
Abstract
Vitamin D has long been established as an elemental factor of bone physiology. Beyond mineral metabolism, the expression of the vitamin D receptor has been identified throughout the cardiovascular (CV) system. Experimental studies showed beneficial effects of vitamin D on heart and vessels, but vitamin D intoxication in animals also led to hypercalcemia and vascular calcification. Our knowledge has been extended by epidemiological studies that showed that 25-hydroxyvitamin D (25(OH)D) levels are inversely associated with an increased CV risk itself, but also with established CV risk factors, such as arterial hypertension, endothelial dysfunction and atherosclerosis. Conversely, randomized controlled trials could not document significant and consistent effects of vitamin D supplementation on CV risk or events. Potential explanations may lie in differences in reference ranges or the possibility that low vitamin D in CV disease is only an epiphenomenon. In the latter case, the key question is why low 25(OH)D levels are such a strong predictor of health. While we wait for new data, the current conclusion is that vitamin D is a strong risk marker for CV risk factors and for CV diseases itself.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
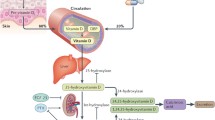
Vitamin D is a secosteroid hormone, synthesized mainly in the skin upon ultraviolet-B (UVB) radiation [1]. Its storage form is 25-hydroxyvitamin-D (25(OH)D) which has an approximate half-life of 2 to 3 weeks. The active metabolite, 1,25-dihydroxy-vitamin D (1,25[OH]2D) is only active for a couple of hours (up to 27 h maximally). The assessment of the vitamin D status is therefore based on the measurement of 25(OH)D [1]. Recommendations for supplementation currently rely on the positive effects of vitamin D on bone health [1–3]. Dietary standards and recommendations for the general population in the EU and the USA therefore mainly apply to the prevention of rickets, osteomalacia and fractures [4–6]. However except for children, randomized trials (RCTs) regarding vitamin D effects on fractures are inconsistent [7–15]. Therefore a broadly accepted consensus neither exists on the vitamin D dose nor on target 25(OH)D levels that should be achieved in clinical practice for cardiovascular disease (CVD) prevention [16–19]. Nevertheless, vitamin D supplementation - also in form of food fortification programs - is increasingly implemented [1, 18, 19]. The main aim of food fortification programs is the prevention of vitamin D deficiency and its adverse consequences [1–6, 16–19]. Vitamin D deficiency can affect up to 50% of a population depending on the geographical region, season and definition [19–23]. The high prevalence rates of vitamin D deficiency may be attributable to several factors including increased age, nutrition and lifestyle but also ethnicity [1, 20, 21]. Independent of latitude a higher prevalence rate of 25(OH)D has been described in African Americans, people of Indian descent and others with darker skin color. Likely because vitamin D3 (cholecalciferol) production is limited by melanin, but socioeconomic differences and prevalence of obesity may be confounding factors. Furthermore, the overall influence of ethnic origin may be less than the influence of age [20, 21]. Children, adolescents and elderly are at similar high risk for vitamin D deficiency and insufficiency [1, 20, 21]. This is thought to be partly due to inadequate exposure to sunlight, reduced dietary intake of vitamin D and changes in the synthesis capacity in the skin [1, 4, 9]. Pregnancy and lactation are also conditions associated with a high risk of vitamin D deficiency [1, 4]. The aforementioned risk groups constitute a vast proportion of the population in developed society so that vitamin D deficiency may represent a public health issue [1–6]. A broad range of studies demonstrated that vitamin D deficiency is a risk marker for several pathologic conditions and that it is associated with all-cause and cardiovascular mortality [1, 10, 20–23]. The United States Institute of Medicine (IOM) has defined vitamin D deficiency at 25(OH)D concentration lower than ≤30 nmol/L, inadequacy lies between 30 and 50 nmol/L and sufficiency at 50–75 nmol/L. Concentrations between 75 and 125 nmol/L have been inconsistently associated with harm whereas potential toxicity is possible to occur at concentrations ≥125 nmol/L [4].
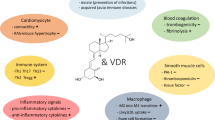
The scientific rationale to study the role vitamin D in cardiovascular diseases is the identification of the vitamin D receptor (VDR) and vitamin D metabolizing enzymes in different parts of the CV system [3]. It is crucial to evaluate whether it indeed has clinically relevant effects on the CV system [24–30]. Besides, it must be addressed whether vitamin D supplementation with commonly used doses satisfies the safety assumptions.
In this umbrella review, we summarize the literature on vitamin D, CV risk factors and CVD. The main focus is on large cohort studies, meta-analyses and systematic reviews of epidemiological studies. We begin with a short historical perspective and then summarize the effects of VDR activation on cardiovascular risk factors in particular on arterial hypertension [27], on the vasculature and the heart. In the third part we review observational studies, including Mendelian randomization studies. Finally, we discuss the ongoing research on vitamin D and current hypotheses, excluding randomized controlled trials as they are discussed extensively elsewhere [1, 10, 16, 19, 21, 24–26].
2 Historical overview
Historically Vitamin D is known to cure rickets a (childhood) bone disease, which was [is] especially prevalent in first few life years. Consequently, vitamin D food fortification was installed in the 1930s and 1940s to eradicate or at least largely decrease the incidence rate of rickets [28, 29]. A major concern was the suspicion in the 1950’s that vitamin D food fortification was leading to hypercalcemia or even supravalvular aortic stenosis. Indeed it has to be noted that animal studies have repeatedly shown that larger doses of vitamin D can cause vascular calcification and hypercalcemia [28–31]. The processes linking hypervitaminosis-D to vascular calcifications is likely to be related to deposition of calcium, phosphate and mineral complexes. The mechanism seems to be the induction of an osteochondrogenic phenotype in vascular smooth muscle cells [31]. However, clinical and epidemiologic data suggest that within broad ranges, vitamin D supplementation is safe. From a contemporary perspective, it is more likely that the children of the original cases-series with vitamin D intoxication actually suffered from Williams syndrome. This chromosomal anomaly is characterized by a disturbed calcium metabolism and such individuals are prone to adverse effects by vitamin D supplementation. Nevertheless vitamin D food fortification was subsequently largely banned due to fear of vitamin D overdosing and intoxication [28, 29]. After further progress in the field associations between higher vitamin D intake and increased cardiovascular risk were postulated in the middle of the 1970s. However, the majority of studies in the late 1970s could not detect any significant associations between 25(OH)D concentrations and cardiovascular events [32–35]. The view of vitamin D was radically changed by Robert Scragg in 1981 [36]. He raised the hypothesis that the increased incidence of CV events in winter months might be partly attributable to lower sunlight exposure and reduced vitamin D synthesis in the skin [36]. In 2008 publications from large community studies on an inverse association between 25(OH)D and CV events triggered an enormous rise in research activity on this topic over the last few years [4–6, 10, 16, 22–26, 28, 37–40].
3 Vitamin D receptor and key enzymes of vitamin D in the cardiovascular system
Experimental studies have identified the VDR and vitamin D metabolizing enzymes (i.e. 1-alpha hydroxylase and 24-hydroxylase) in cells of the CV system [3, 41–44]. This supports the hypothesis of a causal relationship between vitamin D and CVD. However, it has to be kept in mind that the contemporary literature is not fully consistent. There are – although in contrast to the vast majority - some studies published which were unable to detect VDR expression in cardiovascular tissues [41–44].
3.1 Knock out models
Mice with a systemic knock-out of the VDR or of 1-alpha hydroxylase develop cardiovascular anomalies. CVD in VDR knock-out animals is characterized by myocardial hypertrophy, increased activity of the renin-angiotensin-aldosterone system (RAAS), protein homeostasis, lipid metabolism, cellular stress response, arterial hypertension, increased thrombogenicity and changes in the vascular function [3, 41–46]. More so, it has been shown in mice models that a cardiomyocyte-specific knockout of the VDR causes myocardial hypertrophy largely independent of the effects on the RAAS and blood pressure [47]. An endothelial cell-specific VDR knock-out was associated with altered vascular function. This altered vascular function attended by increased sensitivity to angiotensin 2 of endothelial cells and a reduction in endothelial NO synthase expression [48, 49]. Interestingly, mice developed insulin resistance and glucose intolerance following a skeletal muscle-specific VDR knock-out [50]. Studies in LDL receptor knock-out mice found increased vascular calcification and atherosclerosis in the case of additional vitamin D deficiency. In detail the authors described for a reduction in 25(OH)D equal or above 50% that there was an increase by 50% in vascular calcifications [51]. Data from this animal model suggest that vitamin D deficiency may induce or promote a transdifferentiation of vascular cells into osteoblast-like cells what would then explain the excessive calcification. Interestingly, vitamin D treatment was able to reverse the changes [51, 52]. Independent of LDL diet-induced 25(OH)D deficiency was also associated with increased vascular calcification intensity in apolipoprotein E knockout mice [53]. In summary, the VDR knock-out models consistently indicated a causative role between vitamin D and vascular health.
