
Abstract
Vitamin D is necessary for bone health but may also have many extra-skeletal effects. The vitamin D endocrine system has major effects on gene and protein expression in many cells and tissues related to the cardiovascular system. In addition, many preclinical studies in animals with vitamin D deficiency or genetically silenced expression of the vitamin D receptor or vitamin D metabolizing enzymes suggest that the absence of vitamin D action may result in cardiovascular events. This includes dysfunctions of endothelial cells, thereby accelerating the process of atherosclerosis, hypertension or abnormal coagulation, ultimately resulting in higher risks for all major cardiovascular or cerebrovascular events. A wealth of observational studies in different parts of the world have fairly consistently found a strong association between a poor vitamin D status and surrogate markers or hard cardiovascular events. A few Mendelian randomization studies did, however, not find a link between genetically lower serum 25OHD concentrations and cardiovascular events. Finally, many RCTs could not demonstrate a consistent effect on surrogate markers, and a limited number of RCTs did so far not find whatever effect on hard cardiovascular endpoints such as myocardial ischemia or infarction, stroke, or cardiovascular death. In conclusion, preclinical data generated a plausible hypothesis of a link between vitamin D status and extra-skeletal events, including cardiovascular endpoints. Whether the vitamin D endocrine system is redundant for the human vascular system or whether the RCTs have not been optimally designed to answer the research question is thus not yet settled.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Osteoporosis is a frequent disease of the elderly ultimately resulting in major fractures in about a third of all women and somewhat less in men. Cardiovascular events (cardiac ischemia and infarction, stroke, and peripheral arterial diseases) are also affecting a very large percentage of all older men and women. Vitamin D deficiency is also very frequent in old age. Therefore, it is no surprise that these three conditions affect the same elderly subjects. The question is whether this is just because of coincidence or whether one disease or condition is affecting the others. Osteoporosis and cardiovascular diseases may have frequent common pathogenic mechanisms such as oxidative stress [1], sex steroid deficiency, or chronic inflammation. Vitamin D deficiency is also frequently found in both diseases. Therefore, the question whether vitamin D deficiency may accelerate both diseases is important, as prevention of vitamin D deficiency is feasible at low risk and low cost. In this review, I will briefly discuss the association between osteoporosis and cardiovascular diseases and mainly focus on whether vitamin D deficiency (or excess) may accelerate or aggravate cardiovascular diseases.
Several cross-sectional, retrospective, and prospective studies have found an association between osteoporosis and cardiovascular diseases [2]. Indeed, carotid wall thickness [3, 4] or coronary artery calcium burden [5] were higher in patients with osteoporosis compared with controls. Similarly, the risk of coronary artery stenosis was higher in patients with osteoporosis than in controls [6]. Inversely, the presence of osteoporosis significantly increased the risk of coronary artery disease in Caucasians [7] as well as in Koreans [8]. Two independent studies found an inverse correlation between BMD and either ischemic heart disease [9] or silent brain infarction [10]. Males with a previous history of myocardial infarction also had a lower BMD compared to healthy subjects [11]. Similarly, the prevalence of cardiovascular diseases was much higher in women with osteoporosis than in subjects with a normal BMD [12]. The strongest arguments for a link between osteoporosis and cardiovascular diseases come from prospective studies. A selection of relevant studies is shown in Table 1. The overall conclusion is that low BMD, osteopenia, or osteoporosis is associated with higher future risks of cardiovascular events (stroke, myocardial infarction) or that the presence of peripheral arterial disease or aortic calcifications increases future loss of BMD or higher risk of vertebral fractures. A systematic review and meta-analysis of 25 observational and comparative studies of vascular abnormalities and decreased BMD dealing with 10,299 subjects concluded that the OR of atherosclerotic vascular abnormalities was significantly 2.23-fold higher in patients with a low BMD and that the risk was higher if BMD was lower [24].
A poor vitamin D status is associated with increased bone turnover, higher bone loss, and ultimately higher risk of osteoporosis and fractures [25]. Vitamin D deficiency is also associated with many extra-skeletal diseases including vascular risks and events [26,27,28]. In view of the above-described association between bone loss and cardiovascular diseases, it is logical to evaluate the possible role of vitamin D deficiency as one of the pathogenic mechanisms explaining the joint risk of both diseases in older age.
Preclinical data linking the vitamin D endocrine system with cardiovascular system
There are several excellent reviews dealing with various aspects of the action of the vitamin D endocrine system on the cardiovascular system, and the implications thereof for cardiovascular risk factors and events [26, 29,30,31,32].
Vitamin D and endothelial cells
The vitamin D receptor and the enzymes necessary for the activation of 25OHD into 1,25(OH)2D or their inactivation into 24R-hydroxylated metabolites are expressed in several cells of the vascular wall such as the endothelial cells and vascular smooth muscle cells [33]. In addition, these cells respond to the presence of 1,25(OH)2D by regulation of a number of crucial genes with a favorable overall effect (Table 2). The mechanisms involved include the local production of a potent vasodilator, nitric oxide, or NO, by stimulation of the endothelial inducible NO synthetase [34]. At the same time, 1,25(OH)2D decreases the production of vasoconstrictors by inhibition of COX-1 [35]. 1,25(OH)2D also decreases the production of superoxides in the vascular wall by inhibition of p22(phox) and NADPH oxidase subunit, thereby decreasing the production of H2O2 [36]. Sirtuin-1 is also stimulated by 1,25(OH)2D, or at least 1,25(OH)2D can block the downregulation of Sir-1 by H2O2. This strengthens its inhibitory effects on oxidative stress and atherogenesis (Table 2). Finally, 1,25(OH)2D inhibits apoptosis of endothelial cells in vitro, by inhibition of oxidative stress, inhibition of release of cytochrome C from mitochondria, inhibition of caspase activity, and apoptosis/autophagy-related genes [37] (Table 2).
All these in vitro effects are difficult to reproduce in vivo in animals or humans, but one study demonstrated that 1,25(OH)2D can promote the re-endothelialization of the carotic artery after experimental injury in diabetic mice [35]. Selective deletion of Vdr in endothelial cells increased the sensitivity of the vascular wall to angiotensin-2, thereby increasing systemic blood pressure [40].
Vitamin D and cardiac muscle/heart
Cardiomyocytes are responsive to 1,25(OH)2D as shown in vitro by increased calcium uptake and increased contractility and improved relaxation [47, 48] (Table 3). Older ex vivo studies in hearts from vitamin D-deficient chicks showed more rapid myocardial dysfunction and decreased ATP storage compared with hearts from vitamin D replete chickens [54] (Table 3). Similarly, hearts from vitamin D-deficient rats showed cardiomegaly but not due to larger cardiac muscle but due to expansion of the extracellular fluid and collagen content [55]. This is in line with reduced systolic function found in mice with systemic Cyp27b1 deletion [53] (Table 3).
1,25(OH)2D also suppressed Rcan1 expression in neonatal cardiomycytes. This gene codes for a calcineurin inhibitory protein, calcipressin-1. This action on the calcineurin/NFAT/Rcan1 pathway may explain its antihypertrophic effects [56]. Indeed, Vdr deletion in cardiomyocytes causes myocardial hypertrophy and fibrosis [56]. These hearts also show a prolonged fetal gene program with increased atrial natriuretic peptide and actin expression, very similar to the prolonged immature gene profile found in skeletal muscle of global Vdr null mice [57].
