
Abstract
Purpose of Review
This manuscript orients the reader to the underlying motivations of environmental biomarker development for human population studies and provides the foundation for applying these novel biomarkers in future research. In this review, we focus our attention on the DNA methylation–based biomarkers of (i) smoking, among adults and pregnant women, (ii) lifetime cannabis use, (iii) alcohol consumption, and (iv) cumulative exposure to lead.
Recent Findings
Prior environmental exposures and lifestyle modulate DNA methylation levels. Exposure-related DNA methylation changes can either be persistent or reversible once the exposure is no longer present, and this combination of both persistent and reversible changes has essential value for biomarker development. Here, we present available biomarkers representing past and cumulative exposures using individual DNA methylation profiles.
Summary
In the present work, we describe how the field of environmental epigenetics can leverage machine learning algorithms to develop exposure biomarkers and reduce problems of misreporting exposures or limited access technology. We emphasize the crucial role of the individual DNA methylation profiles in those predictions, providing a summary of each biomarker, and highlighting their advantages, and limitations. Future research can cautiously leverage these DNA methylation–based biomarkers to understand the onset and progression of diseases.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
DNA methylation, typically characterized by the addition of methyl-groups to cytosine nucleotides followed by guanine bases (CpGs), is the most widely studied biomarker of epigenetic programming in human population studies [1]. DNA methylation levels are relatively stable and are inherited during cellular division. In addition, DNA methylation changes are critically relevant to human health given DNA methylation’s known ability to alter gene expression thereby impacting phenotypic expression including the potential manifestation and progression of diseases [2]. For these reasons, they have been explored as precursors of diseases and all-cause mortality [3,4,5].
Research demonstrates that DNA methylation profiles can be modulated by prior environmental exposures and lifestyle. Exposure to air pollutants, heavy metals, and smoking has each been associated with DNA methylation changes in adults and children [6,7,8,9,10,11]. Those DNA methylation alterations, once established, can persist in the absence of the initial environmental or lifestyle factors which induced them. Indeed, DNA methylation, having a clear mechanism for post-mitotic inheritance, has the potential to retain the signature of exposures after many years and, due to its inter-individual stability over long time-span, offers an inherent biological mechanism for cells to remember alterations associated with environmental exposures [12, 13]. Some alterations have even been shown to persist across tissue types, on intra and inter-individual levels, and over extended periods of time. For instance, prenatal maternal smoking exposure has been robustly and consistently associated with DNA methylation at specific loci among offspring at birth and in early childhood [10]. Moreover, those smoke-related changes in DNA methylation profiles persisted in exposed offspring for decades after prenatal exposure [14]. Similar findings have been made with respect to mercury [15]. Prenatal blood mercury levels were associated with cord blood DNA methylation changes and with persistent whole blood DNA methylation disruptions in the same loci during early and mid-childhood [15].
Given that exposure concentrations and timing impact how DNA methylation is modulated, DNA methylation incorporates not only information about lifestyle and environmental factors, but also about the timing and consistency of the exposures. Occasional smokers have different DNA methylation profiles than never smokers and even regular/current smokers [16••]. Further, smoking cessation can result in partial restoration of pre-smoking methylation levels [17,18,19]. Therefore, exposure-related DNA methylation changes can either be persistent (i.e., stable changes) or reversible (i.e., return to prior state) once the exposure is no longer present. This combination of both persistent and reversible changes has important value for biomarker development.
The most commonly used technology for DNA methylation analysis in humans are high-density microarrays to measure individual CpG methylation levels at approximately a million CpG sites for each DNA sample. Several platforms—Illumina Infinium HumanMethylation27 BeadChip Array (Illumina27K), Illumina Infinium HumanMethylation450 BeadChip Array (Illumina450K), Illumina Infinium MethylationEPIC BeadChip microarray (IlluminaEPIC)—have been used in epigenome-wide association studies (EWAS) in which DNA methylation levels at each CpG site are analyzed individually. EWAS has been increasingly employed to uncover biological mechanisms that underlie extrinsic environmental stimuli and adverse health outcomes [20,21,22]. Because DNA methylation is sensitive to external factors and can show changes at multiple CpG sites in response to environmental toxicants, multiple changes can be combined to build a composite estimator of environmental exposures and lifestyle factors. These composite estimators are unique because they go beyond simple loci associations. Rather, they represent biomarkers with immense potential to reconstruct exposure in practical situations when exposure data is truly unavailable or the timing for biomarker collection has passed.