3.2 Molecular and (patho-)physiologic effects
Based on the observations that animals with knock-outs of the VDR or 1-alpha hydroxylase suffer from cardiovascular disease, over one-hundred studies aimed to elucidate the mechanism behind this observation [29]. In view of the classic understanding of vitamin D as a master regulator of mineral metabolism, current evidence suggests that vitamin D affects mainly calcium and phosphate homeostasis in cells and the interstitial space [54]. Another additive explanation/mechanism would be the elevation of parathyroid hormone (PTH) occurring as a consequence of vitamin D deficiency [16]. It is well established that vitamin D is able to suppress PTH by direct effects on the parathyroid gland. Epidemiological evidence has indicated that PTH itself is a risk factor for CVD and predicts CV events [55]. The mechanism might be based on PTH-induced calcium overloading of cardiomyocytes with subsequent proarrhythmic actions, myocardial hypertrophy, but also aldosterone secretion [55–62]. This data suggests that to a certain point that both excess, as well as deficiency of vitamin D, can induce vascular calcification through a complex interplay with phosphate, calcium, and lipoproteins [63–69]. High serum phosphate levels even seem to be a prerequisite for vitamin D-induced vascular calcification, at least in animal (knock-out) models [68–76].
3.3 Vitamin D and fibrosis
Other experiments have raised the hypothesis that active vitamin D and phosphate is able to stimulate the secretion of fibroblast growth factor-23 (FGF-23) by osteocytes [39, 40, 44]. Though FGF-23 is an increasingly accepted marker of myocardial and renal fibrosis, the clinical relevance of the interaction between vitamin D and FGF-23 remains to be elucidated [71].
4 Observational studies
Observational studies have consistently reported an association between 25(OH)D concentrations and increased CV risk [39, 40]. More so, studies have described the association between 25(OH)D with traditional cardiovascular risk factors, such as arterial hypertension, hyperlipidaemia and diabetes mellitus (Table 1). However, even strong associations may, despite broad adjustments in statistical models, always be subject to residual confounding, collinearity and reverse causation [78]. The most commonly incriminated confounding factors – which are hard to measure and thus hard to adjust for - are limited physical activity, sun exposure and diet. Another possible confounding factor is Inflammation [79], as it lowers 25(OH)D concentrations by increasing 24-hydroxlyase expression and by decreasing vitamin D binding protein (DBP) [78, 79]. Therefore, some authors suggested that vitamin D deficiency may be just a general marker of poor health [78].
4.1 Rickets and cardiovascular diseases
There are four case reports of nutritional rickets and severe heart failure. Treatment of these children with vitamin D and calcium lead to a recovery of myocardial function [80, 81]. Pathologic insights come from one report of an autopsy from a similar case. It showed a dilated hypertrophic heart, with an increase in interstitial fibrosis in the subendocardial regions [80]. Somehow contradicting evidence comes from patients with hereditary 1,25(OH)2D resistant rickets who do not experience major cardiovascular problems (at least not under adequate treatment) [82, 83].
4.2 Hypertension
The idea that vitamin D affects blood pressure (BP) has been derived from observations that low UVB exposure is associated with increased risk of arterial hypertension [36, 84]. This is supported by reports that BP is lower during the summer compared to winter, although heat and UVA exposure could be obvious confounders. Other authors demonstrated that the prevalence of arterial hypertension increases with increasing distance from the earth’s equator, but ethnic and genetic differences also limit the interpretation of these results [26, 85, 86]. In the meta-analysis by Kunutsor et al. [84] comprising 283,537 participants, the relative risk (RR) for incidence of hypertension with 95% confidence interval (95% CI) in the top versus the bottom tertile of 25(OH)D was 0.70 (0.58 to 0.86). Although there was no significant association between vitamin D intake and presence of hypertension [84]. Nevertheless, it is of interest that in 2006 the American Heart Association issued a scientific statement regarding the effects of diet and dietary supplements on high blood pressure that did not include vitamin D as a relevant supplement to reduce BP [87]. In the very same statement, though, dietary calcium up to two grams per day was indicated to induce a minimal reduction on systolic and diastolic blood pressure, below 1 and 2 mmHg, respectively. These recommendation are based on the current lack of proof for a causal relationship between vitamin D alone and hypertension [20, 26].
4.3 Other cardiovascular risk factors
Several meta-analyses confirmed that 25(OH)D deficiency is an independent risk factor for developing type 2 diabetes mellitus, whether it is not clear if there is an association between vitamin D and metabolic syndrome [88–93]. Vitamin D deficiency has also been discussed as a risk factor for type 1 diabetes, but the data has been inconclusive [92, 94, 95]. Obesity is strongly associated with lower vitamin D concentrations [96, 97]. This might be explained by the studies describing that vitamin D is sequestered in adipose tissue, but other factors such inactivity, inflammation or simply volumetric dilution may be alternative explanations [98, 99]. In regard to the lipid profile, observational studies described a clear association, in particular with high triglycerides and low high-density lipoprotein (HDL) cholesterol [98, 99]. Other cardiovascular risk factors such as inflammation and chronic kidney disease are also associated with vitamin D deficiency [100–107].
4.4 Cardiovascular diseases
Meta-analyses of observational studies have consistently found that 25(OH)D deficiency (and insufficiency) is associated with an increased risk of cardiovascular mortality (Table 2) and cardiovascular events (i.e. myocardial infarction, heart failure and stroke) [107–119]. One meta-analysis including 65,994 patients reported a RR of 1.03 (95%CI 1.00–1.06) per 25 nmol/L decrement in 25(OH)D levels for CVD [109]. But specific for atherosclerotic coronary artery diseases (CAD) there are less consistent data so far [40, 120, 121]. Degerud et al. and Alsancak et al. did not observe any correlation of 25(OH)D and percent lumen loss as assessed with coronary angiography, although their sample size might have been too small [122, 123]. In 2014 Verdoia and co-workers reported an association in 1045 patients between 25(OH)D deficiency with CAD [124]. In line a Korean study reported an association between obstructive coronary artery disease (as assessed by CT angiography) and Vitamin D insufficiency [125]. In a cohort of typ 2 diabetic patients from Denmark Joergensen et al. reported that severe vitamin D deficiency (defined as 25(OH)D < 12.5 nmol/L, n = 19) was associated with increased coronary artery diseases, unfortunately this study suffers from inconsistent application of varying imaging techniques and sample size problems [126]. Even though some authors have claimed certainty about the role of 25(OH)D in CAD development [127], the absence of clinical trials make such allegations currently groundless. Nevertheless, existing evidence points to a possible role of vitamin D deficiency in CAD, raising the necessity of a randomized controlled trial in such a cohort of patients. In summary, the majority of studies concluded that an increased risk of CVD exists in participants with vitamin D deficiency (< 50 nmol/L), only a minority of studies reported on a U-shaped association [128–132]. Zittermann et al. observed that those with the highest 25(OH)D levels had actually low 1,25(OH)2D. They hypothesized that a reduced metabolism of 25(OH)D might explain this observation and that is could be responsible for the increased risk seen at high 25(OH)D concentrations [128]. Although extremely high 25(OH)D concentrations are considered toxic [132], epidemiologic studies did not detect an increased cardiovascular risk at these supranormal 25(OH)D levels. However, such high concentrations of 25(OH)D remain of concern and should to be avoided.
4.5 Myocardial infarction and stroke:
The majority of studies has proposed an association between 25(OH)D and myocardial infarction, stroke and sudden cardiac death [34, 133–138]. Some studies claimed that there is an association between low 25(OH)D and a higher risk of atrial fibrillation and venous thromboembolism [139, 140]. Overall the data on the topic is not consistent [141, 142]. Evidence on carotid atherosclerosis and endothelial dysfunction is also sparse [143–148]. Nevertheless a recent subgroup analysis of our own randomized controlled trial raised the awareness of a possible improvement of endothelial function in patients with arterial hypertension and vitamin deficiency [149].
