldwide [1, 2]. Both preterm birth and the common comorbidity of low birth weight (LBW) are risk factors for chronic kidney disease (CKD), which can be detected even in school aged children [3,4,5,6,7,8]. Preterm neonates frequently develop evidence of acute kidney injury (AKI), more common than in both the adult and pediatric intensive care unit populations [9]. However, the long-term renal effects following AKI in preterm neonates have not been fully studied. Here, we review the data regarding preterm birth, AKI, and CKD and identify areas for future research.
Normal human kidney development and nephrogenesis
Human kidney development begins during the third gestational week and continues until full term birth [10, 11]. During the nephrogenic period, there are three fetal kidneys: the pronephros, mesonephros, and metanephros. The first two fetal kidneys form, then involute, while the third, the metanephros, becomes the definitive kidney. At the fifth gestational week, a mesonephric duct outgrowth known as the ureteric bud invades the metanephros. Reciprocal interactions between the ureteric bud and metanephric mesenchyme result in nephron formation [12, 13]. Secreted metanephric mesenchyme-derived signals promote ureteric bud branching [14]. As nephrons are induced at the tip of each branch, this branching morphogenesis determines the final number of nephrons [13, 15, 16]. At the tip of each branch, the cap mesenchyme houses nephron progenitor cells [17,18,19]. There is a tightly regulated balance between the factors that promote differentiation and maintenance of the progenitor cell populations [17,18,19,20]. In mouse models, premature differentiation depletes progenitors and leads to hypoplastic kidneys [17, 18]. In humans, the deepest nephrons mature first, with nephrogenesis continuing in the outer nephrogenic zone.
The normal window of nephrogenesis closes near 36 weeks gestation and results in ~1,000,000 nephrons in each human kidney [7]. However, nephron number varies widely from an observed number of 200,000 to 2,700,000 per kidney [7]. Nephron number increases exponentially between 19 and 28 weeks (Fig. 1). Indeed, almost two-thirds of human total nephrons form during the third trimester [21]. This highlights the potential impact of preterm birth on nephron endowment, particularly birth prior to 28 weeks gestation [8].

Hinchliffe et al. [24] examined the both kidneys from 11 spontaneous second trimester abortions or stillbirths spanning the gestational ages of 15–40 weeks. Glomerular number was estimated using the dissector method, a stereologic approach. At 15 weeks glomerular number was estimated to be 15,000 and increased to 740,000 by 40 weeks, demonstrating the exponential increase in nephron formation in the third trimester. Several studies have demonstrated the cessation of human nephrogenesis at ~32–36 weeks [40, 88, 89].
The mechanisms underlying the large variation in human nephron number and the driving forces that lead to the cessation of nephrogenesis are largely unknown. Moreover, clinical studies to investigate the impact of preterm birth on renal development are limited in several ways: (1) inability to assess the tissue of the kidney, (2) lack of methods to detect the progression, disruption, or cessation of nephrogenesis, and (3) lack of a gold standard to validate urinary biomarkers, which may indicate developmental phases or injury. Therefore, we turn to animal models recognizing that they too have limitations. Baboons in many ways are an ideal model to study the effect of preterm birth. Neonatal baboons can be delivered and resuscitated in a manner similar to humans with administration of parental nutrition, mechanical ventilation, nephrotoxic medications, and antenatal steroids [22]. However, the cost and resources necessary for the success of this model limit its widespread use and prolonged follow-up of these animals. Therefore, much of the research community uses rodent species that are more cost-effective and allow for long-term studies. However, they may not reflect the human condition of prematurity as accurately. Many animals, including rodents and rabbits, have a variable duration of postnatal nephrogenesis [23] differing from human renal development whereby nephrogenesis is complete at the end of gestation [24].
Though oxygen toxicity is traditionally viewed as an imbalance between the production of reactive oxygen species (ROS) [25] and deficiency in antioxidants, the impact of excessive oxygen exposure is more complicated [26]. Fetal development occurs in a low oxygen tension environment and the pathways governing organogenesis are highly sensitive to changes in oxygen tension. Prematurely born infants are especially susceptible to the effects of hyperoxia due to developmental deficits in antioxidant capacity and an impaired ability to upregulate antioxidant production [26, 27]. Room air represents “hyperoxia” for premature infants and most require additional supplemental oxygen due to deficiencies in lung function. ROS are strongly implicated in the pathogenesis of diseases of prematurity including bronchopulmonary dysplasia, retinopathy of prematurity, neurodevelopmental deficits, and necrotizing enterocolitis [27].
The hypoxia-inducible factor (HIF) family of proteins govern proper organogenesis and oxygen-mediated disruption of HIFs is strongly implicated in lung maldevelopment in BPD [28]. Neither the mechanisms for postnatal nephrogenesis nor the signals for cessation of nephrogenesis are known. What is clear, however, is that altered oxygen tension negatively impacts renal development. For example, rat pups exposed to 80% O2 in the neonatal period have impaired nephrogenesis with a smaller nephrogenic zone width, smaller glomerular diameter with more apoptotic cells, but no difference in glomerular number [29]. These effects on nephrogenesis appeared to be mediated by HIF-1α. Hyperoxia-mediated reductions in kidney HIF-1α expression, nephrogenic zone width, and glomerular diameter were rescued in rats upon pharmacologic HIF-1α stabilization [30]. Longer term studies in the perinatal rat hyperoxia model [31] revealed a decrease in creatinine clearance in females but not at 5 months. Though this functional impairment was no longer apparent at 11 months of age, histologic and microstructural differences were present. Male rats exposed to perinatal hyperoxia developed glomerular crescents while females did not. The authors speculated these differences may be attributable to greater inflammatory and apoptotic effects of hyperoxia in males, but not females, as is the case in perinatal hyperoxic lung injury [32, 33]. Nonetheless, there is a tremendous gap in knowledge regarding the impacts of oxidative stress and altered oxygen tension on renal development and long-term outcomes in infants born prematurely.
