
Abstract
Introduction
Vitamin D is classically involved in maintaining bone and mineral health, but it has been shown to exert many extraskeletal functions, including pleiotropic effects on cardiovascular system.
Materials and method
This review aims to summarize evidences in literature about vitamin D and cardiovascular outcome.
Results and conclusions
Calcitriol or 1,25(OH)2D, the active hormone, binds to the specific nuclear receptor VDR, which is expressed in rat and human heart and vasculature and has effects on myocardiocytes, smooth cells, and endothelial cells. 25-Hydroxy-vitamin D (25OHD) represents the biomarker of vitamin D levels and reflects vitamin D status. There is consistent evidence that low serum 25OHD levels are associated with increased risk of cardiovascular diseases, including hypertension, coronary artery disease, ischemic heart disease, heart failure, stroke, and type 2 diabetes. Randomized-controlled trials and Mendelian randomization studies so far have not succeeded in proving a benefit of vitamin D supplementation. However, the latter investigations are affected by some methodological limitations, and therefore, it is still unclear if vitamin D deficiency has a causative role in cardiovascular diseases or is rather a marker of poor health in chronic disease.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Vitamin D is a steroid hormone and a crucial regulator of skeletal and calcium homeostasis. In the last 10 years, vitamin D has been shown to produce relevant extraskeletal effects, particularly on the cardiovascular system [1,2,3,4]. These data are supported by preclinical experiments, claiming that vitamin D receptor and enzymes modulating vitamin D actions are expressed in the heart. Moreover, 1,25-dihydroxy-vitamin D (1,25(OH)2D) or calcitriol, the active vitamin D metabolite, exerts interesting effects on endothelial cells, vascular smooth cells, macrophages, and cardiomyocytes [5]. Clinical evidence also supports the link between vitamin D and cardiovascular system: low levels of 25-hydroxy-vitamin D (25OHD), the biomarker of vitamin D status, are associated with increased risk of cardiovascular diseases [6] including hypertension, coronary artery disease, ischemic heart disease, heart failure (HF), and stroke [7,8,9,10,11]. On the other hand, the beneficial effect of vitamin D replacement has not been demonstrated so far in randomized-controlled trials (RCTs) and Mendelian randomization studies, so that the meaning of the association between a low vitamin D status and cardiovascular diseases remains to be established.
The aim of this paper is to summarize the most recent evidences about the role of vitamin D on cardiovascular outcomes, including pre-clinical data, association studies and available results from randomized clinical trials, and Mendelian randomization studies.
Cardiovascular effects of 1,25(OH)2D
In the early 1980s, Robert Scragg proposed the hypothesis that the increase in cardiovascular diseases occurring in winter might be the consequence of low serum 25OHD levels, due to the reduced efficacy of sunlight radiations during that season [12]. This idea turned on a great interest in the potential cardiovascular benefits of vitamin D supplementation, leading to several publications in this field. However, the physiological role of vitamin D in the cardiovascular system is still unclear.
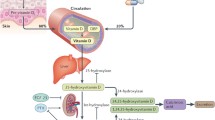
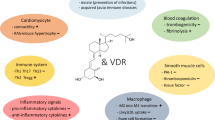
1,25(OH)2D binds to the specific nuclear receptor VDR, which is expressed in rat and human heart and has a potential role as a modulator of cardiac hypertrophy and failure. 1,25(OH)2D is also directly involved in calcium-dependent cellular processes, including synthesis of calcium-binding proteins, activation of adenylate cyclase, rapid activation of voltage-dependent calcium channels, and sarcoplasmic reticulum calcium uptake and release. Notably, altered intracellular handling of ionized calcium has been shown to be a major determinant of contractile impairment in patients with HF [13] (Fig. 1).

Summary of the known effects of Vitamin D on cardiomyocytes and vasculature, as well as that of its systemic effects on cardiovascular risk factors
Vascular effects of 1,25(OH)2D have also been demonstrated; indeed, 1,25(OH)2D can modulate the growth of smooth cells and endothelial cells [14]. In endothelial cells, 1,25(OH)2D also directly stimulates nitric oxide (NO) synthase [15] and inhibits COX 1 [16], leading to vasodilation. Moreover, 1,25(OH)2D may reduce oxidative stress decreasing superoxide production in the vascular wall [17] (Fig. 1).
