
Abstract
Despite advances in the management of type 2 diabetes mellitus (T2DM), one-third of patients with diabetes do not achieve the desired glycemic goal. Considering this inadequacy, many agents that activate glucokinase have been investigated over the last two decades but were withdrawn before submission for marketing permission. Dorzagliatin is the first glucokinase activator that has been granted approval for T2DM, only in China. As overstimulation of glucokinase is linked with pathophysiological disturbances such as fatty liver and cardiovascular issues and a loss of therapeutic efficacy with time. This review aims to highlight the benefits of glucokinase activators vis-à-vis the risks associated with chronic enzymatic activation. We discuss the multisystem disturbances expected with chronic activation of the enzyme, the lessons learned with glucokinase activators of the past, the major efficacy and safety findings with dorzagliatin and its pharmacological properties, and the status of other glucokinase activators in the pipeline. The approval of dorzagliatin in China was based on the SEED and the DAWN trials, the major pivotal phase III trials that enrolled patients with T2DM with a mean glycosylated hemoglobin of 8.3–8.4%, and a mean age of 53–54.5 years from multiple sites in China. Patients with uncontrolled diabetes, cardiac diseases, organ dysfunction, and a history of severe hypoglycemia were excluded. Both trials had a randomized double-blind placebo-controlled phase of 24 weeks followed by an open-label phase of 28 weeks with dorzagliatin. Drug-naïve patients with T2DM with a disease duration of 11.7 months were enrolled in the SEED trial while the DAWN trial involved patients with T2DM with a mean duration of 71.5 months and receiving background metformin therapy. Compared with placebo, the decline in glycosylated hemoglobin at 24 weeks was more with dorzagliatin with an estimated treatment difference of − 0.57% in the SEED trial and − 0.66% in the DAWN trial. The desired glycosylated hemoglobin (< 7%) was also attained at more than two times higher rates with dorzagliatin. The glycemic improvement was sustained in the SEED trial but decreased over 52 weeks in the DAWN trial. Hyperlipidemia was observed in 12–14% of patients taking dorzagliatin versus 9–11% of patients receiving a placebo. Additional adverse effects noticed over 52 weeks with dorzagliatin included an elevation in liver enzymes, hyperuricemia, hyperlacticacidemia, renal dysfunction, and cardiovascular disturbances. Considering the statistically significant improvement in glycosylated hemoglobin with dorzagliatin in patients with T2DM, the drug may be given a chance in treatment-naïve patients with a shorter disease history. However, with the waning therapeutic efficacy witnessed in patients with long-standing diabetes, which was also one of the potential concerns with previously tested molecules, extended studies involving patients with chronic and uncontrolled diabetes are needed to comment upon the long-term therapeutic performance of dorzagliatin. Likewise, evidence needs to be generated from other countries, patients with organ dysfunction, a history of severe hypoglycemia, cardiac diseases, and elderly patients before extending the use of dorzagliatin. Apart from monitoring lipid profiles, long-term safety studies of dorzagliatin should involve the assessment of serum uric acid, lactate, renal function, liver function, and cardiovascular parameters.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Considering the unmet need to achieve the desired glycemic goal, glucokinase activators such as dorzagliatin may be considered in the diabetes armamentarium. |
When administered, extended surveillance is required for adverse effects such as liver dysfunction, dyslipidemia, hyperuricemia, hyperlacticacidemia, renal dysfunction, and a rise in blood pressure. |
Extended studies involving patients of different ethnicities and patients with chronic and uncontrolled diabetes mellitus and organ dysfunction are needed to comment upon the long-term therapeutic performance of dorzagliatin. |
1 Introduction
Type 2 diabetes mellitus (T2DM) is characterized by progressive beta-cell dysfunction, insulin resistance, and ensuing hyperglycemia. As per 2021 estimates of the International Diabetes Federation, around 10.5% of the global adult population aged 20–79 years have diabetes, and nearly 1.5 million deaths per year are attributed to the disease [1, 2]. Diabetes is often accompanied by other components of the metabolic syndrome such as hypertension, obesity, dyslipidemia, and fatty liver. Patients with diabetes are at increased risk of cardiovascular diseases such as heart failure and ischemic heart disease, which are also the major cause of mortality in diabetes [3]. Apart from this, microvascular complications in the form of nephropathy, retinopathy, and neuropathy add to the disease burden and poor quality of life.
The drugs used in the management of diabetes address the hyperglycemic component with a target of achieving glycated hemoglobin (HbA1c) < 7%. Metformin and sulfonylureas are the mainstays of oral therapy in patients with newly diagnosed diabetes, though many patients eventually require insulin therapy. With claims of cardiovascular benefit, newer drugs such as injectable glucagon-like peptide-1 (GLP-1) agonists and sodium-glucose cotransporter-2 inhibitors are also being preferred as the initial therapy in patients with diabetes. However, despite recent advances in the treatment, more than one-third of patients with diabetes do not achieve the desired HbA1c goal [4,5,6]. Additionally, insulin, sulfonylureas, and insulin-promoting agents produce weight gain and hypoglycemia, which contributes significantly to cardiovascular mortality [6,7,8,9]. The need for frequent insulin injections and adverse effects such as urinary infections with sodium-glucose cotransporter-2 inhibitors, and gastrointestinal problems with GLP-1 analogs are other limiting factors that affect treatment adherence and interfere with the achievement of glycemic goals [10,11,12,13,14].
Considering these issues, glucokinase (GCK) located in the liver and the pancreas and known to have crucial roles in glucose homeostasis was being viewed as a potential target in diabetes management. Though the research on GCK activators (GKAs) started in the late 1990s, it took more than two decades for a molecule to reach the approval stage. Many compounds were investigated in pre-clinical and clinical phases but failed to get approval from regulatory bodies or were withdrawn by the manufacturers before filing for licensing owing to safety and efficacy concerns [15,16,17]. Dorzagliatin is the only GKA that has received approval for diabetes therapy in China in 2022. Through this review, we discuss the physiological roles of GCK in human metabolism, the link between GCK defect and hyperglycemia, and the clinical-pharmacological properties of dorzagliatin that led to its approval. The shortcomings noticed with previous GKAs, the negative side of GCK overactivation, and therapeutic parameters to be monitored with dorzagliatin are also highlighted, along with other GKAs in the pipeline.