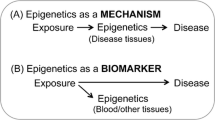
Thus far, developed biomarkers have identified combinations of CpGs that reflect exposure levels and allow for some reconstruction of the time of exposure. Predictive CpGs are first identified with screening approaches to select the sites most strongly associated with the exposure. These screens usually entail a locus-by-locus epigenetic-wide association analysis, and then use those sites in machine learning algorithms, such as elastic nets or LASSO (least absolute shrinkage and selection operator) regressions [23, 24]. Both elastic nets and LASSO regressions evaluate the association of all sites additively and linearly with the exposure in the same model. These approaches also gain further information from features (CpGs) that, when taken individually, are not statistically significant, but when taken jointly contribute to improving the exposure prediction [23, 24]. Presently available biomarkers are blood, cord blood, or buccal derived [9••, 16••, 25••, 26••]. Moreover, they mostly focus on stigmatized exposures, such as lifetime smoking, smoking during pregnancy, or lifetime cannabis use, which may be under-reported. Still, some attention has been given to less-stigmatized exposures with major public health implications, such as chronic lead exposure. Here, we report on the available environmental DNA methylation biomarkers (Fig. 1), emphasizing limitations and advantages.

Characteristics of the DNA methylation–based biomarkers of environmental exposures. The asterisk indicates four alcohol consumption markers were created; here, the most accurate one was reported. Parentheses refer to the population in which the markers were generalized.
Biomarkers of Tobacco Exposure in Adults and in Pregnancy
The motivation for a DNA methylation–based biomarker of tobacco smoking is primarily driven by well-established data demonstrating that individuals under-report or purposefully misclassify their smoking status given perceived social stigmas [9••, 16••]. Remedying this issue of misclassification has many important consequences including better tailoring of services to clinically serve patients and reducing the potential for confounding by smoking in research studies. Numerous studies have identified CpG sites associated or causally linked with smoking. However, a recent smoking DNA methylation predictor tool, EpiSmokEr, was developed as a smoking status prediction tool [16••]. Utilizing a training dataset of peripheral blood cell DNA methylation from 474 Finnish adults and self-reported smoking data, the authors employed a machine learning algorithm (multinomial LASSO regression) to construct a classifier tool from 121 CpG sites corresponding to 92 genes. Gene ontology analysis of these genes demonstrated some enrichment for general processes including DNA binding and skeletal development. Relevant genes included multiple zinc finger proteins (ZNF555, ZNF641, and ZNF808), cartilage oligomeric matrix protein (COMP), and insulin-like growth factor 2 (IGF2). This tool is able to calculate probabilities of an individual being a never, former, or current smoker with the status with the highest probability being reported as the predicted smoking status [16••]. On average, the tool identifies current smokers with a sensitivity of 81% and a specificity of 85%. Never smokers were identified with a sensitivity and specificity of 94% and 57% respectively. The tool performed the poorest in the former smokers demonstrating a sensitivity of 18% and a specificity of 96% [16••]. Three external datasets, originating from different populations, were then used for the biomarker validation.
In addition to its ability to determine the most probable smoking classification, EpiSmokEr is also promising as a biomarker for three additional reasons. First, it can be applied to an individual sample or a population of samples. Second, unlike existing classifiers, it does not require the prior determination of specific thresholds in each individual dataset being examined to assign or predict smoking status. Previous classifiers have required researchers to first determine prediction thresholds in the non-smokers of their dataset before applying the classifiers to all individuals [27, 28]. Even newer potential classifiers, including those that appear more promising for cost saving because they utilize methylation of a single CpG site like cg05575921 (AHRR), have not arrived at a consensus on prediction thresholds that would allow for widespread classifier application [29•]. In contrast, EpiSmokEr can be applied to all individuals comprising a dataset directly. Finally, it demonstrated utility in tissues other than blood cells (e.g., buccal cells and peripheral blood mononuclear cells). Despite these positive aspects, some limitations may remain given its poor performance in former smokers. However, as the authors mention, performance among former smokers can not only be thought of as a deficit in the tool, but it more greatly reflects difficulties that come from both the detection and definition of former smokers [16••]. Unlike current and never smokers, former smokers across different studies are often a heterogeneous category. There are some individuals that barely meet the threshold of former smoker, while other individuals report smoking cessation for decades. In these scenarios, one can imagine former smokers with recent cessation may have a DNA methylation profile more similar to current smokers while the former smoker with a decade of cessation has a profile more similar to never smokers. One can further complicate the smoking classifications by noting that a current occasional smoker may be misclassified as a never smoker as was observed with the EpiSmokEr study [16••]. These issues with misclassification, particularly for individuals with complex smoking behavioral habits, highlight another major caveat of the study: self-reports were utilized as the gold standards to assess tool performance. It is possible that if the researchers utilized a composite self-report and biological measure (e.g., serum cotinine, which is a good marker of short-term tobacco smoke exposure) when developing their gold standards and training their tool, the overall performance would be improved.