4.6 Heart failure
The first clinical observations raising the possibility of a link between vitamin D and heart failure were case reports of children with rickets and heart failure, which resolved after Vitamin D substitution [80, 81]. This launched research into potential CV interaction of vitamin D, but until recently only a few studies have claimed that there is an associations between 25(OH)D and ventricular function [133–136, 150–155]. The observational data coming from the MESA cohort showed no association between 25(OH)D and parameters of LV function as assessed by MRI [150]. The findings were reproduced by a sub-study of AGES [151]. Similarly, LV mass index by echocardiography was also not associated with 25(OH)D in the Hoorn study [152]. It may be interpreted that this further refutes a large influence of vitamin D on blood pressure [26, 36, 84, 87]. Nevertheless all three studies confirmed an association between PTH and parameters of systolic LV function and/or LV mass [150, 151, 153]. In regard to interventional data, two meta-analysis aimed to assess the existing trial evidence. Ford and co-workers published the results of the RECORD trial together with a meta-analyis of existing RCT’s [137]. The overall results of meta-analysis that included 21 trials with a total of 13,033 patients were negative, although the trial itself found a statistically significant reduction in heart failure (adjusted HR: 0.75; 95% CI: 0.58, 0.97; P = 0.027) in 5292 patients [137]. The differences between included trials hamper conclusions in regard to heart failure. Especially as dose (between 400 IU and 500,000 IU of vitamin D per dose) and concomitant calcium supplementation vary heavily between the included studies. Another difference is the baseline 25(OH)D values and the age, but no specific analysis was conducted in such subgroups. A secondary analysis from the women’s health initiative of vitamin D plus calcium effects on heart failure incidence report a beneficial effect in the subgroup of women without diabetes, hypertension or coronary artery diseases [156]. In a very small (23 patients) RCT Dalbeni et al. described an improvement of LVEF with 4000 IU/daily vitamin D [157]. The recently published VINDICATE trial reported in 229 patients with chronic heart failure and vitamin D deficiency (< 50 nmol/L) improvement of LV function and dimension by vitamin D treatment (4000 IU/day) [158]. Although the primary endpoint (6 min walk test) was missed, the results support a causal role of vitamin D and heart failure. In summary, epidemiologic evidence did not find a consistent association of vitamin D deficiency and heart failure in adults, although some trials have indicated a possible effect.
5 Mendelian randomization studies
The inherent weakness of observational studies is their inability to demonstrate a causal interference between two factors. This limitation might be overcome by Mendelian randomization studies. In a nutshell, the presence or absence of certain genetic polymorphisms of a particular gene should be distributed randomly in the community and therefore their association with CV phenotypes allow implicitly assumptions on causality. A detailed discussion of advances and limitations of Mendelian randomization studies can be found elsewhere [159, 169]. Several genetic studies investigating single nucleotide polymorphisms (SNPs) of the VDR gene and cardiovascular outcomes, unfortunately, failed to provide evidence for a causal role of vitamin D for CV risk [160–168]. Genome-wide association studies (GWAS) identified four genetic loci that are associated with serum 25(OH)D levels [166–168]. The identified loci are involved in the biosynthesis of vitamin D at all major steps, and include synthesis of the precursor 7-dehydrocholesterol reductase (DHCR7), 25-hydroxylation of vitamin D (25-hydroxylase), transport (DBP) and in the degradation (i.e. 24-hydroxylase) [167–172]. As 25(OH)D is largely produced in the skin by sunlight exposure and to some degree determined by dietary intake, all known genetic polymorphisms explain only ~1–4% of the variation in serum 25(OH)D concentrations [167]. Results from so far published Mendelian randomization studies refute the hypothesis of vitamin D deficiency as a causal risk factor for obesity and high C-reactive protein (CRP) concentrations [172]. Less consistent data exist in regard to type 2 diabetes and lipid levels [173–179]. Studies on blood pressure are also not settling. The largest study in this field reported that a 10% increase in genetically determined 25(OH)D levels was associated with a change of < 1 mmHg in systolic and diastolic BP, and an 8.1% decreased odds ratio of hypertension [177]. Though this should not be mistakenly interpreted as effect size in individual patients with vitamin D deficiency, RCTs already supported the assumption of small effects of vitamin D supplementation on BP [25, 26, 178].
Mendelian randomization studies were unable to uniformly link vitamin D associated loci with coronary artery disease, myocardial infarction, stroke or cardiovascular mortality [40, 180–182]. These results, provisionally argue against a causal effect of vitamin D deficiency on cardiovascular diseases.
6 Future perspective
Though some large RCTs will soon be concluded and will expand existing knowledge, unfortunately, most of the ongoing vitamin D RCTs are limited by the inclusion of participants regardless of their 25(OH)D serum levels and some allow vitamin D supplement intake in the placebo group. This is a major limitation because the effects of vitamin D supplementation may be profoundly stronger in participants with very low starting 25(OH)D levels (Fig. 1) [193]. More so, in some trials the used immuno-assays for the determination of 25(OH)D may be of concern [183]. Some studies including critically ill patients even reported a mortality reduction of up to 50% (relative risk) but limited to the patients with 25(OH)D level < 30 nmol/L [184]. Similar subgroup analyses of individuals with vitamin D deficiency are expected to be reported in upcoming trials, but have important limitations, such as loss of power and more so they are usually not accepted by regulatory agencies. From a physiological point of view, the routine supplementation of a hormone without deficiency is at best unlikely to exert beneficial effect (in the worst case, harm might even be done) [15, 185–194]. Finally, we like to point out that if at the end we have to conclude that vitamin D is not causally responsible to CVD, we still would have to identify the confounding factors that are responsible for the strong association between vitamin D and CVD. This should be then a valuable clinical-therapeutic target, given the strong epidemiologic association.

Dose-response trend of hazard ratios of death from all causes by standardized 25-hydroxyvitamin D concentrations. Colours referring to the IOM definition of vitamin D deficiency, inadequacy and sufficiency [4]. Hazard ratios [blue line with 95% confidence interval as the dotted blue lines] are referring to the 25-hydroxyvitamin D concentration of 83.4 nmol/L. Reproduced with permission from reference under the Creative Commons CC0 licensing [193].
7 Conclusions
Severe vitamin D deficiency is a major risk factor for osteomalacia and fractures, but might also adversely affect the cardiovascular system. It is not completely clear whether and at which concentrations vitamin D, as it is present in community-dwelling adults, is relevant for cardiovascular outcomes. In general, observational studies reported strong associations between low 25(OH)D concentrations and increased cardiovascular risk, arterial hypertension, dyslipidemia and endothelial dysfunction. Some RCTs have detected effects in patients with low (< 50 nmol/L) and very low vitamin D levels (< 30 nmol/L) [40, 184]. Even though supported by the large majority of cell-based models and VDR knockout animals, data from Mendelian randomization studies and larger RCTs, do not consistently support the assumption of a causal relationship between vitamin D and CVD. Interestingly the vitamin D trials often aim to focus und the hardest clinical endpoints, such as all-cause or organ specific mortality [11, 58, 84, 188, 189]. This may be an overambitious goal. Bone health or even small reductions in cardiovascular endpoints ought to be enough to support the introduction of preventive measures in the general population and high-risk subgroups. Beside, often the increase in 25(OH)D is commonly used as an surrogate for the successful supplementation of vitamin D. Unfortunately there is little evidence to discuss potential other approaches such as the reduction in PTH or time within “normal range”. It is unclear if these variables would correlate better with physiologic effects [24]. Therefore, before final conclusion can be drawn, there is an unmet need for adequately powered RCTs focussing on patients with low to very low 25(OH)D levels. In the case of neutral findings in future RCTs, the question remains which unidentified factor confounded the epidemiological results and if this factor would then turn into a therapeutic target to reduce the burden of CVD.
References
Holick MF. Vitamin D deficiency. N Engl J Med. 2007;357:266–81.
Bouillon R, Suda T. Vitamin D: calcium and bone homeostasis during evolution. Bonekey Rep. 2014;3:480.
Bouillon R, Carmeliet G, Verlinden L, van Etten E, Verstuyf A, Luderer HF, et al. Vitamin D and human health: lessons from vitamin D receptor null mice. Endocr Rev. 2008;29:726–76.
Ross AC, Manson JE, Abrams SA, Aloia JF, Brannon PM, Clinton SK, et al. The 2011 report on dietary reference intakes for calcium and vitamin D from the Institute of Medicine: what clinicians need to know. J Clin Endocrinol Metab. 2011;96:53–8.
Cashman KD, Kiely M. Recommended dietary intakes for vitamin D: where do they come from, what do they achieve and how can we meet them? J Hum Nutr Diet. 2014;27:434–42.
German Nutrition Society. New reference values for vitamin D. Ann. Nutr. Metab. 2014;60:241–6.
H.A. Bischoff-Ferrari, W.C. Willett, E.J. Orav, P. Lips, P.J. Meunier, R.A. Lyons, et al. A pooled analysis of vitamin D dose requirements for fracture prevention. N Engl J Med 367 (2012) 40–49.
Bolland MJ, Grey A. A case study of discordant overlapping meta-analyses: vitamin d supplements and fracture. PLoS One. 2014;9:e115934.
Avenell A, Mak JC, O'Connell D. Vitamin D and vitamin D analogues for preventing fractures in post-menopausal women and older men. Cochrane Database Syst Rev. 2014;4:CD000227.
Bolland MJ, Grey A, Gamble GD, Reid IR. The effect of vitamin D supplementation on skeletal, vascular, or cancer outcomes: a trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014;2:307–20.
Reid IR, Bolland MJ, Grey A. Effects of vitamin D supplements on bone mineral density: a systematic review and meta-analysis. Lancet. 2014;383:146–55.