In addition to potential hyperoxia, preterm neonates are exposed to a host of medications depending on their clinical course and local clinical practice. The commonly cited high risk nephrotoxins include aminoglycosides, amphotericin, acyclovir, diuretics, and nonsteroidal anti-inflammatory drugs. As recently reviewed by Murphy et al. [34], nephrotoxins increase the risk of neonatal AKI. Beyond the risk for AKI, nephrotoxin exposure may have direct and permanent effects on the developing kidney. Several animal studies have demonstrated a reduction in nephron number following neonatal exposures to gentamicin or indomethacin [35,36,37]. In preterm baboons exposed to ibuprofen, their nephrogenic zone was reduced by 27% as compared with controls [38]. However, those that were untreated had more abnormal glomeruli than those treated with ibuprofen. This demonstrates the complex interactions whereby preterm birth complications (such as patent ductus arteriosus and nephrotoxic treatments) can both adversely impact nephrogenesis. In a recently published rabbit study [39], neonatal rabbits were exposed to 4 days of both gentamicin and indomethacin during active nephrogenesis. This resulted in histologically significant AKI but with no differences in serum creatinine concentrations between the groups at 6 weeks of age. When the kidneys were evaluated using cationic ferritin enhanced MRI, a unique circumferential lesion of glomerular loss was detectable. Histologically the glomeruli were shrunken, abnormally shaped, and lacked surrounding tubules. The deposition of contrast agent was sporadic and sparse, and associated with podocyte effacement on electron microscopy signaling significant microstructural changes not detectable by traditional metrics of renal function.
Consistent with these animal studies, preterm birth alters nephrogenesis in human studies. Autopsy studies of neonates who die weeks to months after preterm birth have demonstrated that nephrogenesis continues for up to 40 days after preterm birth [40]. However, postnatal nephrogenesis may come at a cost: glomerular maturation is accelerated with a decreased nephrogenic zone diameter. In addition, structural glomerular anomalies were present including a decrease in glomerular density (GD) (fewer glomeruli/cortical area) and abnormal glomeruli (dilated Bowman’s capsule with shrunken glomerular tufts).
Together, these animal and human studies support hyperoxia and nephrotoxin exposures as potential contributors to adverse effects of preterm birth on nephrogenesis and long-term kidney outcomes. As human kidney tissue is limited and subject to bias and multiple confounders, additional animal studies are needed to elucidate the mechanisms which contribute to poor long-term outcomes in the kidney following preterm birth.
Long-term outcomes of renal function in preterm neonates
Preterm birth alone is a potential risk factor for CKD as a result of its adverse effect on nephrogenesis and nephron number (Fig. 2). CKD is a major public health epidemic that has high associated economic costs, recently estimated at over 20 billion dollars per year [41]. Children with CKD experience decreased quality of life, have impaired neurocognitive development with deficits in specific areas, incur higher rates of cardiovascular disease, and develop end stage renal disease (ESRD) at a rate of 13 times higher by the age of 40 [41,42,43,44]. CKD risk increases with both low nephron number and LBW [45,46,47]. In epidemiologic studies, individuals born weighing <2.5 kg face a 40–60% increased risk for CKD [48, 49]. In the longest follow-up study published to date, Crump et al. found preterm birth to be associated with a two- to threefold increased risk for CKD at 30 years of age [3]. In a similar study of the same population, they also found a 2.45-fold increased risk of hypertension by the age of 18–29 years in former extremely LBW infants [50]. In follow-up studies of a shorter duration, hypertension, decreased glomerular filtration, and albuminuria can be detected in children as young as 2 years of age as well as in adolescents born preterm [51, 52]. Beyond CKD, preterm birth may impact the development of adulthood hypertension and associated cardiovascular comorbidities. The connection between preterm birth and adult hypertension is complex, but the mechanisms are beginning to be understood. In 2018, Paquette et al. compared 23-year-old young adults born preterm (≤29 weeks) with term matched controls and found the former preterm group had smaller kidneys, higher urine albumin-to-creatinine ratio, and higher blood pressure with similar estimated glomerular filtration rates [53]. In addition, they found higher circulating levels of angiotensin 1 in preterm subjects and changes in renin and angiotensin peptides, which correlated with blood pressure in former preterm adults, but not full term born subjects. These findings in conjunction with a number of other studies suggest that the renin angiotensin system is altered by preterm birth and may lead to higher rates of hypertension in adolescence and adulthood.

There are many unique exposures incurred by preterm neonate that lead to an enhanced risk of acute kidney injury as well as chronic kidney disease. Prior to birth, a hostile intrauterine environment can affect branching and nephron development. Following preterm birth, there may be acceleration and early cessation of normal nephron development. In addition, disruption of normal nephron development may be related to nephrotoxic exposures from NICU environment. Preterm neonates face high rates of acute kidney injury which may lead to nephron loss. The low nephron endowment and nephron loss leads to risk for glomerular hyperfiltration and additional damage to residual glomeruli. After leaving the NICU, exposures including obesity, high sodium diet, and hypertension may be additional stressors that accelerate the progression to chronic kidney disease in the preterm population.
In addition to the overall increased risk of CKD, preterm birth induced defects in glomerulogenesis may contribute to childhood onset glomerular disease. Case reports and small case series document a podocytopathy related to preterm birth with LBW [54,55,56,57]. In these children and adolescents born preterm, the most common histopathologic lesion is focal segmental glomerulosclerosis (FSGS) [54]. Interestingly, in children with nephrotic syndrome, LBW correlates with decreased GD and increased glomerular volume [58]. In children with nephrotic syndrome, LBW is associated with reduced response to steroids and more relapses [58]. Similarly, in other glomerular disease studies, those born preterm and/or with LBW had worse outcomes [57, 59, 60]. The fact that only some children develop glomerular disease suggests interactions of preterm birth with gene or environmental effects [61].
