
Abstract
Transthyretin-related cardiac amyloidosis is a disease that is poorly understood and challenging to manage; however, pharmacotherapeutic developments in recent years has raised awareness and promoted earlier diagnosis. Transthyretin protein is a thyroid hormone-binding protein which transports thyroxine from the bloodstream to the brain, and cardiac involvement is either acquired or hereditary. This protein can misfold and lead to abnormal fibril formation, causing the extracellular deposition of ATTR into the heart. Further fibril deposition leads to irreversible organ dysfunction. Disease-modifying pharmacological therapies can be divided into three categories: ATTR stabilizers (diflunisal, tafamidis, and AG10), ATTR silencers (patisiran and inotersen), and ATTR disruptors (doxycycline and ursodeoxycholic acid and polyphenol (-)-epigallocatechin gallate). This manuscript reviews these emerging pharmacological treatments available and under study for ATTR cardiac amyloidosis.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
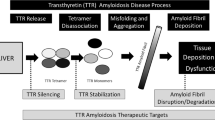
The amyloidoses are classified as a group of diseases related to protein misfolding wherein uncontrolled fibrillogenesis result in amyloid deposits. Over thirty proteins have been identified as being amyloidogenic. These proteins deposit into tissues and organs locally or systemically, including the liver, central nervous system, skin, and heart. These misfolded proteins disrupt the normal function of the organs, thus inducing a range of symptoms dependent upon the pattern of protein trafficking and deposition (Fig. 1) [1, 2].

The TTR amyloid cascade—transthyretin (TTR) is produced in the liver and first dissociates into a monomer which undergoes conformational changes to become misfolded monomeric intermediate. These intermediates can self-assemble into a fibril which can deposit in tissues
Immunoglobulin light chain, also known as AL or primary amyloidosis, and transthyretin amyloidosis, also known as ATTR, are the two most commonly diagnosed types of cardiac amyloidosis [3]. Transthyretin (TTR) is a homotetramer protein primarily produced in the liver as well as in the choroid plexus. Its function is to transport thyroxine and retinol protein [4, 5]. A genetic mutation or age-related factors may destabilize the tetramer causing the quaternary structure to form unstable monomers, leading to monomer misfolding and amyloid fibril formation. These fibrils then deposit into the heart or the nervous system, thus, initiating organ dysfunction [6].
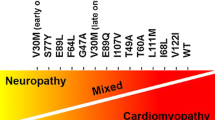
ATTR is further broken down into two subtypes: mutant transthyretin (ATTRm) and wild type transthyretin (ATTRwt). ATTRwt is a genetically unaltered protein that is amyloidogenic and is considered to be sporadic in nature with symptom manifestation later in life. ATTRm arises from a single point genetic mutation leading to TTR misfolding. Over 100 different TTR mutations have been identified. These mutations can give rise to a variety of symptoms predominantly from neurologic and cardiac involvement [1].
Prior to the development of novel therapies, the treatment of amyloidosis was focused on symptom management depending upon which organ was affected. Conventional heart failure medications including calcium channel blockers, beta blockers, and angiotensin-converting enzyme inhibitors are poorly tolerated due to hypotension and negative chronotropy/inotropy [7, 8]. With recent research, newer disease-modifying therapies have shown promising results (Table 1). This review will offer an overview of current therapies for transthyretin-related cardiac amyloidosis.
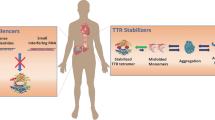
Stabilizers
The TTR protein is in equilibrium between the stable tetramer and thermodynamically unstable monomers. Dissociation of the tetramer allows the monomers to misfold and form amyloid fibrils. Therefore therapies that promote stabilization of the tetramer conformation were developed to prevent amyloid formation.