Vitamin D and systemic effects related to the cardiovascular system
Vdr or Cyp27b1 mice develop systemic hypertension mainly mediated by activation of the renin-angiotensin (RAS) system [52, 58, 59]. These mice have higher (renal) renin expression compared to wild-type mice with therefore higher serum concentrations of angiotensin II and aldosterone. These effects can be reversed in cyp27b1 null mice by administration of 1,25(OH)2D. In Vdr null mice, an angiotensin blocker can correct the systemic hypertension [52]. This phenotype cannot be corrected by a rescue diet normalizing serum calcium homeostasis, indicating that it is the direct consequence of the failure of the vitamin D endocrine system. In vitro studies have clearly shown that the renin gene is under the negative control of VDR by blocking the activity of the cAMP responsive element in the renin promotor [60]. Vitamin D-deficient rats also develop hypertension, independent of hypocalcemia and correctable by 1,25(OH)2D or a low calcemic vitamin D analog [61].
Vdr null mice display increased (stimulated) thrombogenicity and impaired fibrinolysis [62]. The in vitro data on thrombogenicity were largely confirmed in normocalcemic vdr null mice, raised on a rescue diet [62]. These mice indeed showed increased platelet aggregation, reduced NO synthetase expression, and decreased liver expression of antithrombin and decreased expression of thrombomodulin in several tissues. After lipopolysaccharide injection, the vdr null mice showed exacerbated multi-organ thrombus formation [62].
The vitamin D endocrine system also has important anti-inflammatory and immune effects with indirect repercussions on the cardiovascular system. In vitro studies of dendritic cells, macrophages, or immune cells in general have shown that 1,25(OH)2D decreases the expression of pro-inflammatory genes (e.g., IL1, IL6, IL23, TNFα, and IFNy), while upregulating anti-inflammatory genes (e.g., IL4 and IL10) [63,64,65]. In addition, 1,25(OH)2D is a potent inhibitor of Th1–17 cells and modifies the phenotype of dendritic and related cells into more tolerogenic pathways, including an increase in T-reg cells [65], thereby probably decreasing the risk of atherosclerosis [31].
The effects of the vitamin D system on serum lipids are more complex. Rats with transgenic generalized overexpression of Cyp24a1 develop a remarkable phenotype of high serum lipids (all lipids) and accelerated atherosclerotic lesions in the aorta. This phenotype can be greatly enhanced by a high-fat high-cholesterol diet. These rats also have marked albuminuria [66]. The pathogenic mechanism is unknown, but serum 1,25(OH)2D was not altered and despite the transgenic expression of Cyp24a1, serum 24,25(OH)2D concentration was lower than in controls. Other studies linking the vitamin D system with serum lipids and adipocyte development generated variable results. Mice with deletion of vdr or cyp27b1 are lean and resistant to diet-induced obesity due to enhanced energy expenditure, oxygen consumption, and higher than normal uncoupling protein expression [67]. In humans, however, a low vitamin D status is fairly consistently associated with obesity and this species difference remains unexplained. In vitro effects of 1,25(OH)2D on fat cell metabolism and development also generated inconsistent results [67].
In a cross-sectional study of healthy adults, a strong positive correlation was found between serum 25OHD and HDL or apolipoprotein A-1 (p < 0.001), whereby variations in serum 25OHD may explain up to 10% of the variation of these lipid concentrations. Similarly, a 6-week treatment with intravenous 1,25(OH)2D of patients with chronic renal failure resulted in a significant increase in serum apoprotein A1 [68]. This is in strong contrast with a study reporting a negative regulation of the apolipoprotein A-1 by 1,25(OH)2D in a human hepatoma cell line [69].
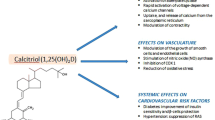
From this overview, it is clear that the vitamin D endocrine system has an effect on many genes and proteins, cells and tissues involved in cardiovascular physiology. Overall, the VDR action seems to be beneficial for optimal cardiovascular function (Fig. 1). 1,25(OH)2D stimulates in vascular smooth muscle cells the production of matrix gla protein, a potent inhibitor of extraskeletal calcification, and thus could potentially have a favorable effect [70]. In vivo, however, excess vitamin D (whether too high 1,25(OH)2D or large excess of 25OHD) may stimulate the transdifferentation of smooth muscle cells into osteoblast like cells, and accelerate vascular calcification and atheromatosis [71]. This is in line with the observation that calcitriol treatment significantly increased aortic expression of the calcification genes Runx2 and Pit-1 [72]. High-dose vitamin D has been used frequently to induce severe aortic calcifications in rats and to test preventive interventions [73].

The vitamin D endocrine system and potential targets in the cardiovascular system
The majority of the symptoms due to vitamin D toxicity are related to hypercalcemia (neurological and gastrointestinal symptoms) and acute impairment of kidney function, but many children nevertheless also show signs of extraskeletal vascular calcifications [74].
Clinical data linking the vitamin D endocrine system with cardiovascular risks and events
Observational studies
A large number of observational studies found a fairly consistent association between a low vitamin D status and cardiovascular risks, hypertension, and cardiovascular events, including ischemic cardiac events, cardiomyopathy, congestive heart failure, stroke, and cardiovascular mortality [29, 30, 75,76,77,78]. Therefore, I will only discuss the main messages here without detailed overview of all individual studies. The major and best known studies are summarized in Table 4, and the major meta-analyses dealing with CV endpoints or CV mortality are summarized in Table 5.
In the Framingham cohort of 1739 subjects followed for 5.4 years, serum 25OHD concentrations below 15 ng/ml were associated with a 62% higher risk for cardiovascular diseases compared to subjects with better vitamin D status (even after correction for identifiable other risk factors) [79]. Very similar results were generated in the Copenhagen City Heart study as subjects with the lowest vitamin D status (1–4th percentile versus those in percentile 50–100) had a 64% higher risk of myocardial infarction or fatal CV event during a 29-years follow-up [82]. Similar results on the risk of myocardial infarction were found in the Health Professional Follow up Study of more than 18,000 subjects: Those with serum 25OHD below 15 ng/ml (measured or calculated) versus > 30 ng/ml had a 2.09-fold higher risk [81]. In the NHANES III study, subjects with serum 25OHD below 12 ng/ml, followed for 18 years, had a 36% higher risk of CV mortality compared with subjects with serum 25OHD between 20 and 30 ng/ml [83]. Also incident hypertension is higher in men as well as in women with baseline normotension and serum 25OHD below 15 ng/ml compared with the ones with serum 25OHD above 30 ng/ml. The overall RR of hypertension during a 4–18 years follow-up period associated with low serum 25OHD was 3.18 and highly significant [80] (Table 4). In a multi-ethnic study, a poor vitamin D status was associated with higher risks of all types of CV diseases in Whites and Chinese but not in Blacks and Hispanics [85]. Using the large Intermountain Healthcare system of a general healthcare population, Anderson et al. [78] found a strong association between serum 25OHD and cardiovascular risks and events (Table 4). This was highly significant whether vitamin D deficiency was defined as serum 25OHD below 30 ng/ml or as serum 25OHD between 16 and 30 ng/ml. Based on NHANES III data on more than 15,000 US adults followed for 9 years, the subjects with the lowest quartile of serum 25OHD (< 14 ng/ml) displayed a higher risk of cardiovascular death in comparison with all others subjects (Black or whites) [88]. Kerstenbaum looked at a lower number of subjects without cardiovascular disease at baseline; over a follow-up of 14 years, those with lower 25OHD concentrations (< 10 ng/ml) had a higher risk for cardiovascular death and myocardial infarction [89] (Table 4).