One gene implicated in risk for kidney disease following preterm birth is APOL1 [62]. The APOL1 gene encodes apolipoprotein L1, a circulating component of innate immune response to Trypanosoma. Trypanosoma is the cause of African sleeping sickness [63]. These parasites engulf APOL1 bound in a lipid protein complex; APOL1 contributes to pore formation, lysis, and death of the parasite. However, some Trypanosoma are resistant to APOL1-induced lysis. Two gene variants in APOL1 (known as G1 and G2) overcome resistant Trypanosoma; carriage of either G1 or G2 leads to a survival advantage. However, carriage of any two APOL1 alleles (G1,G1; G1,G2; or G2,G2) is deleterious and increases risk for kidney failure. APOL1 gene variants account for 70% of the excess renal disease risk in those of African descent [62]. While APOL1 gene variants are common, not all carriers develop kidney disease and “second hits” throughout the lifespan may be required to develop a renal phenotype [62]. The majority of APOL1-attributable kidney disease presents as adult-onset “hypertensive nephropathy” or FSGS, but a subset present with childhood onset disease. African American children with APOL1 related FSGS had higher than expected rates of preterm birth, suggesting preterm birth may be a “second hit” leading to early onset kidney disease [64]. Further study of the interaction of preterm birth with APOL1 gene variants on risk for CKD is warranted.
AKI in preterm neonates
While the effects of preterm birth on long-term renal function in this population are becoming clear, the attributable risk of neonatal AKI has been understudied. Before addressing the long-term impact of AKI, it is important to highlight the impact of AKI on preterm neonates in the NICU. The definition of AKI in neonates has evolved significantly over the past decade. In 2013 an expert panel of neonatologists and pediatric nephrologists convened at the National Institute of Health to discuss challenges and research opportunities in the field of neonatal AKI [65]. Experts in attendance agreed that there were limitations to adapting a pediatric AKI definition to use in neonates, given the shortcomings of serum creatinine concentration as a measure of kidney function in the early postnatal period. However, the group proposed a consensus AKI definition modified from the Kidney Disease Improving Global Outcomes (KDIGO) pediatric definition to be used in neonates (Table 1). This definition was meant to standardize staging of AKI in neonates so that short and long-term outcomes could be correlated to severity of AKI. Since this workshop, there has been an explosion of neonatal AKI research, particularly in the preterm neonatal population, with over 100 publications on this topic in the past 5 years.
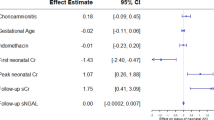
However, the adoption of this definition has not been universal and there is still much to be learned from other definitions of neonatal AKI. For example, Strunk et al. defined AKI using specific gestational age groups [66]. Based on a study of 1461 patients, the authors correlated increased risk of mortality with peak NICU serum creatinine cutoffs of 1.6, 1.1, and 1.0 mg/dL for gestational age groups 24–27, 28–29, and 30–32 weeks, respectively, (AKI-C definition). Although this definition is more specific to the broad range of gestational ages in the preterm population, long-term studies are needed to validate a meaningful definition of AKI.
With an increased focus on neonatal AKI research, immediately following the NIH workshop of neonatal AKI, the Neonatal Kidney Collaborative (NKC) was formed [67]. The NKC conducted the largest study focused on neonatal AKI, titled the Assessment of Worldwide Acute Kidney injury Epidemiology in Neonates (AWAKEN), which provides the most robust estimates of AKI in sick preterm neonates [9]. In this multinational 24 center study, 4273 neonates were screened and 2189 “high risk” neonates (those requiring intravenous fluids for at least 48 h) were enrolled in AWAKEN. These neonates were evaluated for AKI based on the modified neonatal KDIGO AKI definition (Table 1). The results showed the following incidence of AKI per gestational age group: 22–28 weeks: 52%, 29–35 weeks: 18%, ≥36 weeks: 37%. Not only did this study demonstrate that AKI was more common than previously documented, it also showed an independent association of AKI with mortality (adjusted OR 4.6, 95% CI 2.5–8.3; p < 0.0001) and length of stay (adjusted parameter estimate 8.8 days, 95% CI 6.1–11.5; p < 0.0001). Multiple analyses of the AWAKEN study have detailed other interesting findings in relation to AKI and preterm birth. The risk factors for early (≤7 days) and late AKI in the NICU are unique and these two categories of AKI have different causes and outcomes that need to be approached independently [68, 69]. In those born 22–28 gestational weeks, neonates with AKI were more likely to develop intraventricular hemorrhage (IVH), with an adjusted odds of 1.87 (95% CI 1.08–3.23) to develop IVH [70]. In addition to AKI risk factors and comorbid conditions, a secondary analysis of the AWAKEN study found an association between caffeine and lower prevalence of AKI [71], similar to a previously published small single center study [72]. In the AWAKEN cohort, 675 premature infants were evaluated and AKI occurred less frequently in neonates who received caffeine than those who did not. The number that would need to be treated to prevent one case of AKI in extremely preterm neonates was only 3.1. The relationship between AKI and fluid balance and bronchopulmonary dysplasia in preterm neonates has also been evaluated in the AWAKEN cohort [73, 74]. It is clear that the AWAKEN study has improved our understanding of which vulnerable neonates develop AKI and when it occurs in the NICU. However, it has also created new questions regarding the influence of AKI on the short and long-term kidney outcomes of these vulnerable former preterm patients and highlights the need for standardized surveillance for AKI.
