
Abstract
Purpose of review
Transthyretin cardiac amyloidosis is an underdiagnosed, undertreated disease which is associated with significant morbidity and mortality. This review will discuss the recent advancements in novel therapies for transthyretin amyloidosis.
Recent findings
In recent phase 3 clinical trials, transthyretin stabilizers (tafamidis) and transthyretin silencers (patisiran and inotersen) have proven to be effective therapies for various forms of transthyretin amyloidosis.
Summary
Understanding the recent and upcoming clinical trials for transthyretin amyloidosis will be important for improving the management of this challenging disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Cardiac amyloidosis remains a deadly and often underdiagnosed disease [1]. Through a variety of mechanisms, precursor proteins can misfold and aggregate, leading to amyloid fibril formation and deposition in the myocardium. Gradual amyloid accumulation can cause restrictive cardiomyopathy, arrhythmias, and sudden death [2]. The two most common types of cardiac amyloidosis occur as a result of misfolded immunoglobulin light chain (AL) or transthyretin (ATTR). Over the past decade, significant advances have been made in treating AL amyloidosis. Plasma cell-directed therapies, such as proteasome inhibitors (e.g., bortezomib) and monoclonal antibodies (e.g., daratumumab), can often profoundly reduce the deleterious plasma cell clone and thus the production of the amyloid-causing light chain [3,4,5]. These interventions can make a large difference in outcomes in patients at all stages of the disease, including those with significant cardiac involvement.
In contrast, until recently, there have been extremely limited therapies for ATTR amyloidosis. ATTR amyloidosis can arise from misfolding of wild type (wt) or mutant (m) transthyretin. ATTRwt cardiomyopathy is increasingly recognized as an important cause of heart failure with preserved ejection and is likely the most prevalent form of cardiac amyloidosis [6]. ATTRm amyloidosis typically occurs due to a single point mutation in transthyretin, has an autosomal dominant inheritance pattern, and can manifest with cardiac and/or neurologic involvement. ATTRm amyloidosis confers significant disease burden in specific subpopulations, such as African Americans, in whom 3–4% carry the V122I transthyretin mutation [7, 8].
The past few years has seen the rapid translation of extensive preclinical studies into effective treatments for ATTR cardiomyopathy and polyneuropathy as demonstrated by several recent positive phase 3 randomized controlled clinical trials. This review will explore these promising medications for ATTR amyloidosis and highlight additional therapies currently under development (Tables 1 and 2).
Transthyretin stabilizers
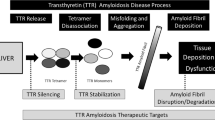
Transthyretin is a protein predominantly synthesized by the liver and functions as a transporter of thyroxine and retinol-binding protein. Its native structure consists of a homotetramer. Through incompletely described mechanisms, this structure can destabilize and dissociate into transthyretin monomers. These monomers are thermodynamically unstable and have an increased propensity to misfold, aggregate, and ultimately form amyloid fibrils. Consequently, tetramer dissociation represents the rate-limiting step in transthyretin amyloid formation. Comprehensive biochemical studies have identified several small molecules that are able to bind to the native transthyretin tetramer with high specificity and thus prevent tetramer dissociation and amyloid fibril formation (Fig. 1). The subsequent section will discuss the increasing clinical evidence that transthyretin stabilizers are effective in treating ATTR amyloidosis.

Therapeutic targets for transthyretin amyloidosis.
Tafamidis
Tafamidis is a small molecule stabilizer that was initially identified through a chemical library screen of benzoxazoles [9]. In vitro studies showed that tafamidis stabilizes the transthyretin tetramer under physiologic conditions and across a range of transthyretin mutations [10]. The first large clinical trial of tafamidis was a phase 2/3 study of patients with ATTR polyneuropathy. Patients (n = 125) were randomized to tafamidis 20 mg or placebo, and the primary endpoint was change in 2 polyneuropathy impairment clinical scores at 18 months. In the prespecified, intention-to-treat analysis, there was a statistically non-significant (p = 0.068) trend toward to more treatment responders in the tafamidis arm compared with placebo. Of note, a significant proportion of participants were on the liver transplant list at the time of study enrollment; 21% of patients underwent liver transplantation during the study and thus were counted as non-responders in the prespecified analysis. This high discontinuation rate significantly reduced the study’s power to detect a statistical difference. Based on this trial, tafamidis was approved for treating ATTR polyneuropathy in Europe, Japan, and parts of Latin America, but not the USA [11].
