
Abstract
Purpose of Review
There is interest in evaluating the developmental origins of health and disease (DOHaD) which emphasizes the role of prenatal and early-life environments on non-communicable health outcomes throughout the life course. The ability to rigorously assess and identify early-life risk factors for later health outcomes, including those with childhood onset, in large population samples is often limited due to measurement challenges such as impractical costs associated with prospective studies with a long follow-up duration, short half-lives for some environmental toxicants, and lack of biomarkers that capture inter-individual differences in biologic response to external environments.
Recent Findings
Epigenomic patterns, and DNA methylation in particular, have emerged as a potential objective biomarker to address some of these study design and exposure measurement challenges. In this article, we summarize the literature to date on epigenetic changes associated with specific prenatal and early-life exposure domains as well as exposure mixtures in human observational studies and their biomarker potential. Additionally, we highlight evidence for other types of epigenetic patterns to serve as exposure biomarkers.
Summary
Evidence strongly supports epigenomic biomarkers of exposure that are detectable across the lifespan and across a range of exposure domains. Current and future areas of research in this field seek to expand these lines of evidence to other environmental exposures, to determine their specificity, and to develop predictive algorithms and methylation scores that can be used to evaluate early-life risk factors for health outcomes across the life span.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
The developmental origins of health and disease (DOHaD) hypothesis is rooted in understanding the role of the early-life environment on health outcomes across the lifespan. This hypothesis stems from evidence built over decades demonstrating that environments encountered in utero and during early life are associated with future non-communicable disease risk [1,2,3]. Most DOHaD studies to date have focused on growth, nutrition, stress, and other exposures that are feasible to measure using questionnaire or medical or other record-based data due to practical challenges with prospectively collecting exposure data from the prenatal and early-life windows and health outcomes in childhood, adolescence, and adulthood. Additional challenges to rigorously evaluating DOHaD include the short biologic half-life of many existing environmental toxicants (e.g., cotinine, phthalates, bisphenol A), self-report or external measures may not capture inter-individual differences in biologic response to an exposure or the “internal dose” experienced, and harmonization across studies due to different exposure measures. For these reasons, our ability to fully assess and identify prenatal and early-life risk factors associated with disease across the life course is limited. Thus, there is a need for effective molecular biomarkers that can be used to predict an individual’s exposure history and are harmonizable across studies to identify early-life exposures associated with future disease risk.
Ideal biomarkers are specific, long-lasting in the body, and reflect the dose of exposure. Additionally, they must be present in easily accessible biospecimens [4]. Contrary to mechanistic studies that would require examination of an affected tissue to determine the relationship between an exposure and a health outcome, assessment of an affected tissue is not required to establish an effective biomarker. However, given that many molecular modifications that are often used as biomarkers are necessarily different across tissue types, this must be accounted for and considered when establishing and implementing biomarkers, and when looking across tissue types. Additionally, many substances have short half-lives; thus, more long-lasting molecular changes that are specific to an exposure can be a more accurate representation of an individual’s past exposure. Thus, the ease and accessibility of the measurement matrix is critical, and tissues such as blood, cord blood, and saliva, are often used in studies of biomarkers. These features of a biomarker are especially important when considering how the environment encountered during critical windows of development, such as early life, can influence future health and disease. Epigenetic modifications provide a possible measure of prenatal and childhood exposures that could be used to fill this purpose.
Epigenetic modifications are mitotically heritable components of cellular material that do not result in an underlying change to the DNA sequence [5]. Multiple types of epigenetic modifications exist including DNA methylation, histone modifications, non-coding RNAs, and chromatin structure. These modifications play critical roles in cellular functions and organismal development and are reversible. Studies have shown that many of these modifications differ by environmental contexts and undergo drastic changes particularly during developmental periods [5].
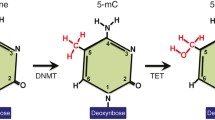
DNA methylation is the best-studied epigenetic modification. DNA methylation is characterized by the addition of a methyl or hydroxymethyl group at the 5’ carbon position of a cytosine base when a cytosine is followed by a guanine in the nucleotide sequence [6]. We refer to this as a CpG site. Over the past decade, evidence has mounted showing DNA methylation is responsive to prenatal and early-life environments, is often specific to a particular exposure, and can persist across the life course and is detectable later in life[4], making it a useful biomarker of exposure.
Other epigenetic modifications may also have utility as biomarkers including post-translational modifications of histones (PTMs) and non-coding RNAs (ncRNAs) [7, 8]. Inside the cell, DNA is packaged into chromatin by wrapping 147 base pairs around an octamer of histone proteins [7]. Covalent modifications such as methylation, acetylation, ubiquitylation, and phosphorylation can be made to the N- or C-terminal tails of histones to regulate DNA compaction and accessibility to transcriptional machinery [7]. It is well-established that PTMs change in response to the environment [9]. Relative to DNA methylation and PTMs, ncRNAs are a more recently studied type of epigenetic modification. While ncRNAs do not encode genes, they do possess unique, and diverse functions inside of the cell, though we are learning quickly that their content can change in response to environmental cues in an exposure and temporally specific manner [8].
In this review, we will provide a summary of the current literature describing how prenatal and early-life environmental exposures, across several exposure domains, have been shown to impact epigenetic patterns, measured from birth through adulthood, in human observational studies. Our exposure domains include environmental toxicants, human behaviors, and social environments experienced during the prenatal, perinatal, and early childhood windows. We also summarize emerging evidence for epigenetic patterns to capture exposure mixtures and statistical tools to enable exposure prediction and algorithms to build methylation scores that can be utilized as an exposure dosimeter/measure of current and/or past exposure.
Methods
The two main databases queried for this literature review were PubMed and Google Scholar. We restricted most of our search to the last 5 years (2017–2022, inclusive). For each search, we used combinations of keywords, examples of which are provided below. For exposures, we used keywords that referred to the broad class of exposure (e.g., “Pesticide,” “Metal,” “Phthalates,” “Smoking,” “Socioeconomic Position”) as well as individual toxicants or factors (e.g., “Lead,” “Folic Acid,” “BPA,” “Stress,” “Tobacco”). We used these terms in combination with the exposure window (e.g., “Maternal,” “in utero,” “childhood”), and the measurement window (e.g., “birth,” “childhood,” “adolescence,” “adulthood”). We also used terms specific to the tissue type (e.g., “cord blood,” “placenta,” “saliva,” “blood,” “buccal cell”) that the epigenetic measurement was performed in. Terms specific to the type of epigenetic modification included “methylation,” “chromatin,” “histone,” “microRNA,” and “ncRNA,” and “mixtures” was included when identifying studies of mixtures. An example of a complete search is as follows: “‘prenatal’ ‘maternal smoking’ ‘DNA Methylation’ ‘cord blood’.” For a comprehensive list of search terms used, please see Supplementary File 1.
Results
We queried the literature to identify reports of DNA methylation changes, present in any biospecimen type at birth, infancy (< 1 year of age), childhood, adolescence, or in adulthood, associated with prenatal and/or early-life exposure to environmental factors with a focus on studies from 2017 to 2022. We did not assess inter- or transgenerational inheritance, nor did we summarize studies in animal or in vitro models because we focus on relevance to human studies seeking to utilize epigenetic biomarkers in epidemiology framework. We report these findings in Table 1, organized by the following exposure domains: (1) toxicant environmental exposures which included metals, endocrine disrupting compounds (EDCs), air pollutants, persistent organic pollutants (POPs), and pesticides; (2) behavioral including smoking (tobacco and nicotine), alcohol, illicit substance use, and diet and nutrition; and (3) social environmental factors including stress, socioeconomic position, and adverse childhood experiences (ACEs). Tables 2 and 3 report studies of early-life exposure across the same domains that identify methylation changes resulting from exposure to environmental mixtures, or that report changes in epigenetic material other than DNA methylation, respectively.
DNA Methylation Changes Associated with Specific Environmental Factors
Most studies across all environmental exposures are focused on the effects of prenatal exposure on methylation changes in tissues collected at birth (Table 1). More than 20 studies assessed the impact of maternal metal exposure on DNA methylation changes at birth, though just two studies examined the impact of childhood metal exposure on the methylome [95, 96], identifying a gap in research that should be addressed. One study specifically assessed the persistent effects of prenatal mercury exposure on DNA methylation across the life course through repeated biospecimen measures and demonstrated that blood methylation changes associated with prenatal mercury exposure persisted into early and mid-childhood [10••].
As shown in Table 1, more than 15 human observational studies conducted in the last 5 years have documented DNA methylation changes in tissues collected from birth, childhood, or adolescence related to prenatal exposure to EDCs including phthalates [33,34,35,36,37,38, 45, 46], bisphenols [33, 39,40,41,42,43, 47, 49, 50, 239, 240], and flame retardants [48]. Though most of the literature is focused on prenatal exposure to these chemicals (Table 1), work from the Spanish INMA-Granada birth cohort identified peripheral blood DNA methylation changes at the brain-derived neurotrophic factor (BDNF) locus in adolescents with childhood bisphenol A (BPA) exposure [100•].
A recent epigenome-wide meta-analysis was among the first to identify several differentially methylated CpG sites in newborns that are significantly associated with prenatal particulate air pollution exposure [51•]. These CpG sites were annotated to genes previously implicated in lung-related health outcomes [51•]. Exposure to air pollution during childhood is associated with methylation changes in children themselves (Table 1), but we were unable to identify studies that examined whether DNA methylation patterns associated with prenatal or childhood exposure to air pollution could be detected in adolescent or adult biospecimens.
Similar to the other classes of environmental toxicants, most of the persistent organic pollutant (POP) literature focused on prenatal exposure and methylation patterns at birth [74,75,76,77,78,79,80,81,82,83,84,85,86,87] (Table 1). In these studies, maternal urine or serum was assayed for detection of PFAS [75, 76, 81,82,83, 87], PAHs [77, 78, 80], and PCBs [74, 79, 84, 85] to determine exposure levels during this critical window. One longitudinal study identified significant associations between prenatal PCB and polychlorinated dibenzofuran (PCDF) exposure levels and blood DNA methylation patterns at 20 CpG sites in early adulthood [88•]. A second recent study assessed the impact of childhood PFAS exposure on blood DNA methylation levels in school-age children; the authors identified 12 differentially methylated CpG sites and 7 differentially methylated genomic regions associated with higher levels of PFAS exposure [107].
We identified only six studies in the last 5 years that assessed the relationship between prenatal or childhood pesticide exposure and DNA methylation patterns (Table 1). Four of those studies assessed cord blood methylation [89,90,91,92] and one examined child blood methylation [93]. Recent work from the Child Health and Development Study detected DNA methylation changes at genes involved in pubertal development and breast cancer susceptibility in adulthood that were associated with prenatal DDT exposure [94•]. These results provide initial evidence to support that DNA methylation patterns, from birth through adulthood, reflect prenatal pesticide exposure and have the potential to serve as a pesticide exposure biomarker.
Across all exposure types, additional, more robust studies are needed to determine whether methylation changes resulting from a particular exposure are 1) specific to the type of chemical or toxicant; 2) persistent and thus consistently detectable across life stages; and 3) detectable across tissue types from easily accessible biological specimens. Methylation changes that meet these criteria would serve as useful biomarkers of exposure and could help inform an individual’s future health and disease risk.
DNA Methylation Changes Associated with Human Behaviors
There is strong evidence to support the utility of DNA methylation as a biomarker of prenatal smoking exposure [108,109,110,111,112,113, 114••, 115,116,117,118,119,120,121,122,123,124,125,126,127, 129,130,131,132, 133••, 134] (Table 1). Multiple studies have identified consistent significant differential methylation in the aryl hydrocarbon receptor repressor (AHRR) gene [110,111,112,113, 117, 122]. Given the reproducibility of this finding, methylation changes at a specific locus within this gene are established and accepted as a biomarker of fetal exposure to tobacco smoke. Studies from the Avon Longitudinal Study of Parents and Children (ALSPAC) have identified persistent methylation changes across the life course associated with prenatal smoking exposure that are independent of postnatal and personal smoking exposures [241, 242••]. Unsurprisingly, there were no studies on the effects of childhood smoking on methylation patterns given tobacco smoking in children is rare. We did however identify one study assessing the effect of childhood secondhand smoke exposure on blood DNA methylation, though no significant associations were detected[128].