Reese et al. (2017) employed such a composite measure of serum cotinine measurements and survey data when constructing their biomarker of sustained maternal smoking during pregnancy [9••]. In training (N = 1057) and test (N = 221) datasets composed of samples from a Norwegian mother and child cohort, they defined “sustained smoking during pregnancy” as maternal serum cotinine > 56.8 nmol/L at about 18 weeks or self-reported later in pregnancy (17 or 30 weeks). Using cord blood samples from their cohort, they performed genome-wide linear regression with the sustained smoking variable as the dichotomous predictor and the log ratios of the DNA methylation data as the response variable [9••]. To accommodate for randomness of the LASSO approach, the authors performed it 100 times on resampled datasets with the same sample size and selected a robust subset of CpGs that appeared in all iterations. Following this iterative LASSO regression procedure, they identified 28 CpGs that were used to build their final score. Of these 28 CpGs, only one was shared with the 121 CpGs that make up EpiSmokEr: cg05575921 (AHRR). Notably, AHRR is one gene whose DNA methylation status has been shown to be robustly associated with cigarette smoking [30]. The remaining 27 CpGs belonged to variety of genes including CYP1A1 and HIVEP2. Furthermore, gene ontology analysis of the genes associated with these CpG sites did not demonstrate any functional or biological process enrichment. Overall, the score performed well in the training data (i.e., sensitivity = 80%, specificity = 98%, accuracy = 96%) but demonstrated diminished sensitivity in the test data (i.e., sensitivity = 58%, specificity = 97%, accuracy = 91%) [9••]. Notably, the training and test data used in the study were acquired at two different time points 2 years apart. The authors tout this as evidence that their score is robust, but admit that generalizability of their measure may be limited by the homogenous composition of their Norwegian cohort [9••]. Additional important limitations include the score being built on the Illumina Methylation platforms—albeit the authors suggest that score is translatable by pyrosequencing the specific loci—and an inability to outperform cotinine given that it was utilized in training stages of biomarker development. Despite these limitations, there remains much promise in utilizing and improving composite measures for DNA methylation–based biosensing.
Biomarker of Lifetime Cannabis Use
In the USA, cannabis use is highly prevalent although it is often stigmatized. Due to increased legalization for both medical and recreational purposes in a growing number of US states, the number of Americans using cannabis is expected to continue to increase [26••]. Previously, epidemiological studies had limited opportunities to evaluate the health effects of cannabis use, and often resulted in inconsistent findings [31, 32]. Additionally, the urinary metabolites, commonly used as cannabis biomarkers, measure only acute exposures, with limited possibilities to increase the window for detection and evaluate consequences of lifetime exposure [33]. However, human cannabinoid pharmacokinetic processes are dynamic, change over time, and are affected by the frequency and magnitude of drug exposure [34]. Cannabis remains in the body more than 5 days, and cannabis exposure reflecting a cumulative and long-term consistent consumption could be a better predictor of adverse subsequent chronic health outcomes [34]. Therefore, an effort was conducted to build a novel epigenetic biomarker for lifetime (ever/never) cannabis use, using whole blood DNA methylation measured with the Illumina450K. The authors discovered that the combination of DNA methylation at three CpGs serves as a unique classifier for individuals with lifetime cannabis use [26••]. These three CpGs (cg15973234, cg04685163, and cg03765885) respectively lie within the CEMIP gene, 10 base pairs away from the DLGAP2 transcription start site and within an intergenic region in chromosome 2 and were negatively associated to lifetime cannabis use. DNA methylation level of cg15973234 was not correlated with reported cannabis-associated SNPs and none of those sites were never previously associated to smoking or alcohol consumption, suggesting that the findings were unlikely to be genetically driven and more likely related to the cannabis exposure. The authors leveraged 1730 non-Hispanic white women from the Sister Study in a LASSO regression and validated results in 853 women [26••]. The lifetime cannabis biomarker provided good accuracy (AUC = 0.79 (95% confidence interval [0.76, 0.82])) for the classification of women with lifetime cannabis use and it can now be used in future epidemiological studies to evaluate long-term health effects of cannabis [26••].
