
Abstract
Increased hepatic glucose output, the primary liver dysregulation associated with Type 2 diabetes mellitus (T2DM), is not directly or effectively targeted by the currently available classes of glucose-lowering medications except metformin. This unmet need might be addressed through activation of a specific enzyme-member of the hexokinase family, namely glucokinase (GK). GK serves as a “glucose-sensor” or “glucose receptor” in pancreatic cells, eliciting glucose-stimulated insulin secretion, and as glucose “gate-keeper” in hepatocytes, promoting hepatic glucose uptake and glycogen synthesis and storage. GK activation by small molecules present an alternative approach to restore/improve glycaemic control in patients with T2DM. GK activators (GKAs) may increase insulin secretion from the pancreas and promote glycogen synthesis in the liver, and hence reduce hepatic glucose output. Despite several setbacks in their development, interest in the GKA class has been renewed, particularly since the introduction of a novel, dual-acting full GKA, dorzagliatin, and a novel hepatoselective molecule, TTP399. In this article we provide an overview of the role, efficacy, safety and future developments of GKAs in the management of T2DM.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Background
Type 2 diabetes mellitus (T2DM) is a complex, multifactorial disease that challenges patients, clinicians and health-care systems [1]. A combination of genetic and environmental factors results in chronic hyperglycaemia as well as a cascade of inflammatory and oxidative processes, leading to vascular complications, which are the primary cause of morbidity and mortality in T2DM. The evolving understanding of the complex multifactorial pathogenesis of T2DM [2] challenge the previously established, “simplistic” notion of a disease characterised by insulin deficiency and resistance and shift the focus onto the islet cells (β, α-cells and others) and their interplay with gut, brain, kidney and the liver [3].
When considering the increasing prevalence of T2DM globally, its progressive natural history and the still large proportion of patients not achieving optimal glycaemic control [4], the need for novel treatment strategies and targets [5] becomes even more imperative. This has resulted in the introduction of new therapies which target several of the more recently researched pathogenetic pathways associated with the gut, brain and kidney. These include glucagon-like peptide-1 receptor agonists (GLP1-RAs) and sodium-glucose co-transporter-2 (SGLT2) inhibitors [6, 7].
Nevertheless, increased hepatic glucose output, the primary liver dysregulation associated with T2DM, is not directly or effectively targeted by any of the currently available classes of glucose-lowering medications except metformin [8]. This unmet need might be addressed through activation of a specific enzyme-member of the hexokinase family, namely glucokinase (GK). In this article, we will provide an overview of the role and future developments of GK activators (GKA) in the management of T2DM.
2 Overview of the Role of Glucokinase (GK) in Glucose Homeostasis
Whilst glucose homeostasis is complex, it can be simplified as resulting from the net effect of a two-hormone competition between insulin and glucagon. The latter is secreted by α islet cells of the pancreas and secures provision of energy by maintaining euglycaemia in the fasting state. This is achieved by promoting gluconeogenesis (de novo glucose production from amino acids and fat) and glycogenolysis (glycogen breakdown and glucose release from the liver), thus increasing hepatic glucose output. Free fatty acid released from adipose tissue also provides a complementary mechanism serving a similar purpose. On the other hand, insulin, secreted by β islet cells, produces a glucose-lowering effect in the fed state by promoting glucose utilisation in the periphery (mainly skeletal muscle and adipose tissue) and hepatic glucose uptake by switching liver into a “glycogen synthesis mode”. Increased glucose (in the fed state) in parallel inhibit gluconeogenesis and glycogenolysis [9].
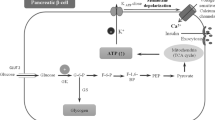
GK serves as a “glucose-sensor” [10] in pancreatic cells, eliciting glucose-stimulated insulin secretion, and as glucose “gate-keeper” in hepatocytes, promoting hepatic glucose uptake and glycogen synthesis and storage. When GK is activated, its preferred substrate glucose is phosphorylated (with magnesium adenosine triphosphate) to glucose-6-phosphate (G6P). Glucose-6-phosphate both activates glycogen synthase and is a substrate for glycogen synthesis [11].
GK biochemical properties and kinetics serve this dual role [12], since the low affinity for glucose (K0.5 of ~ 7–8 mmol/L) restrains GK activity when glucose is in the physiological range [13]. GK is not inhibited by G6P (its end product secretion from the β-cells) and has a sigmoidal saturation curve with glucose (non-Michaelis–Menten kinetics) showing an inflection point of 4–5 mmol/L, which is close to the threshold of insulin secretion. This ensures a graded response to fluctuations in glucose levels and, when glucose is close to the physiological threshold for glucose-stimulated insulin secretion (5 mM), glucokinase activity enters a plateau phase [13].