The AKI to CKD timeline and the impact of CKD
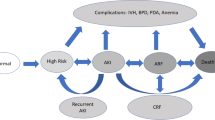
Although we now define AKI and CKD as two distinct syndromes, AKI and CKD likely represent a continuum; patients with a history of AKI are at risk for developing CKD and those with current CKD are at higher risk for developing AKI [75]. A working group at the 16th Acute Dialysis Quality Initiative sought to address this issue and brought forth several important concepts to address [76]. First, in order to recover from any episode of AKI, repair must occur, but not all repair is equal. In adaptive repair, the injury resolves and recovery of renal structures is complete within 90 days; while in maladaptive repair there is an irreversible reduction in kidney function associated with changes in renal structure [77]. The interval between AKI and CKD has been named acute kidney disease (AKD) in the adult literature [76]. However, this 90-day interval is likely very different in preterm neonates compared with adults. When approaching the question of “does AKI result in CKD in former preterm infants?”, it is important to recognize that progression of AKI to CKD is not an acute event, but a gradual process reflected by signs (proteinuria, hypertension, and hyperfiltration) accumulating prior to the ultimate diagnosis of CKD. In Fig. 3, we provide a schematic of factors that may play a role in the transition from AKI to AKD to CKD and the role of the immature kidney in this process. For former preterm infants, this timeline may be different than for children or adults who experience AKI. It is therefore critical to initiate early screening of blood pressure and kidney function in infants at risk so that measures can be taken to delay or prevent CKD development and progression.

Postnatal kidney injury disrupts the normal developmental maturation of the kidney. Postnatal kidney injury then leads to a state that has been termed “acute kidney disease.” At this stage, if the immature kidney can undergo adaptive repair it may recover from acute kidney disease without developing chronic kidney disease. However, if maladaptive repair occurs, this leads to tubular damage, vascular damage, glomerular damage, and interstitial fibrosis ultimately ending in chronic kidney disease. The potential etiology of each process is listed in parentheses, while signs of chronic kidney disease specific to each process are displayed in the sub-boxes.
AKI in children and adults leads to CKD
Although the long-term kidney outcomes of pediatric and adult studies may not fully apply to preterm neonates, it is important to understand the pathophysiology of AKI and the long-term renal outcomes in these groups. The adult literature contains two large systematic reviews. Coca et al. found that compared with adults without AKI, survivors of AKI have a tenfold increased risk of CKD and a threefold risk of ESRD [78]. More recently in an evaluation of over two million participants, adults with AKI had a 2.7-fold increased risk of new or progressive CKD and 4.8-fold risk of ESRD with an increasing risk with higher stages of AKI [79]. Although smaller in number, retrospective studies in the pediatric population have demonstrated similar findings. Mammen et al. [80] evaluated survivors of AKI in the pediatric intensive care unit 1–3 years after discharge and found that 10% of the 126 children had developed CKD. Importantly, an additional 46% were “at risk for CKD” defined as an eGFR of 60–90 or >150 mL/min/1.73 m2. In a retrospective study examining pediatric patients with nephrotoxic AKI [81], the overall CKD prevalence was 33% at >6 months follow-up: 69% had proteinuria, 38% had hypertension, and 23% had an eGFR < 90 mL/min/1.73 m2. Two prospective follow-up studies of AKI in children have added to our understanding of the development of CKD after AKI. “TRIBE AKI” and “FRAIL-AKI” included children with and without AKI after cardiac surgery and found significant rates of renal dysfunction. However, the diagnosis of AKI did not predict the development of CKD [82, 83]. The FRAIL AKI study did however reveal that several inflammatory urinary biomarkers were higher in the AKI group suggesting that there may be an underlying inflammatory process in those without the traditional signs of CKD. These results highlight the need for well-designed, prospective studies directly comparing pediatric AKI populations with similar populations without AKI using standard CKD definitions at multiple time points over an extended period of time with further validation of urinary biomarkers.
Does AKI in preterm neonates lead to CKD?
There is strong evidence from adult studies supporting a link between AKI and CKD, and suggestive evidence from smaller pediatric studies. But what is the evidence of CKD in former preterm infants exposed to AKI? In 2003, 20 former preterm children who had a diagnosis of renal failure in the NICU (defined as peak creatinine >2.0 mg/dL for 48 h and/or urine output < 0.5 mL/kg/h for 24 h) underwent kidney evaluation [84]. The AKI definition in this study would correspond to stage 2–3 in the modified neonatal KDIGO AKI definition. The mean age at follow-up was 7.5 years, but the range was wide (3.2–18.5 years). Almost half of the patients had a low eGFR < 90 mL/min/1.73 m2. The low eGFR group was significantly older, but otherwise similar to the normal eGFR group. Most patients were exposed to indomethacin, gentamicin, and loop diuretics and had complicated NICU courses. Of note, NICU care has changed since that time with an emphasis on reduced antibiotic exposure and early extubation with non-invasive ventilation—two important factors that may affect kidney development and function.
Three more recent publications examined CKD rates in preterm populations that were cared for between 2003 and 2011 (Table 2). First, Maqsood et al. evaluated 222 former extremely LBW (ELBW; <1000 g) at 2–3 years of age [85]. Twenty three had severe AKI and 87 had stage 1 AKI during their NICU course as defined by the modified neonatal KDIGO definition. Although the authors reported no difference in prevalence of hypertension or serum creatinine concentration between the AKI and no AKI groups, 44% of their patients did not have data available. Bruel et al. examined children born <33 weeks at a mean age of 6.6 years and utilized the gestational age serum creatinine cutoffs to define AKI (AKI-C) [86]. They found no differences in eGFR or microalbuminuria, but did report that the kidney volume was smaller in the AKI-C group (57 vs. 68 mL, p = 0.04). Finally, the Follow-up of Aki in Neonates during Childhood Years study utilized the modified neonatal KDIGO definition and evaluated former very LBW (VLBW; <1500 g) preterm infants at an age of 3–7 years [87]. Harer et al. found that of the 34 infants, 20 experienced AKI (n = 12, stage 2/3 AKI) and those with a history of AKI were more likely to have renal dysfunction (composite outcome of hypertension, proteinuria or low eGFR < 90 mL/min/1.73 m2) compared with the no AKI group (65% vs. 14%, RR: 4.5 (CI: 1.2–17.1)). In each these studies of children born preterm, renal dysfunction (reduced eGFR) was common and detected at an early age (Harer: 26%, Bruel: 23%, and Maqsood: 4% of patients). However, all of these studies were small and performed in single centers. Regardless of these limitations, they suggest a potential relationship between AKI in preterm neonates and increased risk for CKD. Large prospective, multicenter studies designed to follow patients through early childhood and into adolescence are needed to define the attributable risk of AKI in the development of CKD in preterm neonates.