Five studies assessed the associations between prenatal exposure to alcohol and methylation patterns at birth [136,137,138,139,140], and just three studies examined methylation changes in children prenatally exposure to alcohol (Table 1). Candidate gene methylation studies in placenta analyzed imprinting at the IFG2/H19 locus [139]. In children prenatally exposed to alcohol, persistent methylation changes were detected in saliva [141], blood [141], and buccal cells [142••, 143]. No longitudinal studies assessed the effects of this early-life exposure on the methylome in adolescence or adulthood. Additionally, we did not identify studies assessing the effect of childhood alcohol use on methylation across the life course likely given the limited consumption of alcohol by children.
Few studies have assessed maternal substance use during pregnancy and DNA methylation changes. This may be due, in part, to the illicit nature of these substances leading to reporting biases by mothers, and perhaps other social confounding factors that may make it more difficult for pregnant mothers experiencing substance use issues to seek care and/or be enrolled in studies. We identified six studies that assessed the impact of exposure to opioids, cocaine, methamphetamine, and cannabis on neonatal DNA methylation patterns (Table 1). Severe maternal addiction to crack cocaine was shown to predict oxytocin receptor (OXTR) gene DNA methylation patterns in newborns [145], and babies born to opioid-dependent mothers sustained on methadone treatment had increased buccal cell methylation at the Opioid Receptor Mu 1 (OPRM1) gene [146, 148]. We identified just one epigenome-scale association study that identified DNA methylation changes in placenta that differed in opioid exposed versus unexposed infants [144]. Children with prenatal methamphetamine exposure had hypermethylated hydroxysteroid 11-beta dehydrogenase 2 (HSD11B2) buccal cell DNA relative to those without exposure [149].
As shown in Table 1, studies of prenatal diet and nutrition status were primarily associated with methylation changes at birth [150,151,152,153,154,155,156,157,158,159,160,161,162,163,164]. Dietary fat intake during pregnancy was associated with imprinted gene status in newborns [151], while folic acid supplementation for the entire duration of pregnancy was associated with methylation change at genes involved in neurodevelopment at birth [154]. A nested studying within the Aberdeen Folic Acid Supplementation Trial assessed methylation changes in adults who were prenatally exposed to two different doses of folic acid supplementation. Methylation changes at individual CpG sites, as well as across regions of the genome, were identified in buccal cells from adult offspring, demonstrating the persistence of the effects of this prenatal exposure [169••]. Further, comparing methylation changes from low and high dose-offspring identified a dose–response relationship between folic acid supplementation and the degree of methylation change, emphasizing the specificity of methylation change to the prenatal exposure [169••]. Given the widespread use of vitamins and supplements during pregnancy, studies should assess methylation changes across multiple life stages and should compare methylation changes across tissue types to establish effective biomarkers of prenatal nutrient status.
DNA Methylation Changes Associated with Social Environmental Factors
Prenatal exposure to several different stressors has been associated with changes in DNA methylation patterns collected at birth (Table 1). Placental methylation was measured in 92 samples from the FinnBrain Birth Cohort, whose mothers were screened for psychosocial stress and depressive symptoms during early pregnancy [179]. Maternal depressive symptoms were associated with DNA methylation at 2833 CpG sites [179]. In one study, the relationship between prenatal exposure to stress from a natural disaster and persistent methylation changes in adolescents was identified in participants from Project Ice Storm [243].
Inequities in socioeconomic position (SEP) are associated with poor health outcomes in individuals. SEP can be influenced by factors including education, occupation, income level, neighborhood, and race or ethnicity [244]. While SEP is comprised of multiple factors, several studies assessed the influence of individual SEP characteristics on DNA methylation patterns at birth [183••, 184•, 185, 188] (Table 1). An EWAS from the Extremely Low Gestational Age Newborns (ELGAN) determined that marital status, receiving supplemental nutrition assistance, education level, and health insurance status were each independently associated with placental DNA methylation at birth [188]. Higher levels of cord blood methylation at the repetitive element LINE-1 were associated with living in the most impoverished neighborhood in a Mexican–American birth cohort of 241 maternal-infant pairs [185]. Women from the Project Viva cohort who were married or cohabitating with a partner had infants with lower levels of cord blood methylation. This prospective study followed infants through early and mid-childhood and demonstrated significant associations between maternal education and global DNA methylation at both stages of life [183••]. The ALSPAC cohort performed an epigenome-scale association study of DNA methylation changes at birth, childhood, and adolescence associated with markers of SEP during pregnancy and found that maternal education was associated with cord blood methylation at birth and methylation changes in adolescence [184•].
While the relationship between adverse childhood experiences (ACEs) and childhood and adolescent methylation was assessed in seven studies, most of the literature in this domain assessed the long-lasting effect of ACEs on methylation changes in adulthood (Table 1). At both time points, altered methylation of NR3C1 was identified as being associated with ACEs [196, 197, 199]. Genome-wide studies on the effects of ACEs on the adult methylome were performed together on blood samples from ALSPAC and buccal cells from the MRC National Survey of Health and Development (NSHD) study [200••]. Nine DMRs were significantly associated with a specific ACE measure across both NSHD and ALSPAC, including one that was associated with a cumulative measure of adversity [200••]. It is notable that these DMRs were replicated between two different cohorts and across tissue types—blood or buccal—suggesting that methylation changes resulting from childhood adversity are stable over time, specific to the exposure, and are detectable across tissues.
DNA Methylation Patterns Associated with Exposure to Mixtures
Biomarkers that reflect exposure to mixtures can be used as a reduced complexity measure to capture the complex mixtures of chemicals that individuals are exposed to daily. This is especially important given that the mixture of chemicals that individuals regularly encounter may influence health outcomes differently as a mixture than they do as individual chemicals. Although most studies to date have focused on identifying DNA methylation patterns related to single exposures, recent statistical and exposome measurement advances have allowed researchers to begin to assess the impacts of environmental mixtures on DNA methylation patterns [245, 246]. As detailed below, mixtures studies to date have largely focused on chemical toxicant mixtures, some studies have examined multiple social exposures, and to our knowledge, no studies have examined behavioral exposure mixtures in relation to DNA methylation. Social exposure mixtures were primarily defined by composite SEP, a summative score that accounts for the individual socioenvironmental factors that comprise SEP. Future studies should continue to use newly developed statistical methods (such as Bayesian kernel machine regression) to evaluate environmental mixtures and its effect on the epigenome.
Associations between prenatal exposure to a 16-metal mixture and cord blood DNA methylation were assessed using the Bayesian kernel machine regression (BKMR) model[213••] (Table 2). A BKMR statistical model allows the joint effect of mixtures on methylation to be estimated in a way that is superior to standard regression approaches as it has features to account for the correlation structure of environmental mixtures, allowing it to overcome challenges from traditional analysis methods such as collinearity and high dimensionality [246]. No significant associations were observed between the metal mixtures and DNA methylation [213••]. Another study from the Norwegian Mother, Father, and Child Cohort assessed the effect of prenatal exposure to a mixture of 12 metals and essential elements on cord blood methylation in 631 mother–child pairs [214] (Table 2). Two-way interactions between exposures were investigated using elastic net regression, and the joint effects of the mixture on DNA methylation were assessed using quantile g-computation, a novel analysis approach that allows for an unbiased inference on the effects of mixtures [245]. Elastic net regression identified significant associations between cord blood methylation and maternal selenium, cobalt, and mercury exposure, individually [214]. However, despite the application of two novel statistical approaches to identifying effects of exposure mixtures, no associations between prenatal metals mixtures and DNA methylation were observed.
The effect of prenatal exposure to mixtures of EDCs on DNA methylation has primarily been assessed in placenta. However, two studies took candidate gene approaches, investigating the relationship between mixtures of xenoestrogens on cord blood methylation at repetitive elements [215], as well as the effects phthalate and phenol exposures on imprinted genes in the placenta [216] (Table 2). For placenta, epigenome-scale association approaches identified methylation changes in the placenta that were associated with prenatal exposure to a mixture of xenoestrogens [217] or other POPs mixtures [79, 85, 86] (Table 2). In one study, multi-pollutant principal component analysis identified significant associations between a mixture of 57 POPs measured in maternal serum and placental DNA methylation at the insulin-line growth factor 2 (IGF2) imprinted locus [85]. Another multi-pollutant study used principal component analysis to evaluate the association between prenatal exposure to multiple POPs and DNA methylation in the placenta [86]. These results demonstrate that exposure to POPs mixtures is associated with DNA methylation changes, supporting the potential for DNA methylation patterns to serve as a biomarker of POPs exposure mixtures.
In addition to environmental mixtures, studies have focused on the effect of composite early-life SEP—a way to assess the cumulative impact of the factors that contribute to socioeconomic status and adversity—on DNA methylation across the life course [186, 187] (Table 2). Work from the NEST cohort demonstrated that hypermethylation of the DLK1/MEG3 imprinting locus in cord blood samples was associated with lower composite SEP, where SEP was assessed as a principal components factor of six individual SEP measures [218]. In addition to work from Project Viva looking at how individual SEP factors associated with global DNA methylation, this study also detected locus specific methylation changes at birth and in childhood that were associated with prenatal composite SEP. Low prenatal SEP was associated with DNA methylation of the gene Leucine Rich Repeat Neuronal 4 (LRRN4) at birth, and those methylation changes persisted into early childhood [183••]. Studies of the association between childhood composite SEP and DNA methylation identified methylation changes in children themselves [189], as well as persistent changes to the methylome in blood [190, 191] and adipose tissues (Table 2). A study from the Cebu Longitudinal Health and Nutrition Survey—a pregnancy cohort of women from the Philippines—identified epigenome-wide leukocyte methylation changes in adults that were associated with their composite SEP in childhood [191].
In sum, there is evidence showing DNA methylation changes are associated with exposure to EDC and POPs mixtures, as well as composite SEP. As next steps, it will be important to determine whether these changes are persistent over time and may have utility in helping us understand how these mixtures may contribute to adverse health outcomes, especially in the context of how they may compare to the effects of individual chemical toxicants on health outcomes. Future research is also needed to determine the specificity of these DNA methylation changes and to investigate other types of exposure mixtures, including across other exposure domains and windows of exposure.
Posttranslational Histone Modification and Noncoding RNA Changes Associated with Environmental Exposures
To date, studies investigating DNA methylation changes associated with prenatal and early-life exposures have largely focused on DNA methylation. This is mainly due to the ease and stability of collecting this measure in epidemiology samples, commercially available cost-effective measurement methods, and because we understand it best at a biologic level. However, other types of epigenetic material, such as posttranslational modifications of histones (PTMs), and noncoding RNAs (ncRNAs), have also been shown to differ by environmental context and play important roles in development and health. As our understanding of how these molecular modifications change in response to the environment expands, we highlight a largely unmet opportunity to consider whether non-methylation-based epigenetic changes may have utility as an exposure biomarker in human epidemiologic studies. Below, and in Table 3, we summarize the evidence to date showing prenatal and early-life exposure associations with PTMs and ncRNAs.
We found limited evidence of any studies investigating the association between prenatal metal exposure and PTMs at any developmental period. We identified one study that examined the relationship between prenatal cadmium exposure and long noncoding RNA (lncRNA) expression in placenta[221•] (Table 3). There was a significant relationship between prenatal cadmium concentrations and expression of four lncRNAs in placenta [221•]. Bioinformatic analysis demonstrated that MIR22HG, the lncRNA whose placental expression was most significantly associated with prenatal cadmium exposure, was associated with gene modules involved in organ development and cellular respiration.
Prenatal exposure to phthalates and phenols has been associated with placental ncRNA expression [222,223,224,225] (Table 3). A study that co-enrolled women from the Harvard Epigenetic Birth Cohort and the Predictors of Preeclampsia Study assessed the relationship between exposure to multiple EDCs and placental micro RNA (miRNA)expression [225]. Maternal urine was collected to measure phthalate and phenol metabolites, and targeted expression of 29 miRNAs was measured via real-time quantitative reverse transcription polymerase chain reaction [225]. Of the 29 miRNAs assessed, three had significant associations with urinary phthalate or phenol metabolite levels [225]. Other studies identified associations between prenatal phthalate exposure and placental lncRNA expression [222, 223]. Additional comprehensive studies are needed to establish ncRNA biomarkers of early-life EDC exposure across multiple tissue types and life stages. Future work should also assess the impact of EDC exposure on PTMs.