Although the authors adjusted all models for several confounders, technical variables, and cell type proportions, limiting the spurious signals, and reduced the overfitting of the model with cross-validation, a few study characteristics (age, sex, race/ethnicity, and development of breast cancer) may limit the generalizability of the results to the overall US population. Among the limitations, the authors acknowledged the missing information about other illicit drugs or environmental exposures, which may have impacted the results. Also, all participants self-reported on lifetime cannabis use, which may have led to underreporting and misclassification, leading results to bias towards the null hypothesis. The authors validated results by using an internal random split sample validation approach. Although validation should be always attempted for any of those epigenetic-derived biomarkers, internal random split sample validation may have led to severely optimistic performance estimates and results should be confirmed with further studies [35].
Biomarkers of Alcohol Consumption
Alcohol abuse is one of the leading causes of death and disability worldwide [36]. Diagnosis and treatment of alcohol-related diseases is limited by the lack of reliable measures of alcohol intake [37]. Alcohol consumption is mostly self-reported during hospitalization and in observational studies, but its social implications lead to under- or misreporting of this exposure. Biological biomarkers of heavy drinking have been previously created, but they are far from ideal [38••]. Alcohol consumption has been suggested to alter global and site-specific DNA methylation, which in turn can affect gene expression levels. Liu et al. (2018) have identify blood-derived DNA methylation biomarkers of heavy alcohol drinking and explored the functional implications of alcohol-related differential methylation by evaluating its association with gene expression in blood [38••]. A total of 13,317 participants from population-based cohorts (9643 European and 2423 African ancestries) of the Cohorts for Heart and Aging Research in Genomic Epidemiology Consortium plus (CHARGE+) Consortium were included in this analysis. Heavy drinkers, ≥ 42 g per day in men and ≥ 28 g per day in women, represented 2–17% of participants across studies. Four biomarkers of alcohol were created by using a four-step procedure. First, the authors performed an inverse-weighted random-effects meta-analysis, condensing results from EWAS analysis of eight cohorts with European ancestry. A total of 333 CpG sites had p values below 5 × 10−6 in the meta-analysis and were included in both Illumina450 and Illumina EPIC assays. All of them were combined in a LASSO regression using the largest training cohort (2427 European ancestry participants with 8% of heavy drinkers). The LASSO approach regressed alcohol intake in grams and the residuals of linear regressions linking methylation levels of those significant CpGs to sociodemographic confounders. Four sets of CpGs (5, 23, 78, and 144) were selected to discriminate heavy drinkers from light- and non-drinkers. All sets were validated in individual cohort populations of both European and African ancestries and results were generalized using monocyte-derived DNA samples from a population cohort including European, African, and Hispanic ancestries. The most parsimonious set of 5 CpGs allows to discriminate between heavy and non-drinkers, and between heavy and light drinkers with a good accuracy (over 80% and 65% respectively) in all validating cohorts. The biomarkers composed by 23, 78, and 144 CpGs had even better performances [38••]. As a biomarker, the selected 144 CpGs performed better than common clinical variables and biomarkers in discriminating current heavy alcohol drinking. The authors have also shown that whole-blood epigenetic changes were associated with gene expression in whole blood [38••]. Namely, methylation alterations in GABA receptor genes were significantly associated with the expression levels of genes involved in immune function [39]. The authors acknowledge that results stratified by ancestry lack of similarities of many CpG sites and larger studies with multiple ethnicities and a broader age range are needed. The authors also mentioned that differences can be driven by heterogeneity in alcohol consumption across different population cohorts [38••].