2 Physiological Roles of GCK
2.1 GCK in the Liver and Pancreas
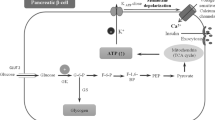
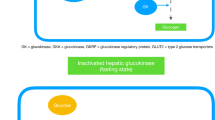
Glucokinase, also known as hexokinase IV, is present mainly in the liver and beta cells of the pancreas. In the latter, the enzyme acts as a ‘glucose sensor’ or ‘glucose receptor’ and brings about glucose phosphorylation. This leads to the production of ATP and inhibition of ATP-sensitive K+ channels, which in turn promotes insulin release by opening calcium channels (Fig. 1) [15]. The enzyme is termed a ‘gatekeeper’ in liver cells where glucose phosphorylation culminates in glycogen synthesis [15, 18]. Various conformations of the enzyme have been identified such as super open, open, and closed, where the super open state implies the inactive form of the enzyme, which has to be converted to closed and open forms to be activated [17]. The enzyme does not follow Michaelis–Menten kinetics and is not saturated by the product glucose-6-phosphate. It has a lower affinity for glucose compared with other hexokinases present in major body sites. The substrate concentration at which the reaction velocity of GCK is half the maximum (K0.5) is 7–8 mmol/L [15, 16]. Consequently, at lower blood glucose levels, the enzyme stays in inactive super open forms and is activated to open and closed forms in the fed state. The enzyme carries a further lower affinity for glucose in the liver, thus necessitating the presence of a higher concentration of glucose in hepatocytes for full activation. Apart from the organ-specific role, the regulation of GCK is also different in the liver. The enzyme at low blood glucose levels stays in the nucleus of hepatocytes in a bound state with GCK regulatory protein (GKRP) and is released from the nucleus after dissociation from GKRP at higher levels of blood glucose. In the pancreas, the enzyme is also present in alpha cells where it regulates glucagon release at higher blood glucose levels. The enzyme is activated by glucose and fructose-1-phosphate and inhibited by fructose-6-phosphate [18].

Physiological roles of glucokinase (GCK) and pathophysiological consequences of GCK overactivation (created with BioRender.com under license). AMP adenosine monophosphate, ATP adenosine triphosphate, CoA coenzyme A, GKRP glucokinase regulatory protein, GLP glucagon-like peptide, GMP guanosine monophosphate, IMP inosine monophosphate, PRPP phosphoribosyl pyrophosphate, TG triglyceride
2.2 GCK in the Brain and Intestine
Glucokinase in trace amounts is present in hypothalamic neurons where it acts as a glucose sensor. Coordination between the hypothalamus and the autonomic nervous system plays a crucial role in maintaining the body’s metabolism. Knowledge of connections between the hypothalamus and the autonomic nervous system is, however, scant. Glucose signaling in these ‘glucose excited’ cells matches that of pancreatic beta cells. Increased glucose-6-phosphate stimulates neurotransmitter release by enhancing calcium entry into the neurons. Some neurons, however, are inhibited by glucose through signaling involving adenosine monophosphate-activated protein kinase and cystic fibrosis transmembrane conductance regulator channels [19, 20]. However, which neurons in the hypothalamus are ‘glucose excited’ or ‘glucose inhibited’ is poorly understood [18]. Glucokinase expression has been noted in neuropeptide Y, proopiomelanocortin, and gamma amino butyric acid neurons of the hypothalamus. The GCK stimulation in the latter inhibits glucagon release from the vagus nerve [21]. Glucokinase activity has been documented in pituitary extracts, follicle-stimulating hormone, and luteinizing hormone cells, and the enzyme can influence glucose metabolism through modulation of sex steroids [18]. Considering the glucose regulatory complexities between the brain and periphery, the neuronal effects of GCK stimulation are an interesting area to be explored.
Glucokinase has also been documented in enteroendocrine L cells where it is believed to modulate incretin release. However, the role of GCK as a glucose sensor in intestinal cells has been challenged in some studies [22, 23]. Incretin release from the intestinal cells might be independent of GCK [23] and glucose homeostasis at present is believed to be determined predominantly by the hepatic and pancreatic GCK [15].
2.3 Role of GCK in Beta-Cell Proliferation
In addition to stimulating insulin release, GCK might trigger the proliferation of beta cells and can prevent beta-cell apoptosis [16], though the evidence is limited to a few pre-clinical studies. Mice with insufficiency of GCK have impaired beta-cell mass and reduced expression of insulin receptor substrate-2 (IRS2) [16]. Glucokinase activation by a small molecule-based GKA has been shown to upregulate IRS2 and enhance the levels of pancreatic duodenal homeobox (pdx1), a transcriptional activator required for beta-cell proliferation [24]. Glucokinase haplo-insufficient mice also show reduced expression of phosphoinositide-dependent kinase 1 and cyclin D2 genes, which is improved by GKAs [24, 25]. In db/db mice, which is a classic animal model of T2DM and insulin resistance, no effect of GKAs was seen on the expression of Pdx1 and IRS2. Interestingly, GKA preceded by the administration of exendin-4, a GLP-1 analog, enhanced Pdx1 and IRS2 gene expression. In this line, the combined use of GKA and GLP-1 analogs has been suggested as an efficient approach for improving insulin release and beta-cell mass [25].
3 GCK and Diabetes Link
Glucokinase dysfunction in diabetes has been validated through genetic studies and animal models of diabetes and obesity. Heterozygous loss-of-function mutation in the GCK gene is responsible for maturity-onset diabetes of young (MODY2) and homozygous loss of function causes permanent neonatal diabetes [26]. Activating mutations in the GCK gene, though less common, cause persistent hyperinsulinemic hypoglycemia in infants and adults [27, 28]. Most of these mutations lie in the allosteric site away from the substrate binding site of GCK [16].