Prenatal gas and particulate air pollution exposure was associated with altered expression of a cluster of miRNAs in cord blood [226], while studies of childhood air pollution exposure detected changes in miRNA expression in serum [228•] and saliva [229] (Table 3). We found just one study that identified PTM changes in cord blood that were associated with prenatal exposure to ambient air pollution [227] (Table 3). This recent work assessed histone H3 methylation in cord blood from 609 mother-newborn pairs and found significant changes associated with PM2.5 and black carbon exposure during pregnancy [227].
As with the DNA methylation data available for early-life exposure to pesticides, epidemiologic studies on the effects of pesticide exposure on PTMs or ncRNAs were limited, and population-based studies of the epigenetic consequences of exposure to a pesticide mixture were sparse. We identified one recent study that examined the relationship between in utero exposure to chlordecone and its impact on PTMs in cord blood [92] (Table 3). A prospective study in the French West Indies collected cord blood samples at birth for PTM analyses, and maternal plasma was assayed for chlordecone concentration. Quantitative immunofluorescence analysis of PTMs in the nuclei of cord blood cells and found significant decreases in H3K4me3 and H3K9me3 levels in cord blood from chlordecone exposed individuals relative to the unexposed controls [92]. This work combined their epidemiologic work with in vitro approaches to experimentally validate their findings and assess the relationship between histone occupancies and changes in gene expression [92].
While studies on the impact of smoking on the methylome are extensive, there is a dearth of epidemiologic literature on the effects of prenatal smoking on PTMs or ncRNAs. Work from the LINA mother–child cohort assessed the role of maternal smoking on chromatin architecture and PTMs in children around four years of age [232••] (Table 3). Chromatin immunoprecipitation sequencing (ChIP-Seq) was performed in blood for six mother–child pairs (three who smoked, three who did not). Two activating and two repressive histone modifications were assessed, and the authors identified a shift from a repressive to an activate chromatin state in cord blood from children prenatally exposed to maternal smoking relative to controls [232••]. Though their sample size was small, this represents one of the few studies that has looked at the effect of prenatal smoking on PTMs, and their findings suggest that additional work is warranted. One study of miRNAs assessed expression of two microRNAs in 441 cord blood samples from a prospective mother–child study of prenatal tobacco smoke exposure [230] (Table 3). They found altered miR-223 expression in cord blood following prenatal tobacco smoke exposure, that was further associated with low numbers of regulatory T cells at birth, and increased allergy risk in childhood [230]. A second study of maternal cigarette smoking assessed the expression of four miRNAs in 25 placentas samples [231] (Table 3). Despite a smaller sample size, authors found that expression of three of the four miRNAs—miR-16, miR-21, and mi-146a—was downregulated in placentas from smoke-exposed individuals compared to controls [231].
Two studies identified associations between prenatal alcohol exposure and microRNA content at birth [233, 234] (Table 3). Plasma extracellular circulating miRNAs were isolated from infants from the Cape Town cohort of mothers and infants [234]. Alcohol-consuming mothers drank on average 1–2 days/week during pregnancy. Offspring miRNA expression was altered in plasma from 2-week-old and 6.5-month-old infants. Confirmatory factor analysis identified three-factor models that explained the variance in expression of miRNAs associated with prenatal alcohol expression at both time points[234].
Micro-RNAs (miRNAs) and histone modification changes related to childhood trauma have been detected in adults, and thus may serve as a useful biomarker [247] (Table 3). Peripheral blood measures of miR-15a showed elevated levels of expression in adults with childhood trauma compared to age and gender matched individuals with no history of early-life trauma or stress [236] (Table 3). More recently, downregulation of miRNA 125b-1-3p was observed in blood from adults that experienced childhood trauma compared to those who did not [237] (Table 3). Related work on the epigenetic eraser, histone-deacetylase 1 (HDAC1) demonstrated elevated levels of HDAC1 expression in patients with schizophrenia who experienced early-life stress compared to patients who did not [238]. Finally, analysis of six PTMs in the amygdala of postmortem brain samples showed changes in the histone landscape in individuals who experienced severe childhood trauma compared to controls [235] (Table 3).
Development of Epigenetic Prediction Tools
In line with demonstrating the utility of DNA methylation as an exposure biomarker, studies have built epigenetic prediction tools that use DNA methylation measures to estimate an individual’s current and previous exposures. These tools are extremely powerful because they provide an opportunity to accurately determine whether an individual incurred an exposure, or to what extent they were exposed to a chemical or stressor, when that data is otherwise unattainable. Additionally, aggregate methylation score tools can have several advantages over traditional exposure ascertainment measures because (1) they include a collection of methylation loci to represent a particular exposure or multiple exposures which can provide greater statistical efficiency over individual CpGs, and (2) they may capture inter-individual biologic responses to similar measures of external exposures, i.e., serve as an “exposure dosimeter” [248, 249].
There are two main approaches to working with methylation-based predictive tools. The first is based on the development and implementation of a methylation score. Here, CpG sites that were previously determined to be significantly associated with an environmental exposure are used to build a single numeric score that is a weighted sum of the selected CpG methylation levels, often using methylation data from an epigenome-scale analysis. This score is then used in linear regression models to determine an association with an exposure of interest [248, 249]. A study from the Norwegian Mother and Child Cohort Study (MoBa) developed and tested a numeric DNA methylation score using 28 CpG sites associated with sustained smoke exposure. [114••]. When applied to a test dataset of cord blood samples, their numeric methylation score was able to predict prenatal smoke exposure with a greater than 90% specificity [114••]. A methylation score of prenatal smoke exposure that persisted into adulthood was developed from the ALSPAC cohort. DNA methylation changes at 568 CpG sites that were previously demonstrated to be associated with prenatal smoke exposure in cord blood were used to create a DNA methylation score to successfully predict whether adults sustained prenatal smoke exposure in utero (AUC 0.69) [133••]. A second score from the same study was derived from 19 CpG sites from children’s peripheral blood that were associated with prenatal smoke exposure. This score was even more successful at predicting prenatal smoke exposure in adult blood samples (AUC 0.72), despite being derived from childhood biospecimens rather than newborn tissues [133••].
In addition to score-based prediction tools that can be used as biomarkers of exposure, machine learning approaches can be used to detect patterns in methylation data in an unsupervised manner that can ultimately be used to predict an individual’s exposure status [248, 249]. Work from the Study to Explore Early Development (SEED) was among the first to use predictive tools to determine whether DNA methylation changes at 26 CpG sites associated with prenatal smoke exposure in cord blood could similarly predict prenatal smoke exposure in childhood [250••]. Using a support vector machine classifier, with tenfold cross validation, prenatal smoke exposure was predicted in children with a greater than 86% accuracy when it was compared to maternal reports of smoking during pregnancy [250••]. Machine learning techniques have also been used to predict fetal alcohol exposure in children and adolescents. DNA methylation changes at 648 CpG sites initially identified in buccal cells from children and adolescents with fetal alcohol spectrum disorder (FASD) were used in a gradient boosting machine learning model to generate an algorithm capable of predicting FASD status in an independent set of individuals [142••]. When tested on buccal cell samples from children and adolescents, the algorithm was able to predict dichotomous FASD status with a specificity of 75% [142••].
Discussion
There are potential advantages to using epigenetic biomarkers instead of traditional external exposure measures. First, epigenetic changes can reflect an internal dosimeter or represent the biologic response to exposure which could explain inter-individual variation. Second, epigenetic changes that are specific to an exposure can address exposure misclassification such as incorrect self-report or other reporting errors or biases. Third, epigenetic analyses of environmental mixtures can overcome statistical concerns and difficulties modeling multi-exposures.
Epigenetic biomarkers need to be assessed in easily accessible tissues [4]. At birth, this typically includes cord blood and placenta tissues. We saw that the majority of studies presented in this review were from cord blood samples collected at birth, from which epigenetic measures were generated. Later in life, saliva and saliva-derived buccal cells, and whole blood, represent two tissue types that can be easily sampled across different stages of life to measure epigenetic biomarkers of exposure. This is consistent with the tissues that were commonly sampled in childhood, adolescence, and adulthood in the studies that we reviewed here. The work summarized in this review provided excellent examples of the utility of epigenetic biomarkers of early-life exposure that are detectable across tissue types and life stages, that are specific to the exposure of interest, and that are predictive of disease outcomes. This emphasizes the effectiveness of epigenetic exposure biomarkers and motivates future research to continue to use exposure biomarkers to identify potential interventions that could mitigate disease risk.
Literature on epigenetic biomarkers of early-life exposure is overwhelmingly focused on DNA methylation. This was true across all exposure domains, exposure timepoints, and measurement timepoints included in this review. However, there were a limited number of observational studies that followed participants from birth through adolescence or adulthood, with repeated sampling of biospecimens to measure methylation changes over time. One question that remains is whether persistent methylation changes associated with prenatal exposures were similarly identifiable following childhood exposures. The data presented here is somewhat inconclusive. For example, childhood metal exposure was associated with methylation changes at novel loci that had not previously been identified following prenatal exposures [95, 96]. Conversely, prenatal and childhood exposures to stress were associated with DNA methylation changes at the NR3C1 locus [243, 251,252,253,254,255], demonstrating that methylation changes here seemed to be more specific to the type of exposure rather than the developmental window that the exposure occurred. Similarly, childhood exposure to BPA was associated with BDNF methylation [100•], and work in animal models found that gestational BPA exposure in mice was associated with persistent DNA methylation changes in offspring of Bdnf, and that boys with high levels of BPA exposure in utero had aberrant BDNF methylation in cord blood [100•, 256]. Contrary to the metals studies, this suggests that perhaps for BPA exposure, the epigenetic effects are specific to the chemical exposure and not necessarily the timing of the exposure. Finally, childhood air pollution was associated with differential methylation of nitric oxide synthase genes, a finding that was similarly detectable in adults with exposure to air pollution [257, 258]. To establish effective biomarkers, rigorous investigations are needed to identify whether methylation patterns associated with environmental influences are more specific to a class of chemicals or a developmental window of exposure.
While the importance of DNA methylation studies in advancing the field of epigenetics cannot be overstated, we were surprised to see the shortage of studies that assessed how early-life exposures during the in utero and childhood time periods were associated with non-methylation epigenetic changes, particularly histone PTMs and ncRNAs. In these instances, work in animal models far surpasses epidemiologic studies [247, 259, 260]. However, those studies provide a strong scientific rationale for investigating the effects of exposure on epigenetic patterns in population-based work. The studies we did find highlight associations between PTMs [7, 89, 92, 227, 235] and ncRNAs [221•, 223, 226, 229, 230, 237] and exposures. Thus, future studies should focus on development and implementation of non-methylation-based biomarkers of early-life exposure.
Recent advances in the development and implementation of epigenetic prediction tools have demonstrated the ability for composite DNA methylation changes to reconstruct an individual’s exposure history when that data is otherwise unavailable. Once developed and tested for accuracy and reliability, these predictive tools can be applied to observational studies to characterize an individual’s exposure history, and to better understand their potential future disease risk. Multiple studies have demonstrated the utility of these numeric methylation scores and machine learning predictive algorithms at predicting prenatal exposures across the life course, and across tissue types [114••, 133••, 142••, 250••]. However, additional methylation tools are needed to comprehensively predict early-life exposures across the three domains covered in this review. Further, these tools should be used to advance the development of biomarkers of the exposome to better understand an individual’s comprehensive exposure history and future disease risk.