Systemic effects of vitamin D on blood pressure can be explained by the role of 1,25(OH)2D as a negative regulator of the renin-angiotensin system (RAS) (Fig. 1). Indeed, in vitro studies demonstrated that 1,25(OH)2D inhibits the cAMP responsive elements in the renin promotor gene, decreasing renin gene expression [18]. These data are supported by the other recent results, showing that several nuclear receptors, including VDR, liver X receptor (LXR), and peroxisome proliferators-activated receptor (PPAR), regulate renin expression via specific elements in the renin promoter [13].
Strong support for the involvement of vitamin D in the pathogenesis of cardiovascular diseases comes from observations performed in vitamin D receptor knockout mice (VDR–/–). These mice develop typical signs of HF, including activation of the RAS system, cardiac hypertrophy, high blood pressure, and increased levels of atrial natriuretic peptide. This phenotype is not modified by restoring serum calcium levels through a calcium-rich diet, suggesting a profound alteration in the endocrine system [2]. Furthermore, the development of hypertension in VDR–/– mice can be corrected by administration of ACE inhibitors, but only as long as vitamin D levels are sufficient [1,2,3].
Additional, indirect endocrine effects of vitamin D on the cardiovascular system may be mediated troughs non-specific anti-inflammatory and immunomodulating actions. 1,25(OH)2D has been shown to reduce the expression of pro-inflammatory genes (TNFα, IL1, and IL6) [19] and to stimulate anti-inflammatory genes (IL10) (Fig. 1). Moreover, 1,25(OH)2D inhibits TH1-17 cells and affects dendritic cell functions [20].
One more aspect to be considered is the potential role of secondary hyperparathyroidism in the setting of vitamin D deficiency. Indeed, a prevalence of 10–33% of secondary hyperparathyroidism is reported among patients with hypovitaminosis D and high PTH levels have been associated with hypertension, arrhythmias, and vascular calcifications [21,22,23]. Further studies are needed to clarify the specific role of calcitriol and PTH on cardiovascular outcomes in this setting.
Observational studies
Several longitudinal cohort studies demonstrated the association of vitamin D deficiency with cardiovascular events, namely HF, stroke, myocardial infarction, cardiomyopathy, and with cardiovascular mortality. In the Framingham Heart Study, published in 2008, low serum 25OHD levels (< 15 ng/mL) were associated with 60% increase in the risk of developing cardiovascular events, even after adjustment for the most important cardiovascular risk factors [24]. Using baseline data from the Third National Health and Nutrition Examination Survey (NHANES 1988–1994), including more than 15,000 subjects, Fiscella et al. showed that low levels of serum 25OHD (< 14 ng/mL) were associated with a higher risk of cardiovascular death, particularly in black people [25]. More recently, data from NHANES 2001–2010 have been published: in a cohort of 7674 subjects, and serum levels of 25OHD > 29 ng/mL were associated with a lower risk of cardiometabolic risk compared to levels of 25OHD < 17 ng/mL, after adjusting for several confounding factors [26]. In the European study, Copenaghen City Heart, conducted on more than 10,000 subjects with a follow-up up to 29 years, the lower percentiles of 25OHD were associated with an increased risk of myocardial infarction (+ 64%) and mortality for cardiovascular diseases (+ 81%) [27]. The association between hypovitaminosis D and cardiovascular risk seemed to be valid either for men, as demonstrated by the Health Professional Follow-Up Study, and for women, as showed in a large Finnish study [28, 29]. In a small Italian study on 200 healthy women (19–50 years), serum 25OHD levels were directly correlated with serum HDL cholesterol and inversely correlated with intima–media thickness, suggesting a cardiovascular protective role for high serum 25OHD levels [30]. Among cardiovascular risk factors, obesity has a central role: several studies have been showed a clear association between low levels of vitamin D and obesity (BMI > 30 kg/m2) [31]. This association could be due to the sequestration of the lipophilic hormone in the adipose tissue [32] and/or to a complex interplay between adipocytes and vitamin D, as suggested by some authors [33]. Indeed, there are some evidences that 1,25(OH)2D can inhibit peroxisome proliferator-activated receptor γ, thus modulating adipogenesis, but still further studies are needed to address the effects of vitamin D on adipose tissue and relation between hypovitaminosis D and obesity [34].
