
Abstract
Purpose of Review
Elevations in circulating branched chain amino acids (BCAAs) have gained attention as potential contributors to the development of insulin resistance and diabetes.
Recent Findings
Epidemiological evidence strongly supports this conclusion. Suppression of BCAA catabolism in adipose and hepatic tissues appears to be the primary drivers of plasma BCAA elevations. BCAA catabolism may be shunted to skeletal muscle, where it indirectly leads to FA accumulation and insulin resistance, via a number of proposed mechanisms.
Summary
BCAAs have an important role in the development of IR, but our understanding of how plasma BCAA elevations occur, and how these elevations lead to insulin resistance, is still limited.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Branched chain amino acids (BCAAs) are fundamental building blocks of mammalian life, comprising ~ 20% of the amino acids incorporated into protein [1]. All three BCAAs, leucine, isoleucine, and valine, are essential, and thus must be ingested. Plants, fungi, and bacteria synthesize BCAAs. BCAAs are thus generally abundant, and BCAA deficiency is rare. Mammals contain large amounts of BCAAs, mostly in muscle protein. Excess protein intake does not, however, lead to equivalent increase in muscle protein; excess protein intake thus obligatorily leads to its catabolism, in contrast to carbohydrates and fats that can be stored as glycogen and adipose tissue, respectively. A tightly regulated system has thus evolved to control BCAA catabolism.
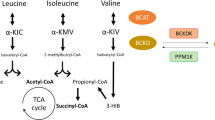
The first two steps of BCAA catabolism are shared by all three BCAAs (Fig. 1). The initial transamination to branched chain keto acids (BCKAs) by BCAA transaminase (BCAT) is followed by decarboxylation and dehydrogenation by the BCAA dehydrogenase complex (BCKDH) [2, 3]. The BCKDH complex is rate-limiting for BCAA catabolism, and is tightly regulated by inhibitory phosphorylation by BCKDK and reciprocal dephosphorylation (activating) by PPM1K [4,5,6]. Subsequent steps of BCAA catabolism are unique to each BCAA. Valine ultimately yields propionyl-CoA that enters the TCA, and is thus potentially gluconeogenic, while leucine yields acetyl-CoA and is thus ketogenic, and isoleucine yields both propionyl-CoA and acetyl-CoA. The BCKDH complex resides on the inner side of the inner membrane of mitochondria, and all BCAA catabolism thus occurs inside the mitochondrial matrix.

Biochemical pathways of BCAA catabolism. See text for details. Leu leucine, Ile isoleucine, Val valine, A-KG alpha-ketoglutarate, Glu glutamate, aKIC alpha keto isocaproate, aKMV alpha keto methyl valerate, aKIV alpha keto isovalerate, BCAT branched chain amino acid aminotransferase, BCKDK branched chain alpha keto acid dehydrogenase kinase, BCKDH branched chain alpha keto acid dehydrogenase, PPM1K protein phosphatase, Mg2+/Mn2+ dependent 1 K, CoA coenzyme A, CO, carbon dioxide. 3-HIB 3-hydroxyl isobutyrate, OAA oxaloacetate, TCA tricarboxylic acid cycle
Do Elevations in BCAAs Cause Insulin Resistance?
Elevations in plasma levels of BCAAs have been noted to associate with obesity and diabetes since the 1960s, and have been repeatedly reported since then [7,8,9,10]. More recently, unbiased metabolomics profiling of obese insulin resistant versus lean insulin sensitive subjects revealed that elevations in BCAAs were in fact the metabolic signature most strongly associated with insulin resistance [11,12,13,14,15,16,17]. The association has often assumed to be consequence rather than cause of insulin resistance, reflecting inability to properly suppress protein breakdown in the context of insulin resistance. However, several recent lines of evidence have strongly suggested that elevations in BCAAs contribute to insulin resistance:
-
1.
Infusions of BCAAs in humans acutely worsen insulin sensitivity, as typically measured by hyperinsulinemic euglycemic clamps [18,19,20,21]. Similarly, co-administration of BCAAs with high-fat diet (HFD) to rodents generally worsens the ensuing insulin resistance [11]. Conversely, low BCAA diets generally improve insulin resistance [22•, 23, 24]. Importantly, in all these rodent models, the presence of lipids or HFD was critical, and BCAAs alone had little or no effect, strongly suggesting that mechanistically, BCAAs interact with lipids in some way to promote insulin resistance. In general, accumulation of incompletely esterified or oxidized lipid species can be demonstrated in rodent models of insulin resistance or diabetes, usually in skeletal muscle [11, 22•, 25•]. These species include acyl-carnitines, acyl-CoAs, and diacylglycerols (DAG), although which species primarily drives insulin resistance remains actively debated.