There are unique advantages and disadvantages to the methylation score and machine learning approaches to predictive modeling of biomarker development. For example, one disadvantage to using DNA methylation scores is that the algorithm requires a mean methylation value for the “exposed” and “unexposed” groups. Thus, it cannot be used to predict exposure status when that data is otherwise unavailable. One could use the mean values that the score was initially developed with in the test dataset, but the inter-experiment conditions, such as the presence of batch effects and other laboratory variables, could greatly impede the accuracy of this approach. However, an advantage to using methylation scores is that they can reflect an internal dosimeter of an individual’s exposure when quantifying an exposure in a biospecimen might not be feasible. Given that methylation scores are weighted, a higher score, for example, might be reflective of a higher level of exposure to a certain toxicant earlier in life. Machine learning approaches do not have this same capability. The output from these predictive algorithms is typically dichotomous, i.e., “yes” or “no,” for a particular exposure or outcome of interest, so no individual metrics about the magnitude of an exposure would be ascertained in that case. However, machine learning is advantageous given that once the algorithm is trained, no exposure history data is needed to predict the binary outcome of whether an individual is or is not exposed. Investigators need to carefully consider the data they have available, and the scientific question that they are trying to address, when choosing an approach to developing biomarkers through predictive modeling.
Conclusions
In summary, the evidence provided in this review strongly supports epigenomic biomarkers of exposure are detectable across the lifespan across a range of exposure domains. Of note, we did not address the transgenerational effects of early-life exposure on the epigenome, or how inheritance of an aberrant epigenetic profile might be associated with disease risk in subsequent generations. Studies have begun to develop biomarkers for intergenerational and transgenerational exposures, and future work should be supported in this area. We have identified promising new directions that would address current gaps in research. These include the ability to capture an epigenetic biomarker of the exposome through mixtures analysis, the derivation and implementation of non-methylation biomarkers of exposure, and the development and use of epigenetic prediction tools. Together, these research avenues will help advance our understanding of how exposures early in life shape health and disease across a generation.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Gillman MW. Developmental origins of health and disease. N Engl J Med. 2005;353(17):1848–50.
Gluckman PD, Hanson MA, Cooper C, et al. Effect of in utero and early-life conditions on adult health and disease. N Engl J Med. 2008;359(1):61–73.
Barker DJ. The origins of the developmental origins theory. J Intern Med. 2007;261(5):412–7.
Ladd-Acosta C. Epigenetic signatures as biomarkers of exposure. Curr Environ Health Rep. 2015;2(2):117–25.
Dolinoy DC, Jirtle RL. Environmental epigenomics in human health and disease. Environ Mol Mutagen. 2008;49(1):4–8.
Greenberg MVC, Bourc’his D. The diverse roles of DNA methylation in mammalian development and disease. Nat Rev Mol Cell Biol. 2019;20(10):590–607.
Zhou VW, Goren A, Bernstein BE. Charting histone modifications and the functional organization of mammalian genomes. Nat Rev Genet. 2011;12(1):7–18.
Watson CN, Belli A, Di Pietro V. Small non-coding RNAs: new class of biomarkers and potential therapeutic targets in neurodegenerative disease. Front Genet. 2019;10:364.
Dai HWZ. Histone modification patterns and their responses to environment. Curr Environ Health Rep. 2014;1:11–21.
•• Cardenas A, Rifas-Shiman SL, Agha G, et al. Persistent DNA methylation changes associated with prenatal mercury exposure and cognitive performance during childhood. Sci Rep. 2017;7(1):288. (Study assessed the persistent effects of prenatal mercury exposure on DNA methylation across multiple life stages.)
Bozack AK, Rifas-Shiman SL, Coull BA, et al. Prenatal metal exposure, cord blood DNA methylation and persistence in childhood: an epigenome-wide association study of 12 metals. Clin Epigenetics. 2021;13(1):208.
Park J, Kim J, Kim E, et al. Prenatal lead exposure and cord blood DNA methylation in the Korean Exposome Study. Environ Res. 2021;195: 110767.
Herrera-Moreno JF, Estrada-Gutierrez G, Wu H, et al. Prenatal lead exposure, telomere length in cord blood, and DNA methylation age in the PROGRESS prenatal cohort. Environ Res. 2022;1(205): 112577.
Cardenas A, Rifas-Shiman SL, Godderis L, et al. Prenatal exposure to mercury: associations with global DNA Methylation and hydroxymethylation in cord blood and in childhood. Environ Health Perspect. 2017;125(8): 087022.
Montes-Castro N, Alvarado-Cruz I, Torres-Sanchez L, et al. Prenatal exposure to metals modified DNA methylation and the expression of antioxidant- and DNA defense-related genes in newborns in an urban area. J Trace Elem Med Biol. 2019;55:110–20.
Wu S, Hivert MF, Cardenas A, et al. Exposure to low levels of lead in utero and umbilical cord blood DNA methylation in project viva: an epigenome-wide association study. Environ Health Perspect. 2017;125(8): 087019.
Lozano M, Yousefi P, Broberg K, et al. DNA methylation changes associated with prenatal mercury exposure: A meta-analysis of prospective cohort studies from PACE consortium. Environ Res. 2022;204(Pt B): 112093.
Koh EJ, Yu SY, Kim SH, Lee JS, Hwang SY. Prenatal exposure to heavy metals affects gestational age by altering DNA methylation patterns. Nanomaterials (Basel). 2021;11(11):2871. https://doi.org/10.3390/nano11112871.
Heiss JA, Tellez-Rojo MM, Estrada-Gutierrez G, et al. Prenatal lead exposure and cord blood DNA methylation in PROGRESS: an epigenome-wide association study. Environ Epigenet. 2020;6(1):dvaa014.
Rygiel CA, Dolinoy DC, Perng W, et al. Trimester-specific associations of prenatal lead exposure with infant cord blood DNA methylation at birth. Epigenet Insights. 2020;13:2516865720938669.
Rygiel CA, Dolinoy DC, Bakulski KM, et al. DNA methylation at birth potentially mediates the association between prenatal lead (Pb) exposure and infant neurodevelopmental outcomes. Environ Epigenet. 2021;7(1):dvab005.
Bozack AK, Cardenas A, Quamruzzaman Q, et al. DNA methylation in cord blood as mediator of the association between prenatal arsenic exposure and gestational age. Epigenetics. 2018;13(9):923–40.
House JS, Hall J, Park SS, et al. Cadmium exposure and MEG3 methylation differences between Whites and African Americans in the NEST Cohort. Environ Epigenet. 2019;5(3):dvz014.
Kaushal A, Zhang H, Karmaus WJJ, et al. Genome-wide DNA methylation at birth in relation to in utero arsenic exposure and the associated health in later life. Environ Health. 2017;16(1):50.
Nishizawa-Jotaki S, Sakurai K, Eguchi A, et al. Association between mercury in cord serum and sex-specific DNA methylation in cord tissues. J Dev Orig Health Dis. 2021;12(1):124–31.
Montrose L, Goodrich JM, Morishita M, Kochmanski J, Klaver Z, Cavalcante R, Lumeng JC, Peterson KE, Dolinoy DC. Neonatal Lead (Pb) Exposure and DNA methylation profiles in dried bloodspots. Int J Environ Res Public Health. 2020;17(18):6775. https://doi.org/10.3390/ijerph17186775.
Appleton AA, Jackson BP, Karagas M, et al. Prenatal exposure to neurotoxic metals is associated with increased placental glucocorticoid receptor DNA methylation. Epigenetics. 2017;12(8):607–15.
Tian FY, Everson TM, Lester B, et al. Selenium-associated DNA methylation modifications in placenta and neurobehavioral development of newborns: an epigenome-wide study of two U.S. birth cohorts. Environ Int. 2020;137:105508.
Gliga AR, Engstrom K, Kippler M, et al. Prenatal arsenic exposure is associated with increased plasma IGFBP3 concentrations in 9-year-old children partly via changes in DNA methylation. Arch Toxicol. 2018;92(8):2487–500.
Cediel Ulloa A, Gliga A, Love TM, et al. Prenatal methylmercury exposure and DNA methylation in seven-year-old children in the Seychelles Child Development Study. Environ Int. 2021;147: 106321.
Rygiel CA, Goodrich JM, Solano-Gonzalez M, et al. Prenatal lead (Pb) exposure and peripheral blood DNA methylation (5mC) and hydroxymethylation (5hmC) in Mexican Adolescents from the ELEMENT Birth Cohort. Environ Health Perspect. 2021;129(6):67002.
Bozack AK, Boileau P, Wei L, et al. Exposure to arsenic at different life-stages and DNA methylation meta-analysis in buccal cells and leukocytes. Environ Health. 2021;20(1):79.
Montrose L, Padmanabhan V, Goodrich JM, et al. Maternal levels of endocrine disrupting chemicals in the first trimester of pregnancy are associated with infant cord blood DNA methylation. Epigenetics. 2018;13(3):301–9.
Solomon O, Yousefi P, Huen K, et al. Prenatal phthalate exposure and altered patterns of DNA methylation in cord blood. Environ Mol Mutagen. 2017;58(6):398–410.
Miura R, Ikeda-Araki A, Ishihara T, et al. Effect of prenatal exposure to phthalates on epigenome-wide DNA methylations in cord blood and implications for fetal growth: The Hokkaido Study on Environment and Children’s Health. Sci Total Environ. 2021;20(783): 147035.
Huang LL, Zhou B, Ai SH, et al. Prenatal phthalate exposure, birth outcomes and DNA methylation of Alu and LINE-1 repetitive elements: A pilot study in China. Chemosphere. 2018;206:759–65.
Chen CH, Jiang SS, Chang IS, et al. Association between fetal exposure to phthalate endocrine disruptor and genome-wide DNA methylation at birth. Environ Res. 2018;162:261–70.
Tindula G, Murphy SK, Grenier C, et al. DNA methylation of imprinted genes in Mexican-American newborn children with prenatal phthalate exposure. Epigenomics. 2018;10(7):1011–26.
Junge KM, Leppert B, Jahreis S, et al. MEST mediates the impact of prenatal bisphenol A exposure on long-term body weight development. Clin Epigenetics. 2018;10:58.
McCabe CF, Padmanabhan V, Dolinoy DC, et al. Maternal environmental exposure to bisphenols and epigenome-wide DNA methylation in infant cord blood. Environ Epigenet. 2020;6(1):dvaa021.
Huang YF, Chang CH, Chen PJ, Lin IH, Tsai YA, Chen CF, Wang YC, Huang WY, Tsai MS, Chen ML. Prenatal bisphenol a exposure, DNA methylation, and low birth weight: A pilot study in Taiwan. Int J Environ Res Public Health. 2021;18(11):6144. https://doi.org/10.3390/ijerph18116144.
Miura R, Araki A, Minatoya M, et al. An epigenome-wide analysis of cord blood DNA methylation reveals sex-specific effect of exposure to bisphenol A. Sci Rep. 2019;9(1):12369.
Song X, Zhou X, Yang F, et al. Association between prenatal bisphenol a exposure and promoter hypermethylation of CAPS2, TNFRSF25, and HKR1 genes in cord blood. Environ Res. 2020;190: 109996.
Mattonet K, Nowack-Weyers N, Vogel V, et al. Prenatal exposure to endocrine disrupting chemicals is associated with altered DNA methylation in cord blood. Epigenetics. 2021;16:1–18.
Jedynak P, Tost J, Calafat AM, et al. Pregnancy exposure to phthalates and DNA methylation in male placenta - an epigenome-wide association study. Environ Int. 2022;160: 107054.
Grindler NM, Vanderlinden L, Karthikraj R, et al. Exposure to phthalate, an endocrine disrupting chemical, alters the first trimester placental methylome and transcriptome in women. Sci Rep. 2018;8(1):6086.
Song X, Wang Z, Zhang Z, et al. Differential methylation of genes in the human placenta associated with bisphenol A exposure. Environ Res. 2021;200: 111389.
Zhao Y, Song Q, Ge W, et al. Associations between in utero exposure to polybrominated diphenyl ethers, pathophysiological state of fetal growth and placental DNA methylation changes. Environ Int. 2019;133(Pt B): 105255.
Engdahl E, Svensson K, Lin PD, et al. DNA methylation at GRIN2B partially mediates the association between prenatal bisphenol F exposure and cognitive functions in 7-year-old children in the SELMA study. Environ Int. 2021;156: 106617.