Diflunisal
Diflunisal has long been recognized as a bioavailable nonsteroidal anti-inflammatory drug (NSAID) with TTR stabilizing properties [13,14,15]. When administered at a dose of 250 mg twice daily, it was found to stabilize TTR in patients with confirmed familial amyloid polyneuropathy (FAP) [16]. A multi-center, randomized-controlled study examined the effect of diflunisal on disease progression in 130 patients with FAP over the course of 24 months. The treatment arm experienced significant symptom improvement as measured by the Neuropathy Impairment Score plus 7 nerve test (NIS+7) and the quality of life (QOL) questionnaire. There was an average score reduction of 60% and overall improved score in the QOL questionnaire. The drug was well tolerated, and overall adverse events were not significantly different between the two groups [17]. A long-term open-label study demonstrated that the clinical effects of diflunisal were sustained even after 2 years, with slower progression of symptoms over time [18].
Limited data is available for the treatment of diflunisal for ATTR cardiomyopathy. One single-center, observational study demonstrated the tolerability of diflunisal in patients with cardiomyopathy, including both ATTRwt and ATTRm. Over the course of 10 to 11 months, ventricular thickness, function, and biomarkers were unchanged [19]. However, treating heart failure patients with NSAIDs such as diflunisal may lead to fluid retention and worsen renal function. Therefore, adaption of this therapy has been limited.
Tafamidis
Tafamidis meglumine is a highly bioavailable, non-NSAID benzoxazole derivative which selectively binds TTR [20, 21]. Tafamidis was first studied in patients with early FAP and demonstrated modest efficacy. Patients on treatment maintained a higher concentration of the transthyretin tetramer in the stabilized conformation. However, there were no changes in neuropathy or QOL assessments. Secondary findings suggested treatment delayed disease progression and improved nutritional status [22, 23]. In 2011, based on these lukewarm results, tafamidis was approved for the treatment of stage 1 FAP only in Europe.
In an open-label phase II trial, patients with ATTRwt or ATTRm cardiomyopathy received 20 mg of tafamidis daily. The majority of patients enrolled (96.7%) were classified as NYHA I or II. Tafamidis stabilized the TTR tetramer in both ATTRwt and ATTRm without significant changes in biochemical, electrocardiogram, or echocardiogram parameters [24].
The Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR-ACT) was a global, phase III study in which patients with ATTRm or ATTRwt were randomized in a 2:1:2 ratio to receive 80 mg tafamidis, 20 mg tafamidis, or placebo. The tafamidis arm demonstrated a significant reduction in all-cause mortality and cardiovascular-related hospitalizations when compared with the placebo arm at the end of the 30-month period. Survival analysis suggested mortality benefits started near 18 months after initiating treatment. In the treatment arm, functional 6-min walk tests and QOL metrics demonstrated a decline in disease progression beginning as early as 6 months [25].
In a subgroup analysis, the benefit was muted in patients with more severe heart failure symptoms at the time of randomization. The authors speculated that the limited response was due to the severe progression of disease. In patients with less severe heart failure symptoms (NYHA class I or II) at baseline, there was a significant reduction in rates of cardiovascular-related hospitalizations as well as all-cause mortality [25].
In 2019, the U.S. Food and Drug Administration (FDA) approved tafamidis for the treatment of ATTR cardiomyopathy. The cost of a year’s supply of the drug currently exceeds $200,000 annually and is a barrier for treatment for many patients.
AG10
AG10 is a highly potent and selective TTR stabilizer that was first identified using a high-throughput chemical library screen of molecules that have high binding affinity and specificity for TTR [9, 26]. The tolerability and efficacy of AG10 in symptomatic heart failure patients with ATTR cardiomyopathy has been studied in a phase II trial. A total of 49 patients were enrolled (29% ATTRm, 70% NYHA II) and the drug was well-tolerated. At the conclusion of the trial, no serious adverse events could be directly attributed to AG10 [27]. The Efficacy and Safety of AG10 in Subjects with Transthyretin Amyloid Cardiomyopathy (ATTRIBUTE-CM) in a randomized, multicenter, placebo-controlled phase III trial that is ongoing (NCT03860935).
Silencers
In recent years, novel therapies have sought to target the protein synthesis of transthyretin in the liver.
Inotersen
Anti-sense oligonucleotides (ASOs) are short, modified oligonucleotides that are specifically designed to bind ribonucleic acid (RNA) and alter protein expression. Inotersen is an ASO that binds TTR messenger RNA and triggers degradation via nuclear ribonuclease H1 (RNaseH1). It was initially studied in cynomolgus monkeys during which investigators found a dose-dependent reduction of TTR serum concentrations [28]. In healthy volunteers, an 80% dose-dependent reduction of TTR serum concentrations was observed by day 29 of treatment [29].