GK in the liver is maintained as an inactive complex with an endogenous inhibitor, the glucokinase regulatory protein (GKRP) when glucose concentration is less than ~ 10 mM and this confers even lower affinity for glucose in hepatic compared to pancreatic β-cells, in practice, activated only during the postprandial state to serve its function of increasing hepatic glucose uptake [14, 15] (Fig. 1). Thus, GKRP acts as a competitive inhibitor with respect to glucose at the hepatic cell, sequestering GK in the presence of low and dissociating from GK in the presence of raised glucose concentrations.

Glucokinase activity in the hepatic cell
GK is not only expressed in pancreatic β- or hepatic cells, but also in entero-endocrine cells, neurons, pancreatic α- and δ-cells, and cells in the anterior pituitary [16]. Yet, its major role in glucose homeostasis is by eliciting insulin secretion (pancreatic β-cells glucose phosphorylation by GK is the rate-limiting step) and promoting glucose uptake and glycogen synthesis (hepatic cells).
The net effect of the above GK-mediated pathways is blood glucose lowering in both direct (insulin-mediated) and indirect ways. Therefore, it seems plausible to hypothesise that control over GK activity might constitute a novel way of intervention on glucose homeostasis, since GK activation leads to glucose lowering and GK activity is low in patients with T2DM [17]. This hypothesis is reinforced by studies of a discrete genetic subgroup of diabetes, known as maturity-onset diabetes of the young type 2 (MODY2). People with MODY2 carry inactivating, heterozygous mutations in the glucokinase gene (present in ~1 in 1000 of the population, but most remain undiagnosed) [18], usually manifest a benign form of (mainly fasting) hyperglycaemia and with low risk of microvascular complications [19], reflecting a defective “glucose-sensing” ability [18]. However, when compound heterozygosity or homozygosity is present, a more severe form of permanent neonatal diabetes may develop [20]. On the other hand, activating mutations in the glucokinase gene do rarely occur and lead to congenital hyperinsulinaemic hypoglycaemia with heterogenous phenotypic manifestations [21]. The above in vivo evidence, taken together with experiments on mice and human cells [22,23,24,25,26] corroborating the physiologic GK role in glucose homeostasis and the feasibility of its activation, set the scene for the therapeutic rationale behind targeting glucokinase in patients with T2DM. In these patients, the gradual decline in β-cell mass resulting in defective insulin secretion, along with increased hepatic glucose output, constitute two discrete (yet interrelated) pathophysiological derangements in T2DM that could potentially be addressed by a pharmacologic upregulation of GK activity.
3 Overview of GK Activators (GKAs)
Several glucokinase activators have been designed and tested [27, 28] since the introduction of the first agent of this class in 2003 [29]. They are all small molecules able to bind to an allosteric site in the enzyme, stabilising a high-affinity conformation and thus, facilitating GK activation. Interestingly, this is the same region of the enzyme where the majority of activating mutations cluster. These small-cell GKAs can be classified according the chemical structure (carbon-, urea-, 1,2,4-substituted aryl-, 1,3,5-substituted aryl-centred or other) [13]. Another potential classification can refer to the site of action; hepatoselective GKAs have been developed acting with or without disrupting the GK-GKRP interaction in hepatic cells [30, 31], as opposed to systemic GKAs (such as piragliatin or dorzagliatin). Additionally, full GKAs are distinguished from partial GKAs (such as agent PF-04937319). As expected, only a small minority of GKAs have reached the phase of clinical trials (detailed below and summarised in Table 1).
3.1 Clinically Tested GKAs (Pharmacodynamics and Safety)
In a Phase Ib randomised, double-blind, placebo-controlled crossover trial in 15 individuals with mild T2DM, the GKA piragliatin (RO4389620) (25 mg or 100 mg) was found to cause a dose-dependent reduction of glucose levels in both fasting and fed states, mainly mediated through a generalised enhancement of β-cell function [32]. In a similar, short-duration (8-day), dose-escalating (up to 200 mg) study in 59 patients with T2DM, it was reported that piragliatin showed rapid, dose-dependent reduction of fasting and postprandial plasma glucose [33].