Conclusion
This review highlights the current evidence for associating preterm birth with CKD and preterm birth with neonatal AKI to CKD. The studies presented in this article focused primarily on kidney outcomes during early childhood, but did not address other important health indicators. It will be important moving forward to determine how AKI affects other long-term outcomes of prematurity. For example, does a history of prematurity and AKI increase the risk of poor neurodevelopmental outcomes compared with children without an AKI history? Moreover, several studies have documented higher blood pressure in former preterm infants, but AKI was not considered as a comorbidity or modifying factor. To begin to answer these questions, long-term follow-up studies of former preterm neonates are needed that include both short- and long-term kidney outcomes. Without more data on renal function in those born preterm during childhood, adolescence, and adulthood, the long-term effects of neonatal AKI and preterm birth will remain unknown. With the recent enhanced focus on preventing long-term morbidities of preterm birth, now is the time to recognize AKI as both a major NICU issue and a potentially serious contributor to progressive CKD through the life course.
References
Purisch SE, Gyamfi-Bannerman C. Epidemiology of preterm birth. Semin Perinatol. 2017;41:387–91.
Chawanpaiboon S, Vogel JP, Moller AB, Lumbiganon P, Petzold M, Hogan D, et al. Global, regional, and national estimates of levels of preterm birth in 2014: a systematic review and modelling analysis. Lancet Glob Health. 2019;7:e37–46.
Crump C, Sundquist J, Winkleby MA, Sundquist K. Preterm birth and risk of chronic kidney disease from childhood into mid-adulthood: national cohort study. BMJ. 2019;365:l1346.
Luyckx VA, Brenner BM. Clinical consequences of developmental programming of low nephron number [published online ahead of print, 2019 Oct 6]. Anat Rec. 2019. https://doi.org/10.1002/ar.24270.
Robertson CC, Gillies CE, Putler RKB, Ng D, Reidy KJ, Crawford B, et al. An investigation of APOL1 risk genotypes and preterm birth in African American population cohorts. Nephrol, Dial, Transplant. 2017;32:2051–8.
Brophy PD, Charlton JR, Bryan Carmody J, Reidy KJ, Harshman L, Segar J, et al. Chronic kidney disease: a life course health development perspective. In: Halfon N, Forrest CB, Lerner RM, Faustman EM, editors. Handbook of life course health development. Cham: Springer; 2018. p. 375-401.
Charlton JR, Springsteen CH, Carmody JB. Nephron number and its determinants in early life: a primer. Pediatr Nephrol. 2014;29:2299–308.
Carmody JB, Charlton JR. Short-term gestation, long-term risk: prematurity and chronic kidney disease. Pediatrics. 2013;131:1168–79.
Jetton JG, Boohaker LJ, Sethi SK, Wazir S, Rohatgi S, Soranno DE, et al. Incidence and outcomes of neonatal acute kidney injury (AWAKEN): a multicentre, multinational, observational cohort study. Lancet Child Adolesc Health. 2017;1:184–94.
Rosenblum S, Pal A, Reidy K. Renal development in the fetus and premature infant. Semin Fetal Neonatal Med. 2017;22:58–66.
Avner ED, Harmon WE, Niaudet P, Yoshikawa N, Emma F, Goldstein SL, et al. Pediatric nephrology. 7th ed. Berlin, Heidelberg: Springer; 2016. p. 1.
McMahon AP. Development of the mammalian kidney. Curr Top Dev Biol. 2016;117:31–64.
Blake J, Rosenblum ND. Renal branching morphogenesis: morphogenetic and signaling mechanisms. Semin Cell Dev Biol. 2014;36:2–12.
Costantini F. Genetic controls and cellular behaviors in branching morphogenesis of the renal collecting system. Wiley Interdiscip Rev Dev Biol. 2012;1:693–713.
Reidy KJ, Rosenblum ND. Cell and molecular biology of kidney development. Semin Nephrol. 2009;29:321–37.
Short KM, Smyth IM. The contribution of branching morphogenesis to kidney development and disease. Nat Rev Nephrol. 2016;12:754–67.
Oxburgh L. Kidney nephron determination. Annu Rev Cell Dev Biol. 2018;34:427–50.
O’Brien LL. Nephron progenitor cell commitment: striking the right balance. Semin Cell Dev Biol. 2019;91:94–103.
Becherucci F, Lazzeri E, Lasagni L, Romagnani P. Renal progenitors and childhood: from development to disorders. Pediatr Nephrol. 2014;29:711–9.
Kopan R, Chen S, Little M. Nephron progenitor cells: shifting the balance of self-renewal and differentiation. Curr Top Dev Biol. 2014;107:293–331.
Abitbol CL, DeFreitas MJ, Strauss J. Assessment of kidney function in preterm infants: lifelong implications. Pediatr Nephrol. 2016;31:2213–22.
Gubhaju L, Sutherland MR, Yoder BA, Zulli A, Bertram JF, Black MJ. Is nephrogenesis affected by preterm birth? Studies in a non-human primate model. Am J Physiol Renal Physiol. 2009;297:F1668–77.
Cebrian C, Borodo K, Charles N, Herzlinger DA. Morphometric index of the developing murine kidney. Dev Dyn. 2004;231:601–8.