The role of GCK mutations in T2DM is unestablished at present. However, there is some evidence of defective hepatic GCK in diabetes from pre-clinical and clinical studies [29,30,31,32]. As GCK causes insulin release from pancreatic beta cells, selective disruption of a pancreas-specific GCK isoform reduces insulin secretion in response to glucose [25]. In animal models of obesity and insulin resistance, GCK activity may initially be increased but decreases subsequently with the development of diabetes [32]. Low levels of GCK in the cytoplasm because of poor dissociation from GKRP have been demonstrated in Zucker diabetic fatty rats [33]. Defective GCK activity not only hampers hepatic glycogen synthesis but inhibits the autoregulatory role of glucose in suppressing endogenous glucose production in hepatocytes [33]. In this context, inhibition of GCK interferes with hepatic glucose uptake and enhances endogenous glucose production. Activation of GCK by sorbitol and fructose-1-phosphate, however, enhances glucose uptake and suppresses endogenous glucose production [33]. Overexpression of GCK increases liver glycogen and imparts resistance to fatty diet-induced diabetes [34]. Likewise, restoration to overcompensation of GCK has been shown to normalize blood glucose in diabetic fatty rats [32, 35]. Thus, pancreatic and hepatic GCK play a crucial role in maintaining glucose homeostasis by modulating pathways related to insulin release, hepatic glucose uptake, glycogen synthesis, and glucose production. Consequently, activation of GCK by small molecule-based GKAs has been endorsed as a potential approach in the treatment of diabetes.
4 Negative Side of GCK Activation
4.1 Metabolic Disturbances of GCK Overactivation
Notwithstanding the usefulness of GCK, increased GCK activity is not devoid of adverse consequences. This is exemplified by the metabolic disturbances seen with the gain-of-function mutations of the GCK gene. Glucokinase expression in the liver is associated with increased expression of fatty acid synthase and acetyl CoA carboxylase, enzymes involved in lipogenesis (Fig. 1). Continuous GCK activation produces pyruvate, which is converted to acetyl CoA, a precursor of fatty acid synthesis. Thus, GCK activation is expected to produce fatty liver [36]. The gain in GCK activity due to a loss-of-function mutation in the GKRP gene (GCKR rs780094 and rs1260326-P446L variant) can cause low blood glucose and high blood triglyceride levels [37, 38]. The P446L variant blunts the fructose-6-phosphate-mediated inhibition of GCK via GKRP, decreases the nuclear localization of GCK, and reduces GKRP-GCK interactions [38, 39]. Interestingly, the same variant has been associated with high blood glucose levels after glucose challenge in some studies [40]. Hyperglycemia, hypertriglyceridemia, insulin resistance, and weight gain are also evident with the long-term expression of GCK [41]. Adenoviral vector-mediated expression of the GCK gene lowers blood glucose but is dose-dependently associated with an increase in plasma triglycerides, lactate, and free fatty acids in animals. A high expression may also cause significant suppression of insulin, the mechanism of which is still unclear [35]. Another ambiguous area is how the suppressed activity of hepatic GCK in diabetes would explain the hypertriglyceridemia of diabetes.
As a corollary to the pre-clinical studies and genetic polymorphism studies, higher rates of hypoglycemia, hypertriglyceridemia, and fatty liver were witnessed in clinical trials of small-molecule GKAs [42,43,44,45,46,47]. However, some heterozygous-activating mutations in GCK genes such as p. Val389Leu cause an aberrant release of insulin and hypoglycemia without causing hypertriglyceridemia and fatty liver, lending hope to the molecules designed for GCK activation [28].
4.2 Other Perturbations Arising from Continuous GCK Activation
Apart from hypoglycemia, hypertriglyceridemia, and the risk of hepatic steatosis, GCK activation can manifest in several other forms. Continuous activation of glycolysis by GCK produces pyruvate and lactic acid (Fig. 1). Elevated lactate levels interfere also with uric acid excretion in the proximal tubules of the kidney [48]. In this regard, the rs1260326 (P446L) variant associated with increased GCK activity has been linked with the modulation of uric acid excretion and hyperuricemia [49]. Further, glucose-6-phosphate participates in the hexose monophosphate shunt, and by increasing the uric acid precursor, phosphoribosyl pyrophosphate enhances uric acid production [50]. Increased GCK reduces the level of adenosine monophosphate and thereby suppresses adenosine monophosphate-activated protein kinase. The latter is a suppressor of urate crystal-induced inflammation in vitro and in vivo [51]. Thus, continuous GCK activation can create a state of hyperuricemia by enhancing the production and interfering with the excretion of uric acid. These pre-clinical findings have further been corroborated by increased trends of uric acid levels witnessed over 52 weeks in the major trials of dorzagliatin, which are discussed in the subsequent sections [52, 53]. Additionally, the risk of gluconeogenesis and insulin resistance by GCK-mediated suppression of adenosine monophosphate-activated protein kinase should be weighed against the benefits of GCK activation. The enhanced levels of adenosine through the hexose monophosphate shunt can produce bradycardia in susceptible patients. Higher rates of bradycardia were evident with another GKA, globalagliatin [45], and are described below in the section on other GKAs.
Mutations in GKRP genes such as rs1260326 T allele and rs1799884 T alleles causing GCK activation have been linked with macroalbuminuria, a fall in glomerular filtration, and end-stage renal disease in Chinese patients with diabetes [54]. The rs1799884 T allele is also associated with high blood pressure. An increase in blood pressure was evident with GKAs such as MK-0941 in early clinical trials [42]. Some other polymorphisms activating GCK such as rs780093 have been linked with an elevated risk of coronary heart disease in older individuals [55]. Other than this, polymorphisms involving the GKRP gene might also influence high-density lipoprotein levels [56]. Thus, pharmacological modulation of GCK can influence metabolic and cardiorenal outcomes. Though no statistically significant differences were observed in pivotal trials of dorzagliatin, numerically higher rates of proteinuria and a small increasing trend of blood creatinine and blood pressure were evident with long-term use of dorzagliatin [52, 53]. An increase in blood urea and uric acid was encountered in a phase I trial of globalagliatin [45]. The clinical significance of these laboratory changes needs to be understood through long-term real-world studies.
