
Abstract
Introduction
The gut microbiome is emerging as an important player in the field of metabolic disorders.
Materials and methods
Currently, several studies are ongoing to determine whether the effect of gut microbiome on obesity, type 2 diabetes, non-alcoholic fatty liver disease, and other metabolic diseases is determined by singular species or rather by a functional role of bacterial metabolism at higher taxonomical level. Deciphering if a single or more species are responsible for metabolic traits or rather microbial metabolic pathways are responsible for effects on host metabolism may help to identify appropriate dietary interventions to support microbial functions according to the prevalent host disease. Furthermore, the combination of metagenomics and metabolomics-based signature might be applied in the future to improve the risk prediction in healthy subjects.
Conclusion
In this review, I will summarize the current findings regarding the role of gut microbiome and metabolites in metabolic disorders to argue whether the current achievements may be translated into clinical practice.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Disorders of the gut microbiome have been connected to a multitude of adverse conditions, including obesity, type 2 diabetes, cardiovascular diseases, inflammatory bowel diseases, anxiety, autism, allergies, and autoimmune diseases [1]. This review will first summarize the basis of metabolic dysbiosis emerged from the analysis of the gut microbiome in the last years, and second, it will discuss some important signals used by the gut microbiome to influence host metabolism especially in metabolic disorders.
The human body and vast number of microbial species including bacteria, archaea, viruses, and unicellular eukaryotes co-evolved in a mutualistic fashion. Over the past decade, growing evidence has shown that the composition of the gut microbiota and its activity might be relevant to maintain metabolic homeostasis [1, 2].
Most of the microorganisms live in the gut, where they benefit from receiving constant supplies of nutrients. Endowed with an immense gene catalog, the gut microbiome profoundly affect human metabolism. First, gut microbiota cooperate to human metabolic pathways, producing essential vitamins (especially the B group); second, gut microorganisms, through fermentation of indigestible carbohydrates, provide the human body with compounds that either are source of energy for the host or activate receptors to coordinate metabolic functions (i.e., short-chain fatty acids and bile acids) [3]. Third, gut microbiota takes part in other relevant physiological processes including regulation of the intestinal mucosal barrier and maturation of the immune system responses to antigens [1].
The gut microbiota in adults is constituted by two major bacterial phyla, Bacteroidetes or Firmicutes, altogether > 90% of the taxa present in the human gut, with a lower abundance of Actinobacteria, Cyanobacteria, Fusobacteria, Proteobacteria, and Verrumicrobia [2].
Therefore, it is not unexpected that the gut microbiome has been proposed to coordinate individual responses to environmental factors and might play a part in the complex penetrance of certain disorders not completely explained by genetics [4]. In fact, It has been recently hypothesized that loss of the microbiota-host equilibrium may explain the onset of “pre-disease” states [pre-diabetes, pre-hypertension, etc.] and thus explain the explosive development of non-communicable chronic diseases (NCDs) such as inflammatory bowel disease, obesity, diabetes, atherosclerosis, and other metabolic/inflammatory disorders [5].
Recent experimental studies suggested that alterations in gut microbial and metabolic composition may be responsible, in part, for induction of systemic chronic inflammation, thus promoting glucose intolerance and cardiovascular disease [6, 7]. For example, both the deregulation of the microbiota-host co-metabolism of bile acids and metabolic endotoxemia may alter glucose tolerance and promote metabolic dysfunction in liver and adipose tissue in experimental models [8,9,10]. Western style diets, that are typically enriched in saturated fats and deprived of fibers, have been shown to affect the gut microbiota structure with downstream detrimental effects on intestinal permeability, lipid accumulation, and inflammatory state [11, 12].
Conversely, triggering the adaptive immune system with commensal gut bacteria protects against insulin resistance and hyperglycemia, suggesting a possible therapeutic role for probiotics that favor regulation of intestinal immunity to protect against dysbiosis [13].
Gut microbiome and NCDs
Analysis of microbiota communities in human oral, gut, and atherosclerotic plaques from individuals with established atherosclerosis showed a reproducible correlation between cardiovascular disease (CVD) and bacterial pathogens, including Chlamydia pneumoniae, Porphyromonas gingivalis, Helicobacter pylori and Aggregatibacter actinomycetemcomitans [14]. Another study found that Collinsella spp. is enriched in subjects with atherosclerosis, while Eubacterium spp. and Roseburia spp. are more abundant among healthy controls [15]. Finally, research over the past decade has uncovered several key microbial metabolites, such as trimethylamine-N-oxide (TMAO), short-chain fatty acids (SCFAs) and secondary bile acids that uniquely affect the progression of CVD [7].
A general consensus exists that also inflammatory bowel disease is associated with compositional and metabolic changes in the intestinal microbiota (dysbiosis) [16]. Increased Enterobacteriaceae, particularly some strains of E. coli and Fusobacterium species as well asreduction in a number of bacterial species, most notably the Bifidobacterium, Lactobacillus, and Faecalibacterium genera, may protect the host from mucosal inflammation via release of microbial metabolites such as short-chain fatty acids [17]. Analysis of gut microbiome in patients affected with multiple sclerosis suggested that proinflammatory Acinetobacter (A.) calcoaceticusis increased, while anti-inflammatory Parabacteroides (P.) distasonis and Butyricimonasare decreased [18].