Hinchliffe SA, Sargent PH, Howard CV, Chan YF, van Velzen D. Human intrauterine renal growth expressed in absolute number of glomeruli assessed by the disector method and Cavalieri principle. Lab Investig. 1991;64:777–84.
Halliwell B. Reactive oxygen species in living systems: source, biochemistry, and role in human disease. Am J Med. 1991;91:14S–22S.
Tipple TE, Ambalavanan N. Oxygen toxicity in the neonate: thinking beyond the balance. Clin Perinatol. 2019;46:435–47.
Ofman G, Tipple TE. Antioxidants & bronchopulmonary dysplasia: beating the system or beating a dead horse? Free Radic Biol Med. 2019;142:138–45.
Ofman G, Tipple TE. Thiol-redox regulation in lung development and vascular remodeling. Antioxid Redox Signal. 2019;31:858–73.
Popescu CR, Sutherland MR, Cloutier A, Benoit G, Bertagnolli M, Yzydorczyk C, et al. Hyperoxia exposure impairs nephrogenesis in the neonatal rat: role of HIF-1alpha. PloS ONE. 2013;8:e82421.
Yzydorczyk C, Comte B, Cambonie G, Lavoie JC, Germain N, Ting Shun Y, et al. Neonatal oxygen exposure in rats leads to cardiovascular and renal alterations in adulthood. Hypertension. 2008;52:889–95.
Sutherland MR, Beland C, Lukaszewski MA, Cloutier A, Bertagnolli M, Nuyt AM. Age- and sex-related changes in rat renal function and pathology following neonatal hyperoxia exposure. Physiol Rep. 2016;4:e12887.
Lingappan K, Jiang W, Wang L, Couroucli XI, Barrios R, Moorthy B. Sex-specific differences in hyperoxic lung injury in mice: implications for acute and chronic lung disease in humans. Toxicol Appl Pharm. 2013;272:281–90.
Lingappan K, Jiang W, Wang L, Couroucli XI, Moorthy B. Sex-specific differences in hyperoxic lung injury in mice: role of cytochrome P450 (CYP)1A. Toxicology. 2015;331:14–23.
Murphy HJ, Thomas B, Van Wyk B, Tierney SB, Selewski DT, Jetton JG. Nephrotoxic medications and acute kidney injury risk factors in the neonatal intensive care unit: clinical challenges for neonatologists and nephrologists [published online ahead of print, 2019 Oct 12]. Pediatr Nephrol. 2019. https://doi.org/10.1007/s00467-019-04350-3.
Kent AL, Koina ME, Gubhaju L, Cullen-McEwen LA, Bertram JF, Lynnhtun J, et al. Indomethacin administered early in the postnatal period results in reduced glomerular number in the adult rat. Am J Physiol Renal Physiol. 2014;307:F1105–10.
Gilbert T, Gaonach S, Moreau E, Merlet-Benichou C. Defect of nephrogenesis induced by gentamicin in rat metanephric organ culture. Lab Investig. 1994;70:656–66.
Gilbert T, Lelievre-Pegorier M, Malienou R, Meulemans A, Merlet-Benichou C. Effects of prenatal and postnatal exposure to gentamicin on renal differentiation in the rat. Toxicology. 1987;43:301–13.
Sutherland MR, Yoder BA, McCurnin D, Seidner S, Gubhaju L, Clyman RI, et al. Effects of ibuprofen treatment on the developing preterm baboon kidney. Am J Physiol Renal Physiol. 2012;302:F1286–92.
Charlton JR, Baldelomar EJ, deRonde K, Cathro HP, Charlton NP, Criswell S, et al. Nephron loss detected by MRI following neonatal acute kidney injury in rabbits [published online ahead of print, 2019 Dec 5]. Pediatr Res. 2019. https://doi.org/10.1038/s41390-019-0684-1.
Rodriguez MM, Gomez AH, Abitbol CL, Chandar JJ, Duara S, Zilleruelo GE. Histomorphometric analysis of postnatal glomerulogenesis in extremely preterm infants. Pediatr Dev Pathol. 2004;7:17–25.
Ozieh MN, Bishu KG, Dismuke CE, Egede LE. Trends in healthcare expenditure in United States adults with chronic kidney disease: 2002–11. BMC Health Serv Res. 2017;17:368.
Ruebner RL, Laney N, Kim JY, Hartung EA, Hooper SR, Radcliffe J, et al. Neurocognitive dysfunction in children, adolescents, and young adults with CKD. Am J Kidney Dis. 2016;67:567–75.
Matteucci MC, Chinali M, Rinelli G, Wuhl E, Zurowska A, Charbit M, et al. Change in cardiac geometry and function in CKD children during strict BP control: a randomized study. Clin J Am Soc Nephrol. 2013;8:203–10.
Calderon-Margalit R, Golan E, Twig G, Leiba A, Tzur D, Afek A, et al. History of childhood kidney disease and risk of adult end-stage renal disease. N Engl J Med. 2018;378:428–38.
Hoy WE, Kincaid-Smith P, Hughson MD, Fogo AB, Sinniah R, Dowling J, et al. CKD in aboriginal Australians. Am J Kidney Dis. 2010;56:983–93.
Murai-Takeda A, Kanda T, Azegami T, Hirose H, Inokuchi M, Tokuyama H, et al. Low birth weight is associated with decline in renal function in Japanese male and female adolescents. Clin Exp Nephrol. 2019;23:1364–72.
Kanda T, Takeda A, Hirose H, Abe T, Urai H, Inokuchi M, et al. Temporal trends in renal function and birthweight in Japanese adolescent males (1998-2015). Nephrol, Dial, Transplant. 2018;33:304–10.
Esmeijer K, de Vries AP, Mook-Kanamori DO, de Fijter JW, Rosendaal FR, Rabelink TJ, et al. Low birth weight and kidney function in middle-aged men and women: the Netherlands epidemiology of obesity study. Am J Kidney Dis. 2019;74:751–60.