In a double-blind study the GKA MK-0941 (at doses 10, 20, 30, or 40 mg tid) efficacy and safety was explored in 587 patients with T2DM treated with insulin glargine [mean baseline glycated haemoglobin (HbA1c) of 9%]. By Week 14, a significant 0.8% drop in HbA1c was noted, along with a significant decrease in the 2-h post-meal glucose (approximately ~ 40 mg or 2.2 mmol/L), yet, by 30 weeks, these responses were not sustained [34]. MK-0941 was also associated with significant increases in the incidence of hypoglycaemia, hypertriglyceridemia (up to ~ 20% increase from baseline) and hypertension.
In a dose-ranging, placebo-controlled study in 458 patients with T2DM, the efficacy and safety of GKA AZD1656 was explored as an add-on to metformin. At 4 months, HbA1c showed a significant reduction, comparable to that associated with glipizide, although the glucose-lowering effect diminished over the two-month extension period, along with an increase in triglycerides [35]. The loss of efficacy, observed after the initial glucose-lowering evidence of efficacy, was replicated in a dose-ranging study in 224 Japanese patients with T2DM [36], in which no major safety concerns were noted.
The safety and efficacy of GKA PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin was explored in dose-ranging RCT trials involving a total of 639 adults with T2DM [37], showing equipotency with sitagliptin in glucose-lowering efficacy at 3 months, when administered as a single-dose of 100 mg. Split-dose regimens explored in a subsequent two-week study showed promising results with regards to safety and tolerability [38].
The GKA AMG 151 (ARRY-403) was evaluated in a multi-dosing, randomised, placebo-controlled, 4-week Phase IIa study involving 236 patients with T2DM on metformin. AMG 151 was found to significantly reduce fasting plasma glucose when administered twice daily, but with increased incidence of hypoglycaemia and hypertriglyceridaemia [39].
More recently, a fourth generation GKA, dorzagliatin (Sinogliatin, HMS5552), a novel, dual-acting, allosteric systemic GKA was initially developed on the basis of piragliatin and finally evaluated in a 3-month, Phase 2, dose-ranging, randomised, double-blind, placebo-controlled study involving 258 Chinese patients with T2DM [40]. At Week 12, a significant reduction in HbA1c was noted in patients on dorzagliatin, − 0.8% with 50 mg bid and ~ 1.1% with 75 mg bid, compared to placebo. Dorzagliatin treatment primarily affected the 2‐h postprandial glucose, which fell significantly in contrast to fasting glucose levels. The glucose-lowering effect was even more pronounced in the drug-naïve patients, which was interpreted as evidence supporting early use of this agent. Notably, no reports of drug-related serious adverse events or severe hypoglycaemia were recorded. In a 4-week clinical study in 24 patients with T2DM, dorzagliatin was shown to also improve β-cell function, as assessed by steady and dynamic state parameters [41]. Active research on dorzagliatin is currently ongoing (NCT03173391 and NCT03141073) and long-term durability of efficacy remains to be proven. In addition, the exact mechanisms for the difference in safety profile of dorzagliatin compared to previously GKAs remain to be investigated [42].
Encouraging results have been reported with another novel hepatoselective molecule, TTP399. A salient difference in the development of this agent was the potential compound screening procedure, which required activity in hepatic cell (glycogen and lactate production) and absence of activity in pancreatic cells (stimulation of insulin secretion) in the presence of high glucose concentrations (15 mM). The molecule was shown to bind directly to GK in the same manner as glucose and express its glucose-lowering activity without affecting glucose-dependent GK translocation [31]. The Phase 2b, 6-month RCT (AGATA trial), explored the efficacy and safety of TTP339 administration (at doses of 400 and 800 mg) in comparison to active drug (sitagliptin) and placebo in 190 patients with T2DM on stable dose of metformin [43]. The main finding was the − 0.9% (− 1.5 to − 0.3) placebo-subtracted decrease in HbA1c for the 800 mg dose at six months, a decrease that started to be notable only after 3 months of treatment. Of note, this significant yet relatively late onset of effect, noted in AGATA trial, was different from that observed in a preceding clinical trial (Phase Ib/IIa), in which the hypoglycaemic effect was noted early [31]. No significant adverse events were recorded and importantly, incidence of symptomatic hypoglycaemia was very low and similar to sitagliptin (one event). Notably, treatment with TTP399 resulted in a significant increase in HDL-c, a neutral effect on triglyceride and cholesterol, and a significant weight loss among those weighting over 100 kg.