Bolland MJ, Grey A, Reid IR. Differences in overlapping meta-analyses of vitamin D supplements and falls. J Clin Endocrinol Metab. 2014;99:4265–72.
Bolland MJ, Grey A, Gamble GD, Reid IR. Vitamin D supplementation and falls: a trial sequential meta-analysis. Lancet Diabetes Endocrinol. 2014;2:573–80.
Weaver CM, Alexander DD, Boushey CJ, Dawson-Hughes B, Lappe JM, LeBoff MS, et al. Calcium plus vitamin D supplementation and risk of fractures: an updated meta-analysis from the National Osteoporosis Foundation. Osteoporos Int. 2016;27:367–76.
Bischoff-Ferrari HA, Dawson-Hughes B, Orav EJ, Staehelin HB, Meyer OW, Theiler R, et al. Monthly high-dose vitamin D treatment for the prevention of functional decline: a randomized clinical trial. JAMA Intern Med. 2016;176:175–83.
Holick MF, Binkley NC, Bischoff-Ferrari HA, Gordon CM, Hanley DA, Heaney RP, et al. Evaluation, treatment, and prevention of vitamin D deficiency: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2011;96:1911–30.
Kanis JA, McCloskey EV, Johansson H, Cooper C, Rizzoli R, Reginster JY, et al. European guidance for the diagnosis and management of osteoporosis in postmenopausal women. Osteoporos Int. 2013;24:23–57.
Brouwer-Brolsma EM, Bischoff-Ferrari HA, Bouillon R, Feskens EJ, Gallagher CJ, Hypponen E, et al. Vitamin D: do we get enough? A discussion between vitamin D experts in order to make a step towards the harmonisation of dietary reference intakes for vitamin D across Europe. Osteoporos Int. 2013;24:1567–77.
Reid IR, Bolland MJ. Skeletal and nonskeletal effects of vitamin D: is vitamin D a tonic for bone and other tissues? Osteoporos Int. 2014;25:2347–57.
Rostand SG. Ultraviolet light may contribute to geographic and racial blood pressure differences. Hypertension. 1997;30:150–6.
Rathish N, Arun M. Vitamin D: the “sunshine” vitamin. J Pharmacol Pharmacother. 2012;3:118–26.
Melamed ML, Michos ED, Post W, Astor B. 25-hydroxyvitamin D levels and the risk of mortality in the general population. Arch Intern Med. 2008;168:1629–37.
Pramyothin P, Holick MF. Vitamin D supplementation guidelines and evidence for subclinical deficiency. Curr Opin Gastroenterol. 2012;28:139–50.
Muscogiuri G, Altieri B, Annweiler C, Balercia G, Pal HB, Boucher BJ. Vitamin D and chronic diseases: the current state of the art. Arch Toxicol. 2017;91:97–107.
Kienreich K, Tomaschitz A, Verheyen N, Pieber T, Gaksch M. MR Grübler. S Pilz Vitamin D and cardiovascular disease Nutrients. 2013;5:3005–21.
Kienreich K, Grubler M, Tomaschitz A, Schmid J, Verheyen N, Rutters F, Dekker JM, Pilz S. Vitamin D, arterial hypertension & cerebrovascular disease. Indian J Med Res. 2013;137:669–79.
Poulter NR, Prabhakaran D, Caulfield M. Hypertension Lancet. 2015;386:801–12.
Wacker M, Holick MF. Sunlight and vitamin D: a global perspective for health. Dermatoendocrinol. 2013;5:51–108.
Zittermann A. Vitamin D and cardiovascular disease. Anticancer Res. 2014;34:4641–8.
Kummerow FA. Nutrition imbalance and angiotoxins as dietary risk factors in coronary heart disease. Am J Clin Nutr. 1979;32:58–83.
Drüeke TB, Massy ZA. Role of vitamin D in vascular calcification: bad guy or good guy? Nephrol Dial Transplant. 2012;27:1704–7.
Linden V. Vitamin D and myocardial infarction. BMJ. 1974;3:647–50.
Schmidt-Gayk H, Goossen J, Lendle F, Seidel D. Serum 25-hydroxycalciferol in myocardial infarction. Atherosclerosis. 1977;26:55–8.
Lund B, Badskjaer J, Lund B, Soerensen OH. Vitamin D and ischaemic heart disease. Horm Metab Res. 1978;10:553–6.
Vik B, Try K, Thelle DS, Førde OH. Tromsø heart study: vitamin D metabolism and myocardial infarction. BMJ. 1979;2:176.
Scragg R. Seasonality of cardiovascular disease mortality and the possible protective effect of ultra-violet radiation. Int J Epidemiol. 2012;10:337–41.
Giovannucci E, Liu Y, Hollis BW, Rimm EB. 25-hydroxyvitamin D and risk of myocardial infarction in men: a prospective study. Arch Intern Med. 2008;168:1174–80.
Dobnig H, Pilz S, Scharnagl H, Renner W, Seelhorst U, Wellnitz B, et al. Independent association of low serum 25-hydroxyvitamin d and 1,25-dihydroxyvitamin d levels with all-cause and cardiovascular mortality. Arch Intern Med. 2008;168:1340–9.
Pilz S, Gaksch M, O'Hartaigh B, Tomaschitz A, März W. The role of vitamin D deficiency in cardiovascular disease: where do we stand in 2013? Arch Toxicol. 2013;87:2083–103.
Pilz S, Verheyen N, Grübler MR, Tomaschitz A, März W. Vitamin D and cardiovascular disease prevention. Nat Rev Cardiol. 2016;13:404–17.
Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: metabolism, molecular mechanism of action, and pleiotropic effects. Physiol Rev. 2016;96:365–408.
Wang Y, DeLuca HF. Is the vitamin d receptor found in muscle? Endocrinology. 2011;152:354–63.
Norman PE, Powell JT. Vitamin D and cardiovascular disease. Circ Res. 2014;114:379–93.
Gardner DG, Chen S, Glenn DJ. Vitamin D and the heart. Am J Physiol Regul Integr Comp Physiol. 2013;305:R969–77.
Asano L, Watanabe M, Ryoden Y, Usuda K, Yamaguchi T, Khambu B, Takashima M, Sato S-I, Sakai J, Nagasawa K, Uesugi M. Vitamin D metabolite, 25-hydroxyvitamin D, regulates lipid metabolism by inducing degradation of SREBP/SCAP. Cell Chemical Biology. 2017;24(2):207–17.
Mark KA, Dumas KJ, Bhaumik D, Schilling B, Davis S, Oron TR, Sorensen DJ, Lucanic M, Brem RB, Melov S, Ramanathan A, Gibson BW, Lithgow GJ. Vitamin D promotes protein homeostasis and longevity via the stress response pathway genes skn-1, ire-1, and xbp-1. Cell Rep. 2016;17(5):1227–37.
Chen S, Law CS, Grigsby CL, Olsen K, Hong TT, Zhang Y, et al. Cardiomyocyte-specific deletion of the vitamin D receptor gene results in cardiac hypertrophy. Circulation. 2011;124:1838–47.
Ni W, Watts SW, Ng M, Chen S, Glenn DJ, Gardner DG. Elimination of vitamin D receptor in vascular endothelial cells alters vascular function. Hypertension. 2014;64:1290–8.
Oh J, Riek AE, Darwech I, Funai K, Shao J, Chin K, et al. Deletion of macrophage vitamin D receptor promotes insulin resistance and monocyte cholesterol transport to accelerate atherosclerosis in mice. Cell Rep. 2015;10:1872–86.
Chen S, Villalta SA, Agrawal DK. FOXO1 mediates vitamin D deficiency-induced insulin resistance in skeletal muscle. J Bone Miner Res. 2016;31:585–95.
Schmidt N, Brandsch C, Schutkowski A, Hirche F, Stangl GI. Dietary vitamin D inadequacy accelerates calcification and osteoblast-like cell formation in the vascular system of LDL receptor knockout and wild-type mice. J Nutr. 2014;144:638–46.
Gupta GK, Agrawal T, Rai V, Del Core MG, Hunter 3rd WJ, Agrawal DK. Vitamin D supplementation reduces intimal hyperplasia and restenosis following coronary intervention in atherosclerotic swine. PLoS One. 2016;11:e0156857.
Ellam T, Hameed A, ul Haque R, Muthana M, Wilkie M, Francis SE, et al. Vitamin D deficiency and exogenous vitamin D excess similarly increase diffuse atherosclerotic calcification in apolipoprotein E knockout mice. PLoS One. 2014;9:e88767.
Bikle DD. Vitamin D metabolism, mechanism of action, and clinical applications. Chem Biol. 2014;21(3):319–29.
Weishaar RE, Simpson RU. Involvement of vitamin D3 with cardiovascular function. II. Direct and indirect effects. Am. J. Physiol. 1987;253:E675–83.