HF is a major cardiovascular disease with a poor prognosis. In 2012, Gotsman et al. evaluated the prevalence of vitamin D deficiency in a large cohort of HF patients, and described the seasonal variation in serum 25OHD levels and the impact of vitamin D deficiency and supplementation on mortality. They demonstrated that serum 25OHD D levels are low in the general population and even lower in patients with HF, and that vitamin D deficiency was a significant predictor of reduced survival. Moreover, vitamin D supplementation was associated with an improved outcome in terms of mortality [35]. We recently evaluated an Italian cohort of 261 patients with HF and showed that low serum levels of 25OHD are inversely correlated with the Metabolic Exercise Cardiac Kidney Index (MECKI), a validated score of mortality [36]. Moreover, in a subgroup of these patients, we also evaluated cardiovascular outcomes: patients with HF had mean serum levels of 25OHD statistically lower than healthy subjects (45.2 ± 23.7 nmol/L vs 58.2 ± 24.0 nmol/L, P < 0.001) and a higher prevalence of vitamin D insufficiency (serum 25OHF < 20 ng/mL) (61.1% vs 39.5%, P < 0.001), that was associated with reduced survival [37].
Several meta-analyses published between 2012 and 2018 [7, 27, 38,39,40,41,42] summarized results obtained in a large number of subjects, ranging from 26,916 in the study of Gaksch et al. [42] to more than 180,000 in the study of Zhang et al. [41]. All meta-analyses confirmed a consistent association between a low vitamin D status and cardiovascular endpoints (myocardial infarction, hypertension, and HF) and/or mortality.
Table 1 summarizes the results of the most important observational studies.
Randomized clinical trials (RCT) and vitamin D supplementation in cardiovascular diseases
As discussed above, observational studies show a clear association between a low vitamin D status and cardiovascular diseases, but this is not sufficient to establish a causal relationship. RCTs are required to prove the benefit of vitamin D supplementation on cardiovascular outcomes. Several RCTs are available in the literature, but most of them were not designed to evaluate extra-skeletal endpoints, and their interpretation is not simple.
The Women’ Health Initiative, designed for skeletal primary endpoints, also considered cardiovascular events as secondary endpoints. In this study, more than 36,000 women were randomized to 400 UI of vitamin D plus 1000 mg calcium daily or placebo: there was no difference between the two arms, in term of coronary events and stroke [43]. Subsequently, an analysis of the same database, including only women not already taking vitamin D and calcium at the baseline, found an increased risk for cardiovascular events in the supplementation arm [44]. In the Cochrane review, vitamin D supplementation significantly reduced all-cause mortality compared with placebo or no intervention, but had no significant effect on cardiovascular mortality (risk ratio 0.98, 95% CI 0.90–1.07; n = 47,267) [45].
The VIDA study is a randomized, double-blind, placebo-controlled trial conducted in New Zealand. Adult patients were randomized to cholecalciferol 100,000 UI/monthly or placebo: the results after 3 years of follow-up have recently been published, showing no beneficial effect of vitamin D supplementation on any cardiovascular endpoint [46].
A meta-analysis focused on HF and vitamin D status showed that vitamin D supplementation was associated with a decrease of serum levels of PTH, TNFα, and CRP, even if the beneficial effects on left-ventricular function and exercise tolerance were limited [47].