Numerous other studies, mostly cited in the meta-analyses (Table 5), also reported similar results. Of course, a few studies also generated null results [96, 97]. The main recent meta-analyses on the association of vitamin D status and cardiovascular events are summarized in Table 5. Most analyses dealt with a very large number of subjects as described in the original studies. None of the meta-analyses reported an analysis of individual patient data (only mean study data). Overall, the data are simple to summarize: Subjects with the lowest vitamin D status (whether expressed as percentiles or quartiles or on a more linear basis) have the highest risk for whatever cardiovascular endpoint—either overall cardiovascular disease, myocardial infarction, ischemic heart diseases, ischemic stroke, hypertension, or cardiovascular mortality. The effect size is relatively large as RR in subjects with the lowest vitamin D status is frequently 40% higher than in the ones with a higher serum 25OHD concentration (Table 5). The highest risks are found in subjects with serum 25OHD below 10 ng/ml, and there is frequently a linear trend with higher serum 25OHD concentration. Several studies, however, also suggested that serum 25OHD concentrations above ~ 25 ng/ml do not further improve the outcome (e.g., as described in [77, 94]). Such observational data, however, are not suitable to define an absolute threshold above which no further risk reductions can be expected. In addition except for one study [90], all studies used serum 25OHD assays that were not standardized according to NIST or NIST equivalent absolute concentrations.
Conclusion
Most of the very large number of observational studies (whether cross-sectional or long-term prospective) conclude that subjects with a low vitamin D status have a higher risk of cardiovascular events. The risk increases with lower 25OHD status, but there seems to be a plateau of about 25 ng/ml above which the risks do not change significantly. As cardiovascular events are frequent and the relative risk potentially attributable to low 25OHD is high, potential gain from vitamin D supplementation, if proven, might be very substantial.
Mendelian randomization studies
Observational studies are usually considered as hypothesis generating, whereas randomized controlled trials should provide the definitive proof of causality and define strategies for prevention of diseases. However, long-term prospective studies may study a very large number of subjects (100,000) for a very long time (several decades), exceeding by far the potential scope of RCTs but still are potentially confounded by hard to correct confounding factors. Mendelian randomization studies can bring together the combination of very large study subjects and very long (~lifetime) follow-up studies while avoiding interfering other factors. Indeed, serum 25OHD concentrations are under strong hereditary control as concluded by twin studies and a large number of GWAS studies have so far identified about 7–8 genes that predispose subjects to higher or lower lifetime serum 25OHD concentrations [98]. Based on up to a limited number of these genes, related to vitamin D metabolism or transport, one can predict about 5% of the variation in serum 25OHD concentrations in Caucasians and Chinese populations [98]. Several large consortia have combined their data on gene polymorphisms involved in serum 25OHD concentrations and a variety of outcomes including cardiovascular events [28].
Two large MR studies looked at a link between genetically low serum 25OHD concentrations and cardiovascular diseases in general. A large Canadian study [99] dealing with more than 80,000 subjects and using 4 SNPs identified by the Sunlight consortium found no relationship. Similarly, in a very large European study dealing with more than 90,000 subjects and using SNPs in 2 genes could not reveal a link with CV diseases. This study is remarkable because measured serum 25OHD was linked with ischemic heart diseases: The HR for this outcome was significantly increased (1.82) in subjects with the lowest quartile of serum 25OHD when compared with the highest quartile [100]. A much smaller Canadian study using only SNPs in one gene also did not find a causal link but that may well be due to underpowering of the study [101].
One MR study revealed a positive result. Vimaleswaran [102] reported that a 10% genetically higher serum 25OHD is associated with 0.3 mmHg lower diastolic and systolic blood pressure and lower risk for hypertension. The study dealt with more than 140,000 Danish subjects and looked at only 2SNPs located in the DHCR7 and CYP2R1 genes, which code for key enzymes responsible for the metabolism of vitamin D. The study is important because of its large size in a homogenous population. The effect size is, however, small, and 10% difference in serum 25OHD is beyond the effect size of the SNPs; thus the study should be considered as supportive but not as a final proof of causality. An extension of this study as to include other SNPs with greater impact on serum 25OHD is desirable. If confirmed, it may have major implications for vitamin D supplementation policy at a population level.
One MR study describes a rather mysterious effect of fillagrin gene polymorphism on higher serum 25OHD concentrations and a better serum lipid profile [103]. They did not study known SNPs related to 25OHD status but found that fillagrin mutation or polymorphism resulted in 10% higher measured serum 25OHD (possibly related to higher UVB induced efficacy in vitamin D production) and better lipid profile (higher HDL, lower LDL and lower VLDL, and triglycerides). This effect of fillagrin on vitamin D synthesis or 25OHD status was not described before nor detected in several very large GWAS studies. Therefore, this study should be interpreted as a potentially novel pathway explaining the large variability of serum 25OHD concentrations and its relationship with serum lipids.
Finally, one large MR study also addressed a novel potential mechanism linking vitamin D status with CV events. The authors studied more than 100,000 Danish subject in search for genes explaining the variation in non-fasting remnant cholesterol and LDL/HDL concentrations [104]. They found that the polymorphisms in genes predisposing to higher cholesterol remnants were associated with lower (measured) serum 25OHD. A doubling of the cholesterol remnant concentrations implied a 15% lower serum 25OHD concentration. The study also reported that measured serum remnant cholesterol concentration (rather than geneticaly predicted) was negatively associated with serum 25OHD. The same genes predisposing for higher remnant cholesterol concentrations were already known to be associated with higher cardiovascular events and low-grade chronic inflammation [104]. Inversely, the genes known to predispose to lower serum 25OHD had no significant effect on serum lipid concentrations. If confirmed in other studies, this may imply that some genes increase cholesterol remnants, a well-known risk factor for cardiovascular diseases, and for unknown reasons, also decrease serum 25OHD, thereby creating a (potentially non-causal) link between vitamin D status and CV diseases [104]. This may also imply that lower serum 25OHD is just a surrogate marker of higher remnant cholesterol concentrations and higher CV risks without being directly and causally linked with these diseases.
Randomized controlled trials
Several randomized controlled trials have evaluated the effects of vitamin D supplementation (with our without calcium) on surrogate cardiovascular endpoints such as vascular stiffness, hypertension (diastolic and systolic blood pressure), and hard endpoints such as myocardial infarction, stroke, coronary revascularisation, cardiovascular, or cerebrovascular death.
Surrogate end points
Endothelial dysfunction can be used as a surrogate marker for more complex cardiovascular endpoints. One meta-analysis looking at flow-mediated dilation as measured by post-occlusion hyperemia did not detect a positive effect of prior vitamin D supplementation [105]. A more recent meta-analysis looking at 16 publications dealing with 1177 participants also concluded that vitamin D supplementation did not significantly improve endothelial function (except for a minor benefit in diabetic patients) [106].
Motivated by the role of the vitamin D endocrine system on renin-angiotensin, many (more than 40) RCTs have looked at the effects of vitamin D supplementation on blood pressure using very different study designs. The interpretations of several meta-analyses are not uniform. An early meta-analysis of Witham et al. [107] concluded, based on 11 RCTs, that vitamin D supplementation of subjects with mild hypertension generated a modest but significant reduction in systolic blood pressure. No effect was seen in subject who were normotensive at baseline. Similarly, Wu et al. [108] concluded, based on the results of 4 RCTs involving 429 participants, that vitamin D supplementation generated a modest reduction in systolic (− 2.5 mmHg) but not in diastolic blood pressure. Pittas [109] concluded in a meta-analyses of 10 trials that supplementation nonsignificantly reduced systolic blood pressure (weighted mean difference [WMD] − 1.9; 95% CI − 4.2, 0.4 mmHg) and did not affect diastolic blood pressure (WMD − 0.1; 95% CI − 0.7, 0.5 mmHg). Beveridge [110] evaluated 46 trial in a trial-level meta-analysis and found no effect of vitamin D supplementation on systolic or diastolic blood pressure (effect size between 0 and 0.1 mmHg). The same group also evaluated 27 trials for which individual patient (n = 3092) data were available and came to the same conclusion: no effect on blood pressure overall nor in subgroups [110].