Gut microbiome in metabolic disorders
Morbid obesity, diabetes, and non-alcoholic fatty liver disease are very inter-connected disorders due to the effect of insulin resistance [19] and the gut microbiota [20]. Obesity was the first metabolic disorder, where altered ecology and structure of the gut microbiome were involved [21,22,23,24].
Studies in which germ-free mice were transplanted with feces from monozygotic twins discordant for body mass index demonstrate that the implanted gut microbiome transfers most of the metabolic features from the donor. A general scenario in obesity consider an increase in Firmicutesas well as loss of Bacteroidetes, particularly species such as Bacteroides spp. which are able to transfer the lean phenotype in functional experiments. Transplantation of gut microbiome from monozygotic twins discordant for BMI resulted in a transmission of the original phenotype in germ-free mice. Cohousing mice transplanted with either the lean microbiomeor the obese microbiome resulted in a prevention of obesity due to the invasion of specific Bacteroidetesfrom the lean microbiome to the obese microbiome. This elegant experiment provided evidence that some species belonging to Bacteroides have a significant impact on metabolism. Intriguingly, metagenomics revealed that the acquisition of the lean phenotype was associated with increased expression of genes regulating bacterial metabolism belonging to BCAA degradation pathway, consistently with the reduced cecal levels of BCAA and reduction of BCAA in the host [25].
In type 2 diabetes mellitus a similar adjustment in Firmicutes/Bacteroidetes ratio has been proposed [26,27,28], although it is unclear what is the effect of the interplay among diet, obesity and insulin resistance on these changes [3, 29]. Intestinal bacterial species such as Roseburia, Eubacterium halii and Faecalibacterium prausnitzii are generally decreased, while Lactobacilli gasseri, Streptococcus mutans and E. coli are increased in subjects with type 2 diabetes. Data from metagenome-wide association studies (MGWAS) revealed that the degree of dysbiosis in patients with Type 2 diabetes is moderate when compared with Inflammatory Bowel Disease (IBD). Nevertheless MGWAS confirmed that butyrate-producing bacteria including Roseburia intestinalis and Roseburia inulinivorans as well as Clostridiales are decreased in type 2 diabetes, while opportunistic pathogens such as Bacteroides caccae, Clostridium hathewayi, Clostridium ramosum, and Clostridium symbosium are increased [30]. Interestingly, the application of MGWAS markers as T2DM classifier reached an AUROC of 0.81, suggesting that gut-microbiota-based T2D index could be used to identify subsets of the population that are at high risk for progressing to clinically defined T2D[30]. A similar approach was tested in Swedish people, where increases in Lactobacillus species were correlated with metabolic control markers such as fasting glucose and HbA1c, while decreases in Clostridium species were negatively correlated with fasting glucose, HbA1c, insulin, c-peptide and triglycerides and positively with HDL [31]. Intriguingly, a recent metagenomics study suggested that HDL is the most influenced, among several CVD biomarkers,by the gut microbiome [32]. The correlation of Clostridiales species with HDL and triglycerides is also suggestive for a role of the gut microbiome in shaping the so-called atherogenic or diabetic dyslipidemia [31].
Other metagenomics studies revealed a major effect of metformin on the gut microbiome. First, a comparison of T2DM patients treated or untreated with metformin with controls and T1DM patients allowed to identify unique characteristics of T2D microbiome independent from metformin or hyperglycemia phenotype. In fact, this study provided evidence that gene richness is decreased in type 2 diabetes, but increased in type 1 diabetes. Interestingly, decrease in Roseburia and some Clostridiales butyrate producers were reduced in T2D microbiome independently from metformin [33]. A randomized placebo-controlled study revealed that metformin but not calorie restriction had rapid effects on the composition and function of gut microbiota in parallel with improved metabolic control (Hba1c and fasting glucose) [34]. Both studies converge on Escherichia and Intestini bacter as substrate of metformin although it is unclear whether theyare result of modified bacteria–bacteria interactions or of other physiological and/or environmental changes within the gut upon metformin treatment [33, 34].
A general emerging picture is that metabolic alterations are associated with low richness of microbial gene which is paralleled by reduced metabolic functions in the bacteria [35, 36].
Non-alcoholic fatty liver disease is another metabolic disorder interplaying with obesity and type 2 diabetes [19, 20, 37]. Metagenomics studies revealed a striking association between low microbial gene richness and hepatic steatosis as well as other metabolic variables such as HDL, LDL and insulin resistance [37]. Data from the FLORINASH Consortium provided evidence of a slight shift of the fecal microbiome in patients with steatosis to one that is more similar to that found in the human small intestine and oral cavity. For example, patients with steatosis had fewer Lachnospiraceae and Ruminococcaceae, which are responsible for butyrate production, and were enriched in Acidaminococcus, Escherichia spp. and Bacteroides spp. which were associated with insulin resistance [38].
How the gut microbiome signals to affect host metabolism
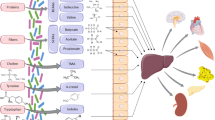
The gut microbiome is the pivotal regulator of nutrients processing in mammals. Its actions also generate essential metabolites not generated by the host such as vitamins and other absorbable compounds. Recent meta-analysis showed no major differences between lean and obese people at phylum level suggesting that metabolic dysbiosis might not derive from changes in microbiome taxonomic but rather depend on loss of metagenomics genomics heterogeneity on genes transcripts codifying for enzymes regulating metabolic functions [39]. In the second part of this review we will focus on changes in gut microbiota organization that lead to generation of bacterial metabolite end-products especially that may act via an interaction with membrane or nuclear receptors.