Finally, several other GKAs, ADV-1002401, TMG-123 and LY2608204 (globagliatin), have entered the clinical trial stage, yet published results are still lacking.
3.2 Challenges Encountered with GKAs
Both efficacy and safety issues raised important concerns with the use of older generation GKAs. Amongst them, the risk of hypoglycaemia, fatty liver induction, dyslipidaemia, and diminishing long term efficacy appear to be the most significant.
In fact, the occurrence of hypoglycaemia and dyslipidaemia, resulting from over-stimulating pancreatic and hepatic GK respectively were perceived as potential risks from the early phases of GKAs development [12, 44]. Acute, (disproportionate to the stimulus) insulin release, resulting from an exaggerated response to glucose could naturally occur as a consequence of GK activation and was always a plausible risk. The occurrence of hypoglycaemic episodes was particularly noted with piragliatin and MK-0941. To address this risk, partial activators that maintained a greater degree of dependency on glucose levels to minimize the risk of activation at low glucose concentrations [27] were developed. Hepatoselective agents were also designed and tested. The risk of hypoglycaemic events was lower with the partial GKA PF-04937319 [37]. With regards to the pathophysiological processes associated with GK activation that may lead to dyslipidaemia, it is postulated that excessive G6-P accumulation, resulting from hepatic GK overstimulation, activates glycolysis, through the mediation of fructose 2,6-bisphosphate, which show a parallel to G6-P increase and act via feedforward allosteric activation mechanisms. This effect (activation of glycolysis) finally lead to the accumulation of acetyl-CoA (converted from pyruvate), which finally leads to an increase influx to fatty acids (via malonyl-CoA) and triglycerides or increased hepatic de-novo lipogenesis [12]. This is consistent with the reported first stage for non-alcoholic fatty liver disease (NAFLD), which can range from simple steatosis to steatohepatitis [45]. While the latter may require chronic exposure to develop, the relatively acute effect of hypertriglyceridemia was especially noted with MK-0941. Whereas the increase in triglyceride levels (< 20%) may not be considered as pronounced, especially when compared to the one induced by a high-carbohydrate low-fat diet [12], it stills remains unwanted in patients with type 2 diabetes who are already prone to develop dyslipidaemia, hypertension and NAFLD [42].
Interestingly, the initial, promising glucose-lowering efficacy, noted early in the first few weeks with the introduction of a GKA, was not sustained over the course of the clinical studies. Instead, this brief window of efficacy was followed by a rapid decline of efficacy during chronic exposure to the GKA agent. This was observed with both MK-0941 and AZD1656 after approximately four months. The duration of clinical studies with GKA PF-04937319 and AMG 151 did not exceed three and one month respectively. The reasons behind this secondary failure are poorly understood. The study population administered MK-0941 possibly had no minimum critical β-cell mass left (this being necessary for GKA to act), since they had long-standing diabetes and were already on insulin treatment). This was not the case for AZD1656. It has been suggested that hepatic de novo lipogenesis or plasma hyperlipidaemia could be a direct cause for GKA failure, but this hypothesis is not universally accepted [12]. The above pattern of initial response followed by rapid decline of efficacy may contrast to the one noted in the case of hepatoselective TTP399, the glucose-lowering efficacy of which is manifested only after 3 months of treatment in AGATA trial (but not in the earlier phase trials).
Toxicity of GKAs on β-cells has also been hypothesized on the basis of histological reports in mouse models showing double-strand breaks in the deoxyribonucleic acid, likely accounting for activation of the p53 tumour suppressor and resulting β-cell death, following genetic activation of β-cell glucokinase [13, 46]. In fact, this activation resulted in a rapid decrease in blood glucose, which was not sustained, analogous to the observed trajectory in human clinical studies. Another hypothesis, proposed by Agius [12], focuses on the alleged two opposing effects of GKA on hepatic GK. The first stage is already described and occurs early when GK-induced insulin secretion corrects any abnormality in insulin/glucagon ratio and promotes GK/GKRP dissociation and hepatic GK activation. The second effect eventually sets in, when G6P and down-stream phosphate-ester intermediates of glucose metabolism accumulate to an extent that repress GK gene [47], offsetting any initial stimulatory effect induced by GKA [48], rendering their effect clinically unimportant, thus practically neutralising them. Whatever the cause for GKA diminishing efficacy over time this, in practice, may inhibit development, maturation and approval of future GKA agents.