-
2.
Elevations in plasma BCAA levels can be detected in people more than 10 years before developing diabetes. This was first demonstrated in an unbiased metabolomics profiling of banked blood samples from 189 subjects in the Framingham Heart Study who subsequently developed diabetes, compared to 189 controls who did not develop diabetes [26]. In this study, BCAAs were the signature most strongly associated with later development of diabetes. At the time of the initial blood sampling, none of the subjects had diabetes or detectable insulin resistance. Similar findings have been reported in an independent study [27], although not in a third study [28].
-
3.
A genetic Mendelian randomization study identified a locus near PPM1K that was associated with BCAA levels with genome-wide significance, and that in turn also correlated with incidence of diabetes, strongly suggesting that the former leads to the latter [29]. However, other genetic studies have identified similar close relationships, but suggested opposite direction of causality [30, 31]. Importantly, genetic studies may be undermined by the fact that genetic variants affect all tissues, while BCAA oxidation may have opposing effects in different tissues (see below).
-
4.
Pharmaceutical activation of BCKDH, leading to oxidative disposal of BCAA and reversal of BCAA elevations in the Zucker rat model of obesity and diabetes, leads to marked improvement in insulin resistance [32••]. However, this study also identified an alternate explanation for improvement in insulin resistance (see below).
Although none of these studies individually provides undeniable proof of causality, in aggregate, the studies provide robust support for the hypothesis that chronic elevations in BCAAs in humans and in rodent models contribute to the development of insulin resistance and diabetes. As noted, insulin resistance conversely likely predisposes to BCAA elevations by failing to fully suppress protein breakdown, thus potentially activating a vicious cycle.
The increasing evidence that BCAA elevations contribute to insulin resistance has led to concerted ongoing investigations by numerous labs into how this process occurs mechanistically. Two overarching questions arise: how do plasma BCAA elevations occur? And how do elevated BCAA levels lead to insulin resistance?
How Do Elevations in BCAAs Arise?
The best evidence to date argues for the inhibition of BCAA catabolism in specific tissues. Which tissues primarily contribute to BCAA catabolism under normal conditions is, surprisingly, controversial. Studies to date have largely relied on ex vivo measurements of BCAA catabolism, using tissue sections or extracts [1, 33]. These approaches, however, do not incorporate the numerous regulatory factors that exist in vivo, including physiological availability of substrates, presence of product inhibitors like NADH and acetyl-CoA, and subcellular localization and compartmentalization of key enzymes. More definitive studies using heavy isotope steady-state infusion studies in awake mice have recently demonstrated that in fact most tissues actively oxidize BCAAs, with the largest contribution likely from skeletal muscle and liver [34••]. Similar studies in humans have not been performed. In this context, adipose tissue and liver have been proposed to be the primary contributors to BCAA elevations (Fig. 2).
-
1.
Adipose tissue. Adipose-specific expression of Glut4 in mice led to systemic insulin resistance, decreased mRNA expression of BCAA catabolic genes, and a strong increase in circulating BCAA levels [35]. BCAA catabolic genes are also suppressed in adipose tissue in other models of obesity and insulin resistance [36], and in humans with diabetes and insulin resistance [37, 38]. Conversely, in humans, bariatric surgery-induced weight loss is associated with both reduced plasma BCAA and rises in adipose BCAT and BCKDH expression [39, 40]. Thus, a concerted suppression of BCAA catabolism in adipose tissue has been proposed as a cause of systemic elevations of BCAAs. ER stress, inflammation, and tissue hypoxia may all contribute to this suppression [41, 42], and TZDs, well-established insulin sensitizers, reverse this suppression in rats and humans [36, 43]. However, an important limitation of these models is that adipose tissue oxidizes only minimal quantities of BCAAs, likely less than 5% of whole-body oxidation even in obese models [34], suggesting that the increase in circulating BCAA levels must have additional contributors.
-
2.
Hepatic tissue. BCKDH activity is also decreased in the liver of obese and diabetic mice, although the mechanism differs from that in adipose tissue: BCKDK expression is increased, leading to increased inhibitory phosphorylation of BCKDH [22•, 32••, 39, 44]. Expression of BCKDK appears to be in part under regulation of ChREBP-beta, a carbohydrate-responsive transcription factor sensitive in particular to ingestion of dietary carbohydrates (including fructose), thus potentially linking modern high-fructose diets to elevations in plasma BCAAs [32••]. Adiponectin and other hormones may also modulate liver BCKDH activity [44], as well as neuronal circuits in response to brain insulin [45].