Choi YJ, Lee YA, Hong YC, et al. Effect of prenatal bisphenol A exposure on early childhood body mass index through epigenetic influence on the insulin-like growth factor 2 receptor (IGF2R) gene. Environ Int. 2020;143: 105929.
• Gruzieva O, Xu CJ, Yousefi P, et al. Prenatal particulate air pollution and DNA Methylation in newborns: an epigenome-wide meta-analysis. Environ Health Perspect. 2019;127(5):57012. (A recent epigenome-wide meta-analysis that was among the first to identify several differentially methylated CpG sites in newborns that are significantly associated with prenatal particulate air pollution exposure.)
Wang C, Plusquin M, Ghantous A, et al. DNA methylation of insulin-like growth factor 2 and H19 cluster in cord blood and prenatal air pollution exposure to fine particulate matter. Environ Health. 2020;19(1):129.
Zhou G, He T, Huang H, et al. Prenatal ambient air pollution exposure and SOD2 promoter methylation in maternal and cord blood. Ecotoxicol Environ Saf. 2019;15(181):428–34.
Isaevska E, Fiano V, Asta F, et al. Prenatal exposure to PM10 and changes in DNA methylation and telomere length in cord blood. Environ Res. 2022;19(209): 112717.
Feng F, Huang L, Zhou G, et al. GPR61 methylation in cord blood: a potential target of prenatal exposure to air pollutants. Int J Environ Health Res. 2022;32(2):463–72.
Liu X, Ye Y, Chen Y, et al. Effects of prenatal exposure to air particulate matter on the risk of preterm birth and roles of maternal and cord blood LINE-1 methylation: A birth cohort study in Guangzhou, China. Environ Int. 2019;133(Pt A): 105177.
Gruzieva O, Xu CJ, Breton CV, et al. Epigenome-wide meta-analysis of methylation in children related to prenatal NO2 air pollution exposure. Environ Health Perspect. 2017;125(1):104–10.
Ladd-Acosta C, Feinberg JI, Brown SC, et al. Epigenetic marks of prenatal air pollution exposure found in multiple tissues relevant for child health. Environ Int. 2019;126:363–76.
Cho HJ, Lee SH, Lee SY, et al. Mid-pregnancy PM2.5 exposure affects sex-specific growth trajectories via ARRDC3 methylation. Environ Res. 2021;200:111640.
Li Z, Yang M, Duan L, et al. The neonatal PROC gene rs1799809 polymorphism modifies the association between prenatal air pollutants exposure and PROC promoter methylation. Environ Sci Pollut Res Int. 2022;29(10):14575–83.
Isaevska E, Moccia C, Asta F, et al. Exposure to ambient air pollution in the first 1000 days of life and alterations in the DNA methylome and telomere length in children: A systematic review. Environ Res. 2021;193: 110504.
Wang Y, Perera F, Guo J, et al. A methodological pipeline to generate an epigenetic marker of prenatal exposure to air pollution indicators. Epigenetics. 2022;17(1):32–40.
Yang SI, Lee SH, Lee SY, et al. Prenatal PM2.5 exposure and vitamin D-associated early persistent atopic dermatitis via placental methylation. Ann Allergy Asthma Immunol. 2020;125(6):665-673 e1.
Maghbooli Z, Hossein-Nezhad A, Adabi E, et al. Air pollution during pregnancy and placental adaptation in the levels of global DNA methylation. PLoS ONE. 2018;13(7): e0199772.
Abraham E, Rousseaux S, Agier L, et al. Pregnancy exposure to atmospheric pollution and meteorological conditions and placental DNA methylation. Environ Int. 2018;118:334–47.
Cai J, Zhao Y, Liu P, et al. Exposure to particulate air pollution during early pregnancy is associated with placental DNA methylation. Sci Total Environ. 2017;31(607–608):1103–8.
Saenen ND, Vrijens K, Janssen BG, et al. Lower placental leptin promoter methylation in association with fine particulate matter air pollution during pregnancy and placental nitrosative stress at birth in the ENVIRONAGE Cohort. Environ Health Perspect. 2017;125(2):262–8.
Nawrot TS, Saenen ND, Schenk J, et al. Placental circadian pathway methylation and in utero exposure to fine particle air pollution. Environ Int. 2018;114:231–41.
Engstrom K, Mandakh Y, Garmire L, et al. Early pregnancy exposure to ambient air pollution among late-onset preeclamptic cases is associated with placental DNA hypomethylation of specific genes and slower placental maturation. Toxics. 2021;9(12):338. https://doi.org/10.3390/toxics9120338.
Neven KY, Saenen ND, Tarantini L, et al. Placental promoter methylation of DNA repair genes and prenatal exposure to particulate air pollution: an ENVIRONAGE cohort study. Lancet Planet Health. 2018;2(4):e174–83.
Deyssenroth MA, Rosa MJ, Eliot MN, et al. Placental gene networks at the interface between maternal PM2.5 exposure early in gestation and reduced infant birthweight. Environ Res. 2021;199:111342.
Merid SK, Bustamante M, Standl M, et al. Integration of gene expression and DNA methylation identifies epigenetically controlled modules related to PM2.5 exposure. Environ Int. 2021;146:106248.
Bakulski KM, Fisher JD, Dou JF, et al. Prenatal Particulate matter exposure is associated with saliva DNA methylation at age 15: applying cumulative DNA methylation scores as an exposure biomarker. Toxics. 2021;9(10):262. https://doi.org/10.3390/toxics9100262.
Kobayashi S, Sata F, Miyashita C, et al. Gender-specific association of exposure to non-dioxin-like polychlorinated biphenyls during pregnancy with methylation levels of H19 and long interspersed nuclear element-1 in cord blood in the Hokkaido study. Toxicology. 2017;1(390):135–45.
Starling AP, Liu C, Shen G, et al. Prenatal exposure to per- and polyfluoroalkyl substances, umbilical cord blood DNA methylation, and cardio-metabolic indicators in newborns: The Healthy Start Study. Environ Health Perspect. 2020;128(12): 127014.
Miura R, Araki A, Miyashita C, et al. An epigenome-wide study of cord blood DNA methylations in relation to prenatal perfluoroalkyl substance exposure: The Hokkaido study. Environ Int. 2018;115:21–8.
Yang P, Gong YJ, Cao WC, et al. Prenatal urinary polycyclic aromatic hydrocarbon metabolites, global DNA methylation in cord blood, and birth outcomes: A cohort study in China. Environ Pollut. 2018;234:396–405.
Cao C, Jia Z, Shao M, et al. Prenatal exposure to polycyclic aromatic hydrocarbons could increase the risk of low birth weight by affecting the DNA methylation states in a Chinese cohort. Reprod Biol. 2021;21(4): 100574.
Leung YK, Ouyang B, Niu L, et al. Identification of sex-specific DNA methylation changes driven by specific chemicals in cord blood in a Faroese birth cohort. Epigenetics. 2018;13(3):290–300.
Lee J, Kalia V, Perera F, et al. Prenatal airborne polycyclic aromatic hydrocarbon exposure, LINE1 methylation and child development in a Chinese cohort. Environ Int. 2017;99:315–20.
Liu CY, Chen PC, Lien PC, Liao YP. Prenatal perfluorooctyl sulfonate exposure and alu DNA hypomethylation in cord blood. Int J Environ Res Public Health. 2018;15(6):1066. https://doi.org/10.3390/ijerph15061066.
Kingsley SL, Kelsey KT, Butler R, et al. Maternal serum PFOA concentration and DNA methylation in cord blood: A pilot study. Environ Res. 2017;158:174–8.
Kobayashi S, Azumi K, Goudarzi H, et al. Effects of prenatal perfluoroalkyl acid exposure on cord blood IGF2/H19 methylation and ponderal index: The Hokkaido Study. J Expo Sci Environ Epidemiol. 2017;27(3):251–9.
Eguchi A, Nishizawa-Jotaki S, Tanabe H, Rahmutulla B, Watanabe M, Miyaso H, Todaka E, Sakurai K, Kaneda A, Mori C. An altered DNA methylation status in the human umbilical cord is correlated with maternal exposure to polychlorinated biphenyls. Int J Environ Res Public Health. 2019;16(15):2786. https://doi.org/10.3390/ijerph16152786.
Kim S, Cho YH, Lee I, et al. Prenatal exposure to persistent organic pollutants and methylation of LINE-1 and imprinted genes in placenta: A CHECK cohort study. Environ Int. 2018;119:398–406.
Kim S, Cho YH, Won S, et al. Maternal exposures to persistent organic pollutants are associated with DNA methylation of thyroid hormone-related genes in placenta differently by infant sex. Environ Int. 2019;130: 104956.
Ouidir M, Mendola P, Buck Louis GM, et al. Concentrations of persistent organic pollutants in maternal plasma and epigenome-wide placental DNA methylation. Clin Epigenetics. 2020;12(1):103.
• Su KY, Li MC, Lee NW, et al. Perinatal polychlorinated biphenyls and polychlorinated dibenzofurans exposure are associated with DNA methylation changes lasting to early adulthood: Findings from Yucheng second generation. Environ Res. 2019;170:481–6. (Prenatal PCB/PCDF exposure associated with DNA methlyaiton in adulthood - demonstrates ability to measure methylation changes in adults from an early-life exposure.)
Chiu KC, Sisca F, Ying JH, et al. Prenatal chlorpyrifos exposure in association with PPARgamma H3K4me3 and DNA methylation levels and child development. Environ Pollut. 2021;1(274): 116511.
Yu X, Zhao B, Su Y, et al. Association of prenatal organochlorine pesticide-dichlorodiphenyltrichloroethane exposure with fetal genome-wide DNA methylation. Life Sci. 2018;1(200):81–6.
Huen K, Solomon O, Kogut K, et al. PON1 DNA methylation and neurobehavior in Mexican-American children with prenatal organophosphate exposure. Environ Int. 2018;121(Pt 1):31–40.
Legoff L, D'Cruz SC, Bouchekhchoukha K, et al. In utero exposure to chlordecone affects histone modifications and activates LINE-1 in cord blood. Life Sci Alliance. 2021;4(6):e202000944. https://doi.org/10.26508/lsa.202000944.
Declerck K, Remy S, Wohlfahrt-Veje C, et al. Interaction between prenatal pesticide exposure and a common polymorphism in the PON1 gene on DNA methylation in genes associated with cardio-metabolic disease risk-an exploratory study. Clin Epigenetics. 2017;9:35.
• Wu HC, Cohn BA, Cirillo PM, et al. DDT exposure during pregnancy and DNA methylation alterations in female offspring in the Child Health and Development Study. Reprod Toxicol. 2020;92:138–47. (A study of prenatal pesticide exposure and DNA methylation changes in adulthood, demonstrating the ability for methylation changes assocaited with early-life exposures to be detected later in life.)
Gonzalez-Cortes T, Recio-Vega R, Lantz RC, et al. DNA methylation of extracellular matrix remodeling genes in children exposed to arsenic. Toxicol Appl Pharmacol. 2017;15(329):140–7.
Xu L, Huo X, Liu Y, et al. Hearing loss risk and DNA methylation signatures in preschool children following lead and cadmium exposure from an electronic waste recycling area. Chemosphere. 2020;246: 125829.
Wang WR, Chen NT, Hsu NY, et al. Associations among phthalate exposure, DNA methylation of TSLP, and childhood allergy. Clin Epigenetics. 2021;13(1):76.
Bowman A, Peterson KE, Dolinoy DC, et al. Phthalate Exposures, DNA methylation and adiposity in Mexican children through adolescence. Front Public Health. 2019;7:162.
Yang CF, Karmaus WJJ, Yang CC, et al. Bisphenol a exposure, DNA methylation, and asthma in children. Int J Environ Res Public Health. 2020;17(1):298. https://doi.org/10.3390/ijerph17010298.
• Mustieles V, Rodriguez-Carrillo A, Vela-Soria F, et al. BDNF as a potential mediator between childhood BPA exposure and behavioral function in adolescent boys from the INMA-Granada cohort. Sci Total Environ. 2022;10(803): 150014. (Identified DNA methylation changes in adolescents that were associated with childhood BPA exposure.)