Pilz S, Tomaschitz A, Drechsler C, Dekker JM, März W. Vitamin D deficiency and myocardial diseases. Mol Nutr Food Res. 2010;54:1103–13.
Assalin HB, Rafacho BP, dos Santos PP, Ardisson LP, Roscani MG, Chiuso-Minicucci F, et al. Impact of the length of vitamin D deficiency on cardiac remodeling. Circ Heart Fail. 2013;6:809–16.
van Ballegooijen AJ, Reinders I, Visser M, Brouwer IA. Parathyroid hormone and cardiovascular disease events: a systematic review and meta-analysis of prospective studies. Am Heart J. 2013;165:655–64.
Tomaschitz A, Ritz E, Pieske B, Rus-Machan J, Kienreich K, Verheyen N. Aldosterone and parathyroid hormone interactions as mediators of metabolic and cardiovascular disease. Metabolism. 2014;63:20–31.
Fitzpatrick LA, Bilezikian JP, Silverberg SJ. Parathyroid hormone and the cardiovascular system. Curr Osteoporos Rep. 2008;6:77–83.
Rutledge MR, Farah V, Adeboye AA, Seawell MR, Bhattacharya SK, Weber KT. Parathyroid hormone, a crucial mediator of pathologic cardiac remodeling in aldosteronism. Cardiovasc Drugs Ther. 2013;27:161–70.
Peterlik M, Kállay E, Cross HS. Calcium nutrition and extracellular calcium sensing: relevance for the pathogenesis of osteoporosis, cancer and cardiovascular diseases. Nutrients. 2013;5:302–27.
Yousefzadeh P, Shapses SA, Wang X. Vitamin D binding protein impact on 25-Hydroxyvitamin D levels under different physiologic and pathologic conditions. International Journal of Endocrinology. 2014:e981581.
Zebger-Gong H, Müller D, Diercke M, Haffner D, Hocher B, Verberckmoes S, et al. 1,25-dihydroxyvitamin D3-induced aortic calcifications in experimental uremia: up-regulation of osteoblast markers, calcium-transporting proteins and osterix. J Hypertens. 2011;29:339–48.
Lomashvili KA, Wang X, O'Neill WC. Role of local versus systemic vitamin D receptors in vascular calcification. Arterioscler Thromb Vasc Biol. 2014;34:146–51.
Bas A, Lopez I, Perez J, Rodriguez M, Aguilera-Tejero E. Reversibility of calcitriol-induced medial artery calcification in rats with intact renal function. J Bone Miner Res. 2006;21:484–90.
Razzaque MS. The dualistic role of vitamin D in vascular calcifications. Kidney Int. 2011;79:708–14.
Lau WL, Leaf EM, Hu MC, Takeno MM, Kuro-o M, Moe OW, et al. Vitamin D receptor agonists increase klotho and osteopontin while decreasing aortic calcification in mice with chronic kidney disease fed a high phosphate diet. Kidney Int. 2012;82:1261–70.
Aoshima Y, Mizobuchi M, Ogata H, Kumata C, Nakazawa A, Kondo F, et al. Vitamin D receptor activators inhibit vascular smooth muscle cell mineralization induced by phosphate and TNF-α. Nephrol Dial Transplant. 2012;27:1800–6.
Hirata M, Serizawa K, Aizawa K, Yogo K, Tashiro Y, Takeda S, et al. 22-Oxacalcitriol prevents progression of endothelial dysfunction through antioxidative effects in rats with type 2 diabetes and early-stage nephropathy. Nephrol Dial Transplant. 2013;28:1166–74.
Negri AL. Fibroblast growth factor 23: associations with cardiovascular disease and mortality in chronic kidney disease. Int Urol Nephrol. 2014;46:9–17.
Wu-Wong JR, Li X, Chen YW. Different vitamin D receptor agonists exhibit differential effects on endothelial function and aortic gene expression in 5/6 nephrectomized rats. J Steroid Biochem Mol Biol. 2015;148:202–9.
Wu-Wong JR, Noonan W, Nakane M, Brooks KA, Segreti JA, Polakowski JS, et al. Vitamin d receptor activation mitigates the impact of uremia on endothelial function in the 5/6 nephrectomized rats. Int J Endocrinol. 2010;2010:625852.
Dong J, Wong SL, Lau CW, Liu J, Wang YX, Dan He Z, et al. Calcitriol restores renovascular function in estrogen-deficient rats through downregulation of cyclooxygenase-2 and the thromboxane-prostanoid receptor. Kidney Int. 2013;84:54–63.
Andrukhova O, Slavic S, Zeitz U, Riesen SC, Heppelmann MS, Ambrisko TD, et al. Vitamin D is a regulator of endothelial nitric oxide synthase and arterial stiffness in mice. Mol Endocrinol. 2014;28:53–64.
Bozic M, Álvarez Á, de Pablo C, Sanchez-Niño MD, Ortiz A, Dolcet X, et al. Impaired vitamin D signaling in endothelial cell leads to an enhanced leukocyte-endothelium interplay: implications for atherosclerosis development. PLoS One. 2015;10:e0136863.
Beveridge LA, Struthers AD, Kahn F, Jorde R, Scragg R, Macdonald HM, et al. Effect of vitamin D supplementation on blood pressure: a systematic review and meta-analysis incorporating individual patient data. JAMA Intern Med. 2015;175:745–54.
Autier P, Boniol M, Pizot C, Mullie P. Vitamin D status and ill health: a systematic review. Lancet Diabetes Endocrinol. 2014;2:76–89.
Reid D, Toole BJ, Knox S, Talwar D, Harten J, O'Reilly DS, et al. The relation between acute changes in the systemic inflammatory response and plasma 25-hydroxyvitamin D concentrations after elective knee arthroplasty. Am J Clin Nutr. 2011;93:1006–11.
Maiya S, Sullivan I, Allgrove J, Yates R, Malone M, Brain C, et al. Hypocalcaemia and vitamin D deficiency: an important, but preventable, cause of life-threatening infant heart failure. Heart. 2008;94:581–4.
Yilmaz O, Olgun H, Ciftel M, Kilic O, Kartal I, Iskenderoglu NY, et al. Dilated cardiomyopathy secondary to rickets-related hypocalcaemia: eight case reports and a review of the literature. Cardiol Young. 2015;25:261–6.
Tiosano D, Gepstein V. Vitamin D action: lessons learned from hereditary 1,25-dihydroxyvitamin-D-resistant rickets patients. Curr Opin Endocrinol Diabetes Obes. 2012;19:452–9.
Tiosano D, Schwartz Y, Braver Y, Hadash A, Gepstein V, Weisman Y, et al. The renin-angiotensin system, blood pressure, and heart structure in patients with hereditary vitamin D-resistance rickets (HVDRR). J Bone Miner Res. 2011;26:2252–60.
Kunutsor SK, Apekey TA, Steur M. Vitamin D and risk of future hypertension: meta-analysis of 283,537 participants. Eur J Epidemiol. 2013;28:205–21.
Ullah MI, Uwaifo GI, Nicholas WC, Koch CA. Does vitamin D deficiency cause hypertension? Current evidence from clinical studies and potential mechanisms. Int J Endocrinol. 2010;2010:1–11.
Ullah M, Koch C, Tamanna S, Rouf S, Shamsuddin L. Vitamin D deficiency and the risk of Preeclampsia and Eclampsia in Bangladesh. Horm Metab Res. 2013;45(09):682–87.
Appel LJ, Brands MW, Daniels SR, Karanja N, Elmer PJ, Sacks FM. American Heart Association. Dietary approaches to prevent and treat hypertension: a scientific statement from the American Heart Association. Hypertension. 2006;47:296–308.
Song Y, Wang L, Pittas AG, Del Gobbo LC, Zhang C, Manson JE, et al. Blood 25-hydroxy vitamin D levels and incident type 2 diabetes: a meta-analysis of prospective studies. Diabetes Care. 2013;36:1422–8.
Lu M, Xu Y, Lv L, Zhang M. Association between vitamin D status and the risk of gestational diabetes mellitus: a meta-analysis. Arch. Gynecol. Obstet. 2016;293:959–66.
Khan H, Kunutsor S, Franco OH, Chowdhury R. Vitamin D, type 2 diabetes and other metabolic outcomes: a systematic review and meta-analysis of prospective studies. Proc. Nutr. Soc. 2013;72:89–97.
Ju SY, Jeong HS, Kim do H. Blood vitamin D status and metabolic syndrome in the general adult population: a dose-response meta-analysis. J. Clin. Endocrinol. Metab. 2014;99:1053–63.
Pilz S, Kienreich K, Rutters F, de Jongh R, van Ballegooijen AJ, Grübler M, Tomaschitz A, Dekker JM. Role of vitamin D in the development of insulin resistance and type 2 diabetes. Curr Diab Rep. 2013;13:261–70.