-
3.
Skeletal muscle. Interestingly, none of the studies above have detected changes in mRNA or protein expression, or of phosphorylation, of BCAT or BCKDH in skeletal muscle, despite the fact that muscle is the primary sink for glucose after a glucose load, and a predominant site of insulin resistance. To the contrary, elevations of C3 and C5 carnitines have been noted in skeletal muscle of diabetic mice, suggesting that skeletal muscle may in fact be a site of increased BCAA catabolism in states of insulin resistance [46]. This inference was recently strongly supported by the use of heavy isotope steady-state infusion studies in db/db mice, demonstrating a shift in tissue-specific BCAA catabolism away from adipose and hepatic tissues, and towards skeletal muscle [34••]. One recent study did, however, observe suppression of some genes of BCAA catabolism in skeletal muscle of patients with insulin resistance [47].

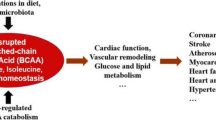
Integration of proposed mechanistic models for BCAA-induced insulin resistance. See text for details. BCKDK branched chain alpha keto acid dehydrogenase kinase, FA fatty acid, ACLY ATP citrate lyase, BCAA branched chain amino acid, Ox oxidation, mTOR mechanistic target of rapamycin, 3-HIB 3-hydroxyl isobutyrate, GDH glutamate dehydrogenase
There are other possible causes for increased BCAA levels. As noted above, mammals cannot synthesize BCAA. The diet may thus contribute, as the overall per capita daily consumption of calories has increased by ~ 20% over the last 50 years. This increase, however, is largely due to increased carbohydrates, rather than protein. Moreover, epidemiological studies are varied, with some studies indicating that a diet high in BCAAs can be associated with circulating BCAAs, while others not [14, 26, 46]. Recent work has also suggested a role for BCAA arising from synthesis by the intestinal microbiome. A small study in four monozygotic twin pairs discordant for obesity showed that obesity-associated microbiota had higher synthesis and lower catabolism of BCAAs [48]. Moreover, transfer of fecal matter from obese twins into germ-free mice led to increased body weight and plasma BCAAs, compared to fecal matter from lean siblings. A more recent study in 277 subjects supported these findings: elevated BCAAs in insulin resistant subjects correlated with a gut microbiome with enriched BCAA biosynthetic capacity, and inoculation of mice with one of these species induced insulin resistance and increased circulating BCAA levels [49]. Thus, microbiota appear able to modulate host circulating BCAA levels. However, none of these studies have demonstrated that plasma BCAA detected in the host were synthesized by the bacteria, leaving the equally likely possibility of indirect effects by the microbiome.
In summary, though diet and microbiome may contribute to elevations in BCAAs, the current data best support the model that suppression of BCAA catabolism in adipose and liver tissues, via still poorly defined mechanisms, leads to increased plasma BCAA levels, in turn shunting BCAA catabolism to skeletal muscle.
How Do Elevated BCAA Levels Lead to Insulin Resistance?
Numerous fundamental questions remain unanswered on this issue, including the following: Do elevations in BCAAs per se promote insulin resistance, or is the increased BCAA catabolism that results from elevated BCAAs to blame? What tissue bed is primarily responsible for promotion of insulin resistance in the face of elevated BCAAs? Which BCAA, if only one, is primarily responsible? We discuss below a number of proposed mechanisms and their implications for these questions (Fig. 2).
-
1.
mTOR. The lysosome-associated mTORC1 complex promotes cellular growth pathways in most cells, and is exquisitely sensitive to metabolic cues, including amino acids [50]. Of these, leucine most potently activates mTORC1, via direct binding to Sestrin2, itself a modulator of the mTORC1-associated GATOR complex [51•, 52•]. Elevated BCAAs can thus be expected to promote mTORC1 association, and indeed BCAA supplementation to rodent diet leads to activation of mTOR and downstream targets [11], and infusions of AAs in humans decreases insulin sensitivity in both muscle and liver, in parallel with activation of mTOR [18,19,20,21]. Moreover, treatment with rapamycin, a specific inhibitor of mTOR, partially reversed the insulin resistance caused by addition of BCAAs to HFD in rodents [11]. However, a primary role for leucine-activated mTOR in driving insulin resistance is not consistent with a number of other observations. For example, while diets supplemented with BCAAs generally worsen insulin resistance [11], diets supplemented with only leucine generally do not, and in fact often promote insulin sensitivity [21, 53,54,55,56]. The latter could be explained by the fact that leucine supplementation activates BCKDH, leading to lower levels of the other two BCAAs. If so, then the observations suggest that isoleucine or valine is primarily responsible for driving insulin resistance, rather than leucine, and thus not mTOR activation (which is primarily mediated by leucine).