These promising data led to a phase III trial that was conducted over 15 months in a FAP cohort. A larger proportion of the patients enrolled (50%) had the Val30Met mutation and most (65%) had evidence of cardiac involvement. Compared with placebo, inotersen delayed progression of neuropathy and improved QOL in FAP patients. The reduction of TTR serum concentrations was maintained through weeks 13–65 of treatment.
Five patients randomized to the inotersen arm died during trial participation. Four of those deaths were consistent with disease progression while one death was associated with intracranial hemorrhage. This death occurred prior to the implementation of weekly lab monitoring. Thrombocytopenia and glomerulonephritis have been associated with inotersen treatment. Over the course of the study, two additional patients developed severe thrombocytopenia with a platelet count of less than 25,000 per mL. These patients returned to baseline after initiating glucocorticoid treatment and discontinuation of the medication. In those randomized to inotersen, three patients developed glomerulonephritis. Renal function in two of those patients returned to baseline after initiation of glucocorticoid treatment while one patient required hemodialysis. As a result of these findings, patients who take inotersen must undergo weekly renal and platelet monitoring [30].
Currently, an open-label extension is ongoing and includes a 5-year follow-up for long-term safety and efficacy for FAP. No studies have focused on patients with primarily cardiac disease (ATTRwt or the Val122Ile mutation); however, a single-center trial is being conducted to assess the tolerability and efficacy of inotersen in patients with transthyretin amyloid cardiomyopathy (NCT03702829).
AKCEA-TTR-LRx (ION-682884) is a newer ASO that requires less frequent drug administration compared with its parent compound, inotersen, in achieving similar reductions in serum TTR concentrations [31]. Early studies suggest fewer complications compared with inotersen and a phase III clinical trial is underway (NCT04136171).
Small Interfering RNA
Small interfering RNA (siRNA) are double-stranded RNA non-coding RNA molecules that silence gene expression by activating an RNA-inducing silencing complex. This complex binds and induces cleavage of a target complementary messenger RNA (mRNA), thereby preventing translation and synthesis of a protein.
Patisiran
Patisiran, a first-generation siRNA, breaks down mRNA in both mutant and wild type TTR, which in turn diminishes TTR protein production. This reduction in manufacturing also translates to a reduction in protein deposition, thus curtailing the progression of symptoms.
A multicenter study, single-blind, phase I study established that patisiran was well-tolerated in healthy volunteers as well as in patients with ATTR. The drug suppressed TTR production at a dose of 0.3 mg per kilogram. By day 28, there was a knockdown of serum TTR concentrations by 86.6% [32].
In a multicenter, open-label, multiple-dose escalation study, the safety and effectiveness of patisiran was studied in 29 patients with FAP. Of note, the majority of these patients were already taking tafamidis (48%) and diflunisal (28%) for this condition. The patients who had received 0.3 mg per kilogram every 3 weeks were able to achieve a 96% knockdown of TTR. This study also reported that 10.3% of study patients experienced mild-to-moderate infusion-related reactions (IRRs). These IRRs included tachycardia, decreased oxygen saturation, dizziness, abdominal pain, bronchospasm, dyspnea, erythema, chills, pallor, pyrexia, and tachypnea [10].
This study was followed by a phase III clinical trial wherein 225 patients with FAP were randomized to patisiran or placebo at a dose of 0.3 mg per kilogram once every 3 weeks. Compared with placebo, the patisiran-treated patients exhibited an 81% reduction in serum concentration of TTR and significant improvements in the mNIS+7 neuropathy assessment and QOL scores. More IRRs were reported in the patisiran cohort (19%) compared with the placebo cohort (9%); however, these reactions were reported to be mild. In subgroup analyses, the incidences of congestive heart failure and arrhythmia were similar in both groups. In the cardiomyopathy subgroup, the treatment arm had a decrease in NT-proBNP concentrations and improvement in cardiac left ventricular (LV) wall thickness (P = 0.02) [33].