Amino acid acid-related signals
Branched-chain amino acids
BCAA are well-known biomarkers for insulin resistance and predictors of incident diabetes and cardiovascular diseases [40, 41]. The effect of BCAA to induce IR is not completely defined although it might involve incomplete BCAA catabolism in adipose and hepatic tissues with consequent plasma BCAA elevations. BCAA catabolism may be shunted to skeletal muscle, where it indirectly leads to FA accumulation and insulin resistance via generation of 3-HIB [42, 43]. In insulin resistant individuals P. copri and B. vulgatus were found significantly associated with increased BCAA biosynthesis and decreased transport genes at fecal metagenomics level [6]. The variation in BCAA microbial genes directly associated with circulating BCAA in the host. These results may explain how and why BCAA increase in insulin resistance and type 2 diabetes [6]. Furthermore, BCAA were found the most associated metabolites to both microbial gene richness, steatosis and insulin resistance in obese women. Among the overlapping genes co-associated with hepatic steatosis and low microbial gene richness, ACADSB (which encodes short/branched-chain acyl-CoA dehydrogenase) and INSR (which encodes the insulin receptor) were the most anti-correlated, suggesting a molecular connection from low MGR to a reduced capacity to respond to insulin and increased circulating BCAA. These observational data were corroborated by functional experiments based on fecal transplantation in which donor microbiota influenced the mouse phenome, showing that the steatosis-associated microbiota influences multiple patterns of association with hepatic triglycerides and circulating BCAAs [38].
Aromatic amino acids and indoles
AAA are a source of microbial metabolites affecting metabolic functions in the host [44].
Recently, we observed phenylacetic acid (PAA) which is a by-product of bacterial AAA metabolism which significantly increases hepatic BCAA utilization and hepatic lipid accumulation in obese women [37]. Therefore, in the context of obesity and insulin resistance the gut microbiota regulates a feedback mechanism to increase host BCAA utilization concurrently to increased BCAA absorption, which, however, prompts liver steatosis [38]. Indolelactate was found associated with insulin resistance and steatosis in experimental models of metabolic syndrome [45], one of the several analyses reporting an association of microbial indoles with metabolic variables [44].
Serum indolepropionate, a metabolite resulting from microbial Trp metabolism in Clostridium Sporogenes, was associated with reduced risk of developing Type 2 diabetes and negatively correlated with low-grade inflammation [46, 47]. It has been suggested the indolepropionate in the host activates Pregnane X Receptor to exert anti-inflammatory effects and may also exert anti-oxidant properties [48].
A reduction in fecal indole-3-acetic (IAA) concentration was found in subjects with obesity and diabetes, showing also increased LPS confirming the concomitance of dysfunctional gut barrier in the same subjects as a marker of gut dysbiosis. The reduction in IAA was also correlated with increased Kynurenine metabolism via increased activity indoleamine 2,3-dioxygenase in the host. Therefore, increased Trp absorption from host may deprivate gut microbiome with the source to generate indole and its derivatives [49].
Nevertheless, we should consider that not all the indole derivatives have beneficial effects on metabolism. In fact, indole compound itself, generated through tryptophanase-expressing bacteria, is first modified via hydroxylation and next by O-sulfation in the liver to generate indoxylsulfate, one of the uremic toxins which is known also to induce insulin resistance in the context of chronic kidney diseases [50].
Fermentation products (short short-chain fatty acids)
The fermentation of dietary fibers in the caecum and in the proximal colon by anaerobic bacteria leads to the generation of short-chain fatty acids [51]. Butyrate, acetate and propionate are the most abundant SCFAs produced by this process. Their conversion into energy for the mucosal cells and after transfer into circulation for the body is well known and the role of SCFAs in metabolic disorders had been resumed recently [44]. There is now consistent agreement that SCFAs play a positive role to improve metabolic homeostasis acting on lipid oxidation [44]. An intervention with propionate at colon levels in overweigh adults resulted in weight and liver steatosis reduction coupled with increased GLP-1 secretion [52].
The effects of SCFAs are in part mediated by G-protein-coupled receptors GPR41, GPR43 and GPR109A. GPR43 (also named FFAR2) is involved in GLP1 secretion, therefore, linking SCFAs to regulation of insulin sensitivity via incretin action [53].
Studies elucidating the bacteria involved in SCFAs generation are still lacking, although Roseburia species are often involved as well as Faecalibacterium praunitszii [6].
A recent contribution found that Firmicutes phylum, specifically those bacteria belonging to the Ruminococcaceae family, positively associated with plasma acetate levels and insulin sensitivity in subjects with obesity [54].