-
2.
Insulin secretion. Chronic hyperinsulinemia has been suggested to be an early contributor to insulin resistance and diabetes, both by exerting constant pressure on the beta-cell leading to early failure, and by exerting pressure on peripheral insulin signaling leading to compensatory desensitization [57]. Elevations in insulin levels, especially post-prandial, occur early in the development of insulin resistance, before changes in glucose levels [58, 59]. BCAAs, in particular leucine, are potent insulin secretagogues, especially under conditions of normal or low glucose levels [60]. Thus, elevated BCAAs may contribute to chronic hyperinsulinemia, potentially promoting compensatory insulin resistance. As with the mTOR hypothesis, however, the model predicts that leucine would be a more potent driver of insulin resistance than the other two BCAAs, while experimentally the opposite appears to be true.
-
3.
Competition for fatty acid oxidation (FAO). The BCAA signatures observed in numerous epidemiological studies are often accompanied by elevations in C3 and C5 acylcarnitines, which are in equilibrium with incompletely oxidized products of BCAA catabolism, including propionyl CoA. These observations thus suggest that BCAA catabolism in insulin resistance is elevated, or at least improperly balanced with combustion of these carnitines, i.e., with TCA cycling. Excess BCAA catabolism may in turn compete with fatty acid beta-oxidation for entry of carbons into the TCA cycle, leading to accumulation of incompletely oxidized lipid intermediates and mitochondrial stress that promote insulin resistance in muscle and liver [46, 61]. This model, however, is difficult to reconcile with the relatively small contribution that BCAAs make to TCA carbons in muscle and liver even under normal physiological conditions [34••]. The model also predicts that levels of acetyl-CoA should increase, which to date has not been demonstrated in insulin resistant states.
-
4.
Glycine. BCAA restriction efficiently reversed muscle insulin resistance in fatty Zucker rats [22•]. Various metabolomics screens in these experiments yielded two prominent observations: BCAA restriction reversed the increase of muscle levels of even chain-length acyl-CoAs in ZF rats, and BCAA restriction similarly rescued the suppressed levels of glycine in ZF rat muscles. Low plasma levels of glycine have been repeatedly noted in human studies of insulin resistance, concordant with the elevations in BCAAs. Glycine can act as an extracellular carrier of incompletely oxidized fatty acids, in the form of acyl-glycines, and indeed levels of acyl-glycines were lower in urine of ZF rats [22•]. These observations led the authors to propose that less glycine in skeletal muscle compounds the accumulation of acyl-CoAs caused by competition between BCAA and fatty acid oxidation, because accumulated acyl-CoAs can less easily escape muscle as acyl-glycines. As above, the model depends on competition of BCAA oxidation with FAO to be sufficient to cause accumulation of acyl-CoAs.
-
5.
3-HIB. All BCAA catabolic products subsequent to the BCKDH step are covalently bound to CoA and thus trapped within the mitochondria, with the single exception of 3-HIB (Fig. 2). 3-HIB can thus escape mitochondria, and be secreted from the cell, likely in some proportion to active BCAA catabolic flux. Secreted 3-HIB stimulates capillary endothelial cells to promote trans-endothelial fatty acid transport, thereby increasing fatty acid flux to skeletal muscle [25•]. Thus, one potential model would suggest that elevated plasma BCAAs lead to increased muscle BCAA catabolism, which promotes 3-HIB secretion and trans-endothelial fatty acid transport, leading to accumulation of lipid species in muscle that drives insulin resistance. Administration of 3-HIB to mice promotes muscle accumulation of diacyl glycerols and insulin resistance [25•], and elevations of 3-HIB in plasma and urine correlate with present and future insulin resistance [62, 63]. 3-HIB is derived from valine. This model is thus consistent with the idea that catabolism of BCAAs, rather than levels per se, drives insulin resistance. The model is also consistent with the observations that leucine alone tends not to promote insulin resistance, while all three BCAAs together do. Interestingly, the effects of valine alone have not been reported, either in humans or animal models. On the other hand, this model is not immediately consistent with the observation that pharmacological inhibition of BCKDK improves insulin sensitivity, because the resulting increase in BCAA catabolism in skeletal muscle would be predicted to increase 3-HIB production, rather than decrease it [32••, 34••].
-
6.