The RECORD study included 5260 participants and concluded that the daily supplementation with 800 UI of cholecalciferol was protective against HF [HRs 95% = 0.75 0.75 (0.58, 0.97)] but not against myocardial infarction or stroke [HR = 0.97 (0.75,1.26), and 1.06 (0.8, 1.32) respectively, 48]. Shleithoff et al. studied a cohort (n = 123) of patients with HF, randomized to daily supplementation with 2000 IU vitamin D (n = 61) or placebo (n = 62) for 9 months, and observed decreased proinflammatory cytokines in the active group, without changes in ventricular function and biochemical markers (LV ejection fraction, LV end-diastolic diameter, VO2max, NT-proBNP) [4]. Using the same dosage, Schroten et al. described a significant decrease of PRA and plasma renin concentration in patients with HF [49]. Dalbeni et al. found that 6 months of vitamin D supplementation significantly improved ejection fraction in elderly patients with HF [50] and Shedeed et al. reported that young patients with HF achieved marked improvement in both cardiac function and inflammatory markers (left-ventricular end-diastolic diameter, LV end-systolic diameter, LV ejection fraction, myocardial performance index, interleukin-10, PTH, interleukin-6, and TNFa) after 12 months of vitamin D analogues [51].
The VINDICATE study, a double-blind, placebo-controlled, trial in HF patients randomized to 4000 IU of vitamin D/daily or placebo for 12 months has shown a statistically significant improvement of left-ventricle (LV) ejection fraction, dimension, and volume in the supplemented group, without any change in the 6-min walk test, that was the primary end-point [52].
Overall, the evidence about the benefit of vitamin D supplementation on cardiovascular health is not conclusive. This may be accounted for by substantial limitations in the experimental design: cardiovascular diseases were not the principal focus of these trials, and in some cases, the cardiovascular endpoints were not pre-specified; different dosage and different preparations of vitamin D were used; serum 25OHD was not always measured at the baseline and/or in the follow-up.
Table 1 summarizes the results of RCTs exploring the effects of vitamin D supplementation on cardiovascular events and/or mortality.
Mendelian randomization studies
Mendelian randomization studies could help to understand the role of vitamin D in cardiovascular diseases, because, with this approach, the effects of the genetic-dependent lifelong vitamin D status are analysed, with limited confounding lifestyle factors. In the Copenaghen Study by Brøndum-Jacobsen et al. 92,416 participants were genotyped using SNPs in the DCHR7 and in the CYP2R1 genes. In that cohort, low serum levels of 25OHD were associated with an increased risk of ischemic heart disease, but there was no evidence that the ischemic risk was due to genetically reduced serum 25OHD levels [53]. Similar conclusions were reached in a large (30,000 subjects) Canadian study, concerning the association of genetically low serum 25OHD levels and an increased risk of coronary artery diseases [54]. At variance, a meta-analysis summarizing the results of 146,581 individuals to evaluate the association between genetically determined 25OHD levels and blood pressure found that increased serum levels of 25OHD were associated with a lower risk of hypertension [55]. Finally, Ooi et al. reported that genetically elevated non-fasting remnant cholesterol was associated with low serum 25OHD, and suggested that vitamin D deficiency could be a marker of the individual atherogenic setting [56].
Conclusions
It is still controversial whether a low vitamin D status has a causative role in cardiovascular diseases and whether vitamin D supplementation could be of benefit. Despite a strong association between hypovitaminosis D and poor cardiovascular outcomes, Mendelian randomization studies do not support a causative role and interventional investigation has not definitely proved a beneficial effect. Vitamin D deficiency could simply be a general marker of poor health in patients with chronic cardiovascular diseases. Many methodological issues are, however, still open, particularly the accuracy of serum 25OHD assays, the cut-off values used to define vitamin D deficiency, and the adequacy of the dosages and reparations used as vitamin D supplementation. Large ongoing RCTs, specifically designed to evaluate vitamin D effects on cardiovascular end-points, will hopefully shed light on the current uncertainties and controversies.