3.3 Future Developments
The minimum requirements for a novel GKA to be considered as a serious alternative to previous GKA agents include clinically relevant and sustained glycaemic efficacy, and low risk of adverse events particularly hypoglycaemia, hepatic steatosis and hypertriglyceridemia. The ideal characteristics for a novel compound would be to address the long-term complications of chronic hyperglycaemia and/or modify the natural course of the disease. Dual-acting full GKA, dorzagliatin and hepatoselective GKA, TTP399 appear to fulfil some of the first set of requirements.
In specific, Scheen provided an indirect comparison of dorzagliatin efficacy with older GKAs [42]. On the basis of HbA1c reduction, dorzagliatin appears to be superior to piragliatin, AZD1656 and PF-04937319 [42]. The placebo‐adjusted ~ − 0.8% decrease in HbA1c with 75 mg dorzagliatin bid was similar to that of MK-0941, although development of the latter was halted, due to safety concerns. The glucose lowering efficacy of AMG151 cannot be compared, since it has been evaluated only over the brief period of one month [39]. Importantly, dorzagliatin-induced reduction in HbA1c, already statistically significant after a month, became progressively more pronounced over the course of the study (both at two and three months), suggesting a sustained effect that might not have reached its maximum by the prespecified study end [42]. This finding is in contrast with the transient glucose-lowering effects reported with older GKAs and suggests that this may be less of an issue with this newer agent. Similarly, TTP399 has shown to be associated with a ~ 0.9% placebo‐adjusted decrease in HbA1c and a glucose-lowering effect that extends (at least) up to 6 months. These glucose-lowering effects of dorzagliatin (− 0.8% decrease in HbA1c) and TTP399 (− 0.9% decrease in HbA1c) may be modest, yet clinically significant, in the similar range with other glucose-lowering medications and adequate for achieving optimal glycaemic control in those patients requiring treatment intensification.
A better safety profile compared with studies of other agents from the class was also noted from the point of view of hypoglycaemia for dorzagliatin, together with no significant change in triglyceride and transaminase levels during the three-month treatment period (one case of eyelid oedema was recorded). With regards to TTP399, treatment also appears to be well tolerated with no symptomatic hypoglycaemia. Moreover, the effect of TTP399 on lipid metabolism appears to be rather favourable, with a modest but significant increase in HDL-c, contrasting with the effects noted inn studies of other agents. Importantly, it should also be noted, along with reports from studies of some of the other GKAs, that dorzagliatin is weight neutral and TTP399 may as well induce weight loss (at least in a subset of patients, with a largely unknown mechanism that remains to be elucidated).
Unfortunately, data extending beyond this three-month period for dorzagliatin and 6-month period for TTP399 are lacking, so that long-term efficacy and safety are still unknown. Also unknown is the long-term net effect on microvascular complications and cardiorenal health. Considering evidence suggesting that common gene variants in GKRP may have a potentially detrimental effect on cardiorenal health, these long-term outcomes are eagerly awaited [49]. Attempting to describe the potential role of GKAs in the therapeutic algorithm in any detail would be premature, especially considering the importance of cardiovascular disease, heart failure and chronic kidney disease in selecting the optimal anti-diabetes agent(s) [50] and the lack of such data with these drugs. Nevertheless, it is reasonable to speculate that GKA are better suited as an early addition to therapy, since their action requires a minimum β-cell mass and they mainly address postprandial hyperglycaemia. Whether this latter property is sufficient for GKAs to displace established second or third-line agents is unlikely unless their novelty of action confers some, as yet, unproven advantage. The reduction in fasting glucagon noted in patients receiving TTP399 may be a potential one. In addition, there may be specific patients (as yet undefined) for whom this type of therapy might be particularly beneficial as part of an individualized approach to management. Clearly much more work needs to be done in this area, including long-term demonstration of safety. This will require clinical trials with longer follow-up, adequate power and carefully selected comparators and outcomes.
4 Conclusions
GK activation by small molecules present an alternative approach to restore/improve glycaemic control in patients with T2DM. GKAs increase insulin secretion and glycogen synthesis and hence reduce hepatic glucose output. Despite several setbacks in their development, interest in the GKA class has been renewed particularly since the introduction of a novel, dual-acting full GKA, dorzagliatin and a novel hepatoselective GKA, TTP399. This is a reflection of overcoming issues of diminishing efficacy and metabolic complications with this class, problems which hindered the development of previous GKAs.