Vikse BE, Irgens LM, Leivestad T, Hallan S, Iversen BM. Low birth weight increases risk for end-stage renal disease. J Am Soc Nephrology. 2008;19:151–7.
Crump C, Sundquist J, Sundquist K. Risk of hypertension into adulthood in persons born prematurely: a national cohort study [published online ahead of print, 2019 Dec 23]. Eur Heart J. 2019;ehz904.
South AM, Nixon PA, Chappell MC, Diz DI, Russell GB, Jensen ET, et al. Renal function and blood pressure are altered in adolescents born preterm. Pediatr Nephrol. 2019;34:137–44.
Horie A, Abe Y, Koike D, Hirade T, Nariai A, Ito T, et al. Long-term renal follow-up of preterm neonates born before 35 weeks of gestation. Pediatrics Int. 2019;61:1244–9.
Paquette K, Fernandes RO, Xie LF, Cloutier A, Fallaha C, Girard-Bock C. et al. Kidney size, renal function, Ang (angiotensin) peptides, and blood pressure in young adults born preterm. Hypertension. 2018;72:918–28.
Ikezumi Y, Suzuki T, Karasawa T, Yamada T, Hasegawa H, Nishimura H, et al. Low birthweight and premature birth are risk factors for podocytopenia and focal segmental glomerulosclerosis. Am J Nephrol. 2013;38:149–57.
Asada N, Tsukahara T, Furuhata M, Matsuoka D, Noda S, Naganuma K, et al. Polycythemia, capillary rarefaction, and focal glomerulosclerosis in two adolescents born extremely low birth weight and premature. Pediatr Nephrol. 2017;32:1275–8.
Hibino S, Abe Y, Watanabe S, Yamaguchi Y, Nakano Y, Tatsuno M, et al. Proteinuria caused by glomerular hypertension during adolescence associated with extremely premature birth: a report of two cases. Pediatr Nephrol. 2015;30:1889–92.
Conti G, De Vivo D, Fede C, Arasi S, Alibrandi A, Chimenz R, et al. Low birth weight is a conditioning factor for podocyte alteration and steroid dependance in children with nephrotic syndrome. J Nephrol. 2018;31:411–5.
Koike K, Ikezumi Y, Tsuboi N, Kanzaki G, Haruhara K, Okabayashi Y, et al. Glomerular density and volume in renal biopsy specimens of children with proteinuria relative to preterm birth and gestational age. Clin J Am Soc Nephrology. 2017;12:585–90.
Konstantelos N, Banh T, Patel V, Vasilevska-Ristovska J, Borges K, Hussain-Shamsy N, et al. Association of low birth weight and prematurity with clinical outcomes of childhood nephrotic syndrome: a prospective cohort study. Pediatr Nephrol. 2019;34:1599–605.
Teeninga N, Schreuder MF, Bokenkamp A, Delemarre-van de Waal HA, van Wijk JA. Influence of low birth weight on minimal change nephrotic syndrome in children, including a meta-analysis. Nephrol, Dial, Transplant. 2008;23:1615–20.
Zidar N, Cavić MA, Kenda RB, Koselj M, Ferluga D. Effect of intrauterine growth retardation on the clinical course and prognosis of IgA glomerulonephritis in children. Nephron. 1998;79:28–32.
Reidy KJ, Hjorten R, Parekh RS. Genetic risk of APOL1 and kidney disease in children and young adults of African ancestry. Curr Opin Pediatr. 2018;30:252–9.
Freedman BI, Limou S, Ma L, Kopp JB. APOL1-associated nephropathy: a key contributor to racial disparities in CKD. Am J Kidney Dis. 2018;72 5(Suppl 1):S8–16.
Ng DK, Robertson CC, Woroniecki RP, Limou S, Gillies CE, Reidy KJ, et al. APOL1-associated glomerular disease among African-American children: a collaboration of the Chronic Kidney Disease in Children (CKiD) and Nephrotic Syndrome Study Network (NEPTUNE) cohorts. Nephrol, Dial, Transplant. 2017;32:983–90.
Zappitelli M, Ambalavanan N, Askenazi DJ, Moxey-Mims MM, Kimmel PL, Star RA, et al. Developing a neonatal acute kidney injury research definition: a report from the NIDDK neonatal AKI workshop. Pediatr Res. 2017;82:569–73.
Strunk T, Bruel A, Rozé J-C, Flamant C, Simeoni U, Roussey-Kesler G, et al. Critical serum creatinine values in very preterm newborns. PLoS ONE. 2013;8:e84892.
Jetton JG, Guillet R, Askenazi DJ, Dill L, Jacobs J, Kent AL, et al. Assessment of worldwide acute kidney injury epidemiology in neonates: design of a retrospective cohort study. Front Pediatrics. 2016;4:68.
Charlton JR, Boohaker L, Askenazi D, Brophy PD, Fuloria M, Gien J, et al. Late onset neonatal acute kidney injury: results from the AWAKEN Study. Pediatr Res. 2019;85:339–48.
Charlton JR, Boohaker L, Askenazi D, Brophy PD, D’Angio C, Fuloria M, et al. Incidence and risk factors of early onset neonatal AKI. Clin J Am Soc Nephrol. 2019;14:184.
Stoops C, Boohaker L, Sims B, Griffin R, Selewski DT, Askenazi D. The association of intraventricular hemorrhage and acute kidney injury in premature infants from the assessment of the worldwide acute kidney injury epidemiology in neonates (AWAKEN) study. Neonatology. 2019;116:321–30.
Harer MW, Askenazi DJ, Boohaker LJ, Carmody JB, Griffin RL, Guillet R, et al. Association between early caffeine citrate administration and risk of acute kidney injury in preterm neonates: results from the AWAKEN study. JAMA Pediatr. 2018;172:e180322.