References
Li YC, Kong J, Wei M et al (2002) 1,25-Dihydroxyvitamin D3 is a negative endocrine regulator of the renin–angiotensin system. J Clin Invest 110:229–238. https://doi.org/10.1172/JCI200215219
Li YC (2003) Vitamin D regulation of the renin–angiotensin system. J Cell Biochem 88:327–331
Rahman A, Hershey S, Ahmed S et al (2007) Heart extracellular matrix gene expression profile in the vitamin D receptor knockout mice. J Steroid Biochem Mol Biol 103:416–419. https://doi.org/10.1016/j.jsbmb.2006.12.081
Schleithoff SS, Zittermann A, Tenderich G et al (2006) Vitamin D supplementation improves cytokine profiles in patients with congestive heart failure: a double-blind, randomized, placebo-controlled trial. Am J Clin Nutr 83:754–759
Wimalawansa SJ (2018) Vitamin D and cardiovascular diseases: causality. J Steroid Biochem Mol Biol 175:29–43. https://doi.org/10.1016/j.jsbmb.2016.12.016
Skaaby T, Husemoen LLN, Pisinger C et al (2013) Vitamin D status and incident cardiovascular disease and all-cause mortality: a general population study. Endocrine 43:618–625. https://doi.org/10.1007/s12020-012-9805-x
Wang L, Song Y, Manson JE et al (2012) Circulating levels of 25Hydroxy-vitamin D and risk of cardiovascular disease: a meta-analysis of prospective studies. Circ Cardiovasc Qual Outcomes 5:819–829. https://doi.org/10.1161/CIRCOUTCOMES.112.967604.Circulating
Lugg ST, Howells PA, Thickett DR (2015) Optimal Vitamin D supplementation levels for cardiovascular disease protection. Dis Mark. https://doi.org/10.1155/2015/864370
Judd SE, Tangpricha V (2009) Vitamin D deficiency and risk for cardiovascular disease. Am J Med Sci 338:40–44. https://doi.org/10.1097/MAJ.0b013e3181aaee91
Kestenbaum B, Katz R, De Boer I et al (2011) Vitamin D, parathyroid hormone, and cardiovascular events among older adults. JAC 58:1433–1441. https://doi.org/10.1016/j.jacc.2011.03.069
Muscogiuri G, Nuzzo V, Gatti A et al (2015) Hypovitaminosis D: a novel risk factor for coronary heart disease in type 2 diabetes? Endocrine. https://doi.org/10.1007/s12020-015-0609-7
Scragg R (1981) Seasonality of cardiovascular disease mortality and the possible protective effect of ultra-violet radiation. Int J Epidemiol 10:337–341
Wang TJ (2016) Vitamin D and cardiovascular disease. Annu Rev Med 67:261–272. https://doi.org/10.1146/annurev-med-051214-025146
Wu-Wong JR, Nakane M, Ma J et al (2006) Effects of Vitamin D analogs on gene expression profiling in human coronary artery smooth muscle cells. Atherosclerosis 186:20–28. https://doi.org/10.1016/j.atherosclerosis.2005.06.046
Andrukhova O, Slavic S, Zeitz U et al (2014) Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol Endocrinol. https://doi.org/10.1210/me.2013-1252
Wong MSK, Delansorne R, Man RYK, Vanhoutte PM (2008) Vitamin D derivatives acutely reduce endothelium-dependent contractions in the aorta of the spontaneously hypertensive rat. AJP Hear Circ Physiol. https://doi.org/10.1152/ajpheart.00116.2008
Hirata M, Serizawa KI, Aizawa K et al (2013) 22-Oxacalcitriol prevents progression of endothelial dysfunction through antioxidative effects in rats with type 2 diabetes and early-stage nephropathy. Nephrol Dial Transplant. https://doi.org/10.1093/ndt/gfs536
Yuan W, Pan W, Kong J et al (2007) 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem. https://doi.org/10.1074/jbc.M705495200
Aihara KI, Azuma H, Akaike M et al (2004) Disruption of nuclear vitamin D receptor gene causes enhanced thrombogenicity in mice. J Biol Chem 279:35798–35802. https://doi.org/10.1074/jbc.M404865200
Harvey NC, Cantorna MT (2013) Vitamin D and the immune system. Diet Immun Inflamm. https://doi.org/10.1533/9780857095749.2.244
Cipriani C, Pepe J, Colangelo L, Minisola S (2018) Vitamin D and secondary hyperparathyroid states. Front Horm Res 50:138–148. https://doi.org/10.1159/000486077