Ji N, Fang M, Baptista A, et al. Exposure to traffic-related air pollution and changes in exhaled nitric oxide and DNA methylation in arginase and nitric oxide synthase in children with asthma. Environ Health. 2021;20(1):12.
Prunicki M, Cauwenberghs N, Lee J, et al. Air pollution exposure is linked with methylation of immunoregulatory genes, altered immune cell profiles, and increased blood pressure in children. Sci Rep. 2021;11(1):4067.
Prunicki M, Stell L, Dinakarpandian D, et al. Exposure to NO2, CO, and PM2.5 is linked to regional DNA methylation differences in asthma. Clin Epigenetics. 2018;10:2. https://doi.org/10.1186/s13148-017-0433-4.
Alvarado-Cruz I, Sanchez-Guerra M, Hernandez-Cadena L, et al. Increased methylation of repetitive elements and DNA repair genes is associated with higher DNA oxidation in children in an urbanized, industrial environment. Mutat Res Genet Toxicol Environ Mutagen. 2017;813:27–36.
Sordillo JE, Cardenas A, Qi C, et al. Residential PM2.5 exposure and the nasal methylome in children. Environ Int. 2021;153:106505.
Zhang X, Biagini Myers JM, Burleson JD, et al. Nasal DNA methylation is associated with childhood asthma. Epigenomics. 2018;10(5):629–41.
Xu Y, Lindh CH, Fletcher T, Jakobsson K, Engström K. Perfluoroalkyl substances influence DNA methylation in school-age children highly exposed through drinking water contaminated from firefighting foam: a cohort study in Ronneby, Sweden. Environ Epigenet. 2022;8(1):dvac004. https://doi.org/10.1093/eep/dvac004.
Howe CG, Zhou M, Wang X, et al. Associations between maternal tobacco smoke exposure and the cord blood [Formula: see text] DNA Methylome. Environ Health Perspect. 2019;127(4):47009.
Fuemmeler BF, Dozmorov MG, Do EK, et al. DNA methylation in babies born to nonsmoking mothers exposed to secondhand smoke during pregnancy: an epigenome-wide association study. Environ Health Perspect. 2021;129(5):57010.
Bergens MA, Pittman GS, Thompson IJB, et al. Smoking-associated AHRR demethylation in cord blood DNA: impact of CD235a+ nucleated red blood cells. Clin Epigenetics. 2019;11(1):87.
Miyake K, Kawaguchi A, Miura R, et al. Association between DNA methylation in cord blood and maternal smoking: The Hokkaido Study on Environment and Children’s Health. Sci Rep. 2018;8(1):5654.
Zhang B, Hong X, Ji H, et al. Maternal smoking during pregnancy and cord blood DNA methylation: new insight on sex differences and effect modification by maternal folate levels. Epigenetics. 2018;13(5):505–18.
Xu R, Hong X, Zhang B, et al. DNA methylation mediates the effect of maternal smoking on offspring birthweight: a birth cohort study of multi-ethnic US mother-newborn pairs. Clin Epigenetics. 2021;13(1):47.
•• Reese SE, Zhao S, Wu MC, et al. DNA methylation score as a biomarker in newborns for sustained maternal smoking during pregnancy. Environ Health Perspect. 2017;125(4):760–6. (Provides an example of the development and implementation of a DNA methylation score to predict prenatal smoke exposure.)
Witt SH, Frank J, Gilles M, et al. Impact on birth weight of maternal smoking throughout pregnancy mediated by DNA methylation. BMC Genomics. 2018;19(1):290.
Miyake K, Miyashita C, Ikeda-Araki A, et al. DNA methylation of GFI1 as a mediator of the association between prenatal smoking exposure and ADHD symptoms at 6 years: the Hokkaido Study on Environment and Children’s Health. Clin Epigenetics. 2021;13(1):74.
Monasso GS, Jaddoe VWV, de Jongste JC, et al. Timing- and dose-specific associations of prenatal smoke exposure with newborn DNA methylation. Nicotine Tob Res. 2020;22(10):1917–22.
Sikdar S, Joehanes R, Joubert BR, et al. Comparison of smoking-related DNA methylation between newborns from prenatal exposure and adults from personal smoking. Epigenomics. 2019;11(13):1487–500.
Gao L, Liu X, Millstein J, et al. Self-reported prenatal tobacco smoke exposure, AXL gene-body methylation, and childhood asthma phenotypes. Clin Epigenetics. 2018 Jul 20;10(1):98.
Cardenas A, Lutz SM, Everson TM, et al. Mediation by Placental DNA Methylation of the Association of Prenatal Maternal Smoking and Birth Weight. Am J Epidemiol. 2019;188(11):1878–86.
Everson TM, Vives-Usano M, Seyve E, et al. Placental DNA methylation signatures of maternal smoking during pregnancy and potential impacts on fetal growth. Nat Commun. 2021;12(1):5095.
Fa S, Larsen TV, Bilde K, et al. Changes in first trimester fetal CYP1A1 and AHRR DNA methylation and mRNA expression in response to exposure to maternal cigarette smoking. Environ Toxicol Pharmacol. 2018;57:19–27.
Rousseaux S, Seyve E, Chuffart F, et al. Immediate and durable effects of maternal tobacco consumption alter placental DNA methylation in enhancer and imprinted gene-containing regions. BMC Med. 2020;18(1):306.
van Otterdijk SD, Binder AM, Michels KB. Locus-specific DNA methylation in the placenta is associated with levels of pro-inflammatory proteins in cord blood and they are both independently affected by maternal smoking during pregnancy. Epigenetics. 2017;12(10):875–85.
Hannon E, Schendel D, Ladd-Acosta C, et al. Variable DNA methylation in neonates mediates the association between prenatal smoking and birth weight. Philos Trans R Soc Lond B Biol Sci. 2019;374(1770):20180120.
Neophytou AM, Oh SS, Hu D, et al. In utero tobacco smoke exposure, DNA methylation, and asthma in Latino children. Environ Epidemiol. 2019;3(3): e048.
Sengupta SM, Smith AK, Grizenko N, et al. Locus-specific DNA methylation changes and phenotypic variability in children with attention-deficit hyperactivity disorder. Psychiatry Res. 2017;256:298–304.
Vives-Usano M, Hernandez-Ferrer C, Maitre L, et al. In utero and childhood exposure to tobacco smoke and multi-layer molecular signatures in children. BMC Med. 2020;18(1):243.
Rauschert S, Melton PE, Burdge G, et al. Maternal smoking during pregnancy induces persistent epigenetic changes into adolescence, independent of postnatal smoke exposure and is associated with cardiometabolic risk. Front Genet. 2019;10:770.
Rauschert S, Melton PE, Heiskala A, et al. Machine learning-based DNA methylation score for fetal exposure to maternal smoking: development and validation in samples collected from adolescents and adults. Environ Health Perspect. 2020;128(9):97003.
Wiklund P, Karhunen V, Richmond RC, et al. DNA methylation links prenatal smoking exposure to later life health outcomes in offspring. Clin Epigenetics. 2019;11(1):97.
Parmar P, Lowry E, Cugliari G, et al. Association of maternal prenatal smoking GFI1-locus and cardio-metabolic phenotypes in 18,212 adults. EBioMedicine. 2018;38:206–16.
•• Richmond RC, Suderman M, Langdon R, et al. DNA methylation as a marker for prenatal smoke exposure in adults. Int J Epidemiol. 2018;47(4):1120–30. (Development of a DNA methylation score in adults for prenatal smoke exposure, as well as the development of a methylation score in children associated with prenatal smoke exposure.)
Tehranifar P, Wu HC, McDonald JA, et al. Maternal cigarette smoking during pregnancy and offspring DNA methylation in midlife. Epigenetics. 2018;13(2):129–34.
Dugue PA, Hodge AM, Wong EM, et al. Methylation marks of prenatal exposure to maternal smoking and risk of cancer in adulthood. Int J Epidemiol. 2021;50(1):105–15.
Sharp GC, Arathimos R, Reese SE, et al. Maternal alcohol consumption and offspring DNA methylation: findings from six general population-based birth cohorts. Epigenomics. 2018;10(1):27–42.
Loke YJ, Muggli E, Saffery R, et al. Sex- and tissue-specific effects of binge-level prenatal alcohol consumption on DNA methylation at birth. Epigenomics. 2021;13(24):1921–38.
Loke YJ, Muggli E, Nguyen L, et al. Time- and sex-dependent associations between prenatal alcohol exposure and placental global DNA methylation. Epigenomics. 2018;10(7):981–91.
Marjonen H, Kahila H, Kaminen-Ahola N. rs10732516 polymorphism at the IGF2/H19 locus associates with a genotype-specific trend in placental DNA methylation and head circumference of prenatally alcohol-exposed newborns. Hum Reprod Open. 2017;2017(3):hox014.
Steane SE, Young SL, Clifton VL, et al. Prenatal alcohol consumption and placental outcomes: a systematic review and meta-analysis of clinical studies. Am J Obstet Gynecol. 2021;225(6):607 e1-607 e22.
Sarkar DK, Gangisetty O, Wozniak JR, et al. Persistent changes in stress-regulatory genes in pregnant women or children exposed prenatally to alcohol. Alcohol Clin Exp Res. 2019;43(9):1887–97.
•• Lussier AA, Morin AM, MacIsaac JL, et al. DNA methylation as a predictor of fetal alcohol spectrum disorder. Clin Epigenetics. 2018;10:5. (Demonstrates use of machine learning approaches to predicting fetal alcohol spectrum disorder in children prenatally exposed to alcohol.)
Frey S, Eichler A, Stonawski V, et al. Prenatal alcohol exposure is associated with adverse cognitive effects and distinct whole-genome DNA methylation patterns in primary school children. Front Behav Neurosci. 2018;12:125.
Radhakrishna U, Vishweswaraiah S, Uppala LV, et al. Placental DNA methylation profiles in opioid-exposed pregnancies and associations with the neonatal opioid withdrawal syndrome. Genomics. 2021;113(3):1127–35.
Baptista T, de Azeredo LA, Zaparte A, et al. Oxytocin receptor exon III methylation in the umbilical cord blood of newborns with prenatal exposure to crack cocaine. Front Cell Dev Biol. 2021;9: 639287.
McLaughlin P, Mactier H, Gillis C, et al. Increased DNA methylation of ABCB1, CYP2D6, and OPRM1 genes in newborn infants of methadone-maintained opioid-dependent mothers. J Pediatr. 2017;190(180–184): e1.
Fransquet PD, Hutchinson D, Olsson CA, et al. Cannabis use by women during pregnancy does not influence infant DNA methylation of the dopamine receptor DRD4. Am J Drug Alcohol Abuse. 2017;43(6):671–7.
Wachman EM, Hayes MJ, Shrestha H, et al. Epigenetic variation in OPRM1 gene in opioid-exposed mother-infant dyads. Genes Brain Behav. 2018;17(7): e12476.
Oni-Orisan OO, Dansereau LM, Marsit CJ, et al. DNA methylation in children with prenatal methamphetamine exposure and environmental adversity. Pediatr Res. 2021;89(5):1152–6.
Pauwels S, Ghosh M, Duca RC, et al. Dietary and supplemental maternal methyl-group donor intake and cord blood DNA methylation. Epigenetics. 2017;12(1):1–10.
Chiu YH, Fadadu RP, Gaskins AJ, et al. Dietary fat intake during early pregnancy is associated with cord blood DNA methylation at IGF2 and H19 genes in newborns. Environ Mol Mutagen. 2021;62(7):388–98.
Geraghty AA, Sexton-Oates A, O'Brien EC, et al. A low glycaemic index diet in pregnancy induces DNA methylation variation in blood of newborns: results from the ROLO randomised controlled trial. nutrients. 2018;10(4):455. https://doi.org/10.3390/nu10040455.
Bianchi M, Alisi A, Fabrizi M, et al. Maternal intake of n-3 polyunsaturated fatty acids during pregnancy is associated with differential methylation profiles in cord blood white cells. Front Genet. 2019;10:1050.
Caffrey A, Irwin RE, McNulty H, et al. Gene-specific DNA methylation in newborns in response to folic acid supplementation during the second and third trimesters of pregnancy: epigenetic analysis from a randomized controlled trial. Am J Clin Nutr. 2018;107(4):566–75.