Hard end points
Table 6 summarizes the main RCTs evaluating directly the effects of vitamin D supplementation on hard endpoints. The large Women’s Health Initiative (WHI) trial has been evaluated twice. First, including all subjects whether or not they were taking already calcium supplementation at baseline, no effect was found on the hazard ratio for myocardial infarction, death from coronary heart disease, or stroke [111]. A reanalysis of the data using only results from women randomized to either vitamin D plus calcium or double placebo excluding those that did already take such supplements at baseline revealed a significant increase of myocardial infarction, coronary revascularization, stroke, or all such cardiovascular events in women assigned to vitamin D and calcium supplements [112] (Table 6). The RECORD trial was designed to study effects on bone but also studied cardiovascular endpoints [115]. Supplementation with 800 IU of vitamin D3 (twice the amount of the WHI study) plus calcium (1 g/day) significantly decreased the risk of heart failure (HR 0.75) but without effect on myocardial infarction or stroke (Table 6). Serum 25OHD was not measured at baseline, nor in the WHI nor in the RECORD trial, and therefore the possible effects of supplementation on subjects with the most severe deficiency could not be evaluated. The most recent VITA trial studied the effect of monthly doses of 100,000 IU of vitamin D3 during 3.3 years in New Zealand adults (after a single loading dose of 200,000 IU). No effect was observed on the hazard ratio of all cardiovascular diseases (Table 6). Whether this negative effect is due to lack of effect of vitamin D in general or due to high intermittent dosing or due to the relatively good vitamin D status at baseline cannot be defined. All these studies have several or divergent limitations, such as including a limited number of subjects with poor vitamin D status at baseline, lack of measurement of serum 25OHD in some, or even all subjects, and many studies used methods for serum 25OHD that lack validation of accuracy.
Several authors have tried repeatedly to evaluate the effects of vitamin D (with or without calcium) supplementation on cardiovascular endpoint by analyzing all existing data by systematic meta-analysis (Table 7).
Pittas [109] concluded that 4 RCTs separately or combined could not demonstrate an effect on cardiovascular outcome. Bolland summarized the results of 3 RCTs [112] and, 3 years later, again of 40 RCTs [114]. From the first meta-analysis, they concluded that vitamin D and calcium negatively influenced major vascular events (myocardial infarction and stroke), but the larger meta-analysis did not show positive nor negative effects (Table 7). Four other meta-analyses [113, 115,116,117] (Table 7) selected different numbers of RCTs, but all concluded that vitamin D supplementation (with or without calcium) did not significantly influence several vascular events.
General discussion and conclusions
As for other extra-skeletal effects, preclinical data strongly suggest that the vitamin D endocrine system may have effects on cells and tissues belonging to the cardiovascular system (Fig. 1). These data include direct effects of 1,25(OH)2D on several genes that regulate endothelial function or blood pressure and hemostasis. In line with these cellular in vitro data, animals with severe vitamin D deficiency or vitamin D resistance display systemic hypertension, cardiac hypertrophy, increased thrombogenicity, and decreased fibrolysis. An important remark, however, is that such data were generated by studying situations of (near) total absence of vitamin D action or in situations of exposure to high concentrations of 1,25(OH)2D or its analogs.
A wealth of observational studies fairly consistently concluded that subjects or patients with poor vitamin D status have a higher risk of cardiovascular risks or events, spanning from endothelial dysfunction, hypertension, ischemic heart diseases, or its clinical consequences, stroke, or cardiovascular death (Table 4). These observational data remain highly significant when subjected to critical meta-analysis, and the effect is not only highly statistically significant but suggest a major effect size of very frequent causes of morbidity or mortality. Of course, all of these cardiovascular diseases have multifactorial origin or pathogenesis, and observational studies may not be able to eliminate or correct for possible confounding factors such as age, gender, physical activity, and exposure to sunlight, obesity, or metabolic syndrome or lipid profile.
The final proof of causality largely depends on the results of Mendelian Randomization or randomized controlled trials. Surprisingly, these MR studies could not detect an association of lifetime genetically lower serum 25OHD and whatever cardiovascular outcome. However, the number of these studies is still limited and the predicted differences in serum 25OHD are small (about 5% difference in serum 25OHD). Larger studies and especially finding additional genetic reasons for differences in serum 25OHD concentrations are needed to validate the conclusion. A fairly large number of intervention studies did not consistently reveal an effect on blood pressure, whether systolic or diastolic. Similarly, RCTs could not reveal an effect on other surrogate markers or even the major cardiovascular events (coronary ischemia, myocardial infarction, stroke, or cardiovascular or cerebrovascular mortality.
What is now the best interpretation of the existing data?
It may well be that the vitamin D endocrine system is just redundant for humans and that the associations are just a reflection of “remaining confounding factors.” This would imply that improvements in vitamin D status would be futile for the prevention of cardiovascular events. In view of the strong preclinical and observational data, such interpretation is rather unlikely.
Another option could be that the very severe vitamin D deficiency as used in preclinical studies that generated the hypothesis of plausible interactions between VDR-1,25(OH)2D and cardiovascular events is not reproduced in humans because such extreme vitamin D deficiency is so rare in humans. Similarly, the beneficial effects of high doses of 1,25(OH)2D or its analogs in preclinical models may not be reproduced in human trials because such very high doses of the active hormone were not reached systemically nor locally. If this option is correct, improvement of the overall vitamin D status is unlikely to reduce the risks of cardiovascular events, unless only subjects with extremely low vitamin D status at baseline are studied or treated for a sufficiently long time.
Finally, it may well be that the duration of the supplementation with vitamin D in the published RCTs is too short to demonstrate an effect. Indeed, the origin of most cardiovascular events has a long trajectory of events such as intimal lesions, exposure to oxidative stress or high pressure, or to inflammatory signals before the clinical onset of symptoms or events. It may thus well be that decades of exposure to low vitamin D status is necessary to promote such long processes of cardiovascular damage and that only living with a better vitamin D status for many decades is effective to prevent its consequences for the cardiovascular system. Mendelian randomization studies should be able to compensate for this long time window of exposure to lower/better vitamin D status, but unfortunately, the present gene polymorphisms are not able to detect more than a 5% difference in serum 25OHD and this difference may well be too small to generate major or detectable differences in outcome.
There are many ongoing studies [118, 119] trying to evaluate the effects of vitamin D supplementation on extra-skeletal effects, including cardiovascular and cerebrovascular events. The design of these studies differs from study to study, as some RCTs include a very large number of subjects and will look at a wide variety of outcomes, whereas other RCTs are much smaller but focus on well-defined outcomes in selected subjects. It is not sure that the design of most ongoing studies can overcome the limitations described above with regard to the selection of the study population, dosage and dosage regimens, and duration of treatment that we think are needed to fully answer the questions and thereby guide clinical treatment options.