Grübler MR, Gaksch M, Kienreich K, Verheyen N, Schmid J, Hartaigh BÓ, et al. Effects of vitamin D supplementation on glycated haemoglobin and fasting glucose levels in hypertensive patients: a randomized controlled trial. Diabetes Obes Metab. 2016;18:1006–12.
Dong JY, Zhang WG, Chen JJ, Zhang ZL, Han SF, Qin LQ. Vitamin D intake and risk of type 1 diabetes: a meta-analysis of observational studies. Nutrients. 2013;5:3551–62.
Rewers M, Ludvigsson J. Environmental risk factors for type 1 diabetes. Lancet. 2016;387:2340–8.
Pereira-Santos M, Costa PR, Assis AM, Santos CA, Santos DB. Obesity and vitamin D deficiency: a systematic review and meta-analysis. Obes Rev. 2015;16:341–9.
Pourshahidi LK. Vitamin D and obesity: current perspectives and future directions. Proc Nutr Soc. 2015;74:115–24.
Challoumas D. Vitamin D supplementation and lipid profile: what does the best available evidence show? Atherosclerosis. 2014;235:130–9.
Jorde R, Grimnes G. Vitamin D and metabolic health with special reference to the effect of vitamin D on serum lipids. Prog Lipid Res. 2011;50:303–12.
Prietl B, Treiber G, Pieber TR, Amrein K. Vitamin D and immune function. Nutrients. 2013;5:2502–21.
Hewison M. An update on vitamin D and human immunity. Clin Endocrinol. 2012;76:315–25.
Liu WC, Wu CC, Hung YM, Liao MT, Shyu JF, Lin YF, et al. Pleiotropic effects of vitamin D in chronic kidney disease. Clin Chim Acta. 2016;453:1–12.
Dusso AS, Tokumoto M. Defective renal maintenance of the vitamin D endocrine system impairs vitamin D renoprotection: a downward spiral in kidney disease. Kidney Int. 2011;79:715–29.
Liu T, Zhong S, Liu L, Liu S, Li X, Zhou T, et al. Vitamin D deficiency and the risk of anemia: a meta-analysis of observational studies. Ren Fail. 2015;37:929–34.
Derakhshanian H, Shab-Bidar S, Speakman JR, Nadimi H, Djafarian K. Vitamin D and diabetic nephropathy: a systematic review and meta-analysis. Nutrition. 2015;31:1189–94.
Michalska-Kasiczak M, Sahebkar A, Mikhailidis DP, Rysz J, Muntner P, Toth PP, et al. Analysis of vitamin D levels in patients with and without statin-associated myalgia - a systematic review and meta-analysis of 7 studies with 2420 patients. Int J Cardiol. 2015;178:111–6.
Lv WS, Zhao WJ, Gong SL, Fang DD, Wang B, Fu ZJ, et al. Serum 25-hydroxyvitamin D levels and peripheral neuropathy in patients with type 2 diabetes: a systematic review and meta-analysis. J Endocrinol Investig. 2015;38:513–8.
Zittermann A, Iodice S, Pilz S, Grant WB, Bagnardi V, Gandini S. Vitamin D deficiency and mortality risk in the general population: A meta-analysis of prospective cohort studies. Am J Clin Nutr. 2012;95:91–100.
Wang L, Song Y, Manson JE, Pilz S, März W, Michaëlsson K, et al. Circulating 25-hydroxy-vitamin D and risk of cardiovascular disease: a meta-analysis of prospective studies. Circ. Cardiovasc. Qual. Outcomes. 2012;5:819–29.
Brøndum-Jacobsen P, Benn M, Jensen GB, Nordestgaard BG. 25-hydroxyvitamin d levels and risk of ischemic heart disease, myocardial infarction, and early death: population-based study and meta-analyses of 18 and 17 studies. Arterioscler Thromb Vasc Biol. 2012;32:2794–802.
Sokol SI, Tsang P, Aggarwal V, Melamed ML, Srinivas VS. Vitamin D status and risk of cardiovascular events: lessons learned via systematic review and meta-analysis. Cardiol Rev. 2011;19:192–201.
Grandi NC, Breitling LP, Brenner H. Vitamin D and cardiovascular disease: systematic review and meta-analysis of prospective studies. Prev Med. 2010;51:228–33.
Pittas AG, Chung M, Trikalinos T, Mitri J, Brendel M, Patel K, et al. Systematic review: Vitamin D and cardiometabolic outcomes. Ann. Intern. Med. 2010;152:307–14.
Tomson J, Emberson J, Hill M, Gordon A, Armitage J, Shipley M, et al. Vitamin D and risk of death from vascular and non-vascular causes in the Whitehall study and meta-analyses of 12,000 deaths. Eur Heart J. 2013;34:1365–74.
Fan H, Yu W, Cao H, Li J, Liu B, Wang J, et al. Meta-analysis of circulating 25-hydroxyvitamin D levels and risk of cardiovascular and all-cause mortality in elderly population. Int J Cardiol. 2014;176:1025–9.
Chowdhury R, Kunutsor S, Vitezova A, Oliver-Williams C, Chowdhury S, Kiefte-de-Jong JC, et al. Vitamin D and risk of cause specific death: systematic review and meta-analysis of observational cohort and randomised intervention studies. BMJ. 2014;348:g1903.
Theodoratou E, Tzoulaki I, Zgaga L, Ioannidis JP. Vitamin D and multiple health outcomes: review of systematic reviews and meta-analyses of observational studies and randomised trials. BMJ. 2014;348:g2035.
Schöttker B, Jorde R, Peasey A, Thorand B, Jansen EH, Groot L, et al. Vitamin D and mortality: meta-analysis of individual participant data from a large consortium of cohort studies from Europe and the United States. BMJ. 2014;348:g3656.
Brøndum-Jacobsen P, Nordestgaard BG, Schnohr P, Benn M. 25-hydroxyvitamin D and symptomatic ischemic stroke: an original study and meta-analysis. Ann Neurol. 2013;73:38–47.
Sogomonian R, Alkhawam H, Jolly J, Vyas N, Ahmad S, Moradoghli Haftevani E, et al. Serum vitamin D levels correlate to coronary artery disease severity: a retrospective chart analysis. Expert Rev Cardiovasc Ther. 2016;14:977–82.
Karohl C, Vaccarino V, Veledar E, Goldberg J, Tangpricha V, Bellasi A, Raggi P. Vitamin D status and coronary flow reserve measured by positron emission tomography: a co-twin control study. J Clin Endocrinol Metabol. 2013;98(1):389–97.
Degerud E, Løland KH, Nygård O, Midttun Ø, Ueland PM, Seifert R, et al. Vitamin D status was not associated with 'one-year' progression of coronary artery disease, assessed by coronary angiography in statin-treated patients. Eur J Prev Cardiol. 2015;22:594–602.
Alsancak Y, Cengel A, Akyel A, Ozkan S, Sezenoz B, Unlu S. S, et al. Relationship between serum vitamin D levels and angiographic severity and extent of coronary artery disease. Eur J Clin Investig. 2015;45:940–8.
Verdoia M, Schaffer A, Sartori C, Barbieri L, Cassetti E, Marino P, Galasso G, De Luca G. Vitamin D deficiency is independently associated with the extent of coronary artery disease. Eur J Clin Investig. 2014;44:634–42.
Lim S, Shin H, Kim MJ, Ahn HY, Kang SM, Yoon JW, et al. Vitamin D inadequacy is associated with significant coronary artery stenosis in a community-based elderly cohort: the Korean longitudinal study on health and aging. J Clin Endocrinol Metab. 2012;97:169–17.
C. Joergensen, MD1, H. Reinhard, A. Schmedes, P.R. Hansen, N. Wiinberg, C. L. Petersen, et al. Vitamin D Levels and Asymptomatic Coronary Artery Disease in Type 2 Diabetic Patients With Elevated Urinary Albumin Excretion Rate. Diabetes Care 35 (2012) 168–172.
Aggarwal R, Akhthar T, Jain SK. Coronary artery disease and its association with vitamin D deficiency. J Midlife Health. 2016;7:56–60.
Zittermann A, Kuhn J, Dreier J, Knabbe C, Gummert JF, Börgermann J. Vitamin D status and the risk of major adverse cardiac and cerebrovascular events in cardiac surgery. Eur. Heart J. 2013;34:1358–64.
Durup D, Jørgensen HL, Christensen J, Tjønneland A, Olsen A, Halkjær J, et al. A reverse J-shaped association between serum 25-hydroxyvitamin D and cardiovascular disease mortality: the CopD study. J Clin Endocrinol Metab. 2015;100:2339–46.
Dror Y, Giveon SM, Hoshen M, Feldhamer I, Balicer RD, Feldman BS. Vitamin D levels for preventing acute coronary syndrome and mortality: evidence of a nonlinear association. J Clin Endocrinol Metab. 2013;98:2160–7.