Tafamidis was recently studied in ATTR cardiomyopathy patients in the Transthyretin Amyloidosis Cardiomyopathy Clinical Trial (ATTR-ACT) [12••]. ATTR-ACT was a relatively large (n = 441) phase 3 trial that randomized patients with ATTR cardiomyopathy to oral administration of placebo, 20 mg of tafamidis, or 80 mg of tafamidis in a 2:1:2 ratio. Of these study patients, 24% had ATTRm and 76% had ATTRwt. The primary endpoint was a composite of all-cause mortality and cardiovascular-related hospitalizations at 30 months. These endpoint components were assessed in a hierarchical manner using the Finkelstein-Schoenfeld method. Both all-cause mortality and cardiovascular-related hospitalizations were significantly lower in the tafamidis group compared with placebo (29.5% vs. 42.9%; 0.48 per year vs. 0.70 per year, respectively).
The reduction in mortality was observed as early as 18 months, and the number needed to treat to prevent death in 30 months was 7.5. In the subgroup analyses, both the 20 and 80 mg doses of tafamidis appeared effective. Tafamidis was well tolerated and had a similar incidence of adverse events compared with placebo. Less severe heart failure symptoms at baseline (i.e., New York Heart Association class 1 or 2) were associated with not only better outcomes but also a larger tafamidis treatment effect, compared with NYHA class 3 symptoms. This finding suggests that tafamidis may be more effective in patients in earlier stages of the disease. Based on these trial results, tafamidis has been approved for use in ATTR cardiomyopathy in Japan and in the USA.
AG10
AG10 is another transthyretin stabilizer currently being studied in ATTR cardiomyopathy patients. AG10 was first identified using a high-throughput chemical library screen of molecules that have high binding affinity and specificity for wild-type transthyretin [13]. Early studies showed that AG10 prevents cardiac cell toxicity induced by the V122I transthyretin. Subsequent in vitro experiments demonstrated that AG10 effectively stabilizes both wild-type and V122I transthyretin in patient serum [14].
A recent phase 2 trial randomized 49 patients with ATTR cardiomyopathy and NYHA class 2 or 3 heart failure to oral administration of placebo, 400 mg twice a day of AG10, or 800 mg twice a day of AG10 [15]. Most patients had ATTRwt (71.4%). Of the ATTRm patients, 78.6% had the V122I mutation. After 28 days of the administration, AG10 was well-tolerated and achieved a high degree of stability for both wild-type and mutant transthyretin. Previous data have suggested that serum transthyretin concentration may be a useful biomarker for ATTR cardiomyopathy. Lower levels are associated with a worse prognosis, and transthyretin stabilizer therapy increases serum transthyretin concentration [16, 17]. In the AG10 trial, both doses of AG10 significantly increase serum transthyretin compared with placebo. Based on these safety and efficacy data, the phase 3, ATTRibute-CM trial was recently launched [18]. The trial aims to enroll 510 patients with ATTR cardiomyopathy who will be randomized to 800 mg twice a day or placebo. The primary endpoint is a change in 6-min walk test at 18 months and all-cause mortality and cardiovascular-related hospitalization at 30 months.
Diflunisal
Diflunisal is a non-steroidal anti-inflammatory drug that was discovered to have transthyretin-stabilizing properties [19, 20]. As a result, diflunisal was repurposed as a stabilizer therapy for ATTR amyloidosis. In a randomized trial of 130 patients with ATTR polyneuropathy, at 2 years, diflunisal was associated with significantly less neurological deterioration compared with placebo [21]. For ATTR cardiomyopathy, diflunisal has only been studied in small, single-center observational studies [22, 23]. Although diflunisal was well-tolerated overall in these small cohorts (n = 13 and n = 23), the risks of long-term non-steroidal anti-inflammatory drug use (e.g., cardiovascular events, kidney injury, and gastrointestinal), particularly for heart failure patients, remain a prominent concern. Given the emergence of tafamidis and AG10, which have benign safety profiles, diflunisal will likely be used less in the future for ATTR cardiomyopathy.