Abbreviations
- VDR:
-
Vitamin D receptor
- MI:
-
Myocardial infarction
- MR:
-
Mendelian randomization
- RCT:
-
Randomized controlled trial
- 25OHD:
-
25-Hydroxyvitamin D
- 1,25(OH)2D:
-
1a,25-Dihydroxyvitamin D
- CYP2R1:
-
Major enzyme responsible for the 25-hydroxylation of vitamin D
- CYP27B1:
-
Enzyme responsible for the 1α-hydroxylation of 25OHD
- CYP24A1:
-
Enzyme responsible for the degradation of 25OHD or 1,25(OH)2D
References
Manolagas SC (2010) From estrogen-centric to aging and oxidative stress: a revised perspective of the pathogenesis of osteoporosis. Endocr Rev 31:266–300
Laroche M, Pecourneau V, Blain H, Breuil V, Chapurlat R, Cortet B, Sutter B, Degboe Y, Committee Gs (2017) Osteoporosis and ischemic cardiovascular disease. Joint Bone Spine 84:427–432
Shaffer JR, Kammerer CM, Rainwater DL, O'Leary DH, Bruder JM, Bauer RL, Mitchell BD (2007) Decreased bone mineral density is correlated with increased subclinical atherosclerosis in older, but not younger, Mexican American women and men: the San Antonio family osteoporosis study. Calcif Tissue Int 81:430–441
Kim SN, Lee HS, Nam HS, Lee HR, Kim JM, Han SW, Park JH, Baik JS, Kim JY, Park JH (2016) Carotid intima-media thickness is inversely related to bone density in female but not in male patients with acute stroke. J Neuroimaging 26:83–88
Barengolts EI, Berman M, Kukreja SC, Kouznetsova T, Lin C, Chomka EV (1998) Osteoporosis and coronary atherosclerosis in asymptomatic postmenopausal women. Calcif Tissue Int 62:209–213
Marcovitz PA, Tran HH, Franklin BA, O'Neill WW, Yerkey M, Boura J, Kleerekoper M, Dickinson CZ (2005) Usefulness of bone mineral density to predict significant coronary artery disease. Am J Cardiol 96:1059–1063
Yesil Y, Ulger Z, Halil M, Halacli B, Yavuz BB, Yesil NK, Kuyumcu ME, Cankurtaran M, Ariogul S (2012) Coexistence of osteoporosis (OP) and coronary artery disease (CAD) in the elderly: it is not just a by chance event. Arch Gerontol Geriatr 54:473–476
Lee HT, Shin J, Min SY, Lim YH, Kim KS, Kim SG, Kim JH, Lim HK (2015) Relationship between bone mineral density and a 10-year risk for coronary artery disease in a healthy Korean population: the Korea National Health and Nutrition Examination Survey 2008-2010. Coron Artery Dis 26:66–71
Paccou J, Edwards MH, Ward KA, Jameson KA, Moss CL, Harvey NC, Dennison EM, Cooper C (2015) Ischemic heart disease is associated with lower cortical volumetric bone mineral density of distal radius. Osteoporos Int 26:1893–1901
Minn YK, Suk SH, Do SY (2014) Osteoporosis as an independent risk factor for silent brain infarction and white matter changes in men and women: the PRESENT project. Osteoporos Int 25:2465–2469
Magnus JH, Broussard DL (2005) Relationship between bone mineral density and myocardial infarction in US adults. Osteoporos Int 16:2053–2062
Ness J, Aronow WS (2006) Comparison of prevalence of atherosclerotic vascular disease in postmenopausal women with osteoporosis or osteopenia versus without osteoporosis or osteopenia. Am J Cardiol 97:1427–1428
Zhou J, Cui X, Jin X, Zhou J, Zhang H, Tang B, Fu M, Herlitz H, Cui J, Zhu H, Sun A, Hu K, Ge J (2014) Association of renal biochemical parameters with left ventricular diastolic dysfunction in a community-based elderly population in China: a cross-sectional study. PLoS One 9:e88638
Szulc P, Samelson EJ, Kiel DP, Delmas PD (2009) Increased bone resorption is associated with increased risk of cardiovascular events in men: the MINOS study. J Bone Miner Res 24:2023–2031
Wiklund P, Nordstrom A, Jansson JH, Weinehall L, Nordstrom P (2012) Low bone mineral density is associated with increased risk for myocardial infarction in men and women. Osteoporos Int 23:963–970
Browner WS, Seeley DG, Vogt TM, Cummings SR (1991) Non-trauma mortality in elderly women with low bone mineral density. Study of Osteoporotic Fractures Research Group. Lancet 338:355–358
Zhou R, Liu D, Li R, Zhou S, Cui M, Chen L, Zhou H (2015) Low bone mass is associated with stroke in Chinese postmenopausal women: the Chongqing osteoporosis study. Cell Biochem Biophys 71:1695–1701
Collins TC, Ewing SK, Diem SJ, Taylor BC, Orwoll ES, Cummings SR, Strotmeyer ES, Ensrud KE, Osteoporotic Fractures in Men Study G (2009) Peripheral arterial disease is associated with higher rates of hip bone loss and increased fracture risk in older men. Circulation 119:2305–2312
von Muhlen D, Allison M, Jassal SK, Barrett-Connor E (2009) Peripheral arterial disease and osteoporosis in older adults: the rancho Bernardo study. Osteoporos Int 20:2071–2078
Kiel DP, Kauppila LI, Cupples LA, Hannan MT, O'Donnell CJ, Wilson PW (2001) Bone loss and the progression of abdominal aortic calcification over a 25 year period: the Framingham heart study. Calcif Tissue Int 68:271–276
Hak AE, Pols HA, van Hemert AM, Hofman A, Witteman JC (2000) Progression of aortic calcification is associated with metacarpal bone loss during menopause: a population-based longitudinal study. Arterioscler Thromb Vasc Biol 20:1926–1931
Schulz E, Arfai K, Liu X, Sayre J, Gilsanz V (2004) Aortic calcification and the risk of osteoporosis and fractures. J Clin Endocrinol Metab 89:4246–4253
Barzilay JI, Buzkova P, Cauley JA, Robbins JA, Fink HA, Mukamal KJ (2018) The associations of subclinical atherosclerotic cardiovascular disease with hip fracture risk and bone mineral density in elderly adults. Osteoporos Int 29:2219–2230
Ye C, Xu M, Wang S, Jiang S, Chen X, Zhou X, He R (2016) Decreased bone mineral density is an independent predictor for the development of atherosclerosis: a systematic review and meta-analysis. PLoS One 11:e0154740
Bouillon R, Carmeliet G (2018) Vitamin D and the skeleton. In: Curr Opin Endocr Metabol Res ehead of print
Wimalawansa SJ (2018) Vitamin D and cardiovascular diseases: causality. J Steroid Biochem Mol Biol 175:29–43
Fuleihan Gel H, Bouillon R, Clarke B, Chakhtoura M, Cooper C, McClung M, Singh RJ (2015) Serum 25-hydroxyvitamin D levels: variability, knowledge gaps, and the concept of a desirable range. J Bone Miner Res 30:1119–1133
Bouillon R, Marcocci C, Carmeliet G et al (2018) Skeletal and extra-skeletal actions of vitamin D: current evidence and outstanding questions. Endocrine Reviews in press
Pilz S, Verheyen N, Grubler MR, Tomaschitz A, Marz W (2016) Vitamin D and cardiovascular disease prevention. Nat Rev Cardiol 13:404–417
Wang TJ (2016) Vitamin D and cardiovascular disease. Annu Rev Med 67:261–272
Norman PE, Powell JT (2014) Vitamin D and cardiovascular disease. Circ Res 114:379–393
Zittermann A (2018) Vitamin D status, supplementation and cardiovascular disease. Anticancer Res 38:1179–1186
Carvalho LS, Sposito AC (2015) Vitamin D for the prevention of cardiovascular disease: are we ready for that? Atherosclerosis 241:729–740