Irwin RE, Thursby SJ, Ondicova M, et al. A randomized controlled trial of folic acid intervention in pregnancy highlights a putative methylation-regulated control element at ZFP57. Clin Epigenetics. 2019;11(1):31.
Bakulski KM, Dou JF, Feinberg JI, et al. Prenatal multivitamin use and MTHFR genotype are associated with newborn cord blood DNA methylation. Int J Environ Res Public Health. 2020;17(24):9190. https://doi.org/10.3390/ijerph17249190.
Huoman J, Martinez-Enguita D, Olsson E, et al. Combined prenatal Lactobacillus reuteri and omega-3 supplementation synergistically modulates DNA methylation in neonatal T helper cells. Clin Epigenetics. 2021;13(1):135.
Murray R, Kitaba N, Antoun E, et al. Influence of maternal lifestyle and diet on perinatal DNA methylation signatures associated with childhood arterial stiffness at 8 to 9 Years. Hypertension. 2021;78(3):787–800.
Polinski KJ, Purdue-Smithe A, Robinson SL, et al. Maternal caffeine intake and DNA methylation in newborn cord blood. Am J Clin Nutr. 2022;115(2):482–91.
Kazmi N, Gaunt TR, Relton C, et al. Maternal eating disorders affect offspring cord blood DNA methylation: a prospective study. Clin Epigenetics. 2017;9:120.
Antoun E, Kitaba NT, Titcombe P, et al. Maternal dysglycaemia, changes in the infant’s epigenome modified with a diet and physical activity intervention in pregnancy: Secondary analysis of a randomised control trial. PLoS Med. 2020;17(11): e1003229.
van Weelden W, Seed PT, Antoun E, et al. Folate and vitamin B12 status: associations with maternal glucose and neonatal DNA methylation sites related to dysglycaemia, in pregnant women with obesity. J Dev Orig Health Dis. 2021;11:1–9.
Caramaschi D, Sharp GC, Nohr EA, et al. Exploring a causal role of DNA methylation in the relationship between maternal vitamin B12 during pregnancy and child’s IQ at age 8, cognitive performance and educational attainment: a two-step Mendelian randomization study. Hum Mol Genet. 2017;26(15):3001–13.
Walker-Short E, Buckner T, Vigers T, et al. Epigenome-wide association study of infant feeding and DNA methylation in infancy and childhood in a population at increased risk for type 1 diabetes. Nutrients. 2021;13(11):4057. https://doi.org/10.3390/nu13114057.
Daniels TE, Sadovnikoff AI, Ridout KK, et al. Associations of maternal diet and placenta leptin methylation. Mol Cell Endocrinol. 2020;5(505): 110739.
Boyle KE, Patinkin ZW, Shapiro ALB, et al. Maternal obesity alters fatty acid oxidation, AMPK activity, and associated DNA methylation in mesenchymal stem cells from human infants. Mol Metab. 2017;6(11):1503–16.
Anderson CM, Gillespie SL, Thiele DK, et al. Effects of maternal vitamin D supplementation on the maternal and infant epigenome. Breastfeed Med. 2018;13(5):371–80.
Geraghty AA, Sexton-Oates A, O'Brien EC, et al. Epigenetic patterns in five-year-old children exposed to a low glycemic index dietary intervention during pregnancy: results from the ROLO Kids Study. Nutrients. 2020;12(12):3602. https://doi.org/10.3390/nu12123602.
•• Richmond RC, Sharp GC, Herbert G, et al. The long-term impact of folic acid in pregnancy on offspring DNA methylation: follow-up of the Aberdeen Folic Acid Supplementation Trial (AFAST). Int J Epidemiol. 2018;47(3):928–37. (Maternal diet during pregnancy was assocaited with saliva DNA methylation later in life, deonstrating persistence of effects.)
Taylor RM, Smith R, Collins CE, et al. Methyl-donor and cofactor nutrient intakes in the first 2–3 years and global DNA methylation at age 4: a prospective cohort study. Nutrients. 2018;10(3):273. https://doi.org/10.3390/nu10030273.
Gallardo-Escribano C, Buonaiuto V, Ruiz-Moreno MI, et al. Epigenetic approach in obesity: DNA methylation in a prepubertal population which underwent a lifestyle modification. Clin Epigenetics. 2020;12(1):144.
Kallak TK, Brann E, Fransson E, et al. DNA methylation in cord blood in association with prenatal depressive symptoms. Clin Epigenetics. 2021;13(1):78.
Montoya-Williams D, Quinlan J, Clukay C, et al. Associations between maternal prenatal stress, methylation changes in IGF1 and IGF2, and birth weight. J Dev Orig Health Dis. 2018;9(2):215–22.
Duis J, Cox OH, Ji Y, et al. Effect of Genotype and maternal affective disorder on intronic methylation of FK506 binding protein 5 in cord blood DNA. Front Genet. 2018;9:648.
Clukay CJ, Hughes DA, Kertes DA, et al. Associations between maternal psychosocial stress, DNA methylation, and newborn birth weight identified by investigating methylation at individual, regional, and genome levels. Hum Biol. 2019;91(2):117–31.
Wu S, Gennings C, Wright RJ, et al. Prenatal stress, methylation in inflammation-related genes, and adiposity measures in early childhood: the programming research in obesity, growth environment and social stress cohort study. Psychosom Med. 2018;80(1):34–41.
Vangeel EB, Pishva E, Hompes T, et al. Newborn genome-wide DNA methylation in association with pregnancy anxiety reveals a potential role for GABBR1. Clin Epigenetics. 2017;9:107.
Milaniak I, Cecil CAM, Barker ED, et al. Variation in DNA methylation of the oxytocin receptor gene predicts children’s resilience to prenatal stress. Dev Psychopathol. 2017;29(5):1663–74.
Lund RJ, Kylaniemi M, Pettersson N, et al. Placental DNA methylation marks are associated with maternal depressive symptoms during early pregnancy. Neurobiol Stress. 2021;15: 100374.
Brunst KJ, Tignor N, Just A, et al. Cumulative lifetime maternal stress and epigenome-wide placental DNA methylation in the PRISM cohort. Epigenetics. 2018;13(6):665–81.
Jahnke JR, Teran E, Murgueitio F, et al. Maternal stress, placental 11beta-hydroxysteroid dehydrogenase type 2, and infant HPA axis development in humans: Psychosocial and physiological pathways. Placenta. 2021;15(104):179–87.
Letourneau N, Ntanda H, Jong VL, et al. Prenatal maternal distress and immune cell epigenetic profiles at 3-months of age. Dev Psychobiol. 2021;63(5):973–84.
•• Laubach ZM, Perng W, Cardenas A, et al. Socioeconomic status and DNA methylation from birth through mid-childhood: a prospective study in Project Viva. Epigenomics. 2019;11(12):1413–27. (Maternal education was assocaited with DNA methylation in cord blood, as well as early and mid-childhood, demonstrating persistence of effects and biomarker utility.)
• Alfano R, Guida F, Galobardes B, et al. Socioeconomic position during pregnancy and DNA methylation signatures at three stages across early life: epigenome-wide association studies in the ALSPAC birth cohort. Int J Epidemiol. 2019;48(1):30–44. (Maternal education was associated with cord blood DNA methylation at birth, and methylation at adolecents, though the changes occurred at different loci across the two timepoints.)
Coker ES, Gunier R, Huen K, et al. DNA methylation and socioeconomic status in a Mexican-American birth cohort. Clin Epigenetics. 2018;10:61.
Ong ML, Tuan TA, Poh J, et al. Neonatal amygdalae and hippocampi are influenced by genotype and prenatal environment, and reflected in the neonatal DNA methylome. Genes Brain Behav. 2019;18(7): e12576.
Provenzi L, Fumagalli M, Giorda R, et al. Maternal sensitivity buffers the association between SLC6A4 methylation and socio-emotional stress response in 3-month-old full term, but not very preterm infants. Front Psychiatry. 2017;8:171.
Santos HP Jr, Bhattacharya A, Martin EM, et al. Epigenome-wide DNA methylation in placentas from preterm infants: association with maternal socioeconomic status. Epigenetics. 2019;14(8):751–65.
Bush NR, Edgar RD, Park M, et al. The biological embedding of early-life socioeconomic status and family adversity in children’s genome-wide DNA methylation. Epigenomics. 2018;10(11):1445–61.
Gouin JP, Zhou QQ, Booij L, et al. Associations among oxytocin receptor gene (OXTR) DNA methylation in adulthood, exposure to early life adversity, and childhood trajectories of anxiousness. Sci Rep. 2017;7(1):7446.
McDade TW, Ryan CP, Jones MJ, et al. Genome-wide analysis of DNA methylation in relation to socioeconomic status during development and early adulthood. Am J Phys Anthropol. 2019;169(1):3–11.
Martins J, Czamara D, Sauer S, et al. Childhood adversity correlates with stable changes in DNA methylation trajectories in children and converges with epigenetic signatures of prenatal stress. Neurobiol Stress. 2021;15: 100336.
Czamara D, Tissink E, Tuhkanen J, et al. Combined effects of genotype and childhood adversity shape variability of DNA methylation across age. Transl Psychiatry. 2021;11(1):88.
Kaufman J, Montalvo-Ortiz JL, Holbrook H, et al. Adverse childhood experiences, epigenetic measures, and obesity in youth. J Pediatr. 2018;202(150–156): e3.
Dunn EC, Soare TW, Zhu Y, et al. Sensitive periods for the effect of childhood adversity on DNA methylation: results from a prospective, longitudinal study. Biol Psychiatry. 2019;85(10):838–49.
Lewis CR, Breitenstein RS, Henderson A, et al. Harsh parenting predicts novel HPA receptor gene methylation and NR3C1 methylation predicts cortisol daily slope in middle childhood. Cell Mol Neurobiol. 2021;41(4):783–93.
Takahashi Y, Kubo R, Sano R, et al. DNA methylation of the NR3C1 promoter region in brains of pediatric victims of physical abuse. Neurocase. 2018;24(5–6):269–75.
Marzi SJ, Sugden K, Arseneault L, et al. Analysis of DNA methylation in young people: limited evidence for an association between victimization stress and epigenetic variation in blood. Am J Psychiatry. 2018;175(6):517–29.
Farrell C, Doolin K, Leary NO, et al. DNA methylation differences at the glucocorticoid receptor gene in depression are related to functional alterations in hypothalamic-pituitary-adrenal axis activity and to early life emotional abuse. Psychiatry Res. 2018;265:341–8.
•• Houtepen LC, Hardy R, Maddock J, et al. Childhood adversity and DNA methylation in two population-based cohorts. Transl Psychiatry. 2018;8(1):266. (Demonstrate DNA methylation changes assocaited with early-life adversity present across two independent cohorts and tissue types.)
Wiegand A, Kreifelts B, Munk MHJ, et al. DNA methylation differences associated with social anxiety disorder and early life adversity. Transl Psychiatry. 2021;11(1):104.
Vangeel EB, Kempke S, Bakusic J, et al. Glucocorticoid receptor DNA methylation and childhood trauma in chronic fatigue syndrome patients. J Psychosom Res. 2018;104:55–60.
Zou Z, Huang Y, Wang J, et al. DNA methylation of IL-4 gene and the association with childhood trauma in panic disorder. Psychiatry Res. 2020;293: 113385.
Mihaljevic M, Franic D, Soldatovic I, et al. The FKBP5 genotype and childhood trauma effects on FKBP5 DNA methylation in patients with psychosis, their unaffected siblings, and healthy controls. Psychoneuroendocrinology. 2021;128: 105205.
Janusek LW, Tell D, Gaylord-Harden N, et al. Relationship of childhood adversity and neighborhood violence to a proinflammatory phenotype in emerging adult African American men: an epigenetic link. Brain Behav Immun. 2017;60:126–35.
He Y, Vinkers CH, Houtepen LC, et al. Childhood adversity is associated with increased KITLG methylation in healthy individuals but not in bipolar disorder patients. Front Psychiatry. 2018;9:743.
Engdahl E, Alavian-Ghavanini A, Forsell Y, et al. Childhood adversity increases methylation in the GRIN2B gene. J Psychiatr Res. 2021;132:38–43.