Gut microbiome microbiome-based therapeutics in metabolic disorders
Several clinical studies compared bariatric surgery techniques such as laparoscopic Roux-en-Y gastric bypass (RYGB) or sleeve gastrectomy (SG) surgery in obese patients with T2D [55, 56]. Meta-analysis of clinical studies suggested that post-operative gut microbiota improved to resemble that of lean subjects although most of these studies had very low numbers of recruited patients and results were often controversial [57]. Metagenomics studies on a Chinese cohort of young obese subjects observed decreased abundance of Bacteroides thetaiotaomicron in obese subjects. B. thetaiotaomicron is a glutamatefermenting commensal and was inversely correlated with serum glutamate concentration. Bariatric surgery intervention partially reversed the decreased gene richness observed in obesity and also reduced the abundance of B. thetaiotaomicron as well as the elevated serum glutamate concentration after 3 months from the intervention [58]. Interestingly, in a proof of concept experiment in mice gavage with B. thetaiotaomicron reduced plasma glutamate concentration and alleviated diet-induced body-weight gain [58]. However, another recent study combining metagenomics to bariatric surgery, in subjects of European ancestry with 12 months observation after the intervention, confirmed improved microbial gene richness but not a full rescue in most of the patients, despite all subjects showed metabolic improvement [59]. Overall, it is still unclear whether the low microbial gene richness characterizing obesity and its metabolic sequels are a consequence or a cause of morbid obesity [60]. The 5 year follow-up of the Florinash study which has been recently completed will be able to answer some of these questions. In type 2 diabetes dietary fibers were shown to selectively modulate a group of SCFA-producing strains and positively affect level of glycated hemoglobin in part via increased secretion of glucagon-like-peptide-1 [GLP-1) [61].
Conclusions
Several efforts and contributions from many laboratories allowed to start a map of gut dysbiosis in metabolic disorders. Metabolic dysbiosis is the product of subtle changes in gut microbiota and Table 1 contains a summary of the most significant results recently reported.
There are still several questions before we may translate research on the gut microbiome to clinical practice in metabolic disorders.
First, it is conceivable that effect of the gut microbiome is profoundly intertwined with the diet and the genetic background. We need more studies with a country focus and an in-depth analysis of the nutritional patterns of recruited subjects. Second, the impact of therapeutics remains to be determined. A recent study found associations among several therapeutic agents used in chronic disorders and gut microbiome, confirming that metformin/T2DM interaction is the most significantly associated with gut microbiome but revealing associations with other endocrine disorders such as hypothyroidism (an interplay between an anti-diabetic drug such as Metformin and the gut microbiome to explain part of the drug effect to improve metabolic control [18, 62,63,64]. Third, most of the gut microbiome species influence host metabolism and immunity via specific molecules and small metabolites (Table 2). Therefore, once a microbial metabolite is involved in a disease either as biomarker or as pathogenic agent we should make more efforts to explore the gut microbiome genetic potential to make specific metabolites.
References
Durack J, Lynch SV (2018) The gut microbiome: Relationships with disease and opportunities for therapy. J Exp Med https://doi.org/10.1084/jem.20180448
Gilbert JA, Blaser MJ, Caporaso JG, Jansson JK, Lynch SV, Knight R (2018) Current understanding of the human microbiome. Nat Med 24(4):392–400
Sonnenburg JL, Bäckhed F (2016) Diet-microbiota interactions as moderators of human metabolism. Nature 535(7610):56–64
Clemente JC, Manasson J, Scher JU (2018) The role of the gut microbiome in systemic inflammatory disease. BMJ 360:j5145
van de Guchte M, Blottière HM, Doré J (2018) Humans as holobionts: implications for prevention and therapy. Microbiome 6(1):81
Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, Forslund K, Hildebrand F, Prifti E, Falony G, Le Chatelier E, Levenez F, Doré J, Mattila I, Plichta DR, Pöhö P, Hellgren LI, Arumugam M, Sunagawa S, Vieira-Silva S, Jørgensen T, Holm JB, Trošt K, MetaHIT Consortium, Kristiansen K, Brix S, Raes J, Wang J, Hansen T, Bork P, Brunak S, Oresic M, Ehrlich SD, Pedersen O (2016) Human gut microbes impact host serum metabolome and insulin sensitivity. Nature 535(7612):376–381