Transthyretin silencers
The liver is the main source of transthyretin synthesis (though small amounts are made by the retina and choroid plexus). In the case of ATTRm amyloidosis, eliminating the production of amyloidogenic mutant transthyretin has long been hypothesized as an effective treatment strategy. Indeed, liver transplantation has been used to replace the organ which produces mutant transthyretin with one that synthesizes wild-type transthyretin. This form of “gene therapy” can be effective in halting disease progression in ATTR polyneuropathy, but confers significant short- and long-term risks associated with solid-organ transplantation [24]. Recently, oligonucleotide drugs have emerged as a non-invasive form of gene therapy [25]. These drugs act by promoting the degradation of messenger RNA (mRNA), thereby reducing protein expression. Moreover, the liver appears to be a particularly accessible target for these therapies. For these reasons, oligonucleotide drugs may be well suited for treating diseases originating from the liver, such as ATTR amyloidosis [26] (Fig. 1).
Patisiran
Patisiran is a small interfering RNA (siRNA) that binds to the highly conserved 3′ untranslated region of both wild-type and mutant transthyretin mRNA to induce its degradation. This drug is carried in lipid nanoparticles to target drug delivery to hepatocytes [27]. Early clinical studies demonstrated that patisiran reduces serum transthyretin levels in a dose-dependent manner [28, 29]. These data led to the APOLLO study [30••]. This phase 3 trial randomized 225 patients with ATTRm polyneuropathy, in a 2:1 manner, to patisiran or placebo. Patisiran was administered intravenously (0.3 mg/kg) every 3 weeks and was associated with a median reduction in serum transthyretin of 81%. The primary endpoint assessed the change in neurological function using the modified Neuropathy Impairment Score+7 (mNIS+7). A higher mNIS+7 score reflects more severe neurologic impairment, with typical diabetic neuropathy progressing at approximately 3 points/year. Patisiran was associated with a significant improvement in the mNIS+7, and a dramatic difference in outcomes compared to placebo. The absolute difference of nearly 34 points on the mNIS+7 score is larger than with any other therapy to date and achieved a P value of 9.26 × 10−24 compared with placebo. Overall adverse events were similar between the treatment groups. The patisiran arm had a higher frequency of mild to moderate infusion-related reactions and peripheral edema. Based on this trial, patisiran was approved by the FDA for the treatment of patients with ATTRm polyneuropathy.
Patients with ATTRm polyneuropathy often have concomitant cardiomyopathy. In the APOLLO study, 56% of patients (n = 126) had evidence of cardiac involvement. In this prespecified cardiac subgroup, patisiran was associated with decreased left ventricular wall thickness, improved global and regional longitudinal strain, and reductions in NT-proBNP [31, 32]. Future studies will assess the safety and efficacy of RNA interference for ATTR cardiomyopathy populations. The APOLLO-B study will study patisiran in ATTRm and ATTRwt cardiomyopathy patients and will allow for the inclusion of patients receiving a transthyretin stabilizer as part of the standard of care. Vutrisiran is a next-generation transthyretin RNA interference therapy that, in contrast to patisiran, is administered subcutaneously and is administered every 3 months. The HELIOS-A trial, which is a phase 3 study, multicenter study, is currently randomizing ATTRm polyneuropathy patients to vutrisiran or patisiran [33]. The HELIOS-B trial has been announced and is planned to assess vutrisiran in patients with ATTRm or ATTRwt cardiomyopathy.