References
Tahrani AA, Barnett AH, Bailey CJ. Pharmacology and therapeutic implications of current drugs for type 2 diabetes mellitus. Nat Rev Endocrinol. 2016;12(10):566–92. https://doi.org/10.1038/nrendo.2016.86.
Defronzo RA. Banting lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–95. https://doi.org/10.2337/db09-9028.
Schwartz SS, Epstein S, Corkey BE, Grant SF, Gavin JR 3rd, Aguilar RB. The time is right for a new classification system for diabetes: rationale and implications of the beta-cell-centric classification schema. Diabetes Care. 2016;39(2):179–86. https://doi.org/10.2337/dc15-1585.
Khunti K, Ceriello A, Cos X, De Block C. Achievement of guideline targets for blood pressure, lipid, and glycaemic control in type 2 diabetes: a meta-analysis. Diabetes Res Clin Pract. 2018;137:137–48. https://doi.org/10.1016/j.diabres.2017.12.004.
Altaf QA, Barnett AH, Tahrani AA. Novel therapeutics for type 2 diabetes: insulin resistance. Diabetes Obes Metab. 2015;17(4):319–34. https://doi.org/10.1111/dom.12400.
Toulis KA, Hanif W, Saravanan P, Willis BH, Marshall T, Kumarendran B, et al. All-cause mortality in patients with diabetes under glucagon-like peptide-1 agonists: a population-based, open cohort study. Diabetes Metab. 2017;43(3):211–6. https://doi.org/10.1016/j.diabet.2017.02.003.
Toulis KA, Willis BH, Marshall T, Kumarendran B, Gokhale K, Ghosh S, et al. All-cause mortality in patients with diabetes under treatment with dapagliflozin: a population-based, open-cohort study in the health improvement network database. J Clin Endocrinol Metab. 2017;102(5):1719–25. https://doi.org/10.1210/jc.2016-3446.
Tahrani AA, Bailey CJ, Del Prato S, Barnett AH. Management of type 2 diabetes: new and future developments in treatment. Lancet. 2011;378(9786):182–97. https://doi.org/10.1016/S0140-6736(11)60207-9.
Rines AK, Sharabi K, Tavares CD, Puigserver P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov. 2016;15(11):786–804. https://doi.org/10.1038/nrd.2016.151.
Perseghin G. Exploring the in vivo mechanisms of action of glucokinase activators in type 2 diabetes. J Clin Endocrinol Metab. 2010;95(11):4871–3. https://doi.org/10.1210/jc.2010-2049.
Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572–87. https://doi.org/10.1038/nrendo.2017.80.
Agius L. Lessons from glucokinase activators: the problem of declining efficacy. Expert Opin Ther Pat. 2014;24(11):1155–9. https://doi.org/10.1517/13543776.2014.965680.
Nakamura A, Terauchi Y. Present status of clinical deployment of glucokinase activators. J Diabetes Investig. 2015;6(2):124–32. https://doi.org/10.1111/jdi.12294.
Choi JM, Seo MH, Kyeong HH, Kim E, Kim HS. Molecular basis for the role of glucokinase regulatory protein as the allosteric switch for glucokinase. Proc Natl Acad Sci USA. 2013;110(25):10171–6. https://doi.org/10.1073/pnas.1300457110.
Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26(2):88–95. https://doi.org/10.1097/MOL.0000000000000155.
Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of langerhans. Front Physiol. 2019;10:148. https://doi.org/10.3389/fphys.2019.00148.
Agius L. Glucokinase and molecular aspects of liver glycogen metabolism. Biochem J. 2008;414(1):1–18. https://doi.org/10.1042/BJ20080595.
Chakera AJ, Steele AM, Gloyn AL, Shepherd MH, Shields B, Ellard S, et al. Recognition and management of individuals with hyperglycemia because of a heterozygous glucokinase mutation. Diabetes Care. 2015;38(7):1383–92. https://doi.org/10.2337/dc14-2769.
Steele AM, Shields BM, Wensley KJ, Colclough K, Ellard S, Hattersley AT. Prevalence of vascular complications among patients with glucokinase mutations and prolonged, mild hyperglycemia. JAMA. 2014;311(3):279–86. https://doi.org/10.1001/jama.2013.283980.
Amed S, Oram R. Maturity-onset diabetes of the young (MODY): making the right diagnosis to optimize treatment. Can J Diabetes. 2016;40(5):449–54. https://doi.org/10.1016/j.jcjd.2016.03.002.