Andrukhova O, Slavic S, Zeitz U, Riesen SC, Heppelmann MS, Ambrisko TD, Markovic M, Kuebler WM, Erben RG (2014) Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol Endocrinol 28:53–64
Wong MS, Man RY, Vanhoutte PM (2010) Calcium-independent phospholipase a(2) plays a key role in the endothelium-dependent contractions to acetylcholine in the aorta of the spontaneously hypertensive rat. Am J Physiol Heart Circ Physiol 298:H1260–H1266
Hirata M, Serizawa K, Aizawa K, Yogo K, Tashiro Y, Takeda S, Moriguchi Y, Endo K, Fukagawa M (2013) 22-Oxacalcitriol prevents progression of endothelial dysfunction through antioxidative effects in rats with type 2 diabetes and early-stage nephropathy. Nephrol Dial Transplant 28:1166–1174
Uberti F, Lattuada D, Morsanuto V, Nava U, Bolis G, Vacca G, Squarzanti DF, Cisari C, Molinari C (2014) Vitamin D protects human endothelial cells from oxidative stress through the autophagic and survival pathways. J Clin Endocrinol Metab 99:1367–1374
Wong MS, Leisegang MS, Kruse C et al (2014) Vitamin D promotes vascular regeneration. Circulation 130:976–986
Oh J, Weng S, Felton SK, Bhandare S, Riek A, Butler B, Proctor BM, Petty M, Chen Z, Schechtman KB, Bernal-Mizrachi L, Bernal-Mizrachi C (2009) 1,25(OH)2 vitamin d inhibits foam cell formation and suppresses macrophage cholesterol uptake in patients with type 2 diabetes mellitus. Circulation 120:687–698
Yin K, You Y, Swier V, Tang L, Radwan MM, Pandya AN, Agrawal DK (2015) Vitamin D protects against atherosclerosis via regulation of cholesterol efflux and macrophage polarization in hypercholesterolemic swine. Arterioscler Thromb Vasc Biol 35:2432–2442
Torremade N, Bozic M, Panizo S, Barrio-Vazquez S, Fernandez-Martin JL, Encinas M, Goltzman D, Arcidiacono MV, Fernandez E, Valdivielso JM (2016) Vascular calcification induced by chronic kidney disease is mediated by an increase of 1alpha-hydroxylase expression in vascular smooth muscle cells. J Bone Miner Res 31:1865–1876
Wu-Wong JR, Nakane M, Ma J, Ruan X, Kroeger PE (2006) Effects of vitamin D analogs on gene expression profiling in human coronary artery smooth muscle cells. Atherosclerosis 186:20–28
Wakasugi M, Noguchi T, Inoue M, Kazama Y, Tawata M, Kanemaru Y, Onaya T (1991) Vitamin D3 stimulates the production of prostacyclin by vascular smooth muscle cells. Prostaglandins 42:127–136
Bukoski RD, DeWan P, McCarron DA (1989) 1,25 (OH)2 vitamin D3 modifies growth and contractile function of vascular smooth muscle of spontaneously hypertensive rats. Am J Hypertens 2:553–556
Wu-Wong JR, Nakane M, Ma J (2007) Vitamin D analogs modulate the expression of plasminogen activator inhibitor-1, thrombospondin-1 and thrombomodulin in human aortic smooth muscle cells. J Vasc Res 44:11–18
Cardus A, Panizo S, Encinas M, Dolcet X, Gallego C, Aldea M, Fernandez E, Valdivielso JM (2009) 1,25-dihydroxyvitamin D3 regulates VEGF production through a vitamin D response element in the VEGF promoter. Atherosclerosis 204:85–89
Walters MR, Ilenchuk TT, Claycomb WC (1987) 1,25-Dihydroxyvitamin D3 stimulates 45Ca2+ uptake by cultured adult rat ventricular cardiac muscle cells. J Biol Chem 262:2536–2541
Green JJ, Robinson DA, Wilson GE, Simpson RU, Westfall MV (2006) Calcitriol modulation of cardiac contractile performance via protein kinase C. J Mol Cell Cardiol 41:350–359
Rahman A, Hershey S, Ahmed S, Nibbelink K, Simpson RU (2007) Heart extracellular matrix gene expression profile in the vitamin D receptor knockout mice. J Steroid Biochem Mol Biol 103:416–419
O'Connell TD, Weishaar RE, Simpson RU (1994) Regulation of myosin isozyme expression by vitamin D3 deficiency and 1,25-dihydroxyvitamin D3 in the rat heart. Endocrinology 134:899–905
Li Q, Gardner DG (1994) Negative regulation of the human atrial natriuretic peptide gene by 1,25-dihydroxyvitamin D3. J Biol Chem 269:4934–4939
Li YC, Kong J, Wei M, Chen ZF, Liu SQ, Cao LP (2002) 1,25-Dihydroxyvitamin D(3) is a negative endocrine regulator of the renin-angiotensin system. J Clin Invest 110:229–238
Gardner DG, Chen S, Glenn DJ (2013) Vitamin D and the heart. Am J Physiol Regul Integr Comp Physiol 305:R969–R977
Hochhauser E, Barak J, Kushnir T, Navon G, Meyer MS, Edelstein S, Ben Bassat MB, Vidne BA (1989) Mechanical, biochemical, and structural effects of vitamin D deficiency on the chick heart. Angiology 40:300–308
Weishaar RE, Kim SN, Saunders DE, Simpson RU (1990) Involvement of vitamin D3 with cardiovascular function. III. Effects on physical and morphological properties. Am J Phys 258:E134–E142
Chen S, Law CS, Grigsby CL, Olsen K, Hong TT, Zhang Y, Yeghiazarians Y, Gardner DG (2011) Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation 124:1838–1847
Endo I, Inoue D, Mitsui T, Umaki Y, Akaike M, Yoshizawa T, Kato S, Matsumoto T (2003) Deletion of vitamin D receptor gene in mice results in abnormal skeletal muscle development with deregulated expression of myoregulatory transcription factors. Endocrinology 144:5138–5144
Zhou C, Lu F, Cao K, Xu D, Goltzman D, Miao D (2008) Calcium-independent and 1,25(OH)2D3-dependent regulation of the renin-angiotensin system in 1alpha-hydroxylase knockout mice. Kidney Int 74:170–179
Li YC, Qiao G, Uskokovic M, Xiang W, Zheng W, Kong J (2004) Vitamin D: a negative endocrine regulator of the renin-angiotensin system and blood pressure. J Steroid Biochem Mol Biol 89-90:387–392
Yuan W, Pan W, Kong J, Zheng W, Szeto FL, Wong KE, Cohen R, Klopot A, Zhang Z, Li YC (2007) 1,25-Dihydroxyvitamin D3 suppresses renin gene transcription by blocking the activity of the cyclic AMP response element in the renin gene promoter. J Biol Chem 282:29821–29830
Sundersingh F, Plum LA, DeLuca HF (2015) Vitamin D deficiency independent of hypocalcemia elevates blood pressure in rats. Biochem Biophys Res Commun 461:589–591
Aihara K, Azuma H, Akaike M, Ikeda Y, Yamashita M, Sudo T, Hayashi H, Yamada Y, Endoh F, Fujimura M, Yoshida T, Yamaguchi H, Hashizume S, Kato M, Yoshimura K, Yamamoto Y, Kato S, Matsumoto T (2004) Disruption of nuclear vitamin D receptor gene causes enhanced thrombogenicity in mice. J Biol Chem 279:35798–35802
Zhang Y, Leung DY, Richers BN, Liu Y, Remigio LK, Riches DW, Goleva E (2012) Vitamin D inhibits monocyte/macrophage proinflammatory cytokine production by targeting MAPK phosphatase-1. J Immunol 188:2127–2135
Helming L, Bose J, Ehrchen J, Schiebe S, Frahm T, Geffers R, Probst-Kepper M, Balling R, Lengeling A (2005) 1alpha,25-Dihydroxyvitamin D3 is a potent suppressor of interferon gamma-mediated macrophage activation. Blood 106:4351–4358
Vanherwegen AS, Gysemans C, Mathieu C (2017) Regulation of immune function by vitamin D and its use in diseases of immunity. Endocrinol Metab Clin N Am 46:1061–1094