Indirect effects. Recent work has suggested that liver BCKDK, in addition to regulating BCKDH, may also directly phosphorylate and activate ATP citrate lyase (ACLY), a rate-limiting enzyme for fatty acid synthesis [32••]. Like BCKDH, phosphorylation of ACLY is reversed by PPM1K. Thus, elevated BCKDK in the liver may promote fatty acid synthesis directly, thereby promoting steatosis and insulin resistance independently of its effects on BCAA catabolism. How these data relate to elevated BCAAs is not clear however. BCKAs, in particular aKIC (from leucine), allosterically suppress BCKDK activity on BCKDH. If elevated BCKAs also suppressed the activity of BCKDK on ACLY, as predicted, then high BCAAs would be predicted to improve insulin resistance and steatosis, rather than worsen it.
In summary, numerous mechanisms have been proposed to explain how elevations in BCAA promote insulin resistance, but none of them is yet proven. It is important to realize that in all likelihood multiple mechanisms occur simultaneously, perhaps explaining some of the apparent contradictions noted above. Returning to the three questions posed at the beginning of this section: the data thus far best support the notion that increased catabolism, in particular in skeletal muscle, and perhaps not of leucine but rather of isoleucine or valine, is the dominant mechanism contributing to BCAA-related insulin resistance and diabetes risk, but this notion is far from conclusive. Proof will at least require tissue-specific genetic modification of BCAA catabolism in skeletal muscle.
Conclusions
Strong evidence indicates that chronic elevations in BCAAs causally contribute to the development of insulin resistance in humans. How elevations in BCAAs occur remains poorly defined, but current data point to tissue-specific suppression of BCAA catabolism in adipose and hepatic tissues. How elevations in BCAAs cause insulin resistance also remains poorly understood, but current evidence points to excess BCAA catabolism in skeletal muscle, possibly simultaneously suppressing FAO and promoting fatty acid influx, leading to FA accumulation and lipotoxicity. Deeper understanding of either of these processes might identify new targets to get at the heart of insulin resistance.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Harper AE, Miller RH, Block KP. Branched-chain amino acid metabolism. Annu Rev Nutr. 1984;4:409–54.
Ichihara A, Koyama E. Transaminase of branched chain amino acids. I. Branched chain amino acids-alpha-ketoglutarate transaminase. J Biochem. 1966;59(2):160–9.
Paxton R, Harris RA. Isolation of rabbit liver branched chain alpha-ketoacid dehydrogenase and regulation by phosphorylation. J Biol Chem. 1982;257(23):14433–9.
Harris RA, Popov KM, Shimomura Y, Zhao Y, Jaskiewicz J, Nanaumi N, et al. Purification, characterization, regulation and molecular cloning of mitochondrial protein kinases. Adv Enzym Regul. 1992;32:267–84.
Damuni Z, Reed LJ. Purification and properties of the catalytic subunit of the branched-chain alpha-keto acid dehydrogenase phosphatase from bovine kidney mitochondria. J Biol Chem. 1987;262(11):5129–32.
Lu G, Sun H, She P, Youn JY, Warburton S, Ping P, et al. Protein phosphatase 2Cm is a critical regulator of branched-chain amino acid catabolism in mice and cultured cells. J Clin Invest. 2009;119(6):1678–87.
Adibi SA. Influence of dietary deprivations on plasma concentration of free amino acids of man. J Appl Physiol. 1968;25(1):52–7.
Felig P, Marliss E, Cahill GF Jr. Plasma amino acid levels and insulin secretion in obesity. N Engl J Med. 1969;281(15):811–6.
Guasch-Ferre M, Hruby A, Toledo E, Clish CB, Martinez-Gonzalez MA, Salas-Salvado J, et al. Metabolomics in prediabetes and diabetes: a systematic review and meta-analysis. Diabetes Care. 2016;39(5):833–46.
Wurtz P, Soininen P, Kangas AJ, Ronnemaa T, Lehtimaki T, Kahonen M, et al. Branched-chain and aromatic amino acids are predictors of insulin resistance in young adults. Diabetes Care. 2013;36(3):648–55.
Newgard CB, An J, Bain JR, Muehlbauer MJ, Stevens RD, Lien LF, et al. A branched-chain amino acid-related metabolic signature that differentiates obese and lean humans and contributes to insulin resistance. Cell Metab. 2009;9(4):311–26.
Walford GA, Ma Y, Clish C, Florez JC, Wang TJ, Gerszten RE, et al. Metabolite profiles of diabetes incidence and intervention response in the diabetes prevention program. Diabetes. 2016;65(5):1424–33.