Grant WB, Karras SN, Bischoff-Ferrari HA, Annweiler C, Boucher BJ, Juzeniene A, et al. Do studies reporting 'U'-shaped serum 25-hydroxyvitamin D-health outcome relationships reflect adverse effects? Dermatoendocrinol. 2016;8:e1187349.
Zittermann A, Prokop S, Gummert JF, Börgermann J. Safety issues of vitamin D supplementation. Anti Cancer Agents Med Chem. 2013;13:4–10.
Pilz S, März W, Wellnitz B, Seelhorst U, Fahrleitner-Pammer A, Dimai HP, et al. Association of vitamin D deficiency with heart failure and sudden cardiac death in a large cross-sectional study of patients referred for coronary angiography. J Clin Endocrinol Metab. 2008;93:3927–35.
Drechsler C, Pilz S, Obermayer-Pietsch B, Verduijn M, Tomaschitz A, Krane V, et al. Vitamin D deficiency is associated with sudden cardiac death, combined cardiovascular events, and mortality in haemodialysis patients. Eur Heart J. 2010;31:2253–61.
Deo R, Katz R, Shlipak MG, Sotoodehnia N, Psaty BM, Sarnak MJ, et al. Vitamin D, parathyroid hormone, and sudden cardiac death: results from the cardiovascular health study. Hypertension. 2011;58:1021–8.
Naesgaard PA, León De La Fuente RA, Nilsen ST, Woie L, Aarsland T, Brede C, et al. Serum 25(OH)D is a 2-year predictor of all-cause mortality, cardiac death and sudden cardiac death in chest pain patients from northern Argentina. PLoS One. 2012;7:e43228.
Ford JA, MacLennan GS, Avenell A, Bolland M, Grey A, Witham M. RECORD Trial Group. Cardiovascular disease and vitamin D supplementation: trial analysis, systematic review, and meta-analysis. Am J Clin Nutr. 2014;100:746–55.
Sun Q, Pan A, Hu FB, Manson JE, Rexrode KM. 25-hydroxyvitamin D levels and the risk of stroke: a prospective study and meta-analysis. Stroke. 2012;43:1470–7.
Brøndum-Jacobsen P, Benn M, Tybjaerg-Hansen A, Nordestgaard BG. 25-hydroxyvitamin D concentrations and risk of venous thromboembolism in the general population with 18,791 participantsJ Thromb Haemost. 2013;11:423–31.
Folsom AR, Roetker NS, Rosamond WD, Heckbert SR, Basu S, Cushman M, et al. Serum 25-hydroxyvitamin D and risk of venous thromboembolism: the atherosclerosis risk in communities (ARIC) study. J Thromb Haemost. 2014;12:1455–60.
Brodin E, Lerstad G, Grimnes G, Brækkan SK, Vik A, Brox J, et al. Serum levels of vitamin D are not associated with future risk of venous thromboembolism. The Tromsø study. Thromb. Haemost. 2013;109:885–90.
Thompson J, Nitiahpapand R, Bhatti P, Kourliouros A. Vitamin D deficiency and atrial fibrillation. Int J Cardiol. 2015;184:159–62.
Deleskog A, Piksasova O, Silveira A, Gertow K, Baldassarre D, Veglia F, et al. Serum 25-hydroxyvitamin D concentration in subclinical carotid atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:2633–8.
Blondon M, Sachs M, Hoofnagle AN, Ix JH, Michos ED, Korcarz C, et al. 25-hydroxyvitamin D and parathyroid hormone are not associated with carotid intima-media thickness or plaque in the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2013;33:2639–45.
Gepner AD, Colangelo LA, Blondon M, Korcarz CE, de Boer IH, Kestenbaum B, et al. 25-hydroxyvitamin D and parathyroid hormone levels do not predict changes in carotid arterial stiffness: the multi-ethnic study of atherosclerosis. Arterioscler Thromb Vasc Biol. 2014;34:1102–9.
Nsengiyumva V, Fernando ME, Moxon JV, Krishna SM, Pinchbeck J, Omer SM, et al. The association of circulating 25-hydroxyvitamin D concentration with peripheral arterial disease: a meta-analysis of observational studies. Atherosclerosis. 2015;243:645–51.
Zheng Z, Shi H, Jia J, Li D, Lin S. Vitamin D supplementation and mortality risk in chronic kidney disease: a meta-analysis of 20 observational studies. BMC Nephrol. 2013;14:199.
Levin GP, Robinson-Cohen C, de Boer IH, Houston DK, Lohman K, Liu Y, et al. Genetic variants and associations of 25-hydroxyvitamin D concentrations with major clinical outcomes. JAMA. 2012;308:1898–905.
Grübler MR, Gaksch M, Kienreich K, Verheyen ND, Schmid J, Müllner C. Effects of Vitamin D3 on asymmetric- and symmetric dimethylarginine in arterial hypertension. J Steroid Biochem Mol Biol. (2016) In press, Corrected Proof DOI: 10.1016/j.jsbmb.2016.12.014.
Bansal N, Zelnick L, Robinson-Cohen C, Hoofnagle AN, Ix JH, Lima JA, et al. Serum parathyroid hormone and 25-hydroxyvitamin D concentrations and risk of incident heart failure: the multi-ethnic study of atherosclerosis. J Am Heart Assoc. 2014;3:e001278.
van Ballegooijen AJ, Visser M, Cotch MF, Arai AE, Garcia M, Harris TB, et al. Serum vitamin D and parathyroid hormone in relation to cardiac structure and function: the ICELAND-MI substudy of AGES-Reykjavik. J Clin Endocrinol Metab. 2013;98:2544–52.
van Ballegooijen AJ, Snijder MB, Visser M, van den Hurk K, Kamp O, Dekker JM, et al. Vitamin D in relation to myocardial structure and function after eight years of follow-up: the Hoorn study. Ann Nutr Metab. 2012;60:69–77.
van Ballegooijen AJ, Visser M, Kestenbaum B, Siscovick DS, de Boer IH, Gottdiener JS, et al. Relation of vitamin D and parathyroid hormone to cardiac biomarkers and to left ventricular mass (from the cardiovascular health study). Am J Cardiol. 2013;111:418–24.
Pekkanen MP, Ukkola O, Hedberg P, Piira OP, Lepojärvi S, Lumme J, et al. Serum 25-hydroxyvitamin D is associated with major cardiovascular risk factors and cardiac structure and function in patients with coronary artery disease. Nutr Metab Cardiovasc Dis. 2015;25:471–8.
Zittermann A, Ernst JB. Calciotropic and phosphaturic hormones in heart failure. Dis: Nutr. Metab. Cardiovasc; 2017. doi:10.1016/j.numecd.2016.06.007.
M.M. Donneyong, C.A. Hornung, K.C. Taylor, R. N. Baumgartner, J.A. Myers, C.B. Eaton, et. al. Risk of heart failure among postmenopausal women: a secondary analysis of the randomized trial of vitamin D plus calcium of the women's health initiative. Circ Heart Fail 8 (2015) 49–56.
Dalbeni A, Scaturro G, Degan M, Minuz P, Delva P. Effects of six months of vitamin D supplementation in patients with heart failure: a randomized double-blind controlled trial. Nutr Metab Cardiovasc Dis. 2014;24:861–8.
Witte KK, Byrom R, Gierula J, Paton MF, Jamil HA, Lowry JE, et al. Effects of vitamin D on cardiac function in patients with chronic HF: the VINDICATE study. JACC. 2016;67:2593–603.
Davey Smith G, Hemani G. Mendelian randomization: genetic anchors for causal inference in epidemiological studies. Hum Mol Genet. 2014;23:R89–98.
Wang L, Chu A, Buring JE, Ridker PM, Chasman DI, Sesso HD. Common genetic variations in the vitamin D pathway in relation to blood pressure. Am J Hypertens. 2014;27:1387–95.
Tizaoui K, Kaabachi W, Hamzaoui A, Hamzaoui K. Contribution of VDR polymorphisms to type 1 diabetes susceptibility: systematic review of case-control studies and meta-analysis. J Steroid Biochem Mol Biol. 2014;143:240–9.
Li L, Wu B, Liu JY, Yang LB. Vitamin D receptor gene polymorphisms and type 2 diabetes: a meta-analysis. Arch Med Res. 2013;44:235–41.
Zhu B, Zhao HL, Ou C, Huang LS, Li PZ, Lao M. Association of vitamin D receptor BsmI gene polymorphism with the risk of type 2 diabetes mellitus. J Recept Signal Transduct Res. 2014;34:458–62.
Liu Z, Liu L, Chen X, He W, Yu X. Associations study of vitamin D receptor gene polymorphisms with diabetic microvascular complications: a meta-analysis. Gene. 2014;546:6–10.
Mao S, Huang S. Lack of an association between vitamin D receptor BsmI gene polymorphism and the risk of end-stage renal disease: a meta-analysis. Intern Med. 2013;52:2423–30.