Brown JM, Hazen SL (2018) Microbial modulation of cardiovascular disease. Nat Rev Microbiol 16(3):171–181
Wahlström A, Sayin SI, Marschall HU, Bäckhed F (2016) Intestinal crosstalk between bile acids and microbiota and its impact on host metabolism. Cell Metab 24(1):41–50
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R (2007) Metabolic endotoxemia initiates obesity and insulin resistance. Diabetes 56(7):1761–1772
Cani PD, Bibiloni R, Knauf C, Waget A, Neyrinck AM, Delzenne NM, Burcelin R (2008) Changes in gut microbiota control metabolic endotoxemia-induced inflammation in high-fat diet-induced obesity and diabetes in mice. Diabetes 57(6):1470–1481
Serino M, Luche E, Gres S, Baylac A, Bergé M, Cenac C, Waget A, Klopp P, Iacovoni J, Klopp C, Mariette J, Bouchez O, Lluch J, Ouarné F, Monsan P, Valet P, Roques C, Amar J, Bouloumié A, Théodorou V, Burcelin R (2012) Metabolic adaptation to a high-fat diet is associated with a change in the gut microbiota. Gut 61(4):543–553
Amar J, Chabo C, Waget A, Klopp P, Vachoux C, Bermúdez-Humarán LG, Smirnova N, Bergé M, Sulpice T, Lahtinen S, Ouwehand A, Langella P, Rautonen N, Sansonetti PJ, Burcelin R (2011) Intestinal mucosal adherence and translocation of commensal bacteria at the early onset of type 2 diabetes: molecular mechanisms and probiotic treatment. EMBO Mol Med 3(9):559–572
Pomié C, Blasco-Baque V, Klopp P, Nicolas S, Waget A, Loubières P, Azalbert V, Puel A, Lopez F, Dray C, Valet P, Lelouvier B, Servant F, Courtney M, Amar J, Burcelin R, Garidou L (2016) Triggering the adaptive immune system with commensal gut bacteria protects against insulin resistance and dysglycemia. Mol Metab 5(6):392–403
Cani PD, Amar J, Iglesias MA, Poggi M, Knauf C, Bastelica D, Neyrinck AM, Fava F, Tuohy KM, Chabo C, Waget A, Delmée E, Cousin B, Sulpice T, Chamontin B, Ferrières J, Tanti JF, Gibson GR, Casteilla L, Delzenne NM, Alessi MC, Burcelin R, Lindskog Jonsson A, Hållenius FF, Akrami R, Johansson E, Wester P, Arnerlöv C, Bäckhed F, Bergström G (2017) Bacterial profile in human atherosclerotic plaques. Atherosclerosis 263:177–183
Karlsson FH, Fåk F, Nookaew I, Tremaroli V, Fagerberg B, Petranovic D, Bäckhed F, Nielsen J (2012) Symptomatic atherosclerosis is associated with an altered gut metagenome. Nat Commun 3:1245
Ni J, Wu GD, Albenberg L, Tomov VT (2017) Gut microbiota and IBD: causation or correlation? Nat Rev Gastroenterol Hepatol 14(10):573–584
Marchix J, Goddard G, Helmrath MA (2018) Host-gut microbiota crosstalk in intestinal adaptation. Cell Mol Gastroenterol Hepatol 6(2):149–162
Jangi S, Gandhi R, Cox LM, Li N, von Glehn F, Yan R, Patel B, Mazzola MA, Liu S, Glanz BL, Cook S, Tankou S, Stuart F, Melo K, Nejad P, Smith K, Topçuolu BD, Holden J, Kivisäkk P, Chitnis T, De Jager PL, Quintana FJ, Gerber GK, Bry L, Weiner HL (2016) Alterations of the human gut microbiome in multiple sclerosis. Nat Commun 7:12015
Long MT, Fox CS (2016) The Framingham Heart Study—67 years of discovery in metabolic disease. Nat Rev Endocrinol 12(3):177–183
Scorletti E, Byrne CD (2016) Extrahepatic diseases and NAFLD: the triangular relationship between NAFLD, type 2-diabetes and dysbiosis. Dig Dis 34(Suppl 1):11–18
Ley RE, Bäckhed F, Turnbaugh P, Lozupone CA, Knight RD, Gordon JI (2005) Obesity alters gut microbial ecology. Proc Natl Acad Sci USA 102(31):11070–11075
Ley RE, Turnbaugh PJ, Klein S, Gordon JI (2006) Microbial ecology: human gut microbes associated with obesity. Nature 444(7122):1022–1023
Turnbaugh PJ, Hamady M, Yatsunenko T, Cantarel BL, Duncan A, Ley RE, Sogin ML, Jones WJ, Roe BA, Affourtit JP, Egholm M, Henrissat B, Heath AC, Knight R, Gordon JI (2009) A core gut microbiome in obese and lean twins. Nature 457(7228):480–484
Turnbaugh PJ, Ley RE, Mahowald MA, Magrini V, Mardis ER, Gordon JI (2006) An obesity-associated gut microbiome with increased capacity for energy harvest. Nature 444(7122):1027–1031 (PubMed PMID: 17183312)
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, Griffin NW, Lombard V, Henrissat B, Bain JR, Muehlbauer MJ, Ilkayeva O, Semenkovich CF, Funai K, Hayashi DK, Lyle BJ, Martini MC, Ursell LK, Clemente JC, Van Treuren W, Walters WA, Knight R, Newgard CB, Heath AC, Gordon JI (2013) Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science 341(6150):1241214