5 Search Methodology
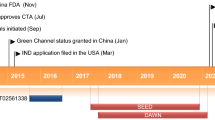
A literature search was performed in PubMed/MEDLINE using ‘HMS5552’ AND ‘diabetes’, ‘dorzagliatin’ AND ‘diabetes’, and ‘Sinogliatin’ AND ‘diabetes’. The authors UK and BKP performed the literature search related to dorzagliatin between February 2023 and mid-April 2023. A search was also performed on ClinicalTrials.gov, using the keywords ‘HMS5552’ or ‘dorzagliatin’. A total of 18 studies were found. Of these, the majority (n = 11) were phase I studies, two were phase II studies, and three studies were phase III clinical trials. Status-wise, the majority (n = 15) have been completed, but results were available for only six. Out of these six studies, the efficacy and safety findings of five studies [52, 53, 57,58,59] are displayed in Table 1. The phase I study by Xu et al. on the pharmacokinetics of dorzagliatin is described separately under the section on pharmacokinetics. For other GKAs, a literature search was performed in MEDLINE using ‘glucokinase’ AND ‘diabetes,’ ‘glucokinase activator’ AND ‘diabetes’ and ‘GKA’ AND ‘diabetes’. A search was performed on ClinicalTrials.gov using ‘diabetes’ in ‘condition or disease’ and ‘glucokinase’ or ‘glucokinase activator ‘or ‘GKA’ in the ‘Other terms’. The authors UK and TJM performed the literature search related to other GKAs between June 2023 and mid-August 2023. To extract the clinical evidence on individual GKAs, a manual reference search was performed from the published review papers, and the names of GKAs retrieved from published reviews were also searched in ClinicalTrials.gov.
6 Findings of Major Clinical Trials on Dorzagliatin (Sinogliatin/ HMS5552)
6.1 SEED Trial (NCT03173391)
This was a phase III, multi-center, randomized, double-blind, placebo-controlled trial of 24 weeks duration, which was followed by an open-label phase lasting for another 28 weeks. The trial focused on the efficacy and safety of dorzagliatin (HMS5552) as monotherapy in drug-naive Chinese adults with T2DM. A total of 463 patients were randomized in a 2:1 ratio to the treatment and placebo arms, respectively. Mean HbA1c of patients was 8.4% at baseline. Change in HbA1c at 24 weeks compared to baseline was the primary efficacy endpoint while the percentage of patients showing a reduction in HbA1c to < 7%, an improvement in fasting plasma glucose (FPG) and postprandial glucose (PPG), and the safety profile of the drug were the secondary measures. A total of 351 patients completed the open-label phase.
Compared with placebo, a statistically significant estimated treatment difference of − 0.57% was evident in HbA1c with dorzagliatin at 24 weeks. A reduction in HbA1c to < 7% was achieved in 42.5% of patients of the dorzagliatin arm and 17.3% of patients of the placebo group. Around 29.4% of patients in the dorzagliatin group and 13.3% in the placebo group showed a reduction in HbA1c to < 7% without hypoglycemia and weight gain. A reduction in HbA1c was evident as early as 4 weeks and was sustained over 52 weeks. The time for HbA1c to decline to < 7% was 12.1 weeks in the dorzagliatin arm and not assessable in the placebo arm. While PPG showed an improvement in the treatment arm, improvement in FPG was less discernible with the GKA. No significant difference was observed in adverse events (AEs) between the dorzagliatin (77%) and placebo (67%) groups. Upper respiratory tract infections (URTIs) and hyperlipidemia were the two most common AEs in both groups. Close to 12% of patients in the dorzagliatin group developed hyperlipidemia. Clinically significant hypoglycemia (less than 3 mmol/L) was noticed in 0.3% of participants in the dorzagliatin group and none in the placebo group. Serious AEs were reported in a few patients (4%), but none was considered related to the drug. A mild decline in renal function, an elevation of serum uric acid and liver enzymes, and a marginal rise in blood pressure were other safety concerns noticed over 52 weeks with dorzagliatin [53].
6.2 DAWN Trial (NCT03141073)
This phase III trial evaluated the efficacy and safety of dorzagliatin in Chinese patients with T2DM with a median duration of illness close to 6 years and who were already taking metformin therapy at 1.5 g twice-daily (BID) dose. Similar to the SEED trial, the trial had a randomized, double-blind, placebo-controlled phase of 24 weeks, followed by an open-label phase of 28 weeks. The dorzagliatin arm had 382 participants and 385 participants were included in the placebo group. Mean HbA1c of patients was 8.3%. A total of 646 participants successfully completed the open-label phase. The primary efficacy endpoint was a decline in HbA1c at 24 weeks compared to baseline. Secondary endpoints included: the percentage of patients with a HbA1c reduction to below 7%, changes in FPG and PPG, and AEs. A statistically significant reduction in HbA1c was evident with dorzagliatin at 24 weeks with an estimated treatment difference of − 0.66%. Compared to 10.7% of patients achieving HbA1c < 7% in the placebo group, a higher percentage (44.4%) of patients in the dorzagliatin group achieved the desired HbA1c. A reduction in HbA1c without hypoglycemia and weight gain was achieved at more than three times higher rates with dorzagliatin. A decline in HbA1c was visible as early as 4 weeks similar to the findings of the SEED trial. Though the trial results mention that glycemic improvement was sustained, the improvement in HbA1c, FPG, and PPBG decreased with time over 52 weeks. Additionally, an improvement in blood glucose was seen more in the post-prandial levels and was less significant in the fasting state. Upper respiratory tract infections and hyperlipidemia were the common AEs, but no significant difference was observed between the groups in overall rates of AEs. Hyperlipidemia and hyperuricemia were noticed respectively in 14% and 10% of patients taking dorzagliatin. Serious AEs were reported in 5% of patients taking the GKA but none was considered as related to the drug. Significant hypoglycemia (< 3 mmol/L) was noticed in 0.8% of participants in the dorzagliatin group and none in the placebo arm. Long-term monitoring showed a mild decline in renal function and an increase in the levels of serum uric acid and liver enzymes, similar to the findings of the SEED trial [52].
6.3 NCT02561338
This was a randomized, double-blind, placebo-controlled, phase II clinical trial that aimed to investigate the efficacy, safety, and tolerability of dorzagliatin in Chinese patients with T2DM. The study was conducted for 12 weeks and involved participants who were either drug naive or treated with metformin or an α-glucosidase inhibitor. Dorzagliatin was tested in four different dose regimens of 75 mg once daily, 100 mg once daily, 50 mg BID, and 75 mg BID. The primary outcome was a reduction in HbA1c at 12 weeks compared with baseline. Secondary outcomes included: the percentage of patients with a HbA1c reduction to below 7%, a decline in FPG and PPG, and AEs. The maximum reduction in HbA1c was observed in the 75-mg BID group with a decrease of 1.12% compared with baseline. Participants achieving HbA1c levels below 7% without weight gain and hypoglycemia were significantly higher in the 50-mg BID and 75-mg BID groups. Though PPG declined significantly, FPG did not show an improvement at any dose of dorzagliatin compared to placebo. Serious AEs were noticed in around 2% of participants of the GKA group but were not related to the drug. Common AEs were hyperuricemia (6–12%), upper respiratory tract infection (2–12%), dizziness (4–8%), and hypoglycemia (4–6%). No clinically significant disturbances were observed in liver function, renal function, and lipid levels (data not mentioned in the published version for lipid levels and renal function) [57].