Inotersen
Inotersen is a 2′-O-methoxyethyl-modified antisense oligonucleotide that degrades transthyretin mRNA and decreases hepatic production of transthyretin protein. In animal studies and in healthy volunteers, inotersen was shown to significantly reduce serum transthyretin concentration in a dose-dependent manner [34]. The recent phase 3 NEURO-TTR trial evaluated the safety and efficacy of inotersen in patients with ATTRm polyneuropathy [35••]. In total, 172 patients were 2:1 randomized to receive weekly subcutaneous injections of 300 mg of inotersen or placebo. Inotersen led to median serum transthyretin reduction of 79%. The primary endpoint was a composite of the mNIS+7 and the Norfolk Quality of Life-Diabetic Neuropathy score. At 66 weeks, the inotersen group had significantly less worsening in both clinical scores compared with placebo, with a difference in mNIS+7 of 19.73 points (P = 4 × 10−8). Notable adverse events in the inotersen group were severe thrombocytopenia (3% of patients), including 1 death due to intracranial hemorrhage, and glomerulonephritis (3%). As a result, the study was modified to include weekly platelet monitoring. Regarding cardiovascular endpoints, in both the overall populations and the subgroups with evidence of concomitant amyloid cardiomyopathy, there was no significant change in left ventricular wall thickness or global longitudinal strain. The NEURO-TTR trial led to FDA approval of inotersen for ATTRm polyneuropathy. A next-generation antisense oligonucleotide, AKCEA-TTR-LRX, is currently being studied in a phase 1/2 trial in healthy volunteers and ATTR polyneuropathy patients [36].
Amyloid fibril antibodies
Both transthyretin stabilizers and silencers primarily aim to inhibit the formation of pre-amyloid species and subsequent amyloid fibril deposition. However, patients with ATTR cardiomyopathy often are diagnosed at a late disease stage when significant end-organ fibril deposition has already occurred. In this unfortunately common situation, therapies that prevent new fibril deposition may be ineffective at reversing the disease burden caused by the existing amyloid deposits. Indeed, in the ATTR-ACT trial, patients with more advanced heart failure (i.e., NYHA class 3) had an attenuated survival benefit from tafamidis compared with NYHA class 1 and 2 patients [12••]. Therefore, there is significant interest in identifying therapies that can disrupt and remove amyloid fibrils already deposited in tissues.
Miridesap/dezamizumab
Extensive preclinical and early clinical studies have examined targeting serum amyloid P component (SAP) as a mechanism of removing amyloid fibrils. SAP is a glycoprotein that is a key constituent of all amyloid fibrils, regardless of the precursor protein. Drugs have been developed to target clearance of circulating SAP as well as to bind SAP in tissue-deposited amyloid fibrils. Miridesap, formerly known as CPHPC, cross-links circulating SAP molecules, causing its clearance by the liver [37, 38]. Dezamizumab is a humanized anti-SAP antibody, which can bind to amyloid deposits in tissues and mediate fibril removal via complement factor fixation and macrophage clearance [39]. An open-label study of 15 patients with various types of amyloidosis showed that one-time administration of miridesap followed by dezamizumab reduced hepatic amyloid deposition as measured by 123I SAP scintigraphy [40]. A subsequent study of 23 patients receiving repeated dosing of these therapies demonstrated amyloid clearance in the liver, kidneys, and spleen [41••]. Of note, only 6 patients in this study had cardiac amyloidosis. There is a theoretical concern that rapid, immune-mediated removal of large amounts of amyloid from the heart may acute exacerbate heart failure and arrhythmias. A recent study aimed to investigate the safety and efficacy of miridesap and dezamizumab combination therapy in patients with ATTR or AL cardiomyopathy [42]. This study was terminated in April 2019 with the investigators citing a “change in benefit/risk profile.”
PRX004
PRX004 is a monoclonal antibody that selectively binds to misfolded conformations of transthyretin via a cryptic epitope [43]. PRX004 is currently being assessed in a phase 1, open-label, dose-escalation study of patients with ATTRm cardiomyopathy and/or neuropathy [44].
Conclusion
Recent breakthroughs have drastically changed the management of ATTR amyloidosis. There have recently been multiple strongly positive phase 3 clinical trials with subsequent FDA approval for tafamidis (ATTR cardiomyopathy) and patisiran and inotersen (ATTRm polyneuropathy). Moreover, additional transthyretin stabilizers and silencers are being studied. The arrival of the first disease-specific therapies for ATTR amyloidosis raises many new clinical management questions. Considerations include developing diagnostic protocols to promote early disease diagnosis, determining the role of combination therapy (e.g., concomitant use of a transthyretin stabilizer and silencer), and identifying biomarkers of treatment response. The field of ATTR amyloidosis will likely continue to evolve rapidly over the next several years.