Although a study with patisiran in ATTR cardiomyopathy has not yet been completed, a phase III clinical trial with revusiran, another siRNA agent, was halted in 2016 due to increased mortality (18:2, revusiran:placebo) in ATTRm cardiomyopathy patients [34]. Though patisiran is a success story for siRNAs, the safety and efficacy of this therapy in patients with cardiac amyloidosis requires further research. Currently the study to Evaluate Patisiran in Participants with Transthyretin Amyloidosis with Cardiomyopathy (APOLLO-B) is in progress in an effort to establish the safety and efficacy of patisiran in patients with ATTRm and ATTRwt cardiomyopathy (NCT03997383).
Vutrisiran is another siRNA that is administered subcutaneously and is currently being evaluated in trials for those with ATTRm. A Study of Vutrisiran (ALN-TTRSC02) in Patients with Hereditary Transthyretin Amyloidosis (HELIOS-A) and A Study to Evaluate Vutrisiran in Patients with Transthyretin Amyloidosis with Cardiomyopathy (HELIOS-B) trials will compare vutrisiran to patisiran in patients with FAP and ATTR cardiomyopathy, respectively (NCT03759379 and NCT04153149).
Fibril Disruptors
Doxycycline and Ursodeoxycholic Acid
In vitro studies have identified the commonly used antibiotic, doxycycline, as a disruptor of amyloid fibrils [35]. Doxycycline has been studied often in combination with tauro-ursodeoxycholic acid (TUDCA) or ursodeoxycholic acid (UDCA), bile acids that have demonstrated an anti-apoptotic and anti-oxidative stress properties [36, 37].
The safety and tolerability of doxycycline and TUDCA was evaluated in a 12-month open-label study. Patients tolerated the medication well without any notable adverse events. Out of 20 patients, only 6 completed the planned 12-month course with 10 completing a 6-month treatment. The researchers reported nonprogression of the NIS-LL score and NYHA class however the study lacked a control group [38].
In a single-center study, 53 patients with cardiac amyloidosis (89% ATTRwt and 11% ATTRm) were given the combination of doxycycline and ursodeoxycholic acid (UDCA). Interestingly, the researchers discovered that cardiac function measured by the global longitudinal strain improved in this cohort [12]. However, 11% of study participants were unable to tolerate the treatment due to photosensitivity or gastrointestinal complaints.
An ongoing phase III trial is investigating the use of doxycycline and TUDCA when compared with standard therapy in those with ATTRm or ATTRwt (NCT03481972).
Polyphenol (-)-epigallocatechin Gallate
Polyphenol (-)-epigallocatechin gallate (EGCG) is the most abundant catechin found in green tea. It has been shown to change the amyloidogenic polypeptides into oligomers [39]. The EGCG compound inhibits TTR aggregation in cell cultures [40]. EGCG not only halts further fibril formation, it also converts existing fibrils into non-fibril conformers in vitro [41].
A single-center observational study evaluated the use of EGCG in patients with ATTR cardiomyopathy. Nineteen patients were followed for 12 months. No increase in LV wall thickness or mass was observed by echocardiography and cardiac magnetic resonance imaging (MRI) [11]. This study lacked a control group and could not draw a definitive connection, but this data is suggestive of EGCG or green tea inhibiting the progression of ATTR cardiomyopathy.
The aforementioned trial was followed by a second observational study in which green tea extract was provided to patients with ATTRwt. In this trial, 25 patients with ATTRwt cardiomyopathy were provided with 1200-mg capsules of green tea extract which contained 600 mg of EGCG. In patients who received a cardiac MRI, researchers observed a 5.9% reduction of LV mass over the 12-month period when compared with the patients’ baselines [42]. Though health benefits and excellent tolerance have been associated with the use of green tea for many years, larger comparative research is needed to further bolster the belief that it can impact patients with cardiac amyloidosis in a substantial way.