Yu N, Donnan PT, Flynn RWV et al (2010) Increased mortality and morbidity in mild primary hyperparathyroid patients. The Parathyroid Epidemiology and Audit Research Study (PEARS). Clin Endocrinol (Oxf). https://doi.org/10.1111/j.1365-2265.2009.03766.x
Pepe J, Diacinti D, Fratini E et al (2016) High prevalence of abdominal aortic calcification in patients with primary hyperparathyroidism as evaluated by Kauppila score. Eur J Endocrinol. https://doi.org/10.1530/EJE-15-1152
Wang TJ, Pencina MJ, Booth SL et al (2008) Vitamin D deficiency and risk of cardiovascular disease. Circulation 117:503–511. https://doi.org/10.1161/CIRCULATIONAHA.107.706127
Fiscella K, Franks P (2010) Vitamin D, race, and cardiovascular mortality: findings from a national US sample. Ann Fam Med 8:11–18. https://doi.org/10.1370/afm.1035.2
Al-Khalidi B, Kimball SM, Rotondi MA, Ardern CI (2017) Standardized serum 25-hydroxyvitamin D concentrations are inversely associated with cardiometabolic disease in US adults: a cross-sectional analysis of NHANES, 2001–2010. Nutr J. https://doi.org/10.1186/s12937-017-0237-6
Brøndum-Jacobsen P, Benn M, Jensen GB, Nordestgaard BG (2012) 25-Hydroxyvitamin D levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arterioscler Thromb Vasc Biol. https://doi.org/10.1161/ATVBAHA.112.248039
Giovannucci E, Liu Y, Hollis BW, Rimm EB (2008) 25-Hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 168:1174–1180. https://doi.org/10.1001/archinte.168.11.1174
Kilkkinen A, Knekt P, Aro A et al (2009) Vitamin D status and the risk of cardiovascular disease death. Am J Epidemiol 170:1032–1039. https://doi.org/10.1093/aje/kwp227
Giovinazzo S, Alibrandi A, Campennì A et al (2017) Correlation of cardio-metabolic parameters with vitamin D status in healthy premenopausal women. J Endocrinol Invest 40:1337–1343. https://doi.org/10.1007/s40618-017-0707-x
Vanlint S (2013) Vitamin D and obesity. Nutrients. https://doi.org/10.3390/nu5030949
Wortsman J, Matsuoka LY, Chen TC et al (2000) Decreased bioavailability of vitamin D in obesity. Am J Clin Nutr 72:690
Cipriani C, Pepe J, Piemonte S et al (2014) Vitamin D and its relationship with obesity and muscle. Int J Endocrinol. https://doi.org/10.1155/2014/841248
Lương KVQ, Nguyễn LTH (2013) The beneficial role of vitamin D in obesity: possible genetic and cell signaling mechanisms. Nutr J. https://doi.org/10.1186/1475-2891-12-89
Gotsman I, Shauer A, Zwas DR et al (2012) Vitamin D deficiency is a predictor of reduced survival in patients with heart failure; Vitamin D supplementation improves outcome. Eur J Heart Fail 14:357–366. https://doi.org/10.1093/eurjhf/hfr175
Saponaro F, Marcocci C, Zucchi R et al (2017) Hypovitaminosis D in patients with heart failure: effects on functional capacity and patients’ survival. Endocrine. https://doi.org/10.1007/s12020-017-1282-9
Saponaro F, Saba A, Frascarelli S et al (2018) Vitamin D measurement and effect on outcome in a cohort of patients with heart failure. Endocr Connect. https://doi.org/10.1530/EC-18-0207
Autier P, Boniol M, Pizot C, Mullie P (2014) Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. https://doi.org/10.1016/S2213-8587(13)70165-7
Tomson J, Emberson J, Hill M et al (2013) Vitamin D and risk of death from vascular and non-vascular causes in the Whitehall study and meta-analyses of 12,000 deaths. Eur Heart J. https://doi.org/10.1093/eurheartj/ehs426
Zhou R, Wang M, Huang H et al (2018) Lower vitamin D status is associated with an increased risk of ischemic stroke: a systematic review and meta-analysis. Nutrients. https://doi.org/10.3390/nu10030277