References and Recommended Reading
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Alexander KM, Orav J, Singh A, Jacob SA, Menon A, Padera RF, et al. Geographic disparities in reported US amyloidosis mortality from 1979 to 2015: potential underdetection of cardiac amyloidosis. JAMA Cardiol. 2018;3(9):865–70.
Falk RH, Alexander KM, Liao R, Dorbala S. AL (light-chain) cardiac amyloidosis: a review of diagnosis and therapy. J Am Coll Cardiol. 2016;68(12):1323–41.
Palladini G, Sachchithanantham S, Milani P, Gillmore J, Foli A, Lachmann H, et al. A European collaborative study of cyclophosphamide, bortezomib, and dexamethasone in upfront treatment of systemic AL amyloidosis. Blood. 2015;126(5):612–5.
Sperry BW, Ikram A, Hachamovitch R, Valent J, Vranian MN, Phelan D, et al. Efficacy of chemotherapy for light-chain amyloidosis in patients presenting with symptomatic heart failure. J Am Coll Cardiol. 2016;67(25):2941–8.
Kaufman GP, Schrier SL, Lafayette RA, Arai S, Witteles RM, Liedtke M. Daratumumab yields rapid and deep hematologic responses in patients with heavily pretreated AL amyloidosis. Blood. 2017;130(7):900–2.
Gonzalez-Lopez E, Gallego-Delgado M, Guzzo-Merello G, de Haro-Del Moral FJ, Cobo-Marcos M, Robles C, et al. Wild-type transthyretin amyloidosis as a cause of heart failure with preserved ejection fraction. Eur Heart J. 2015;36(38):2585–94.
Quarta CC, Buxbaum JN, Shah AM, Falk RH, Claggett B, Kitzman DW, et al. The amyloidogenic V122I transthyretin variant in elderly black Americans. N Engl J Med. 2015;372(1):21–9.
Dungu JN, Papadopoulou SA, Wykes K, Mahmood I, Marshall J, Valencia O, et al. Afro-Caribbean heart failure in the United Kingdom: cause, outcomes, and ATTR V122I cardiac amyloidosis. Circ Heart Fail. 2016;9(9). https://doi.org/10.1161/CIRCHEARTFAILURE.116.003352.
Razavi H, Palaninathan SK, Powers ET, Wiseman RL, Purkey HE, Mohamedmohaideen NN, et al. Benzoxazoles as transthyretin amyloid fibril inhibitors: synthesis, evaluation, and mechanism of action. Angew Chem Int Ed Engl. 2003;42(24):2758–61.
Bulawa CE, Connelly S, Devit M, Wang L, Weigel C, Fleming JA, et al. Tafamidis, a potent and selective transthyretin kinetic stabilizer that inhibits the amyloid cascade. Proc Natl Acad Sci U S A. 2012;109(24):9629–34.
Scott LJ. Tafamidis: a review of its use in familial amyloid polyneuropathy. Drugs. 2014;74(12):1371–8.
•• Maurer MS, Schwartz JH, Gundapaneni B, Elliott PM, Merlini G, Waddington-Cruz M, et al. Tafamidis treatment for patients with transthyretin amyloid cardiomyopathy. N Engl J Med. 2018;379(11):1007–16. This large phase 3 trial showed that tafamidis improved survival and reduce cardiovascular-related hospitalizations in ATTR cardiomyopathy patients. This led to tafamidis being approved as the first drug for ATTR cardiomyopathy.
Alhamadsheh MM, Connelly S, Cho A, Reixach N, Powers ET, Pan DW, et al. Potent kinetic stabilizers that prevent transthyretin-mediated cardiomyocyte proteotoxicity. Sci Transl Med. 2011;3(97):97ra81.
Penchala SC, Connelly S, Wang Y, Park MS, Zhao L, Baranczak A, et al. AG10 inhibits amyloidogenesis and cellular toxicity of the familial amyloid cardiomyopathy-associated V122I transthyretin. Proc Natl Acad Sci. 2013;110(24):9992–7.