Kasuga H, Hosogane N, Matsuoka K, Mori I, Sakura Y, Shimakawa K, Shinki T, Suda T, Taketomi S (2002) Characterization of transgenic rats constitutively expressing vitamin D-24-hydroxylase gene. Biochem Biophys Res Commun 297:1332–1338
Bouillon R, Carmeliet G, Lieben L, Watanabe M, Perino A, Auwerx J, Schoonjans K, Verstuyf A (2014) Vitamin D and energy homeostasis in mice and men. Nat Rev Endocrinol 10:79–87
Lin SH, Lin YF, Lu KC, Diang LK, Chyr SH, Liao WK, Shieh SD (1994) Effects of intravenous calcitriol on lipid profiles and glucose tolerance in uraemic patients with secondary hyperparathyroidism. Clin Sci (Lond) 87:533–538
Wehmeier K, Beers A, Haas MJ, Wong NC, Steinmeyer A, Zugel U, Mooradian AD (2005) Inhibition of apolipoprotein AI gene expression by 1, 25-dihydroxyvitamin D3. Biochim Biophys Acta 1737:16–26
Farzaneh-Far A, Weissberg PL, Proudfoot D, Shanahan CM (2001) Transcriptional regulation of matrix gla protein. Z Kardiol 90(Suppl 3):38–42
Han MS, Che X, Cho GH, Park HR, Lim KE, Park NR, Jin JS, Jung YK, Jeong JH, Lee IK, Kato S, Choi JY (2013) Functional cooperation between vitamin D receptor and Runx2 in vitamin D-induced vascular calcification. PLoS One 8:e83584
McCabe KM, Zelt JG, Kaufmann M, Laverty K, Ward E, Barron H, Jones G, Adams MA, Holden RM (2018) Calcitriol accelerates vascular calcification irrespective of vitamin K status in a rat model of chronic kidney disease with hyperphosphatemia and secondary hyperparathyroidism. J Pharmacol Exp Ther 366:433–445
Shi Y, Lu W, Hou Y, Fu K, Gan F, Liu J (2018) Fibroblast growth factor 21 ameliorates vascular calcification by inhibiting osteogenic transition in vitamin D3 plus nicotine-treated rats. Biochem Biophys Res Commun 495:2448–2455
Vogiatzi MG, Jacobson-Dickman E, De Boer MD, Drugs, Therapeutics Committee of The Pediatric Endocrine S (2014) Vitamin D supplementation and risk of toxicity in pediatrics: a review of current literature. J Clin Endocrinol Metab 99:1132–1141
Ke L, Mason RS, Kariuki M, Mpofu E, Brock KE (2015) Vitamin D status and hypertension: a review. Integrated Blood Press Control 8:13–35
Wang L, Song Y, Manson JE et al (2012) Circulating 25-hydroxy-vitamin D and risk of cardiovascular disease: a meta-analysis of prospective studies. Circ Cardiovasc Qual Outcomes 5:819–829
Leu M, Giovannucci E (2011) Vitamin D: epidemiology of cardiovascular risks and events. Best Pract Res Clin Endocrinol Metab 25:633–646
Anderson JL, May HT, Horne BD, Bair TL, Hall NL, Carlquist JF, Lappe DL, Muhlestein JB, Intermountain Heart Collaborative Study G (2010) Relation of vitamin D deficiency to cardiovascular risk factors, disease status, and incident events in a general healthcare population. Am J Cardiol 106:963–968
Wang TJ, Pencina MJ, Booth SL, Jacques PF, Ingelsson E, Lanier K, Benjamin EJ, D'Agostino RB, Wolf M, Vasan RS (2008) Vitamin D deficiency and risk of cardiovascular disease. Circulation 117:503–511
Forman JP, Giovannucci E, Holmes MD, Bischoff-Ferrari HA, Tworoger SS, Willett WC, Curhan GC (2007) Plasma 25-hydroxyvitamin D levels and risk of incident hypertension. Hypertension 49:1063–1069
Giovannucci E, Liu Y, Hollis BW, Rimm EB (2008) 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med 168:1174–1180
Brondum-Jacobsen P, Benn M, Jensen GB, Nordestgaard BG (2012) 25-hydroxyvitamin d levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arterioscler Thromb Vasc Biol 32:2794–2802
Al-Khalidi B, Kimball SM, Rotondi MA, Ardern CI (2017) Standardized serum 25-hydroxyvitamin D concentrations are inversely associated with cardiometabolic disease in U.S. adults: a cross-sectional analysis of NHANES, 2001-2010. Nutr J 16:16
Melamed ML, Muntner P, Michos ED, Uribarri J, Weber C, Sharma J, Raggi P (2008) Serum 25-hydroxyvitamin D levels and the prevalence of peripheral arterial disease: results from NHANES 2001 to 2004. Arterioscler Thromb Vasc Biol 28:1179–1185
Robinson-Cohen C, Hoofnagle AN, Ix JH, Sachs MC, Tracy RP, Siscovick DS, Kestenbaum BR, de Boer IH (2013) Racial differences in the association of serum 25-hydroxyvitamin D concentration with coronary heart disease events. JAMA 310:179–188
Perna L, Schottker B, Holleczek B, Brenner H (2013) Serum 25-hydroxyvitamin D and incidence of fatal and nonfatal cardiovascular events: a prospective study with repeated measurements. J Clin Endocrinol Metab 98:4908–4915
Kuhn T, Kaaks R, Teucher B, Hirche F, Dierkes J, Weikert C, Katzke V, Boeing H, Stangl GI, Buijsse B (2013) Plasma 25-hydroxyvitamin D and its genetic determinants in relation to incident myocardial infarction and stroke in the European prospective investigation into cancer and nutrition (EPIC)-Germany study. PLoS One 8:e69080
Fiscella K, Franks P (2010) Vitamin D, race, and cardiovascular mortality: findings from a national US sample. Ann Fam Med 8:11–18
Kestenbaum B, Katz R, de Boer I, Hoofnagle A, Sarnak MJ, Shlipak MG, Jenny NS, Siscovick DS (2011) Vitamin D, parathyroid hormone, and cardiovascular events among older adults. J Am Coll Cardiol 58:1433–1441
Gaksch M, Jorde R, Grimnes G, Joakimsen R, Schirmer H, Wilsgaard T, Mathiesen EB, Njølstad I, Løchen ML, März W, Kleber ME, Tomaschitz A, Grübler M, Eiriksdottir G, Gudmundsson EF, Harris TB, Cotch MF, Aspelund T, Gudnason V, Rutters F, Beulens JWJ, van ‘t Riet E, Nijpels G, Dekker JM, Grove-Laugesen D, Rejnmark L, Busch MA, Mensink GBM, Scheidt-Nave C, Thamm M, Swart KMA, Brouwer IA, Lips P, van Schoor NM, Sempos CT, Durazo-Arvizu RA, Škrabáková Z, Dowling KG, Cashman KD, Kiely M, Pilz S (2017) Vitamin D and mortality: individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLoS One 12:e0170791
Tomson J, Emberson J, Hill M, Gordon A, Armitage J, Shipley M, Collins R, Clarke R (2013) Vitamin D and risk of death from vascular and non-vascular causes in the Whitehall study and meta-analyses of 12,000 deaths. Eur Heart J 34:1365–1374
Autier P, Boniol M, Pizot C, Mullie P (2014) Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol 2:76–89
Theodoratou E, Tzoulaki I, Zgaga L, Ioannidis JP (2014) Vitamin D and multiple health outcomes: umbrella review of systematic reviews and meta-analyses of observational studies and randomised trials. BMJ 348:g2035
Zhang R, Li B, Gao X, Tian R, Pan Y, Jiang Y, Gu H, Wang Y, Wang Y, Liu G (2017) Serum 25-hydroxyvitamin D and the risk of cardiovascular disease: dose-response meta-analysis of prospective studies. Am J Clin Nutr 105:810–819
Zhou R, Wang M, Huang H, Li W, Hu Y, Wu T (2018) Lower vitamin D status is associated with an increased risk of ischemic stroke: a systematic review and Meta-analysis. Nutrients 10:27
Alele JD, Luttrell LM, Hollis BW, Luttrell DK, Hunt KJ, Group VS (2013) Relationship between vitamin D status and incidence of vascular events in the veterans affairs diabetes trial. Atherosclerosis 228:502–507