Huffman KM, Shah SH, Stevens RD, Bain JR, Muehlbauer M, Slentz CA, et al. Relationships between circulating metabolic intermediates and insulin action in overweight to obese, inactive men and women. Diabetes Care. 2009;32(9):1678–83.
Tai ES, Tan ML, Stevens RD, Low YL, Muehlbauer MJ, Goh DL, et al. Insulin resistance is associated with a metabolic profile of altered protein metabolism in Chinese and Asian-Indian men. Diabetologia. 2010;53(4):757–67.
Palmer ND, Stevens RD, Antinozzi PA, Anderson A, Bergman RN, Wagenknecht LE, et al. Metabolomic profile associated with insulin resistance and conversion to diabetes in the insulin resistance atherosclerosis study. J Clin Endocrinol Metab. 2015;100(3):E463–8.
Shah SH, Crosslin DR, Haynes CS, Nelson S, Turer CB, Stevens RD, et al. Branched-chain amino acid levels are associated with improvement in insulin resistance with weight loss. Diabetologia. 2012;55(2):321–30.
Perng W, Gillman MW, Fleisch AF, Michalek RD, Watkins SM, Isganaitis E, et al. Metabolomic profiles and childhood obesity. Obesity (Silver Spring). 2014;22(12):2570–8.
Krebs M, Brehm A, Krssak M, Anderwald C, Bernroider E, Nowotny P, et al. Direct and indirect effects of amino acids on hepatic glucose metabolism in humans. Diabetologia. 2003;46(7):917–25.
Krebs M, Krssak M, Bernroider E, Anderwald C, Brehm A, Meyerspeer M, et al. Mechanism of amino acid-induced skeletal muscle insulin resistance in humans. Diabetes. 2002;51(3):599–605.
Tremblay F, Krebs M, Dombrowski L, Brehm A, Bernroider E, Roth E, et al. Overactivation of S6 kinase 1 as a cause of human insulin resistance during increased amino acid availability. Diabetes. 2005;54(9):2674–84.
Harris LLS, Smith GI, Patterson BW, Ramaswamy RS, Okunade AL, Kelly SC, et al. Alterations in 3-Hydroxyisobutyrate and FGF21 metabolism are associated with protein ingestion-induced insulin resistance. Diabetes. 2017;66(7):1871–8.
• White PJ, Lapworth AL, An J, Wang L, RW MG, Stevens RD, et al. Branched-chain amino acid restriction in Zucker-fatty rats improves muscle insulin sensitivity by enhancing efficiency of fatty acid oxidation and acyl-glycine export. Mol Metab. 2016;5(7):538–51. This study demonstrated that dietary restriction of BCAAs promotes insulin sensitivity.
Fontana L, Cummings NE, Arriola Apelo SI, Neuman JC, Kasza I, Schmidt BA, et al. Decreased consumption of branched-chain amino acids improves metabolic health. Cell Rep. 2016;16(2):520–30.
Cummings NE, Williams EM, Kasza I, Konon EN, Schaid MD, Schmidt BA, et al. Restoration of metabolic health by decreased consumption of branched-chain amino acids. J Physiol. 2018;596(4):623–45.
• Jang C, Oh SF, Wada S, Rowe GC, Liu L, Chan MC, et al. A branched-chain amino acid metabolite drives vascular fatty acid transport and causes insulin resistance. Nat Med. 2016;22(4):421–6. This study proposes a novel mechanism whereby excess BCAA catabolism in muscle leads to the secretion of signaling metabolites that promote fatty acid influx into muscle, leading to lipotoxicity and insulin resistance.
Wang TJ, Larson MG, Vasan RS, Cheng S, Rhee EP, McCabe E, et al. Metabolite profiles and the risk of developing diabetes. Nat Med. 2011;17(4):448–53.
Liu J, Semiz S, van der Lee SJ, van der Spek A, Verhoeven A, van Klinken JB, et al. Metabolomics based markers predict type 2 diabetes in a 14-year follow-up study. Metabolomics. 2017;13(9):104.
Merino J, Leong A, Liu CT, Porneala B, Walford GA, von Grotthuss M, et al. Metabolomics insights into early type 2 diabetes pathogenesis and detection in individuals with normal fasting glucose. Diabetologia. 2018;61(6):1315–24.
Lotta LA, Scott RA, Sharp SJ, Burgess S, Luan J, Tillin T, et al. Genetic predisposition to an impaired metabolism of the branched-chain amino acids and risk of type 2 diabetes: a Mendelian randomisation analysis. PLoS Med. 2016;13(11):e1002179.