6.4 Dorzagliatin and the Incretin Axis
The role of GCK in enteroendocrine cells is scarcely explored. In a recently published study on a limited number of patients (n = 15) with obesity and T2DM, dorzagliatin apart from increasing the C-peptide levels also increased the total GLP-1 and the active GLP-1 [60]. Though authors attributed the GLP-1 increasing action as unique to dorzagliatin, larger studies are required to affirm the role of GKAs in enhancing incretin and incretin-mediated pathways.
6.5 Remarks on Clinical Trials of Dorzagliatin
A significant improvement in HbA1c compared with placebo, a statistically insignificant difference in adverse effects such as dyslipidemia, and liver function derangement in the double-blinded phase and the uncommon occurrence of clinically significant hypoglycemia might be the reasons that led to the approval of dorzagliatin as a new therapeutic avenue in the diabetes armamentarium. Whereas activation of pancreatic GCK by dorzagliatin aims to improve the insulin release from pancreatic beta cells, the activation of GCK in the liver improves glucose uptake indirectly and suppresses endogenous glucose production. Additional actions include an improvement in beta-cell mass and an increment in incretin response, both of which need validation from larger future studies.
The pivotal clinical trial of dorzagliatin (SEED trial) was conducted in Chinese patients with T2DM with a disease duration of 11.7 months. The results showed around 2.4 times higher rates of glycemic control (HbA1c < 7%) with dorzagliatin compared with placebo [53]. Desired HbA1c without AEs such as hypoglycemia and weight gain was also attained at more than two times higher rates with dorzagliatin. In both phase III trials, the glycemic improvement was evident as early as the fourth week of the start of therapy with the maximum response coming at week 12. The improvement was sustained over 52 weeks in the SEED trial but waned with time in the DAWN trial that enrolled patients with long-standing diabetes (≈ 6 years) and taking metformin [52]. Reasons for the decline in the therapeutic efficacy of dorzagliatin are not known but perhaps reflect the development of saturation in the activation kinetics of GCK. Suppression of the adenosine monophosphate kinase enzyme might be another factor interfering with the therapeutic performance of the compound. Fasting insulin decreased during the initial 24 weeks with dorzagliatin in the DAWN trial and an improvement in HOMA-β, a measure of beta-cell function, was measured only over the 24 weeks. Additionally, whether the drug performance is affected by the duration of diabetes warrants further evaluation. Secondary drug failure is already a known phenomenon with insulin secretagogues such as sulfonylureas [61, 62]. Longer studies involving patients with newly diagnosed and long-standing diabetes are needed to understand the long-term performance of dorzagliatin with respect to the duration of T2DM. Another striking but common observation was discordance in the improvement of FPG and PPG. An improvement in blood glucose was more evident in PPG [52, 53, 57] with FPG showing only a modest difference.
There were other interesting observations pertaining to efficacy and safety endpoints in phase III trials. The decline in body weight was less compared with placebo and systolic blood pressure showed a small increasing trend over 52 weeks with dorzagliatin in the SEED trial [52, 53]. Though the DAWN trial showed a decline in body weight with dorzagliatin at 52 weeks [52], the reduction in body weight was less compared with placebo in the double-blind phase. A small rising trend in blood pressure was also observed in the placebo group of the DAWN trial after being shifted to dorzagliatin. Apart from this, liver enzymes, serum uric acid, and creatinine levels increased, and glomerular filtration decreased with time.
Broader exclusion criteria were a major shortcoming of the published trials. Patients with baseline liver and kidney disease, anemia, immunosuppression, psychiatric disease, major cardiovascular disease, history of frequent or severe hypoglycemia, and a history of diabetic ketoacidosis or hyperosmolar coma were excluded. Likewise, the representation of elderly patients and frail elderly patients was inadequate. The mean HbA1c of the enrolled patients was around 8.3–8.4% and severe diabetes was inadequately represented. Data specific to these subsets are therefore needed before extrapolating the results to the general population with T2DM.
Both phase III studies failed to provide comparative data against placebo over 52 weeks as patients were switched to the open-label phase after 24 weeks. While no significant lipid abnormalities or liver function derangements were observed compared to placebo, the increasing trends of plasma liver enzymes, triglycerides, uric acid, and creatinine levels and cardiovascular changes in the form of a rise in blood pressure, witnessed over 52 weeks were not highlighted in the major findings. The clinical significance of these safety issues should be ascertained from active surveillance of patients prescribed dorzagliatin in the post-marketing period.
7 Pharmacokinetics of Dorzagliatin
The pharmacokinetic properties of dorzagliatin were tested in a placebo-controlled phase I trial in healthy individuals with a mean age of 24.2 years. The drug was administered in six different doses with a range from 5 to 50 mg. The maximum plasma concentration following a single oral administration of dorzagliatin 50 mg was 582 ng/mL and depending on the doses, the median time to reach the peak plasma concentration varied from 1.25 to 2.5 h. The elimination half-life ranged from 4.48 to 7.51 h in healthy subjects and around 13–15 h in diabetic patients [58, 63]. The drug has a plasma clearance of around 12 L/h. The biliary route seems to be the major route of elimination as less than 11% of the administered drug was excreted in urine. The role of cytochrome P450 enzymes in the metabolism of dorzagliatin is unclear, but minor metabolites are produced by oxidation, hydrolysis, and a reduction of the drug [63]. No change in the volume of distribution, clearance, and elimination half-life of the drug was observed in patients with end-stage renal disease. As such, no dosage modification has been advised in diabetic patients with kidney disease [64].