Biomarkers of Cumulative Lead Exposure
Lead exposure has chronic adverse effects on the cardiovascular, neurocognitive, and renal systems [40]. It is of pressing importance for environmental and occupational health, with more than a million employees, mostly men, working in general industry and construction, suffering from this exposure every year [41]. However, the Flint crisis, which gained much attention starting in 2014, showed that lead is still a traceable heavy metal in both adults and children. Lead exposure can be measured in several tissues: blood, bones, hair, nails, and urine. However, tissues measuring longer, and cumulative lead exposure are more appropriate for evaluating its long-term health effects. Bone lead levels are better biomarkers than lead levels of blood and any other tissues in capturing cumulative lead exposure [42,43,44]. Patella and tibia bone lead levels reflect exposure of 8–20 years [45] and up to 50 years [46], respectively, and require specialized X-ray-fluorescence-spectroscopy available only in a few centers worldwide. Thus, DNA methylation biomarkers have been developed to reflect those cumulative lead exposures and to be applied to other study populations, which do not have access to that technology [25••]. Those biomarkers were discovered in a population of 348 elderly non-Hispanic white men (73 years old on average) from the Normative Aging Study with moderate lead levels (mean ± SD patella, 27 ± 18 μg/g; tibia, 21 ± 13 μg/g). Both lead biomarkers reconstructed individual cumulative lead exposure using blood DNA methylation profiles—obtainable via Illumina450K or IlluminaEPIC assays. Biomarkers for lead in patella and tibia were computed via leave-one-out cross-validation elastic nets in the 80% of the data and included DNA methylation measured in 59 and 138 sites, respectively. Estimated lead levels were well correlated with actual measured values in the remaining validation set (20%) of the data. These methylation-based biomarkers discriminated participants highly exposed (> median) to lead with good accuracy for both biomarkers (patella AUC = 0.79 (95% CI 0.68–0.90) and tibia AUC = 0.75 (95% CI 0.64–0.87)) [25••]. The two sets of CpGs composing the methylation-based lead biomarkers were mapped to genes biologically related to diseases or processes previously associated with lead exposure. Genes included in the patella biomarker were involved in Alzheimer’s disease, while genes in the tibia biomarker were mainly related to nutritional pathways, including low-protein diets previously linked to increased lead absorption. These findings supported the hypothesis that DNA methylation is an intermediate mechanism between lead exposure and the development of chronic and neurocognitive diseases. Using this approach, the authors were able to reflect two cumulative periods of lead exposure [25••]. However, applications of those biomarkers will require caution, due to the limited data on extreme lead values, and no inclusion of women, and children.
Discussion
DNA methylation is a stable biomarker, with unique properties of persistence and reversibility [47]. Its ability to reflect lifestyle behaviors and their changes enables it to simultaneously incorporate information about exposure concentrations and their timing [20,21,22, 25••]. DNA methylation profiles can be measured in several tissues, including whole blood. Unlike many other epigenetic measures (e.g., RNAs), DNA methylation can be easily detected in retrospective archived and appropriately frozen samples without special handling requirements. Together, these characteristics have made DNA a promising candidate biomarker of past or current environmental exposures and enabled it to contribute to the understanding of the onset and progression of diseases [48]. Those methylation-derived exposure biomarkers are indicative of norms or aberrations present at molecular level linked to environmental exposures. So, they can provide more information on the biological mechanism and function associated to human health than biomarkers based on exposure compounds. Still, studies continue to advance the utility of this biomarker as combinations of DNA methylation at several CpGs are now being utilized as biomarkers to reconstruct different exposures.
More specifically, these novel methylation-derived tools can now be applied to observational studies in order to reconstruct exposures and understand the causes of disease onset. Importantly, this may help in measurement error correction and reducing misclassified exposures when the exposure is associated with a stigma or when technology is unavailable [25, 49•]. Ignoring exposure misclassification when evaluating any exposure-disease relationships can lead to false or exaggerated conclusions [49•]. Thus, measurement error correction approaches, including the development of methylation-derived biomarkers, can improve validity of findings and be of enormous public utility.