Mahendran Y, Jonsson A, Have CT, Allin KH, Witte DR, Jorgensen ME, et al. Genetic evidence of a causal effect of insulin resistance on branched-chain amino acid levels. Diabetologia. 2017;60(5):873–8.
Wang Q, Holmes MV, Davey Smith G, Ala-Korpela M. Genetic support for a causal role of insulin resistance on circulating branched-chain amino acids and inflammation. Diabetes Care. 2017;40(12):1779–86.
•• White PJ, RW MG, Grimsrud PA, Tso SC, Yang WH, Haldeman JM, et al. The BCKDH kinase and phosphatase integrate BCAA and lipid metabolism via regulation of ATP-citrate lyase. Cell Metab. 2018;27(6):1281–93 e7. This study demonstrates that lowering BCAAs pharmaceutically can improve insulin resistance. The study also identifies regulation of BCKDK by carbohydrate signaling, and ACLY as a novel target of BCKDK, thereby integrating BCAA, lipid, and carbohydrate metabolism in the liver.
Brosnan JT, Brosnan ME. Branched-chain amino acids: enzyme and substrate regulation. J Nutr. 2006;136(1 Suppl):207S–11S.
•• Neinast MA, Jang C, Hui S, Murashige DS, Chu Q, Morscher RJ, et al. Whole-body metabolic fate of branched chain amino acids in health and insulin resistance. Cell Metab. 2018; in revision. This study provides an integrated and comprehensive description of whole-body BCAA catabolism, using steady-state heavy isotope infusion studies.
Herman MA, She P, Peroni OD, Lynch CJ, Kahn BB. Adipose tissue branched chain amino acid (BCAA) metabolism modulates circulating BCAA levels. J Biol Chem. 2010;285(15):11348–56.
Hsiao G, Chapman J, Ofrecio JM, Wilkes J, Resnik JL, Thapar D, et al. Multi-tissue, selective PPARgamma modulation of insulin sensitivity and metabolic pathways in obese rats. Am J Physiol Endocrinol Metab. 2011;300(1):E164–74.
Pietilainen KH, Naukkarinen J, Rissanen A, Saharinen J, Ellonen P, Keranen H, et al. Global transcript profiles of fat in monozygotic twins discordant for BMI: pathways behind acquired obesity. PLoS Med. 2008;5(3):e51.
Wiklund P, Zhang X, Pekkala S, Autio R, Kong L, Yang Y, et al. Insulin resistance is associated with altered amino acid metabolism and adipose tissue dysfunction in normoglycemic women. Sci Rep. 2016;6:24540.
She P, Van Horn C, Reid T, Hutson SM, Cooney RN, Lynch CJ. Obesity-related elevations in plasma leucine are associated with alterations in enzymes involved in branched-chain amino acid metabolism. Am J Physiol Endocrinol Metab. 2007;293(6):E1552–63.
Lips MA, Van Klinken JB, van Harmelen V, Dharuri HK, t Hoen PA, Laros JF, et al. Roux-en-Y gastric bypass surgery, but not calorie restriction, reduces plasma branched-chain amino acids in obese women independent of weight loss or the presence of type 2 diabetes. Diabetes Care. 2014;37(12):3150–6.
Burrill JS, Long EK, Reilly B, Deng Y, Armitage IM, Scherer PE, et al. Inflammation and ER stress regulate branched-chain amino acid uptake and metabolism in adipocytes. Mol Endocrinol. 2015;29(3):411–20.
Lo KA, Labadorf A, Kennedy NJ, Han MS, Yap YS, Matthews B, et al. Analysis of in vitro insulin-resistance models and their physiological relevance to in vivo diet-induced adipose insulin resistance. Cell Rep. 2013;5(1):259–70.
Sears DD, Hsiao G, Hsiao A, Yu JG, Courtney CH, Ofrecio JM, et al. Mechanisms of human insulin resistance and thiazolidinedione-mediated insulin sensitization. Proc Natl Acad Sci U S A. 2009;106(44):18745–50.
Lian K, Du C, Liu Y, Zhu D, Yan W, Zhang H, et al. Impaired adiponectin signaling contributes to disturbed catabolism of branched-chain amino acids in diabetic mice. Diabetes. 2015;64(1):49–59.
Shin AC, Fasshauer M, Filatova N, Grundell LA, Zielinski E, Zhou JY, et al. Brain insulin lowers circulating BCAA levels by inducing hepatic BCAA catabolism. Cell Metab. 2014;20(5):898–909.
Newgard CB. Interplay between lipids and branched-chain amino acids in development of insulin resistance. Cell Metab. 2012;15(5):606–14.