Ping F, Wang Z, Xiao X. Clinical and enzymatic phenotypes in congenital hyperinsulinemic hypoglycemia due to glucokinase-activating mutations: a report of two cases and a brief overview of the literature. J Diabetes Investig. 2019. https://doi.org/10.1111/jdi.13072.
Doliba NM, Fenner D, Zelent B, Bass J, Sarabu R, Matschinsky FM. Repair of diverse diabetic defects of beta-cells in man and mouse by pharmacological glucokinase activation. Diabetes Obes Metab. 2012;14(Suppl 3):109–19. https://doi.org/10.1111/j.1463-1326.2012.01652.x.
Lu M, Li P, Bandyopadhyay G, Lagakos W, Dewolf WE Jr, Alford T, et al. Characterization of a novel glucokinase activator in rat and mouse models. PLoS One. 2014;9(2):e88431. https://doi.org/10.1371/journal.pone.0088431.
Nakamura A, Shimazaki H, Ohyama S, Eiki J, Terauchi Y. Effect of long-term treatment with a small-molecule glucokinase activator on glucose metabolism, lipid profiles and hepatic function. J Diabetes Investig. 2011;2(4):276–9. https://doi.org/10.1111/j.2040-1124.2011.00104.x.
Nakamura A, Terauchi Y, Ohyama S, Kubota J, Shimazaki H, Nambu T, et al. Impact of small-molecule glucokinase activator on glucose metabolism and beta-cell mass. Endocrinology. 2009;150(3):1147–54. https://doi.org/10.1210/en.2008-1183.
Nakamura A, Togashi Y, Orime K, Sato K, Shirakawa J, Ohsugi M, et al. Control of beta cell function and proliferation in mice stimulated by small-molecule glucokinase activator under various conditions. Diabetologia. 2012;55(6):1745–54. https://doi.org/10.1007/s00125-012-2521-5.
Filipski KJ, Pfefferkorn JA. A patent review of glucokinase activators and disruptors of the glucokinase–glucokinase regulatory protein interaction: 2011–2014. Expert Opin Ther Pat. 2014;24(8):875–91. https://doi.org/10.1517/13543776.2014.918957.
Sarabu R, Berthel SJ, Kester RF, Tilley JW. Novel glucokinase activators: a patent review (2008–2010). Expert Opin Ther Pat. 2011;21(1):13–33. https://doi.org/10.1517/13543776.2011.542413.
Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, et al. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301(5631):370–3. https://doi.org/10.1126/science.1084073.
Pfefferkorn JA. Strategies for the design of hepatoselective glucokinase activators to treat type 2 diabetes. Expert Opin Drug Discov. 2013;8(3):319–30. https://doi.org/10.1517/17460441.2013.748744.
Egan A, Vella A. TTP399: an investigational liver-selective glucokinase (GK) activator as a potential treatment for type 2 diabetes. Expert Opin Investig Drugs. 2019;28(9):741–7. https://doi.org/10.1080/13543784.2019.1654993.
Bonadonna RC, Heise T, Arbet-Engels C, Kapitza C, Avogaro A, Grimsby J, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95(11):5028–36. https://doi.org/10.1210/jc.2010-1041.
Zhi J, Zhai S. Effects of piragliatin, a glucokinase activator, on fasting and postprandial plasma glucose in patients with type 2 diabetes mellitus. J Clin Pharmacol. 2016;56(2):231–8. https://doi.org/10.1002/jcph.589.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34(12):2560–6. https://doi.org/10.2337/dc11-1200.
Wilding JP, Leonsson-Zachrisson M, Wessman C, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2013;15(8):750–9. https://doi.org/10.1111/dom.12088.
Kiyosue A, Hayashi N, Komori H, Leonsson-Zachrisson M, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15(10):923–30. https://doi.org/10.1111/dom.12100.
Amin NB, Aggarwal N, Pall D, Paragh G, Denney WS, Le V, et al. Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes. Diabetes Obes Metab. 2015;17(8):751–9. https://doi.org/10.1111/dom.12474.
Denney WS, Denham DS, Riggs MR, Amin NB. Glycemic effect and safety of a systemic, partial glucokinase activator, PF-04937319, in patients with type 2 diabetes mellitus inadequately controlled on Metformin-A randomized, crossover, Active-Controlled Study. Clin Pharmacol Drug Dev. 2016;5(6):517–27. https://doi.org/10.1002/cpdd.261.