Judge DP, Falk RH, Maurer MS, Shah SJ, Witteles RM, Grogan M, et al. Transthyretin stabilization by AG10 in symptomatic transthyretin amyloid cardiomyopathy. J Am Coll Cardiol. 2019. https://doi.org/10.1016/j.jacc.2019.03.012.
Hanson JLS, Arvanitis M, Koch CM, Berk JL, Ruberg FL, Prokaeva T, et al. Use of serum transthyretin as a prognostic Indicator and predictor of outcome in cardiac amyloid disease associated with wild-type transthyretin. Circ Heart Fail. 2018;11(2):e004000.
Ruberg FL, Maurer MS, Judge DP, Zeldenrust S, Skinner M, Kim AY, et al. Prospective evaluation of the morbidity and mortality of wild-type and V122I mutant transthyretin amyloid cardiomyopathy: the transthyretin amyloidosis cardiac study (TRACS). Am Heart J. 2012;164(2):222–8 e1.
Efficacy and safety of AG10 in subjects with transthyretin amyloid cardiomyopathy. https://ClinicalTrials.gov/show/NCT03860935. Accessed 15 May 2019.
Adamski-Werner SL, Palaninathan SK, Sacchettini JC, Kelly JW. Diflunisal analogues stabilize the native state of transthyretin. Potent inhibition of amyloidogenesis. J Med Chem. 2004;47(2):355–74.
Sekijima Y, Dendle MA, Kelly JW. Orally administered diflunisal stabilizes transthyretin against dissociation required for amyloidogenesis. Amyloid. 2006;13(4):236–49.
Berk JL, Suhr OB, Obici L, Sekijima Y, Zeldenrust SR, Yamashita T, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–67.
Castano A, Helmke S, Alvarez J, Delisle S, Maurer MS. Diflunisal for ATTR cardiac amyloidosis. Congest Heart Fail. 2012;18(6):315–9.
Ikram A, Donnelly JP, Sperry BW, Samaras C, Valent J, Hanna M. Diflunisal tolerability in transthyretin cardiac amyloidosis: a single center’s experience. Amyloid. 2018;25(3):197–202.
Ericzon BG, Wilczek HE, Larsson M, Wijayatunga P, Stangou A, Pena JR, et al. Liver transplantation for hereditary transthyretin amyloidosis: after 20 years still the best therapeutic alternative? Transplantation. 2015;99(9):1847–54.
Buxbaum JN. Oligonucleotide drugs for transthyretin amyloidosis. N Engl J Med. 2018;379(1):82–5.
Dong Y, Siegwart DJ, Anderson DG. Strategies, design, and chemistry in siRNA delivery systems. Adv Drug Deliv Rev. 2019. https://doi.org/10.1016/j.addr.2019.05.004.
Akinc A, Querbes W, De S, Qin J, Frank-Kamenetsky M, Jayaprakash KN, et al. Targeted delivery of RNAi therapeutics with endogenous and exogenous ligand-based mechanisms. Mol Ther. 2010;18(7):1357–64.
Coelho T, Adams D, Silva A, Lozeron P, Hawkins PN, Mant T, et al. Safety and efficacy of RNAi therapy for transthyretin amyloidosis. N Engl J Med. 2013;369(9):819–29.
Suhr OB, Coelho T, Buades J, Pouget J, Conceicao I, Berk J, et al. Efficacy and safety of patisiran for familial amyloidotic polyneuropathy: a phase II multi-dose study. Orphanet J Rare Dis. 2015;10:109.
•• Adams D, Gonzalez-Duarte A, O’Riordan WD, Yang CC, Ueda M, Kristen AV, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. This phase 3 study showed that patisiran improved neuropathy impairment in ATTR polyneuropathy patients and led to patisiran’s FDA approval.
Solomon SD, Adams D, Kristen A, Grogan M, Gonzalez-Duarte A, Maurer MS, et al. Effects of patisiran, an RNA interference therapeutic, on cardiac parameters in patients with hereditary transthyretin-mediated amyloidosis. Circulation. 2019;139(4):431–43.