8 Interactions of Dorzagliatin with Other Oral Antihyperglycemic Agents
Studies assessing the drug interactions between dorzagliatin, and other oral antihyperglycemic agents are currently limited. Dorzagliatin has been tested for efficacy and safety in patients with T2DM uncontrolled on metformin with no mention of pharmacokinetic interactions between the two drugs [52]. Though metformin remains unmetabolized and is less prone to drug interactions, the effect of dorzagliatin on the bioavailability of metformin should be ascertained. In this regard, a phase I trial (NCT02597400) is ongoing. Another trial assessing drug interactions between dorzagliatin and empagliflozin (NCT03790787) is also completed but without results. In a study conducted on a limited number of diabetic patients, pharmacokinetic changes of dorzagliatin addition to sitagliptin were evaluated. No significant effect was observed in the area under the concentration–time curve and the maximum concentration of either drug when used in combination [60]. The pharmacological parameters of dorzagliatin should also be evaluated in the presence of strong cytochrome inducers such as rifampicin and cytochrome inhibitors such as isoniazid, diltiazem, and itraconazole. In this context, some phase I trials have been completed but results are not available (NCT04080596 and NCT04080609).
9 Lessons Learned from GKAs in the Past
The evidence related to the GKAs of the past and those expected to gain marketing approval in the future are described below and in Table 2 [42,43,44,45,46,47, 65,66,67,68,69,70]. Piragliatin, a dual activator of GCK in the liver and beta cells of the pancreas, was the first GKA that made entry into clinical trials in 2010. However, the drug could not progress to the phase of approval owing to high rates of hypoglycemia [47, 69]. Subsequently, many other compounds were developed but only a few of them have produced favorable efficacy and safety results. High rates of hypoglycemia, hypertriglyceridemia, and fatty liver were the main safety concerns with GKAs such as AMG 151/ARRY-403 and MK-0941, which necessitated early termination of clinical trials [42, 43]. MK-0941 also failed to produce a sustained glycemic improvement [42]. In various unpublished trials of MK-0941 in diabetic patients (NCT00824616, NCT00792935, NCT00511472, and NCT00511667), higher rates of hypoglycemia, tremors, dizziness, and gastrointestinal disturbances such as nausea and constipation were evident. A lack of persistent glycemic control and higher rates of hypoglycemia were the reason for the termination of trials of some dual GKAs such as AZD1656 and AZD6370 [17, 44, 65]. Therapeutic failure observed with some of the GKAs has occurred in the settings of long-standing diabetes as patients enrolled were having diabetes for a mean duration varying from 6 to 12 years [42, 44, 65]. Considering that high rates of hypoglycemia and hepatic steatosis are observed with many dual-acting GKAs, some hepatic selective GKAs were developed that spare the GCK of beta cells and have a lower risk of hypoglycemia. In this regard, a lower risk of hypoglycemia and hypertriglyceridemia has been shown with some hepato-selective GKAs such as Pfizer’s PF-04991532 and vTv Therapeutics’ TTP 399 [70]. Dual GKAs have also been categorized as partial activators, which cause a reduction in Km, and full activators, which enhance the Vmax of GCK. Examples of full GKAs include MK-0941 and dorzagliatin while PB-201 (previously PF-04937319), and possibly, AZD1656 activate GKA partially [17, 71]. The safety and efficacy findings of GKAs, however, have not displayed a consistent relationship with the degree of enzymatic activation. For example, PF-04937319, a partial GKA has produced variable results with a low risk of hypoglycemia in some studies to a significantly higher rate of hypoglycemia in other studies [46, 67]. Further, the dose–response relationship of AEs has also not been confirmed and some AEs such as hypertriglyceridemia occurred even at lower doses of the compounds. Though HbA1c showed an improvement, no consistent change and rather, an increasing trend of fasting glucose has been observed with hepato-selective GKAs such as PF-04991532 and dual activators such as PF-04937319, AZD1656, and AZD6370 [44, 46, 66, 72]. In this regard, even dorzagliatin, which is a dual full activator of GCK has failed to produce a significant improvement in fasting blood glucose. That the improvement in HbA1c is largely determined by a decrease in post-prandial and not fasting blood glucose has also been reproduced in a systematic review of limited studies involving four GKAs: PF-04937319, MK-0941, AZD1656, and dorzagliatin [71]. These conflicting safety and efficacy findings necessitate further research to validate the tissue selectivity hypothesis and the extent of enzymatic activation as determinants of clinical outcomes with GKAs.
10 Other GKAs in the Pipeline
Some other GKAs are being tested in various phases of clinical trials. LY 2608204 (Globalagliatin, SY-004, Eli Lilly) has been investigated in many phase I trials. A significantly higher percentage of individuals in the treatment arm developed AEs [45, 73]. Apart from hypoglycemia and hypertriglyceridemia, other events noticed at higher rates included hyperuricemia, sinus bradycardia, and elevations in blood urea [45]. The GKA, TMG-123, has demonstrated a sustained and better glycemic control than metformin or glibenclamide (glyburide) without disturbing liver enzymes and lipid levels in animal studies. Clinical studies on this compound, however, have not been started [74]. PB-201 (previously PF-04937319 by PegBio-Pfizer), a partial activator of GCK as mentioned above, is being investigated in a phase III multicentric trial in China (NCT05102149). This 52-week trial involves treatment-naïve patients with T2DM and aims to compare therapeutic outcomes with PB-201, vildagliptin, and placebo. TTP-399 by vTv Therapeutics, recently named as cadisegliatin, is so far the only liver selective GKA that has shown significant improvement in HbA1c compared with placebo and equivalent to a dipeptidyl peptidase-IV inhibitor. The risk of hypoglycemia and hypertriglyceridemia is also low with TTP-399. Glycemic improvement is, however, noticed after 3 months of drug intake. At a higher dose of 800 mg, this GKA showed additional benefits such as weight loss in individuals with a baseline body weight of ≥ 100 kg, a modest decline in fasting blood glucose, a decline in fasting glucagon, and an increment in high-density lipoprotein-cholesterol [70]. The compound has also been evaluated in patients with type 1 DM (NCT03335371) and results have been favorable in terms of an improvement in HbA1c with lower rates of hypoglycemia [75]. The ongoing trials shall throw more light on the future of TTP-399 and related agents.