Brunkwall L, Orho-Melander M (2017) The gut microbiome as a target for prevention and treatment of hyperglycaemia in type 2 diabetes: from current human evidence to future possibilities. Diabetologia 60(6):943–951
Komaroff AL (2017) The microbiome and risk for obesity and diabetes. JAMA 317(4):355–356
Larsen N, Vogensen FK, van den Berg FW, Nielsen DS, Andreasen AS, Pedersen BK, Al-Soud WA, Sørensen SJ, Hansen LH, Jakobsen M (2010) Gut microbiota in human adults with type 2 diabetes differs from non-diabetic adults. PLoS One 5(2):e9085
Egshatyan L, Kashtanova D, Popenko A, Tkacheva O, Tyakht A, Alexeev D, Karamnova N, Kostryukova E, Babenko V, Vakhitova M, Boytsov S (2016) Gut microbiota and diet in patients with different glucose tolerance. Endocr Connect 5(1):1–9
Qin J, Li Y, Cai Z, Li S, Zhu J, Zhang F, Liang S, Zhang W, Guan Y, Shen D, Peng Y, Zhang D, Jie Z, Wu W, Qin Y, Xue W, Li J, Han L, Lu D, Wu P, Dai Y, Sun X, Li Z, Tang A, Zhong S, Li X, Chen W, Xu R, Wang M, Feng Q, Gong M, Yu J, Zhang Y, Zhang M, Hansen T, Sanchez G, Raes J, Falony G, Okuda S, Almeida M, LeChatelier E, Renault P, Pons N, Batto JM, Zhang Z, Chen H, Yang R, Zheng W, Li S, Yang H, Wang J, Ehrlich SD, Nielsen R, Pedersen O, Kristiansen K, Wang J (2012) A metagenome-wide association study of gut microbiota in type 2 diabetes. Nature 490(7418):55–60
Karlsson FH, Tremaroli V, Nookaew I, Bergström G, Behre CJ, Fagerberg B, Nielsen J, Bäckhed F (2013) Gut metagenome in European women with normal, impaired and diabetic glucose control. Nature 498(7452):99–103
Rothschild D, Weissbrod O, Barkan E, Kurilshikov A, Korem T, Zeevi D, Costea PI, Godneva A, Kalka IN, Bar N, Shilo S, Lador D, Vila AV, Zmora N, Pevsner-Fischer M, Israeli D, Kosower N, Malka G, Wolf BC, Avnit-Sagi T, Lotan-Pompan M, Weinberger A, Halpern Z, Carmi S, Fu J, Wijmenga C, Zhernakova A, Elinav E, Segal E (2018) Environment dominates over host genetics in shaping human gut microbiota. Nature 555(7695):210–215
Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Pedersen HK, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Costea PI, Kultima JR, Li J, Jørgensen T, Levenez F, Dore J, MetaHIT Consortium, Nielsen HB, Brunak S, Raes J, Hansen T, Wang J, Ehrlich SD, Bork P, Pedersen O (2015) Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528(7581):262–266
Wu H, Esteve E, Tremaroli V, Khan MT, Caesar R, Mannerås-Holm L, Ståhlman M, Olsson LM, Serino M, Planas-Fèlix M, Xifra G, Mercader JM, Torrents D, Burcelin R, Ricart W, Perkins R, Fernàndez-Real JM, Bäckhed F (2017) Metformin alters the gut microbiome of individuals with treatment-naive type 2 diabetes, contributing to the therapeutic effects of the drug. Nat Med 23(7):850–858
Cotillard A, Kennedy SP, Kong LC, Prifti E, Pons N, Le Chatelier E, Almeida M, Quinquis B, Levenez F, Galleron N, Gougis S, Rizkalla S, Batto JM, Renault P, ANR MicroObes Consortium, Doré J, Zucker JD, Clément K, Ehrlich SD (2013) Dietary intervention impact on gut microbial gene richness. Nature 500(7464):585–588. https://doi.org/10.1038/nature12480
Le Chatelier E, Nielsen T, Qin J, Prifti E, Hildebrand F, Falony G, Almeida M, Arumugam M, Batto JM, Kennedy S, Leonard P, Li J, Burgdorf K, Grarup N, Jørgensen T, Brandslund I, Nielsen HB, Juncker AS, Bertalan M, Levenez F, Pons N, Rasmussen S, Sunagawa S, Tap J, Tims S, Zoetendal EG, Brunak S, Clément K, Doré J, Kleerebezem M, Kristiansen K, Renault P, Sicheritz-Ponten T, de Vos WM, Zucker JD, Raes J, Hansen T, MetaHIT Consortium, Bork P, Wang J, Ehrlich SD, Pedersen O (2013) Richness of human gut microbiome correlates with metabolic markers. Nature 500:541–546
Radaelli MG et al (2018) NAFLD/NASH in patients with type 2 diabetes and related treatment Options. J Endocrinol Investig 41:509–521
Hoyles L, Fernández-Real JM, Federici M, Serino M, Abbott J, Charpentier J, Heymes C, Luque JL, Anthony E, Barton RH, Chilloux J, Myridakis A, Martinez-Gili L, Moreno-Navarrete JM, Benhamed F, Azalbert V, Blasco-Baque V, Puig J, Xifra G, Ricart W, Tomlinson C, Woodbridge M, Cardellini M, Davato F, Cardolini I, Porzio O, Gentileschi P, Lopez F, Foufelle F, Butcher SA, Holmes E, Nicholson JK, Postic C, Burcelin R, Dumas ME (2018) Molecular phenomics and metagenomics of hepatic steatosis in non-diabetic obese women. Nat Med 24:1070–1080
Sze MA, Schloss PD (2016) Looking for a signal in the noise: revisiting obesity and the microbiome. MBio 7:e01018–e1116