Conclusion
Remarkable advancements have given hope to patients with ATTR, where therapeutic interventions to slow disease progression and improve survival are now available. The recent FDA approvals of tafamidis for ATTR cardiomyopathy and patisiran and inotersen for FAP are landmark achievements. Additional ATTR stabilizers and silencers are on the horizon as clinical trials are actively evaluating their efficacy. This continuous progress offers an opportunity to further explore and understand this complex disease while helping patients amass an improved quality of life.
References
Benson MD, Kincaid JC. The molecular biology and clinical features of amyloid neuropathy. Muscle Nerve. 2007;36(4):411–23.
Claudio R, Giampaolo M, Quarta CC, Letizia R, Simone L, Ornella L, et al. Systemic cardiac Amyloidoses. Circulation. 2009;120(13):1203–12.
Sipe JD, Benson MD, Buxbaum JN, Ikeda S, Merlini G, Saraiva MJM, et al. Amyloid fibril proteins and amyloidosis: chemical identification and clinical classification International Society of Amyloidosis 2016 Nomenclature Guidelines. Amyloid. 2016 Oct 1;23(4):209–13.
Ingbar S. Pre-albumin: a thyroxine-binding protein of human plasma. Endocrinology. 1958;63(2):256–9.
Raz A, Goodman DS. The interaction of thyroxine with human plasma prealbumin and with the prealbumin-retinol-binding protein complex. J Biol Chem. 1969 Jun 25;244(12):3230–7.
Mohammed SF, Mirzoyev SA, Edwards WD, Dogan A, Grogan DR, Dunlay SM, et al. Left ventricular amyloid deposition in patients with heart failure and preserved ejection fraction. JACC: Heart Failure. 2014;2(2):113–22.
Rubinow A, Skinner M, Cohen AS. Digoxin sensitivity in amyloid cardiomyopathy. Circulation. 1981;63(6):1285–8.
Gertz MA, Falk RH, Skinner M, Cohen AS, Kyle RA. Worsening of congestive heart failure in amyloid heart disease treated by calcium channel-blocking agents. Am J Cardiol. 1985;55(13):1645.
Alhamadsheh MM, Connelly S, Cho A, Reixach N, Powers ET, Pan DW, et al. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci Transl Med. 2011;3(97):97.
Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis [Internet]. 2015 Sep 4 [cited 2019 Nov 18];10.
Kristen AV, Lehrke S, Buss S, Mereles D, Steen H, Ehlermann P, et al. Green tea halts progression of cardiac transthyretin amyloidosis: an observational report. Clin Res Cardiol. 2012;101(10):805–13.
Karlstedt E, Jimenez-Zepeda V, Howlett JG, White JA, Fine NM. Clinical experience with the use of doxycycline and ursodeoxycholic acid for the treatment of transthyretin cardiac amyloidosis. J Card Fail. 2019;25(3):147–53.
Hammarström P, Wiseman RL, Powers ET, Kelly JW. Prevention of transthyretin amyloid disease by changing protein misfolding energetics. Science. 2003;299(5607):713–6.
Miller SR, Sekijima Y, Kelly JW. Native state stabilization by NSAIDs inhibits transthyretin amyloidogenesis from the most common familial disease variants. Lab Investig. 2004;84(5):545–52.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–49.
Tojo K, Sekijima Y, Kelly JW, Ikeda S. Diflunisal stabilizes familial amyloid polyneuropathy-associated transthyretin variant tetramers in serum against dissociation required for amyloidogenesis. Neurosci Res. 2006;56(4):441–9.
Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–67.
Sekijima Y, Tojo K, Morita H, Koyama J, Ikeda S. Safety and efficacy of long-term diflunisal administration in hereditary transthyretin (ATTR) amyloidosis. Amyloid. 2015;22(2):79–83.
Castaño A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18(6):315–9.
Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN, et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew Chem Int Ed. 2003;42(24):2758–61.
Bulawa CE, Connelly S, DeVit M, Wang L, Weigel C, Fleming JA, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–34.
Coelho T, Maia LF, Martins da Silva A, Waddington Cruz M, Planté-Bordeneuve V, Lozeron P, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792.
Coelho T, Maia LF, da Silva AM, Cruz MW, Planté-Bordeneuve V, Suhr OB, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–14.