Katz L, Manamley N, Snyder WJ, Dodds M, Agafonova N, Sierra-Johnson J, et al. AMG 151 (ARRY-403), a novel glucokinase activator, decreases fasting and postprandial glycaemia in patients with type 2 diabetes. Diabetes Obes Metab. 2016;18(2):191–5. https://doi.org/10.1111/dom.12586.
Zhu D, Gan S, Liu Y, Ma J, Dong X, Song W, et al. Dorzagliatin monotherapy in Chinese patients with type 2 diabetes: a dose-ranging, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Diabetes Endocrinol. 2018;6(8):627–36. https://doi.org/10.1016/S2213-8587(18)30105-0.
Zhu XX, Zhu DL, Li XY, Li YL, Jin XW, Hu TX, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic beta-cell function in patients with type 2 diabetes: a 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018;20(9):2113–200. https://doi.org/10.1111/dom.13338.
Scheen AJ. New hope for glucokinase activators in type 2 diabetes? Lancet Diabetes Endocrinol. 2018;6(8):591–3. https://doi.org/10.1016/S2213-8587(18)30133-5.
Vella A, Freeman JLR, Dunn I, Keller K, Buse JB, Valcarce C. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Sci Transl Med. 2019. https://doi.org/10.1126/scitranslmed.aau3441.
Matschinsky FM. GKAs for diabetes therapy: why no clinically useful drug after two decades of trying? Trends Pharmacol Sci. 2013;34(2):90–9. https://doi.org/10.1016/j.tips.2012.11.007.
Brouwers M, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med. 2015;21(10):583–94. https://doi.org/10.1016/j.molmed.2015.08.004.
Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Schyr-Ben Haroush R, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab. 2014;19(1):109–21. https://doi.org/10.1016/j.cmet.2013.11.007.
Arden C, Petrie JL, Tudhope SJ, Al-Oanzi Z, Claydon AJ, Beynon RJ, et al. Elevated glucose represses liver glucokinase and induces its regulatory protein to safeguard hepatic phosphate homeostasis. Diabetes. 2011;60(12):3110–20. https://doi.org/10.2337/db11-0061.
Arden C, Tudhope SJ, Petrie JL, Al-Oanzi ZH, Cullen KS, Lange AJ, et al. Fructose 2,6-bisphosphate is essential for glucose-regulated gene transcription of glucose-6-phosphatase and other ChREBP target genes in hepatocytes. Biochem J. 2012;443(1):111–23. https://doi.org/10.1042/BJ20111280.
Simons P, Simons N, Stehouwer CDA, Schalkwijk CG, Schaper NC, Brouwers M. Association of common gene variants in glucokinase regulatory protein with cardiorenal disease: A systematic review and meta-analysis. PLoS One. 2018;13(10):e0206174. https://doi.org/10.1371/journal.pone.0206174.
Davies MJ, D'Alessio DA, Fradkin J, Kernan WN, Mathieu C, Mingrone G, et al. Management of hyperglycaemia in type 2 diabetes, 2018. A consensus report by the American Diabetes Association (ADA) and the European Association for the Study of Diabetes (EASD). Diabetologia. 2018;61(12):2461–98. https://doi.org/10.1007/s00125-018-4729-5.
Acknowledgements
We are grateful to the expert reviewers for their constructive suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
None.
Conflict of Interest
Dr. Tahrani reports grants from National Institute for Health Research UK, personal fees and non-financial support from Boehringer-Ingelheim, personal fees from Bristol-Myers Squibb, personal fees and non-financial support from Eli Lilly, grants from Novo Nordisk, grants from Sanofi Aventis, personal fees and non-financial support from Astra Zeneca, personal fees from Janssen, personal fees and non-financial support from Novo Nordisk, non-financial support from MSD outside the submitted work; Professor Barnett reports personal fees from Novo Nordisk UK, personal fees from MSD, personal fees from Boehringer-Ingelheim, and personal fees from Napp. Dr. Toulis reports travel grant from Novo Nordisk and personal fees and non-financial support from Sanofi Aventis and Astra Zeneca outside the submitted work.
Rights and permissions
About this article

Cite this article
Toulis, K.A., Nirantharakumar, K., Pourzitaki, C. et al. Glucokinase Activators for Type 2 Diabetes: Challenges and Future Developments. Drugs 80, 467–475 (2020). https://doi.org/10.1007/s40265-020-01278-z
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40265-020-01278-z