Chun RF, Liu PT, Modlin RL, Adams JS, Hewison M. Impact of vitamin D on immune function: lessons learned from genome-wide analysis. Front Physiol. 2014;5:151.
Wang TJ, Zhang F, Richards JB, Kestenbaum B, van Meurs JB, Berry D, et al. Common genetic determinants of vitamin D insufficiency: a genome-wide association study. Lancet. 2010;376:180–8.
Ahn J, Yu K, Stolzenberg-Solomon R, Simon KC, McCullough ML, Gallicchio L, et al. Genome-wide association study of circulating vitamin D levels. Hum Mol Genet. 2010;19:2739–45.
Verduijn M, Siegerink B, Jager KJ, Zoccali C, Dekker FW. Mendelian randomization: use of genetics to enable causal inference in observational studies. Nephrol Dial Transplant. 2010;25:1394–8.
Vimaleswaran KS, Berry DJ, Lu C, Tikkanen E, Pilz S, Hiraki LT, et al. Causal relationship between obesity and vitamin D status: Bi-directional mendelian randomization analysis of multiple cohorts. PLoS Med. 2013;10:e1001383.
Leong A, Rehman W, Dastani Z, Greenwood C, Timpson N, Langsetmo L, et al. The causal effect of vitamin D binding protein (DBP) levels on calcemic and cardiometabolic diseases: a mendelian randomization study. PLoS Med. 2014;11:e1001751.
Liefaard MC, Ligthart S, Vitezova A, Hofman A, Uitterlinden AG, Kiefte-de Jong JC, et al. Vitamin D and C-reactive protein: a mendelian randomization study. PLoS One. 2015;10:e0131740.
Afzal S, Brøndum-Jacobsen P, Bojesen SE, Nordestgaard BG. Vitamin D concentration, obesity, and risk of diabetes: a mendelian randomisation study. Lancet Diabetes Endocrinol. 2014;2:298–306.
Ye Z, Sharp SJ, Burgess S, Scott RA, Imamura F, InterAct Consortium, et al. Association between circulating 25-hydroxyvitamin D and incident type 2 diabetes: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2015;3:35–42.
Skaaby T, Husemoen LL, Martinussen T, Thyssen JP, Melgaard M, Thuesen BH, et al. Vitamin D status, filaggrin genotype, and cardiovascular risk factors: a mendelian randomization approach. PLoS One. 2013;8:e57647.
Ooi EM, Afzal S, Nordestgaard BG. Elevated remnant cholesterol in 25-hydroxyvitamin D deficiency in the general population: mendelian randomization study. Circ Cardiovasc Genet. 2014;7:650–8.
Vimaleswaran KS, Cavadino A, Berry DJ, LifeLines Cohort Study investigators, Jorde R, Dieffenbach AK, et al. Association of vitamin D status with arterial blood pressure and hypertension risk: A mendelian randomisation study. Lancet Diabetes Endocrinol. 2014;2:719–29.
Pilz S, Gaksch M, Kienreich K, Grübler M, Verheyen N, Fahrleitner-Pammer A. Effects of vitamin D on blood pressure and cardiovascular risk factors: a randomized controlled trial. Hypertension. 2012;65:1195–201.
Husemoen LL, Skaaby T, Martinussen T, Jørgensen T, Thuesen BH, Kistorp C, et al. Investigating the causal effect of vitamin D on serum adiponectin using a mendelian randomization approach. Eur J Clin Nutr. 2014;68:189–95.
Manousaki D, Mokry LE, Ross S, Goltzman D, Richards JB. Mendelian randomization studies do not support a role for vitamin D in coronary artery disease. Circ Cardiovasc Genet. 2016;9:349–56. doi:10.1161/CIRCGENETICS.116.001396.
Brøndum-Jacobsen P, Benn M, Afzal S, Nordestgaard BG. No evidence that genetically reduced 25-hydroxyvitamin D is associated with increased risk of ischaemic heart disease or myocardial infarction: a mendelian randomization study. Int J Epidemiol. 2015;44:651–61.
Afzal S, Brøndum-Jacobsen P, Bojesen SE, Nordestgaard BG. Genetically low vitamin D concentrations and increased mortality: mendelian randomisation analysis in three large cohorts. BMJ. 2014;349:g6330.
Schmid J, Kienreich K, Gaksch M, Grübler M, Raggam R, Meinitzer A, et al. The importance of assays in vitamin D status classification: a comparison of four automated 25-hydroxyvitamin D immunoassays. Laboratoriumsmedizin. 2013;37:261–8.
Amrein K, Schnedl C, Holl A, Riedl R, Christopher KB, Pachler C, et al. Effect of high-dose vitamin D3 on hospital length of stay in critically ill patients with vitamin D deficiency: The VITdAL-ICU randomized clinical trial. JAMA. 2014;312:1520–30.
Kiely M, Cashman KD, on behalf of the ODIN Consortium. The ODIN project: Development of food-based approaches for prevention of vitamin D deficiency throughout life. Nutr. Bull. 2015;40:235–46.
Kiely M, Black LJ. Dietary strategies to maintain adequacy of circulating 25-hydroxyvitamin D concentrations. Scand J Clin Lab Invest Suppl. 2012;243:14–23.
Cashman KD, Dowling KG, Škrabáková Z, Gonzalez-Gross M, Valtueña J, De Henauw S, et al. Vitamin D deficiency in Europe: pandemic? Am J Clin Nutr. 2016;103:1033–44.
Rejnmark L, Avenell A, Masud T, Anderson F, Meyer HE, Sanders KM, et al. Vitamin D with calcium reduces mortality: patient level pooled analysis of 70,528 patients from eight major vitamin D trials. J Clin Endocrinol Metab. 2012;97:2670–81.
Bjelakovic G, Gluud LL, Nikolova D, Whitfield K, Krstic G, Wetterslev J, et al. Vitamin D supplementation for prevention of cancer in adults. Cochrane Database Syst Rev. 2014;6:CD007469.
Riley RD, Lambert PC, Abo-Zaid G. Meta-analysis of individual participant data: rationale, conduct, and reporting. BMJ. 2010;340:c221.
Sanders KM, Stuart AL, Williamson EJ, Simpson JA, Kotowicz MA, Young D, et al. Annual high-dose oral vitamin D and falls and fractures in older women: a randomized controlled trial. JAMA. 2010;303:1815–22.
Eaton CB, Young A, Allison MA, Robinson J, Martin LW, Kuller LH, et al. Prospective association of vitamin D concentrations with mortality in postmenopausal women: Results from the Women's Health Initiative (WHI). Am J Clin Nutr. 2011;94:1471–8.
Gaksch M, Jorde R, Grimnes G, Joakimsen R, Schirmer H, Wilsgaard T, Mathiesen EB, Njølstad I, Løchen M-L, März W, Kleber ME, Tomaschitz A, Grübler M, Eiriksdottir G, Gudmundsson EF, Harris TB, Cotch MF, Aspelund T, Gudnason V, Rutters F, Beulens JWJ, van ‘t Riet E, Nijpels G, Dekker JM, Grove-Laugesen D, Rejnmark L, Busch MA, Mensink GBM, Scheidt-Nave C, Thamm M, Swart KMA, Brouwer IA, Lips P, van Schoor NM, Sempos CT, Durazo-Arvizu RA, Škrabáková Z, Dowling KG, Cashman KD, Kiely M, Pilz S, Zmijewski M. Vitamin D and mortality: individual participant data meta-analysis of standardized 25-hydroxyvitamin D in 26916 individuals from a European consortium. PLOS ONE. 2017;12(2):e0170791.
Bolland MJ, Bacon CJ, Horne AM, Mason BH, Ames RW, Wang TK, Grey AB, Gamble GD, Reid IR. Vitamin D insufficiency and health outcomes over 5 y in older women. Am J Clin Nutr. 2010;91:82–9.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
For the present review there was no funding.
Conflict of interest
M.R.G. received partial travel support by Amgen Switzerland AG and Synlab Holding Deutschland Germany, GmbH. WM reports grants and personal fees from AMGEN, BASF, Sanofi, Siemens Diagnostics, Aegerion Pharmaceuticals, Astrazeneca, Danone Research, Numares, Pfizer, Hoffmann LaRoche: personal fees from MSD, Alexion; grants from Abbott Diagnostics, all outside the submitted work. W.M. is employed with synlab Holding Deutschland GmbH. Other authors report no potential conflicts of interest in regard to the present work.
Ethical approval
All studies cited in the current manuscript that were performed by the authors and include procedures performed involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards. That includes that informed consent was obtained from all individual participants included in the studies cited hat where performed by any of the authors.
Rights and permissions
About this article

Cite this article
Grübler, M.R., März, W., Pilz, S. et al. Vitamin-D concentrations, cardiovascular risk and events - a review of epidemiological evidence. Rev Endocr Metab Disord 18, 259–272 (2017). https://doi.org/10.1007/s11154-017-9417-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11154-017-9417-0