Welsh P, Doolin O, McConnachie A, Boulton E, McNeil G, Macdonald H, Hardcastle A, Hart C, Upton M, Watt G, Sattar N (2012) Circulating 25OHD, dietary vitamin D, PTH, and calcium associations with incident cardiovascular disease and mortality: the MIDSPAN family study. J Clin Endocrinol Metab 97:4578–4587
Bouillon R (2017) Genetic and racial differences in the vitamin D endocrine system. Endocrinol Metab Clin N Am 46:1119–1135
Manousaki D, Mokry LE, Ross S, Goltzman D, Richards JB (2016) Mendelian randomization studies Do not support a role for vitamin D in coronary artery disease. Circ Cardiovasc Genet 9:349–356
Brondum-Jacobsen P, Benn M, Afzal S, Nordestgaard BG (2015) No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: a Mendelian randomization study. Int J Epidemiol 44:651–661
Leong A, Rehman W, Dastani Z, Greenwood C, Timpson N, Langsetmo L, Berger C, METASTROKE, Fu L, Wong BYL, Malik S, Malik R, Hanley DA, Cole DEC, Goltzman D, Richards JB (2014) The causal effect of vitamin D binding protein (DBP) levels on calcemic and cardiometabolic diseases: a Mendelian randomization study. PLoS Med 11:e1001751
Vimaleswaran KS, Cavadino A, Berry DJ, LifeLines Cohort Study investigators, Jorde R, Dieffenbach AK, Lu C, Alves AC, Heerspink HJ, Tikkanen E, Eriksson J, Wong A, Mangino M, Jablonski KA, Nolte IM, Houston DK, Ahluwalia TS, van der Most P, Pasko D, Zgaga L, Thiering E, Vitart V, Fraser RM, Huffman JE, de Boer RA, Schöttker B, Saum KU, McCarthy M, Dupuis J, Herzig KH, Sebert S, Pouta A, Laitinen J, Kleber ME, Navis G, Lorentzon M, Jameson K, Arden N, Cooper JA, Acharya J, Hardy R, Raitakari O, Ripatti S, Billings LK, Lahti J, Osmond C, Penninx BW, Rejnmark L, Lohman KK, Paternoster L, Stolk RP, Hernandez DG, Byberg L, Hagström E, Melhus H, Ingelsson E, Mellström D, Ljunggren O, Tzoulaki I, McLachlan S, Theodoratou E, Tiesler CM, Jula A, Navarro P, Wright AF, Polasek O, International Consortium for Blood Pressure (ICBP), Cohorts for Heart and Aging Research in Genomic Epidemiology (CHARGE) consortium, Global Blood Pressure Genetics (Global BPGen) consortium, Caroline Hayward, Wilson JF, Rudan I, Salomaa V, Heinrich J, Campbell H, Price JF, Karlsson M, Lind L, Michaëlsson K, Bandinelli S, Frayling TM, Hartman CA, Sørensen TI, Kritchevsky SB, Langdahl BL, Eriksson JG, Florez JC, Spector TD, Lehtimäki T, Kuh D, Humphries SE, Cooper C, Ohlsson C, März W, de Borst MH, Kumari M, Kivimaki M, Wang TJ, Power C, Brenner H, Grimnes G, van der Harst P, Snieder H, Hingorani AD, Pilz S, Whittaker JC, Järvelin MR, Hyppönen E (2014) Association of vitamin D status with arterial blood pressure and hypertension risk: a Mendelian randomisation study. Lancet Diabetes Endocrinol 2:719–729
Skaaby T, Husemoen LL, Martinussen T et al (2013) Vitamin D status, filaggrin genotype, and cardiovascular risk factors: a Mendelian randomization approach. PLoS One 8:e57647
Ooi EM, Afzal S, Nordestgaard BG (2014) Elevated remnant cholesterol in 25-hydroxyvitamin D deficiency in the general population: Mendelian randomization study. Circ Cardiovasc Genet 7:650–658
Stojanovic M, Radenkovic M (2015) Vitamin D versus placebo in improvement of endothelial dysfunction: a meta-analysis of randomized clinical trials. Cardiovasc Ther 33:145–154
Hussin AM, Ashor AW, Schoenmakers I, Hill T, Mathers JC, Siervo M (2017) Effects of vitamin D supplementation on endothelial function: a systematic review and meta-analysis of randomised clinical trials. Eur J Nutr 56:1095–1104
Witham MD, Nadir MA, Struthers AD (2009) Effect of vitamin D on blood pressure: a systematic review and meta-analysis. J Hypertens 27:1948–1954
Wu SH, Ho SC, Zhong L (2010) Effects of vitamin D supplementation on blood pressure. South Med J 103:729–737
Pittas AG, Chung M, Trikalinos T, Mitri J, Brendel M, Patel K, Lichtenstein AH, Lau J, Balk EM (2010) Systematic review: vitamin D and cardiometabolic outcomes. Ann Intern Med 152:307–314
Beveridge LA, Struthers AD, Khan F, Jorde R, Scragg R, Macdonald HM, Alvarez JA, Boxer RS, Dalbeni A, Gepner AD, Isbel NM, Larsen T, Nagpal J, Petchey WG, Stricker H, Strobel F, Tangpricha V, Toxqui L, Vaquero MP, Wamberg L, Zittermann A, Witham MD, D-PRESSURE Collaboration (2015) Effect of vitamin D supplementation on blood pressure: a systematic review and Meta-analysis incorporating individual patient data. JAMA Intern Med 175:745–754
Hsia J, Heiss G, Ren H, Allison M, Dolan NC, Greenland P, Heckbert SR, Johnson KC, Manson JE, Sidney S, Trevisan M, Women's Health Initiative Investigators (2007) Calcium/vitamin D supplementation and cardiovascular events. Circulation 115:846–854
Bolland MJ, Grey A, Avenell A, Gamble GD, Reid IR (2011) Calcium supplements with or without vitamin D and risk of cardiovascular events: reanalysis of the Women's Health Initiative limited access dataset and meta-analysis. BMJ 342:d2040
Elamin MB, Abu Elnour NO, Elamin KB, Fatourechi MM, Alkatib AA, Almandoz JP, Liu H, Lane MA, Mullan RJ, Hazem A, Erwin PJ, Hensrud DD, Murad MH, Montori VM (2011) Vitamin D and cardiovascular outcomes: a systematic review and meta-analysis. J Clin Endocrinol Metab 96:1931–1942
Bolland MJ, Grey A, Gamble GD, Reid IR (2014) The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol 2:307–320
Ford JA, GS ML, Avenell A, Bolland M, Grey A, Witham M, Group RT (2014) Cardiovascular disease and vitamin D supplementation: trial analysis, systematic review, and meta-analysis. Am J Clin Nutr 100:746–755
Wang L, Manson JE, Song Y, Sesso HD (2010) Systematic review: vitamin D and calcium supplementation in prevention of cardiovascular events. Ann Intern Med 152:315–323
Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Wetterslev J, Simonetti RG, Bjelakovic M, Gluud C (2014) Vitamin D supplementation for prevention of mortality in adults. Cochrane Database Syst Rev:CD007470
Scragg R, Stewart AW, Waayer D, Lawes CM, Toop L, Sluyter J, Murphy J, Khaw KT, Camargo CA Jr (2017) Effect of monthly high-dose vitamin D supplementation on cardiovascular disease in the vitamin D assessment study : a randomized clinical trial. JAMA Cardiol 2:608–616
Mao PJ, Zhang C, Tang L, Xian YQ, Li YS, Wang WD, Zhu XH, Qiu HL, He J, Zhou YH (2013) Effect of calcium or vitamin D supplementation on vascular outcomes: a meta-analysis of randomized controlled trials. Int J Cardiol 169:106–111
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author declares to have received lecture fees (last 2 years) from Abiogen, FAES, l’Oreal and Frisenius. He is also co-owner of a university patent on vitamin D analogs licensed to Hybrigenics (France).
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Bouillon, R. Vitamin D and cardiovascular disorders. Osteoporos Int 30, 2167–2181 (2019). https://doi.org/10.1007/s00198-019-05098-0
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s00198-019-05098-0