Minamisawa M, Claggett B, Adams D, Kristen AV, Merlini G, Slama MS, et al. Association of patisiran, an RNA interference therapeutic, with regional left ventricular myocardial strain in hereditary transthyretin amyloidosis: the APOLLO study. JAMA Cardiol. 2019;4:466.
HELIOS-A: A study of vutrisiran (ALN-TTRSC02) in patients with hereditary transthyretin amyloidosis (hATTR Amyloidosis). https://ClinicalTrials.gov/show/NCT03759379. Accessed 15 May 2019.
Ackermann EJ, Guo S, Benson MD, Booten S, Freier S, Hughes SG, et al. Suppressing transthyretin production in mice, monkeys and humans using 2nd-generation antisense oligonucleotides. Amyloid. 2016;23(3):148–57.
•• Benson MD, Waddington-Cruz M, Berk JL, Polydefkis M, Dyck PJ, Wang AK, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. This phase 3 study showed that inotersen slowed disease progression in ATTR polyneuropathy patients and led to inotersen’s FDA approval.
A study to evaluate the safety, tolerability, pharmacokinetics, and pharmacodynamics of ION-TTR-LRx in healthy volunteers and patients with hereditary transthyretin-mediated amyloidosis. https://ClinicalTrials.gov/show/NCT03728634. Accessed 15 May 2019.
Pepys MB, Herbert J, Hutchinson WL, Tennent GA, Lachmann HJ, Gallimore JR, et al. Targeted pharmacological depletion of serum amyloid P component for treatment of human amyloidosis. Nature. 2002;417(6886):254–9.
Gillmore JD, Tennent GA, Hutchinson WL, Gallimore JR, Lachmann HJ, Goodman HJ, et al. Sustained pharmacological depletion of serum amyloid P component in patients with systemic amyloidosis. Br J Haematol. 2010;148(5):760–7.
Bodin K, Ellmerich S, Kahan MC, Tennent GA, Loesch A, Gilbertson JA, et al. Antibodies to human serum amyloid P component eliminate visceral amyloid deposits. Nature. 2010;468(7320):93–7.
Richards DB, Cookson LM, Berges AC, Barton SV, Lane T, Ritter JM, et al. Therapeutic clearance of amyloid by antibodies to serum amyloid P component. N Engl J Med. 2015;373(12):1106–14.
• Richards DB, Cookson LM, Barton SV, Liefaard L, Lane T, Hutt DF, et al. Repeat doses of antibody to serum amyloid P component clear amyloid deposits in patients with systemic amyloidosis. Sci Transl Med. 2018;10(422). This study examines the safety and efficacy of a novel monoclonal antibody, dezamizumab, that stimulates the removal of organ-deposited amyloid fibrils.
Multiple treatment session study to assess GSK2398852 administered following and along with GSK2315698. https://ClinicalTrials.gov/show/NCT03044353. Accessed 15 May 2019.
Higaki JN, Chakrabartty A, Galant NJ, Hadley KC, Hammerson B, Nijjar T, et al. Novel conformation-specific monoclonal antibodies against amyloidogenic forms of transthyretin. Amyloid. 2016;23(2):86–97.
A study of PRX004 in subjects with amyloid transthyretin (ATTR) amyloidosis. https://ClinicalTrials.gov/show/NCT03336580. Accessed 15 May 2019.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Kevin M. Alexander has received an investigator-initiated research grant from Pfizer.
Alessandro Evangelisti declares no potential conflicts of interest.
Ronald M. Witteles has received consulting fees (modest) from Pfizer and Alnylam, and has received clinical trial support from Pfizer, Alnylam, and Eidos.
Human and Animal Rights and Informed Consent
The article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
All authors take responsibility for all aspects of the reliability and freedom from bias of the data presented and their discussed interpretation.
This article is part of the Topical Collection on Heart Failure
Rights and permissions
About this article

Cite this article
Alexander, K.M., Evangelisti, A. & Witteles, R.M. Emerging Therapies for Transthyretin Cardiac Amyloidosis. Curr Treat Options Cardio Med 21, 40 (2019). https://doi.org/10.1007/s11936-019-0743-2
Published:
DOI: https://doi.org/10.1007/s11936-019-0743-2