11 Conclusions
Considering the progressive beta-cell dysfunction of T2DM and the failure to achieve the desired glycemic target in a considerable percentage of patients, activation of GCK seems a justifiable therapeutic avenue. However, the first two decades of GCK activation witnessed unfavorable outcomes in the form of a lack of significant efficacy, inconsistent glycemic improvement, and adverse effects such as hypoglycemia, liver function disturbances, and dyslipidemia. Dorzagliatin, so far, is the only compound that has gained marketing approval in patients with T2DM, and only in China. A significant improvement in HbA1c compared with placebo, a statistically insignificant difference in adverse effects in the double-blinded phase, and the uncommon occurrence of clinically significant hypoglycemia might be the reasons that led to the approval of this molecule. TTP-399 is another compound that inspires hope in diabetes therapeutics. Worth observing, however, is whether an improvement in the glycemic index noticed within weeks of dorzagliatin initiation will persist or wane with time in the context of the duration and severity of diabetes. Currently, it seems, dorzagliatin and other GKAs may be attractive options for the initial period of managing T2DM. Apart from enhancing insulin release and glucose uptake in the liver, other projected roles of dorzagliatin such as the modulation of incretin release should be verified in larger pre-clinical and clinical studies. A non-Chinese population should be recruited to understand ethnicity-related differences in drug disposition and pharmacodynamics. Sufficient evidence needs to be generated from subsets such as elderly patients, individuals with organ dysfunction, cardiac diseases, and uncontrolled diabetes before extending dorzagliatin use to all patients with T2DM. Whether the decline in the therapeutic action of GKAs is related to the suppression of adenosine monophosphate kinase or the duration of diabetes is worth evaluating. Drug interactions between dorzagliatin and other antidiabetic agents as well as with cytochrome P450 enzyme modulators should be evaluated. Long-term safety studies should incorporate the assessment of serum uric acid, lactate, renal function, liver function, and periodic cardiovascular monitoring in addition to blood glucose levels and lipid profiles. The fate of dorzagliatin lies in real-world studies.
References
Diabetes. World Health Organization. https://www.who.int/health-topics/diabetes. Accessed 21 Feb 2024.
International Diabetes Federation. Facts and figures. https://idf.org/about-diabetes/facts-figures/. Accessed 21 Feb 2024.
Ma C-X, Ma X-N, Guan C-H, Li Y-D, Mauricio D, Fu S-B. Cardiovascular disease in type 2 diabetes mellitus: progress toward personalized management. Cardiovasc Diabetol. 2022;21:74.
de Pablos-Velasco P, Parhofer KG, Bradley C, Eschwège E, Gönder-Frederick L, Maheux P, et al. Current level of glycemic control and its associated factors in patients with type 2 diabetes across Europe: data from the PANORAMA study. Clin Endocrinol (Oxf). 2014;80:47–56.
Kazemian P, Shebl FM, McCann N, Walensky RP, Wexler DJ. Evaluation of the cascade of diabetes care in the United States, 2005–2016. JAMA Intern Med. 2019;179:1376–85.
Bar-Tana J. Type 2 diabetes: unmet need, unresolved pathogenesis, mTORC1-centric paradigm. Rev Endocr Metab Disord. 2020;21:613–29.
International Hypoglycemia Study Group. Hypoglycemia, cardiovascular disease, and mortality in diabetes: epidemiology, pathogenesis, and management. Lancet Diabetes Endocrinol. 2019;7:385–96.
Herman ME, O’Keefe JH, Bell DSH, Schwartz SS. Insulin therapy increases cardiovascular risk in type 2 diabetes. Prog Cardiovasc Dis. 2017;60:422–34.
Middleton TL, Wong J, Molyneaux L, Brooks BA, Yue DK, Twigg SM, et al. Cardiac effects of sulfonylurea-related hypoglycemia. Diabetes Care. 2017;40:663–70.
US FDA. FDA revises labels of SGLT2 inhibitors for diabetes to include warnings about too much acid in the blood and serious urinary tract infections. https://www.fda.gov/drugs/drug-safety-and-availability/fda-revises-labels-sglt2-inhibitors-diabetes-include-warnings-about-too-much-acid-blood-and-serious. Accessed 21 Feb 2024.
Gorgojo-Martínez JJ, Mezquita-Raya P, Carretero-Gómez J, Castro A, Cebrián-Cuenca A, de Torres-Sánchez A, et al. Clinical recommendations to manage gastrointestinal adverse events in patients treated with Glp-1 receptor agonists: a multidisciplinary expert consensus. J Clin Med. 2022;12:145.
Drucker DJ. Mechanisms of action and therapeutic application of glucagon-like peptide-1. Cell Metab. 2018;27:740–56.
Uitrakul S, Aksonnam K, Srivichai P, Wicheannarat S, Incomenoy S. The incidence and risk factors of urinary tract infection in patients with type 2 diabetes mellitus using SGLT2 inhibitors: a real-world observational study. Med Basel Switz. 2022;9:59.
Yang H, Choi E, Park E, Na E, Chung SY, Kim B, et al. Risk of genital and urinary tract infections associated with SGLT-2 inhibitors as an add-on therapy to metformin in patients with type 2 diabetes mellitus: a retrospective cohort study in Korea. Pharmacol Res Perspect. 2022;10: e00910.
Toulis KA, Nirantharakumar K, Pourzitaki C, Barnett AH, Tahrani AA. Glucokinase activators for type 2 diabetes: challenges and future developments. Drugs. 2020;80:467–75.
Nakamura A, Terauchi Y. Present status of clinical deployment of glucokinase activators. J Diabetes Investig. 2015;6:124–32.
Ren Y, Li L, Wan L, Huang Y, Cao S. Glucokinase as an emerging anti-diabetes target and recent progress in the development of its agonists. J Enzym Inhib Med Chem. 2022;37:606–15.
Matschinsky FM, Wilson DF. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of Langerhans. Front Physiol. 2019;10:148.
Fournel A, Marlin A, Abot A, Pasquio C, Cirillo C, Cani PD, et al. Glucosensing in the gastrointestinal tract: impact on glucose metabolism. Am J Physiol Gastrointest Liver Physiol. 2016;310:G645–58.
Routh VH. Glucose sensing neurons in the ventromedial hypothalamus. Sensors. 2010;10:9002–25.