Both the LASSO and elastic net approaches, used for the methylation-derived exposure biomarkers, have three main advantages. They are easy to interpret, provide a variable selection emphasizing each CpG site importance, and can be regularized with a small number of hyperparameters to avoid overfitting [23, 24]. For these characteristics, both LASSO and elastic net regressions are well suited for environmental epigenetic analyses; however, their performance depends on the number of CpGs included in the model and the relationship between CpGs and the environmental exposure [23, 24]. Indeed, those approaches are prone to overfit with a large number of CpGs, especially if the CpGs are highly correlated. To limit this issue, all authors screened the CpGs most significantly associated with the environmental exposure with an EWAS strategy before combining them all in the machine learning approach. Those approaches also assume additivity of CpGs, and linearity between each CpG and the exposure and approaches evaluating multiplicative and non-linear relationships can be considered for improving the development of methylation-derived environmental exposure biomarkers.
The excitement to use methylation-derived biomarkers across observational studies has to be balanced with caution. The methylation-derived biomarkers discussed in this text were developed in observational studies—with specific cohort characteristics (i.e., ethnicity/race, sex, age)—and all biomarkers used blood to reconstruct exposures. Hence, applying them to studies with different characteristics or from different tissues might lead to inappropriate exposure reconstruction. Further studies are needed to validate biomarkers under different population characteristics, and open-data sharing policy would facilitate this process. Other tissue types can also be considered, especially if they are closer to the target tissue. For instance, buccal biospecimens, which are easy to collect and bank, may be more appropriate to exposures related to inhalation. Importantly, additional studies are needed to evaluate the interplay relationships between DNA methylation marks in the biomarker prediction and machine learning algorithms not requiring assumptions of additivity and linearity among those DNA methylation marks, may improve the accuracy and prediction of environmental exposures. In moving forward, the utility of such biomarkers will depend on their ability to have consistent results across populations, and in our understanding about the pathways reflected by alterations of those specific CpGs included in each biomarker.
Conclusions
Overall, these novel DNA methylation–based biomarkers are attractive tools to accurately capture past and cumulative environmental exposures, reducing misclassified or missing exposures. Prospective studies with extant methylation data can leverage these novel biomarkers to understand the onset or progression of human diseases. Furthermore, the ability to reconstruct past environmental exposure using extant methylomic data may open the door to novel research questions that would have prospective data on exposure and health, that otherwise would not exist.
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Barros SP, Offenbacher S. Epigenetics: connecting environment and genotype to phenotype and disease. J Dent Res. 2009;88(5):400–8.
Leenen FA, Muller CP, Turner JD. DNA methylation: conducting the orchestra from exposure to phenotype? Clin Epigenetics. 2016;8:92.
Marioni RE, Shah S, McRae AF, et al. DNA methylation age of blood predicts all-cause mortality in later life. Genome Biol. 2015;16(1):25.
Byun HM, Colicino E, Trevisi L, et al. Effects of air pollution and blood mitochondrial DNA methylation on markers of heart rate variability. J Am Heart Assoc Cardiovas Cerebrovas Dis. 2016;5(4):e003218.
Agha G, Mendelson MM, Ward-Caviness CK, Joehanes R, Huan T, Gondalia R, et al. Blood leukocyte DNA methylation predicts risk of future myocardial infarction and coronary heart disease. Circulation. 2019;140(8):645–57.
Wu S, Hivert MF, Cardenas A, Zhong J, Rifas-Shiman SL, Agha G, et al. Exposure to low levels of lead in utero and umbilical cord blood DNA methylation in project viva: an epigenome-wide association study. Environ Health Perspect. 2017;125(8):087019.
Wright RO, Schwartz J, Wright RJ, Bollati V, Tarantini L, Park SK, et al. Biomarkers of lead exposure and DNA methylation within retrotransposons. Environ Health Perspect. 2010;118(6):790–5.
Bitto A, Pizzino G, Irrera N, Galfo F, Squadrito F. Epigenetic modifications due to heavy metals exposure in children living in polluted areas. Curr Genomics. 2014;15(6):464–8.
•• Reese SE, Zhao S, Wu MC, Joubert BR, Parr CL, Håberg SE, et al. DNA methylation score as a biomarker in newborns for sustained maternal smoking during pregnancy. Environ Health Perspect. 2017;125(4):760–6. This manuscript describes of the smoking biomarker during pregnancy.