Carmody JB, Harer MW, Denotti AR, Swanson JR, Charlton JR. Caffeine exposure and risk of acute kidney injury in a retrospective cohort of very low birth weight neonates. J Pediatr. 2016;172:63–8.e61.
Starr MC, Boohaker L, Eldredge LC, Menon S, Griffin R, Mayock D, et al. Acute kidney injury is associated with poor lung outcomes in infants born >/=32 weeks of gestational age. Am J Perinatol. 2020;37:231–40.
Selewski DT, Gist KM, Nathan AT, Goldstein SL, Boohaker LJ, Akcan-Arikan A, et al. The impact of fluid balance on outcomes in premature neonates: a report from the AWAKEN study group. Pediatr Res. 2020;87:550–57.
Chawla LS, Eggers PW, Star RA, Kimmel PL. Acute kidney injury and chronic kidney disease as interconnected syndromes. N Engl J Med. 2014;371:58–66.
Chawla LS, Bellomo R, Bihorac A, Goldstein SL, Siew ED, Bagshaw SM, et al. Acute kidney disease and renal recovery: consensus report of the Acute Disease Quality Initiative (ADQI) 16 Workgroup. Nat Rev Nephrol. 2017;13:241–57.
Basile DP, Bonventre JV, Mehta R, Nangaku M, Unwin R, Rosner MH, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27:687–97.
Coca SG, Singanamala S, Parikh CR. Chronic kidney disease after acute kidney injury: a systematic review and meta-analysis. Kidney Int. 2012;81:442–8.
See EJ, Jayasinghe K, Glassford N, Bailey M, Johnson DW, Polkinghorne KR, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019;95:160–72.
Mammen C, Al Abbas A, Skippen P, Nadel H, Levine D, Collet JP, et al. Long-term risk of CKD in children surviving episodes of acute kidney injury in the intensive care unit: a prospective cohort study. Am J Kidney Dis. 2012;59:523–30.
Menon S, Kirkendall ES, Nguyen H, Goldstein SL. Acute kidney injury associated with high nephrotoxic medication exposure leads to chronic kidney disease after 6 months. J Pediatr. 2014;165:522–7. e522.
Cooper DS, Claes D, Goldstein SL, Bennett MR, Ma Q, Devarajan P, et al. Article follow-up renal assessment of injury long-term after acute kidney injury (FRAIL-AKI). Clin J Am Soc Nephrol. 2015;11:1–9.
Greenberg JH, Zappitelli M, Devarajan P, Thiessen-Philbrook HR, Krawczeski C, Li S, et al. Kidney outcomes 5 years after pediatric cardiac surgery: the TRIBE-AKI study. JAMA Pediatr. 2016;170:1071–8.
Abitbol CL, Bauer CR, Montane B, Chandar J, Duara S, Zilleruelo G. Long-term follow-up of extremely low birth weight infants with neonatal renal failure. Pediatr Nephrol. 2003;18:887–93.
Maqsood S, Fung N, Chowdhary V, Raina R, Mhanna MJ. Outcome of extremely low birth weight infants with a history of neonatal acute kidney injury. Pediatr Nephrol. 2017;32:1035–43.
Bruel A, Rozé J-C, Quere M-P, Flamant C, Boivin M, Roussey-Kesler G, et al. Renal outcome in children born preterm with neonatal acute renal failure: IRENEO—a prospective controlled study. Pediatr Nephrol. 2016;31:2365–73.
Harer MW, Pope CF, Conaway MR, Charlton JR. Follow-up of acute kidney injury in neonates during childhood years (FANCY): a prospective cohort study. Pediatr Nephrol. 2017;32:1067–76.
Ryan D, Sutherland MR, Flores TJ, Kent AL, Dahlstrom JE, Puelles VG, et al. Development of the human fetal kidney from mid to late gestation in male and female infants. EBioMedicine. 2018;27:275–83.
Sutherland MR, Gubhaju L, Moore L, Kent AL, Dahlstrom JE, Horne RS, et al. Accelerated maturation and abnormal morphology in the preterm neonatal kidney. J Am Soc Nephrology. 2011;22:1365–74.
Acknowledgements
The authors would like to acknowledge Drs Robert Chevalier and Frederick Kaskel for their guidance and mentorship.
Funding
KJR is supported by the NIH NIDDK and the Preeclampsia Foundation. JRC is funded by NIH/NIDDK: 1U34DK117128, R01DK111861, R01DK110622.
Author information
Authors and Affiliations
Contributions
All authors have participated in the concept and design, drafting and revising of the manuscript, and they have approved the manuscript as submitted here.
Corresponding author
Ethics declarations
Conflict of interest
JRC—co-owner of Sindri Technologies, LLC. KJR—site PI for unrelated Advicenne and Complexa studies. MWH and TT—none.
Additional information
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Harer, M.W., Charlton, J.R., Tipple, T.E. et al. Preterm birth and neonatal acute kidney injury: implications on adolescent and adult outcomes. J Perinatol 40, 1286–1295 (2020). https://doi.org/10.1038/s41372-020-0656-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1038/s41372-020-0656-7
- Springer Nature America, Inc.
This article is cited by
-
Decreased renal function among children born to women with obstructed labour in Eastern Uganda: a cohort study
BMC Nephrology (2024)
-
Sustained acute kidney injury as an independent risk factor for neurodevelopmental and growth outcomes in a single NICU center
BMC Pediatrics (2024)
-
Recurrent neonatal acute kidney injury: incidence, predictors, and outcomes in the neonatal intensive care unit
Journal of Perinatology (2024)
-
Magnitude and associated factors of acute kidney injury among preterm neonates admitted to public hospitals in Bahir Dar city, Ethiopia 2022: cross-sectional study
BMC Pediatrics (2023)
-
Acute kidney injury decreases pulmonary vascular growth and alveolarization in neonatal rat pups
Pediatric Research (2023)