Ruiz-Canela M, Guasch-Ferré M, Toledo E, Clish CB, Razquin C, Liang L, Wang DD, Corella D, Estruch R, Hernáez Á, Yu E, Gómez-Gracia E, Zheng Y, Arós F, Romaguera D, Dennis C, Ros E, Lapetra J, Serra-Majem L, Papandreou C, Portoles O, Fitó M, Salas-Salvadó J, Hu FB, Martínez-González MA (2018) Plasma branched chain/aromatic amino acids, enriched Mediterranean diet and risk of type 2 diabetes: case-cohort study within the PREDIMED Trial. Diabetologia 61(7):1560–1571
Tobias DK, Lawler PR, Harada PH, Demler OV, Ridker PM, Manson JE, Cheng S, Mora S (2018) Circulating branched-chain amino acids and incident cardiovascular disease in a prospective cohort of US women. Circ Genom Precis Med 11(4):e002157
Arany Z, Neinast M (2018) Branched chain amino acids in metabolic disease. Curr Diabetes Rep 18(10):76. https://doi.org/10.1007/s11892-018-1048-7
Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, Rhee J, Hoshino A, Kim B, Ibrahim A, Baca LG, Kim E, Ghosh CC, Parikh SM, Jiang A, Chu DE, Forman Q, Lecker SH, Krishnaiah S, Rabinowitz JD, Weljie AM, Baur JA, Kasper DL, Arany Z (2016) A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med 22(4):421–426
Brial F, Le Lay A, Dumas ME, Gauguier D (2018) Implication of gut microbiota metabolites in cardiovascular and metabolic diseases. Cell Mol Life Sci 75(21):3977–3990
Mavilio M, Marchetti V, Fabrizi M, Stöhr R, Marino A, Casagrande V, Fiorentino L, Cardellini M, Kappel B, Monteleone I, Garret C, Mauriello A, Monteleone G, Farcomeni A, Burcelin R, Menghini R, Federici M (2016) A role for Timp3 in microbiota-driven hepatic steatosis and metabolic dysfunction. Cell Rep 16(3):731–743
Tuomainen M, Lindström J, Lehtonen M, Auriola S, Pihlajamäki J, Peltonen M, Tuomilehto J, Uusitupa M, de Mello VD, Hanhineva K (2018) Associations of serum indolepropionic acid, a gut microbiota metabolite, with type 2 diabetes and low-grade inflammation in high-risk individuals. Nutr Diabetes 8(1):35
de Mello VD, Paananen J, Lindström J, Lankinen MA, Shi L, Kuusisto J, Pihlajamäki J, Auriola S, Lehtonen M, Rolandsson O, Bergdahl IA, Nordin E, Ilanne-Parikka P, Keinänen-Kiukaanniemi S, Landberg R, Eriksson JG, Tuomilehto J, Hanhineva K, Uusitupa M (2017) Indolepropionic acid and novel lipid metabolites are associated with a lower risk of type 2 diabetes in the Finnish Diabetes Prevention Study. Sci Rep 7:46337
Venkatesh M, Mukherjee S, Wang H, Li H, Sun K, Benechet AP, Qiu Z, Maher L, Redinbo MR, Phillips RS, Fleet JC, Kortagere S, Mukherjee P, Fasano A, Le Ven J, Nicholson JK, Dumas ME, Khanna KM, Mani S (2014) Symbiotic bacterial metabolites regulate gastrointestinal barrier function via the xenobiotic sensor PXR and Toll-like receptor 4. Immunity 41(2):296–310
Laurans L, Venteclef N, Haddad Y, Chajadine M, Alzaid F, Metghalchi S, Sovran B, Denis RGP, Dairou J, Cardellini M, Moreno-Navarrete JM, Straub M, Jegou S, McQuitty C, Viel T, Esposito B, Tavitian B, Callebert J, Luquet S, Federici M, Fernandez-Real JM, Burcelin R, Launay JM, Tedgui A, Mallat Z, Sokol H, Taleb S (2018) Indoleamine 2–3 dioxygenase shapes microbiota to reduce IL-22 and promote metabolic disease. Nat Med. https://doi.org/10.1038/s41591-018-0060-4
Lau WL, Savoj J, Nakata MB, Vaziri ND (2018) Altered microbiome in chronic kidney disease: systemic effects of gut-derived uremic toxins. Clin Sci (Lond) 132(5):509–522
Koh A, De Vadder F, Kovatcheva-Datchary P, Bäckhed F (2016) From dietary fiber to host physiology: short-chain fatty acids as key bacterial metabolites. Cell 165(6):1332–1345
Chambers ES, Viardot A, Psichas A, Morrison DJ, Murphy KG, Zac-Varghese SE, MacDougall K, Preston T, Tedford C, Finlayson GS, Blundell JE, Bell JD, Thomas EL, Mt-Isa S, Ashby D, Gibson GR, Kolida S, Dhillo WS, Bloom SR, Morley W, Clegg S, Frost G (2015) Effects of targeted delivery of propionate to the human colon on appetite regulation, body weight maintenance and adiposity in overweight adults. Gut 64(11):1744–1754
Tolhurst G, Heffron H, Lam YS, Parker HE, Habib AM, Diakogiannaki E, Cameron J, Grosse J, Reimann F, Gribble FM (2012) Short-chain fatty acids stimulate glucagon-like peptide-1 secretion via the G-protein-coupled receptor FFAR2. Diabetes 61(2):364–371
Moreno-Navarrete JM, Serino M, Blasco-Baque V, Azalbert V, Barton RH, Cardellini M, Latorre J, Ortega F, Sabater-Masdeu M, Burcelin R, Dumas ME, Ricart W, Federici M, Fernández-Real JM (2018) Gut microbiota interacts with markers of adipose tissue browning, insulin action and plasma acetate in morbid obesity. Mol Nutr Food Res. https://doi.org/10.1002/mnfr.201700721