Lerin C, Goldfine AB, Boes T, Liu M, Kasif S, Dreyfuss JM, et al. Defects in muscle branched-chain amino acid oxidation contribute to impaired lipid metabolism. Mol Metab. 2016;5(10):926–36.
Ridaura VK, Faith JJ, Rey FE, Cheng J, Duncan AE, Kau AL, et al. Gut microbiota from twins discordant for obesity modulate metabolism in mice. Science. 2013;341(6150):1241214.
Pedersen HK, Gudmundsdottir V, Nielsen HB, Hyotylainen T, Nielsen T, Jensen BA, et al. Human gut microbes impact host serum metabolome and insulin sensitivity. Nature. 2016;535(7612):376–81.
Saxton RA, Sabatini DM. mTOR signaling in growth, metabolism, and disease. Cell. 2017;168(6):960–76.
• Saxton RA, Knockenhauer KE, Wolfson RL, Chantranupong L, Pacold ME, Wang T, et al. Structural basis for leucine sensing by the Sestrin2-mTORC1 pathway. Science. 2016;351(6268):53–8. This manuscript identified the primary molecular sensor for cytoplasmic leucine.
• Wolfson RL, Chantranupong L, Saxton RA, Shen K, Scaria SM, Cantor JR, et al. Sestrin2 is a leucine sensor for the mTORC1 pathway. Science. 2016;351(6268):43–8. This study and the previous one identify Sestrin2 as the molecular sensor for leucine-mediated activation of mTOR signaling.
Nairizi A, She P, Vary TC, Lynch CJ. Leucine supplementation of drinking water does not alter susceptibility to diet-induced obesity in mice. J Nutr. 2009;139(4):715–9.
Macotela Y, Emanuelli B, Bang AM, Espinoza DO, Boucher J, Beebe K, et al. Dietary leucine--an environmental modifier of insulin resistance acting on multiple levels of metabolism. PLoS One. 2011;6(6):e21187.
Guo K, Yu YH, Hou J, Zhang Y. Chronic leucine supplementation improves glycemic control in etiologically distinct mouse models of obesity and diabetes mellitus. Nutr Metab (Lond). 2010;7:57.
Pedroso JA, Zampieri TT, Donato J Jr. Reviewing the effects of L-leucine supplementation in the regulation of food intake, energy balance, and glucose homeostasis. Nutrients. 2015;7(5):3914–37.
Corkey BE. Banting lecture 2011: hyperinsulinemia: cause or consequence? Diabetes. 2012;61(1):4–13.
Cavaghan MK, Ehrmann DA, Polonsky KS. Interactions between insulin resistance and insulin secretion in the development of glucose intolerance. J Clin Invest. 2000;106(3):329–33.
Roberts CK, Hevener AL, Barnard RJ. Metabolic syndrome and insulin resistance: underlying causes and modification by exercise training. Compr Physiol. 2013;3(1):1–58.
Wilson DF, Cember ATJ, Matschinsky FM. Glutamate dehydrogenase: role in regulating metabolism and insulin release in pancreatic beta-cells. J Appl Physiol. 1985;2018
Koves TR, Ussher JR, Noland RC, Slentz D, Mosedale M, Ilkayeva O, et al. Mitochondrial overload and incomplete fatty acid oxidation contribute to skeletal muscle insulin resistance. Cell Metab. 2008;7(1):45–56.
Haufe S, Engeli S, Kaminski J, Witt H, Rein D, Kamlage B, et al. Branched-chain amino acid catabolism rather than amino acids plasma concentrations is associated with diet-induced changes in insulin resistance in overweight to obese individuals. Nutr Metab Cardiovasc Dis. 2017;27(10):858–64.
Mardinoglu A, Gogg S, Lotta LA, Stancakova A, Nerstedt A, Boren J, et al. Elevated plasma levels of 3-Hydroxyisobutyric acid are associated with incident type 2 diabetes. EBioMedicine. 2018;27:151–5.
Funding
The authors are supported by grants from the NIH: T32 GM-07229 (MN), and R01 DK107667, DK114103 (ZA).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Zolt Arany and Michael Neinast declare that they have no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Pathogenesis of Type 2 Diabetes and Insulin Resistance
Rights and permissions
About this article

Cite this article
Arany, Z., Neinast, M. Branched Chain Amino Acids in Metabolic Disease. Curr Diab Rep 18, 76 (2018). https://doi.org/10.1007/s11892-018-1048-7
Published:
DOI: https://doi.org/10.1007/s11892-018-1048-7