Joubert BR, Haberg SE, Nilsen RM, et al. 450K epigenome-wide scan identifies differential DNA methylation in newborns related to maternal smoking during pregnancy. Environ Health Perspect. 2012;120(10):1425–31.
Baccarelli A, Wright RO, Bollati V, Tarantini L, Litonjua AA, Suh HH, et al. Rapid DNA methylation changes after exposure to traffic particles. Am J Respir Crit Care Med. 2009;179(7):572–8.
Ladd-Acosta C. Epigenetic signatures as biomarkers of exposure. Cur Environ Health Rep. 2015;2(2):117–25.
Ladd-Acosta C, Fallin MD. DNA methylation signatures as biomarkers of prior environmental exposures. Cur Epidemiol Rep. 2019;6(1):1–13.
Richmond RC, Suderman M, Langdon R, et al. DNA methylation as a marker for prenatal smoke exposure in adults. Int J Epidemiol. 2018;47(4):1120–30.
Cardenas A, Rifas-Shiman SL, Agha G, Hivert MF, Litonjua AA, DeMeo D, et al. Persistent DNA methylation changes associated with prenatal mercury exposure and cognitive performance during childhood. Sci Rep. 2017;7(1):288.
•• Bollepalli S, Korhonen T, Kaprio J, Anders S, Ollikainen M. EpiSmokEr: a robust classifier to determine smoking status from DNA methylation data. Epigenomics. 2019;11(13):1469–86. This manuscript describes of the smoking biomarker.
Zeilinger S, Kuhnel B, Klopp N, et al. Tobacco smoking leads to extensive genome-wide changes in DNA methylation. PLoS One. 2013;8(5):e63812.
McCartney DL, Stevenson AJ, Hillary RF, Walker RM, Bermingham ML, Morris SW, et al. Epigenetic signatures of starting and stopping smoking. EBioMedicine. 2018;37:214–20.
Joehanes R, Just AC, Marioni RE, Pilling LC, Reynolds LM, Mandaviya PR, et al. Epigenetic signatures of cigarette smoking. Circ Cardiovasc Genet. 2016;9(5):436–47.
Rakyan VK, Down TA, Balding DJ, Beck S. Epigenome-wide association studies for common human diseases. Nat Rev Genet. 2011;12(8):529–41.
Feinberg AP. Epigenomics reveals a functional genome anatomy and a new approach to common disease. Nat Biotechnol. 2010;28(10):1049–52.
Portela A, Esteller M. Epigenetic modifications and human disease. Nat Biotechnol. 2010;28(10):1057–68.
Zou H, Hastie T. Regularization and variable selection via the elastic net. J Royal Statis Soc Ser B. 2005;67(2):301–20.
Tibshirani R. Regression shrinkage and selection via the Lasso. J R Stat Soc Ser B Methodol. 1996;58(1):267–88.
•• Colicino E, Just A, Kioumourtzoglou MA, et al. Blood DNA methylation biomarkers of cumulative lead exposure in adults. J Exposure Sci Environ Epidemiol. 2019. This manuscript describes of the lead exposure biomarkers.
•• Markunas CA, Hancock DB, Xu Z, et al. Epigenome-wide analysis uncovers a blood-based DNA methylation biomarker of lifetime cannabis use. bioRxiv. 2019:620641. This manuscript describes of the cannabis biomarker.
Gao X, Zhang Y, Breitling LP, Brenner H. Relationship of tobacco smoking and smoking-related DNA methylation with epigenetic age acceleration. Oncotarget. 2016;7(30):46878–89.
Nwanaji-Enwerem JC, Cardenas A, Chai PR, et al. Relationships of long-term smoking and moist snuff consumption with a DNA methylation age relevant smoking index: an analysis in buccal cells. Nicotine Tob Res. 2018;21(9):1267–73.
• Philibert R, Dogan M, Beach SRH, et al. AHRR methylation predicts smoking status and smoking intensity in both saliva and blood DNA. Am J Med Genetic B Neuropsychiatr Genet. 2020;183(1):51–60. This manuscript describes the role of a few CpGs in the prediction of smoking status.
Gao X, Jia M, Zhang Y, et al. DNA methylation changes of whole blood cells in response to active smoking exposure in adults: a systematic review of DNA methylation studies. Clin Epigenetics. 2015;7:113.