Campisciano G, Palmisano S, Cason C, Giuricin M, Silvestri M, Guerra M, Macor D, De Manzini N, Crocé LS, Comar M (2018) Gut microbiota characterisation in obese patients before and after bariatric surgery. Benef Microbes 9(3):367–373. https://doi.org/10.3920/BM2017.0152
Pucci A, Batterham RL (2018) Mechanisms underlying the weight loss effects of RYGB and SG: similar, yet different. J Endocrinol Invest. https://doi.org/10.1007/s40618-018-0892-2 ([Epub ahead of print] Review. PubMed PMID: 29730732)
Magouliotis DE, Tasiopoulou VS, Sioka E, Chatedaki C, Zacharoulis D (2017) Impact of bariatric surgery on metabolic and gut microbiota profile: a systematic reviewand meta-analysis. Obes Surg 27(5):1345–1357. https://doi.org/10.1007/s11695-017-2595-8 (Review. PubMed PMID: 28265960)
Liu R, Hong J, Xu X, Feng Q, Zhang D, Gu Y, Shi J, Zhao S, Liu W, Wang X, Xia H, Liu Z, Cui B, Liang P, Xi L, Jin J, Ying X, Wang X, Zhao X, Li W, Jia H, Lan Z, Li F, Wang R, Sun Y, Yang M, Shen Y, Jie Z, Li J, Chen X, Zhong H, Xie H, Zhang Y, Gu W, Deng X, Shen B, Xu X, Yang H, Xu G, Bi Y, Lai S, Wang J, Qi L, Madsen L, Wang J, Ning G, Kristiansen K, Wang W (2017) Gut microbiome and serummetabolome alterations in obesity and after weight-loss intervention. Nat Med 23(7):859–868. https://doi.org/10.1038/nm.4358 (Epub 2017 Jun 19. PubMed PMID:28628112)
Aron-Wisnewsky J, Prifti E, Belda E, Ichou F, Kayser BD, Dao MC, Verger EO, Hedjazi L, Bouillot JL, Chevallier JM, Pons N, Le Chatelier E, Levenez F, Ehrlich SD, Dore J, Zucker JD, Clément K (2018) Major microbiota dysbiosis in severe obesity: fate after bariatric surgery. Gut. https://doi.org/10.1136/gutjnl-2018-316103 ([Epub ahead of print] PubMed PMID: 29899081)
Cani PD (2018) Severe obesity and gut microbiota: does bariatric surgery really reset the system? Gut. https://doi.org/10.1136/gutjnl-2018-316815 ([Epub ahead of print] PubMed PMID: 29991642)
Zhao L, Zhang F, Ding X, Wu G, Lam YY, Wang X, Fu H, Xue X, Lu C, Ma J, Yu L, Xu C, Ren Z, Xu Y, Xu S, Shen H, Zhu X, Shi Y, Shen Q, Dong W, Liu R, Ling Y, Zeng Y, Wang X, Zhang Q, Wang J, Wang L, Wu Y, Zeng B, Wei H, Zhang M, Peng Y, Zhang C (2018) Gut bacteria selectively promoted by dietary fibers alleviate type 2 diabetes. Science 359(6380):1151–1156
Covelli D, Ludgate M (2017) The thyroid, the eyes and the gut: a possible connection. J Endocrinol Invest 40(6):567–576. https://doi.org/10.1007/s40618-016-0594-6 (Epub 2017 Jan 7. Review. PubMed PMID: 28063079)
Jackson MA, Verdi S, Maxan ME, Shin CM, Zierer J, Bowyer RCE, Martin T, Williams FMK, Menni C, Bell JT, Spector TD, Steves CJ (2018) Gut microbiota associations with common diseases and prescription medications in a population-based cohort. Nat Commun 9(1):2655. https://doi.org/10.1038/s41467-018-05184-7 (PubMed PMID: 29985401; PubMed Central PMCID:PMC6037668)
Brechmann T, Sperlbaum A, Schmiegel W (2017) Levothyroxine therapy and impaired clearance are the strongest contributors to small intestinal bacterial overgrowth: Results of a retrospective cohort study. World J Gastroenterol 23(5):842–852. https://doi.org/10.3748/wjg.v23.i5.842 (PubMed PMID: 28223728; PubMed Central PMCID: PMC5296200)
Acknowledgements
This review isbased on the Italian Society of Endocrinology (SIE) career award lecture. M.F. work related to this manuscript was in part funded by EU-FP7 FLORINASH (Health-F2-2009-241913), Ministry of University (MIUR) Progetti di Ricerca di Interesse Nazionale (PRIN) protocol number 2015MPESJS_004, Ministry of Health Ricerca Finalizzata RF-2011-02349921, Fondazione Roma call for Non-Communicable Diseases NCD 2014. The author thanks Dr. Carla Pietrini for editorial assistance.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The author is co-inventor on pending patents held by INSERM Transfert, INSERM, University of Rome Tor Vergata, University of Girona and Imperial College on NAFLD diagnostics and has the right to receive royalty payments for inventions or discoveries related to NAFLD diagnostics.
Ethical approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed consent
No informed consent.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Federici, M. Gut microbiome and microbial metabolites: a new system affecting metabolic disorders. J Endocrinol Invest 42, 1011–1018 (2019). https://doi.org/10.1007/s40618-019-01022-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40618-019-01022-9