Parianen Lesemann FH, Spencer H, Montoya ER, et al. Methylation of oxytocin related genes and early life trauma together shape the N170 response to human faces. Eur Neuropsychopharmacol. 2020;39:19–28.
Carleial S, Natt D, Unternahrer E, et al. DNA methylation changes following narrative exposure therapy in a randomized controlled trial with female former child soldiers. Sci Rep. 2021;11(1):18493.
Peng H, Zhu Y, Strachan E, et al. Childhood trauma, DNA methylation of stress-related genes, and depression: findings from two monozygotic twin studies. Psychosom Med. 2018;80(7):599–608.
Marinova Z, Maercker A, Kuffer A, et al. DNA methylation profiles of elderly individuals subjected to indentured childhood labor and trauma. BMC Med Genet. 2017;18(1):21.
Fiacco S, Gardini ES, Mernone L, et al. DNA methylation in healthy older adults with a history of childhood adversity-findings from the women 40+ healthy aging study. Front Psychiatry. 2019;10:777.
•• Zhang M, Liu C, Li WD, et al. Individual and mixtures of metal exposures in associations with biomarkers of oxidative stress and global DNA methylation among pregnant women. Chemosphere. 2022;293: 133662. (Demosntrates implelemtation of novel statistical approaches to examining the effect of exposure to metal mixtures on DNA methylation.)
Weyde KVF, Olsen AK, Duale N, et al. Gestational blood levels of toxic metal and essential element mixtures and associations with global DNA methylation in pregnant women and their infants. Sci Total Environ. 2021;15(787): 147621.
Vilahur N, Bustamante M, Byun HM, et al. Prenatal exposure to mixtures of xenoestrogens and repetitive element DNA methylation changes in human placenta. Environ Int. 2014;71:81–7.
LaRocca J, Binder AM, McElrath TF, et al. The impact of first trimester phthalate and phenol exposure on IGF2/H19 genomic imprinting and birth outcomes. Environ Res. 2014;133:396–406.
Vilahur N, Bustamante M, Morales E, et al. Prenatal exposure to mixtures of xenoestrogens and genome-wide DNA methylation in human placenta. Epigenomics. 2016;8(1):43–54.
King KE, Kane JB, Scarbrough P, et al. Neighborhood and family environment of expectant mothers may influence prenatal programming of adult cancer risk: discussion and an illustrative DNA methylation example. Biodemography Soc Biol. 2016;62(1):87–104.
Borghol N, Suderman M, McArdle W, et al. Associations with early-life socio-economic position in adult DNA methylation. Int J Epidemiol. 2012;41(1):62–74.
Loucks EB, Huang YT, Agha G, et al. Epigenetic mediators between childhood socioeconomic disadvantage and mid-life body mass index: The New England Family Study. Psychosom Med. 2016;78(9):1053–65.
• Hussey MR, Burt A, Deyssenroth MA, et al. Placental lncRNA expression associated with placental cadmium concentrations and birth weight. Environ Epigenet. 2020;6(1):dvaa003. (Prenatal cadmium exposure was associated with lncRNA expression in the placenta.)
Paquette AG, MacDonald J, Lapehn S, et al. A comprehensive assessment of associations between prenatal phthalate exposure and the placental transcriptomic landscape. Environ Health Perspect. 2021;129(9):97003.
Machtinger R, Zhong J, Mansur A, et al. Placental lncRNA expression is associated with prenatal phthalate exposure. Toxicol Sci. 2018;163(1):116–22.
Zhong J, Baccarelli AA, Mansur A, et al. Maternal phthalate and personal care products exposure alters extracellular placental miRNA profile in twin pregnancies. Reprod Sci. 2019;26(2):289–94.
LaRocca J, Binder AM, McElrath TF, et al. First-trimester urine concentrations of phthalate metabolites and phenols and placenta miRNA expression in a cohort of U.S. women. Environ Health Perspect. 2016;124(3):380–7.
Tsamou M, Nawrot TS, Carollo RM, et al. Prenatal particulate air pollution exposure and expression of the miR-17/92 cluster in cord blood: Findings from the ENVIRONAGE birth cohort. Environ Int. 2020;142: 105860.
Vrijens K, Trippas AJ, Lefebvre W, et al. Association of prenatal exposure to ambient air pollution with circulating histone levels in maternal cord blood. JAMA Netw Open. 2020;3(5): e205156.
• Liu Q, Wang W, Jing W. Indoor air pollution aggravates asthma in Chinese children and induces the changes in serum level of miR-155. Int J Environ Health Res. 2019;29(1):22–30. (Childhood air pollution was associated with miRNA expression hcanges in children, one of the few studies to assess this.)
Vriens A, Nawrot TS, Saenen ND, et al. Recent exposure to ultrafine particles in school children alters miR-222 expression in the extracellular fraction of saliva. Environ Health. 2016;15(1):80.
Herberth G, Bauer M, Gasch M, et al. Maternal and cord blood miR-223 expression associates with prenatal tobacco smoke exposure and low regulatory T-cell numbers. J Allergy Clin Immunol. 2014;133(2):543–50.
Maccani MA, Avissar-Whiting M, Banister CE, et al. Maternal cigarette smoking during pregnancy is associated with downregulation of miR-16, miR-21, and miR-146a in the placenta. Epigenetics. 2010;5(7):583–9.
•• Bauer T, Trump S, Ishaque N, et al. Environment-induced epigenetic reprogramming in genomic regulatory elements in smoking mothers and their children. Mol Syst Biol. 2016;12(3):861. (Example of prenatal smoke exposure associated with persistent changes in lncRNA expression in children.)
Salem NA, Mahnke AH, Wells AB, et al. Association between fetal sex and maternal plasma microRNA responses to prenatal alcohol exposure: evidence from a birth outcome-stratified cohort. Biol Sex Differ. 2020;11(1):51.
Mahnke AH, Sideridis GD, Salem NA, et al. Infant circulating MicroRNAs as biomarkers of effect in fetal alcohol spectrum disorders. Sci Rep. 2021;11(1):1429.
Lutz PE, Chay MA, Pacis A, et al. Non-CG methylation and multiple histone profiles associate child abuse with immune and small GTPase dysregulation. Nat Commun. 2021;12(1):1132.
Volk N, Pape JC, Engel M, et al. Amygdalar MicroRNA-15a Is Essential for Coping with Chronic Stress. Cell Rep. 2016;17(7):1882–91.
Cattane N, Mora C, Lopizzo N, et al. Identification of a miRNAs signature associated with exposure to stress early in life and enhanced vulnerability for schizophrenia: New insights for the key role of miR-125b-1-3p in neurodevelopmental processes. Schizophr Res. 2019;205:63–75.
Bahari-Javan S, Varbanov H, Halder R, et al. HDAC1 links early life stress to schizophrenia-like phenotypes. Proc Natl Acad Sci U S A. 2017;114(23):E4686–94.
Nahar MS, Liao C, Kannan K, et al. In utero bisphenol A concentration, metabolism, and global DNA methylation across matched placenta, kidney, and liver in the human fetus. Chemosphere. 2015;124:54–60.
Goodrich JM, Dolinoy DC, Sanchez BN, et al. Adolescent epigenetic profiles and environmental exposures from early life through peri-adolescence. Environ Epigenet. 2016;2(3):dvw018.
Richmond RC, Simpkin AJ, Woodward G, et al. Prenatal exposure to maternal smoking and offspring DNA methylation across the lifecourse: findings from the Avon Longitudinal Study of Parents and Children (ALSPAC). Hum Mol Genet. 2015;24(8):2201–17.
•• Lee KW, Richmond R, Hu P, et al. Prenatal exposure to maternal cigarette smoking and DNA methylation: epigenome-wide association in a discovery sample of adolescents and replication in an independent cohort at birth through 17 years of age. Environ Health Perspect. 2015;123(2):193–9. (Detected persistent DNA methylation changes associated with prenatal smoke exposure across the life course.)
van der Knaap LJ, Riese H, Hudziak JJ, et al. Glucocorticoid receptor gene (NR3C1) methylation following stressful events between birth and adolescence. The TRAILS study Transl Psychiatry. 2014;8(4): e381.
Wang J, Geng L. Effects of socioeconomic status on physical and psychological health: lifestyle as a mediator. int j environ res public health. 2019;16(2):281. https://doi.org/10.3390/ijerph16020281.
Keil AP, Buckley JP, O’Brien KM, et al. A quantile-based g-computation approach to addressing the effects of exposure mixtures. Environ Health Perspect. 2020;128(4):47004.
Bobb JF, Claus Henn B, Valeri L, et al. Statistical software for analyzing the health effects of multiple concurrent exposures via Bayesian kernel machine regression. Environ Health. 2018;17(1):67.
Thumfart KM, Jawaid A, Bright K, et al. Epigenetics of childhood trauma: long term sequelae and potential for treatment. Neurosci Biobehav Rev. 2022;132:1049–66.
Ladd-Acosta C, Fallin MD. DNA methylation signatures as biomarkers of prior environmental exposures. Curr Epidemiol Rep. 2019;6(1):1–13.
Nwanaji-Enwerem JC, Colicino E. DNA methylation-based biomarkers of environmental exposures for human population studies. Curr Environ Health Rep. 2020;7(2):121–8.
•• Ladd-Acosta C, Shu C, Lee BK, et al. Presence of an epigenetic signature of prenatal cigarette smoke exposure in childhood. Environ Res. 2016;144(Pt A):139–48. (This was the first study to implement machine learning techniques to predict prenatal smoke exposure in children.)
Hompes T, Izzi B, Gellens E, et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J Psychiatr Res. 2013;47(7):880–91.
Oberlander TF, Weinberg J, Papsdorf M, et al. Prenatal exposure to maternal depression, neonatal methylation of human glucocorticoid receptor gene (NR3C1) and infant cortisol stress responses. Epigenetics. 2008;3(2):97–106.
Mansell T, Vuillermin P, Ponsonby AL, et al. Maternal mental well-being during pregnancy and glucocorticoid receptor gene promoter methylation in the neonate. Dev Psychopathol. 2016;28(4pt2):1421–30.
Monk C, Feng T, Lee S, et al. Distress during pregnancy: epigenetic regulation of placenta glucocorticoid-related genes and fetal neurobehavior. Am J Psychiatry. 2016;173(7):705–13.
Stroud LR, Papandonatos GD, Parade SH, et al. Prenatal major depressive disorder, placenta glucocorticoid and serotonergic signaling, and infant cortisol response. Psychosom Med. 2016;78(9):979–90.
Kundakovic M, Gudsnuk K, Herbstman JB, et al. DNA methylation of BDNF as a biomarker of early-life adversity. Proc Natl Acad Sci U S A. 2015;112(22):6807–13.
Zhang Q, Wang W, Niu Y, et al. The effects of fine particulate matter constituents on exhaled nitric oxide and DNA methylation in the arginase-nitric oxide synthase pathway. Environ Int. 2019;131: 105019.
Chen R, Qiao L, Li H, et al. Fine particulate matter constituents, nitric oxide synthase DNA methylation and exhaled nitric oxide. Environ Sci Technol. 2015;49(19):11859–65.
Miguel V, Lamas S, Espinosa-Diez C. Role of non-coding-RNAs in response to environmental stressors and consequences on human health. Redox Biol. 2020;37: 101580.
Dong E, Pandey SC. Prenatal stress induced chromatin remodeling and risk of psychopathology in adulthood. Int Rev Neurobiol. 2021;156:185–215.
Acknowledgements
Rose Schrott is supported by the NIMH Psychiatric Epidemiology Training Program (5T32MH014592-44; PI: Volk, Heather).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Dr. Ladd-Acosta reports receiving consulting fees from the University of Iowa for providing expertise on autism spectrum disorder epigenetics outside of this work. Drs. Schrott and Song declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Early Life Environmental Health
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Schrott, R., Song, A. & Ladd-Acosta, C. Epigenetics as a Biomarker for Early-Life Environmental Exposure. Curr Envir Health Rpt 9, 604–624 (2022). https://doi.org/10.1007/s40572-022-00373-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-022-00373-5