Volkow ND, Baler RD, Compton WM, Weiss SR. Adverse health effects of marijuana use. N Engl J Med. 2014;370(23):2219–27.
Lafaye G, Karila L, Blecha L, Benyamina A. Cannabis, cannabinoids, and health. Dialogues Clin Neurosci. 2017;19(3):309–16.
Andersen AM, Dogan MV, Beach SRH, Philibert RA. Current and future prospects for epigenetic biomarkers of substance use disorders. Genes (Basel). 2015;6(4):991–1022.
Huestis MA. Human cannabinoid pharmacokinetics. Chem Biodivers. 2007;4(8):1770–804.
Steyerberg EW, Harrell FE Jr. Prediction models need appropriate internal, internal-external, and external validation. J Clin Epidemiol. 2016;69:245–7.
Limosin F. Epidemiologic warnings from studies on alcohol use disorders. L'Encephale. 2014;40(2):129–35.
Allen JP. Use of biomarkers of heavy drinking in health care practice. Mil Med. 2003;168(5):364–7.
•• Liu C, Marioni RE, Hedman AK, Pfeiffer L, Tsai PC, Reynolds LM, et al. A DNA methylation biomarker of alcohol consumption. Mol Psychiatry. 2018;23(2):422–33. This manuscript describes of the alcohol consumption biomarkers.
Jin Z, Mendu SK, Birnir B. GABA is an effective immunomodulatory molecule. Amino Acids. 2013;45(1):87–94.
Bakulski KM, Rozek LS, Dolinoy DC, Paulson HL, Hu H. Alzheimer’s disease and environmental exposure to lead: the epidemiologic evidence and potential role of epigenetics. Curr Alzheimer Res. 2012;9(5):563–73.
Holland MG, Cawthon D. Levels ATFoBL. workplace lead exposure. J Occup Environ Med. 2016;58(12):e371–e4.
Weisskopf MG, Proctor SP, Wright RO, et al. Cumulative lead exposure and cognitive performance among elderly men. Epidemiol. 2007;18(1):59–66.
Hu H, Shih R, Rothenberg S, Schwartz BS. The epidemiology of lead toxicity in adults: measuring dose and consideration of other methodologic issues. Environ Health Perspect. 2007;115(3):455–62.
Navas-Acien A, Schwartz BS, Rothenberg SJ, et al. Bone lead levels and blood pressure endpoints: a meta-analysis. Epidemiol. 2008;19(3):496–504.
Hu H, Rabinowitz M, Smith D. Bone lead as a biological marker in epidemiologic studies of chronic toxicity: conceptual paradigms. Environ Health Perspect. 1998;106(1):1–8.
Wilker E, Korrick S, Nie LH, Sparrow D, Vokonas P, Coull B, et al. Longitudinal changes in bone lead levels: the VA normative aging study. J Occup Environ Med. 2011;53(8):850–5.
Handy DE, Castro R, Loscalzo J. Epigenetic modifications: basic mechanisms and role in cardiovascular disease. Circulation. 2011;123(19):2145–56.
Chen BH, Marioni RE, Colicino E, Peters MJ, Ward-Caviness CK, Tsai PC, et al. DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging. 2016;8(9):1844–65.
• Valeri L, Reese SL, Zhao S, Page CM, Nystad W, Coull BA, et al. Misclassified exposure in epigenetic mediation analyses. Does DNA methylation mediate effects of smoking on birthweight? Epigenomics. 2017;9(3):253–65. This manuscript identifies the critical role of misclassified exposure in epidemiology.
Funding
EC was supported by the National Institute on Minority Health and Health Disparities (NIMHD) (R01MD013310) and by the National Institute of Environmental Health Sciences (NIEHS) (P30ES023515, U2CES026444, and UH3OD023337). JCN was supported by a NIH/NIA Ruth L. Kirschstein National Research Service Award (1 F31AG056124-01A1).
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This article is part of the Topical Collection on Environmental Epigenetics
Rights and permissions
About this article

Cite this article
Nwanaji-Enwerem, J.C., Colicino, E. DNA Methylation–Based Biomarkers of Environmental Exposures for Human Population Studies. Curr Envir Health Rpt 7, 121–128 (2020). https://doi.org/10.1007/s40572-020-00269-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40572-020-00269-2