Maurer Mathew S., Grogan Donna R., Judge Daniel P., Mundayat Rajiv, Packman Jeff, Lombardo Ilise, et al. Tafamidis in transthyretin amyloid cardiomyopathy. Circ Heart Fail 2015;8(3):519–526.
Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16.
Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci U S A. 2013;110(24):9992–7.
Judge DP, Heitner SB, Falk RH, Maurer MS, Shah SJ, Witteles RM, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019;74(3):285–95.
Ackermann EJ, Guo S, Benson MD, Booten S, Freier S, Hughes SG, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 2016;23(3):148–57.
Ackermann EJ, Guo S, Booten S, Alvarado L, Benson M, Hughes S, et al. Clinical development of an antisense therapy for the treatment of transthyretin-associated polyneuropathy. Amyloid. 2012;19:43–4.
Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31.
Viney NJ, Tai L-J, Jung S, Yu RZ, Guthrie S, Baker BF, et al. Phase 1 Investigation of a ligand-conjugated antisense oligonucleotide with increased potency for the treatment of transthyretin amyloidosis. J Card Fail. 2019;25(8 Supplement):S80–1.
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29.
Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang C-C, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21.
Judge DP, Kristen AV, Grogan M, Maurer MS, Falk RH, Hanna M, et al. Phase 3 multicenter study of revusiran in patients with hereditary transthyretin-mediated (hATTR) amyloidosis with cardiomyopathy (ENDEAVOUR). Cardiovasc Drugs Ther. 2020;34(3):357–70.
Cardoso I, Saraiva MJ. Doxycycline disrupts transthyretin amyloid: evidence from studies in a FAP transgenic mice model. FASEB J. 2006;20(2):234–9.
Macedo B, Batista AR, Ferreira N, Almeida MR, Saraiva MJ. Anti-apoptotic treatment reduces transthyretin deposition in a transgenic mouse model of familial amyloidotic polyneuropathy. Biochim Biophys Acta (BBA) - Mol Basis Dis. 2008;1782(9):517–22.
Cardoso I, Martins D, Ribeiro T, Merlini G, Saraiva MJ. Synergy of combined doxycycline/TUDCA treatment in lowering Transthyretin deposition and associated biomarkers: studies in FAP mouse models. J Transl Med. 2010;8:74.
Obici L, Cortese A, Lozza A, Lucchetti J, Gobbi M, Palladini G, et al. Doxycycline plus tauroursodeoxycholic acid for transthyretin amyloidosis: a phase II study. Amyloid. 2012a;19:34–6.
Ehrnhoefer DE, Bieschke J, Boeddrich A, Herbst M, Masino L, Lurz R, et al. EGCG redirects amyloidogenic polypeptides into unstructured, off-pathway oligomers. Nat Struct Mol Biol. 2008;15(6):558–66.
Ferreira N, Cardoso I, Domingues MR, Vitorino R, Bastos M, Bai G, et al. Binding of epigallocatechin-3-gallate to transthyretin modulates its amyloidogenicity. FEBS Lett. 2009;583(22):3569–76.
Bieschke J, Russ J, Friedrich RP, Ehrnhoefer DE, Wobst H, Neugebauer K, et al. EGCG remodels mature alpha-synuclein and amyloid-beta fibrils and reduces cellular toxicity. Proc Natl Acad Sci U S A. 2010;107(17):7710–5.
aus dem Siepen F, Bauer R, Aurich M, Buss SJ, Steen H, Altland K, et al. Green tea extract as a treatment for patients with wild-type transthyretin amyloidosis: an observational study. Drug Des Devel Ther. 2015;9:6319–25.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
KBS is a consultant for Akcea Therapeutics and Alnylam Pharmaceuticals. KBS serves on the ATTRIBUTE Trial Steering Committee for Eidos Therapeutics.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
Informed consent was not required for this review.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Medicine
Rights and permissions
About this article

Cite this article
Park, J.H., Cei, L.F. & Shah, K.B. Disease-Modifying Pharmacological Therapies for Transthyretin Cardiac Amyloidosis. SN Compr. Clin. Med. 2, 1607–1613 (2020). https://doi.org/10.1007/s42399-020-00420-y
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s42399-020-00420-y