Zhang R, Li B, Gao X et al (2017) Serum 25-hydroxyVitamin D and the risk of cardiovascular disease: dose–response meta-analysis of prospective studies 1–3. Am J Clin Nutr. https://doi.org/10.3945/ajcn.116.140392
Gaksch M, Jorde R, Grimnes G et al (2017) Vitamin D and mortality: individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLoS ONE. https://doi.org/10.1371/journal.pone.0170791
Hsia J, Heiss G, Ren H et al (2007) Calcium/vitamin D supplementation and cardiovascular events. Circulation 115:846–854. https://doi.org/10.1161/CIRCULATIONAHA.106.673491
Bolland MJ, Grey A, Avenell A et al (2011) Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women’s Health Initiative limited access dataset and meta-analysis. BMJ. https://doi.org/10.1136/bmj.d2040
Bjelakovic G, Gluud LL, Nikolova D et al (2014) Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev 1:CD007470
Scragg R, Stewart AW, Waayer D et al (2017) Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study: a randomized clinical trial. JAMA Cardiol. https://doi.org/10.1001/jamacardio.2017.0175
Jiang W-L, Gu H-B, Zhang Y-F et al (2015) Vitamin D supplementation in the treatment of chronic heart failure: a meta-analysis of randomized controlled trials. Clin Cardiol. https://doi.org/10.1002/clc.22473
Ford JA, MacLennan GS, Avenell A et al (2014) Cardiovascular disease and vitamin D supplementation: trial analysis, systematic review, and meta-analysis. Am J Clin Nutr. https://doi.org/10.3945/ajcn.113.082602
Schroten NF, Ruifrok WPT, Kleijn L et al (2013) Short-term vitamin D3 supplementation lowers plasma renin activity in patients with stable chronic heart failure: an open-label, blinded end point, randomized prospective trial (VitD-CHF trial). Am Heart J. https://doi.org/10.1016/j.ahj.2013.05.009
Dalbeni A, Scaturro G, Degan M et al (2014) Effects of six months of vitamin D supplementation in patients with heart failure: a randomized double-blind controlled trial. Nutr Metab Cardiovasc Dis 24:861–868. https://doi.org/10.1016/j.numecd.2014.02.015
Shedeed SA (2012) Vitamin D supplementation in infants with chronic congestive heart failure. Pediatr Cardiol 33:713–719. https://doi.org/10.1007/s00246-012-0199-6
Witte KK, Byrom R, Gierula J et al (2016) Effects of vitamin D on cardiac function in patients with chronic HF: the VINDICATE study. J Am Coll Cardiol. https://doi.org/10.1016/j.jacc.2016.03.508
Brøndum-Jacobsen P, Benn M, Afzal S, Nordestgaard BG (2015) No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: a Mendelian randomization study. Int J Epidemiol. https://doi.org/10.1093/ije/dyv078
Manousaki D, Mokry LE, Ross S et al (2016) Mendelian randomization studies do not support a role for vitamin d in coronary artery disease. Circ Cardiovasc Genet. https://doi.org/10.1161/CIRCGENETICS.116.001396
Vimaleswaran KS, Cavadino A, Berry DJ et al (2014) Association of vitamin D status with arterial blood pressure and hypertension risk: a mendelian randomisation study. Lancet Diabetes Endocrinol. https://doi.org/10.1016/S2213-8587(14)70113-5
Ooi EM, Afzal S, Nordestgaard BG (2014) Elevated remnant cholesterol in 25-hydroxyvitamin D deficiency in the general population mendelian randomization study. Circ Cardiovasc Genet. https://doi.org/10.1161/CIRCGENETICS.113.000416
Funding
No funding.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval
This manuscript is a review of the literature and does not contain original research either on animal or on human subjects.
Informed consent
For this type of manuscript, informed consent is not required.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Saponaro, F., Marcocci, C. & Zucchi, R. Vitamin D status and cardiovascular outcome. J Endocrinol Invest 42, 1285–1290 (2019). https://doi.org/10.1007/s40618-019-01057-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-019-01057-y