Lamy CM, Sanno H, Labouèbe G, Picard A, Magnan C, Chatton J-Y, et al. Hypoglycemia-activated GLUT2 neurons of the nucleus tractus solitarius stimulate vagal activity and glucagon secretion. Cell Metab. 2014;19:527–38.
Reimann F, Gribble FM. Glucose-sensing in glucagon-like peptide-1-secreting cells. Diabetes. 2002;51:2757–63.
Murphy R, Tura A, Clark PM, Holst JJ, Mari A, Hattersley AT. Glucokinase, the pancreatic glucose sensor, is not the gut glucose sensor. Diabetologia. 2009;52:154–9.
Nakamura A, Togashi Y, Orime K, Sato K, Shirakawa J, Ohsugi M, et al. Control of beta cell function and proliferation in mice stimulated by small-molecule glucokinase activator under various conditions. Diabetologia. 2012;55:1745–54.
Terauchi Y, Takamoto I, Kubota N, Matsui J, Suzuki R, Komeda K, et al. Glucokinase and IRS-2 are required for compensatory β cell hyperplasia in response to high-fat diet-induced insulin resistance. J Clin Investig. 2007;117:246–57.
Hulín J, Škopková M, Valkovičová T, Mikulajová S, Rosoľanková M, Papcun P, et al. Clinical implications of the glucokinase impaired function: GCK MODY today. Physiol Res. 2020;69:995–1011.
Loh WJ, Dacay LM, Tan CSH, Ang SF, Yap F, Lim SC, et al. Glucokinase activating mutation causing hypoglycemia diagnosed late in adult who fasts for Ramadhan. Endocrinol Diabetes Metab Case Rep. 2021;2021:21–0043.
Challis BG, Harris J, Sleigh A, Isaac I, Orme SM, Seevaratnam N, et al. Familial adult onset hyperinsulinism due to an activating glucokinase mutation: implications for pharmacological glucokinase activation. Clin Endocrinol (Oxf). 2014;81:855–61.
Haeusler RA, Camastra S, Astiarraga B, Nannipieri M, Anselmino M, Ferrannini E. Decreased expression of hepatic glucokinase in type 2 diabetes. Mol Metab. 2015;4:222–6.
Caro JF, Triester S, Patel VK, Tapscott EB, Frazier NL, Dohm GL. Liver glucokinase: decreased activity in patients with type II diabetes. Horm Metab Res. 1995;27:19–22.
Basu A, Basu R, Shah P, Vella A, Johnson CM, Nair KS, et al. Effects of type 2 diabetes on the ability of insulin and glucose to regulate splanchnic and muscle glucose metabolism: evidence for a defect in hepatic glucokinase activity. Diabetes. 2000;49:272–83.
Torres TP, Catlin RL, Chan R, Fujimoto Y, Sasaki N, Printz RL, et al. Restoration of hepatic glucokinase expression corrects hepatic glucose flux and normalizes plasma glucose in Zucker diabetic fatty rats. Diabetes. 2009;58:78–86.
Shin J-S, Torres TP, Catlin RL, Donahue EP, Shiota M. A defect in glucose-induced dissociation of glucokinase from the regulatory protein in Zucker diabetic fatty rats in the early stage of diabetes. Am J Physiol Regul Integr Comp Physiol. 2007;292:R1381–90.
Shiota M, Postic C, Fujimoto Y, Jetton TL, Dixon K, Pan D, et al. Glucokinase gene locus transgenic mice are resistant to the development of obesity-induced type 2 diabetes. Diabetes. 2001;50:622–9.
O’Doherty RM, Lehman DL, Télémaque-Potts S, Newgard CB. Metabolic impact of glucokinase overexpression in liver: lowering of blood glucose in fed rats is accompanied by hyperlipidemia. Diabetes. 1999;48:2022–7.
Peter A, Stefan N, Cegan A, Walenta M, Wagner S, Königsrainer A, et al. Hepatic glucokinase expression is associated with lipogenesis and fatty liver in humans. J Clin Endocrinol Metab. 2011;96:E1126–30.
Orho-Melander M, Melander O, Guiducci C, Perez-Martinez P, Corella D, Roos C, et al. Common missense variant in the glucokinase regulatory protein gene is associated with increased plasma triglyceride and C-reactive protein but lower fasting glucose concentrations. Diabetes. 2008;57:3112–21.
Beer NL, Tribble ND, McCulloch LJ, Roos C, Johnson PRV, Orho-Melander M, et al. The P446L variant in GCKR associated with fasting plasma glucose and triglyceride levels exerts its effect through increased glucokinase activity in liver. Hum Mol Genet. 2009;18:4081–8.
Rees MG, Wincovitch S, Schultz J, Waterstradt R, Beer NL, Baltrusch S, et al. Cellular characterisation of the GCKR P446L variant associated with type 2 diabetes risk. Diabetologia. 2012;55:114–22.
Saxena R, Hivert M-F, Langenberg C, Tanaka T, Pankow JS, Vollenweider P, et al. Genetic variation in GIPR influences the glucose and insulin responses to an oral glucose challenge. Nat Genet. 2010;42:142–8.
Ferre T, Riu E, Franckhauser S, Agudo J, Bosch F. Long-term overexpression of glucokinase in the liver of transgenic mice leads to insulin resistance. Diabetologia. 2003;46:1662–8.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, et al. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34:2560–6.
Katz L, Manamley N, Snyder WJ, Dodds M, Agafonova N, Sierra-Johnson J, et al. AMG 151 (ARRY-403), a novel glucokinase activator, decreases fasting and postprandial glycemia in patients with type 2 diabetes. Diabetes Obes Metab. 2016;18:191–5.
Wilding JPH, Leonsson-Zachrisson M, Wessman C, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 in patients with type 2 diabetes mellitus on metformin. Diabetes Obes Metab. 2013;15:750–9.
Zheng S, Shao F, Ding Y, Fu Z, Fu Q, Ding S, et al. Safety, pharmacokinetics, and pharmacodynamics of globalagliatin, a glucokinase activator, in Chinese patients with type 2 diabetes mellitus: a randomized, phase Ib, 28-day ascending dose study. Clin Drug Investig. 2020;40:1155–66.
Amin NB, Aggarwal N, Pall D, Paragh G, Denney WS, Le V, et al. Two dose-ranging studies with PF-04937319, a systemic partial activator of glucokinase, as add-on therapy to metformin in adults with type 2 diabetes. Diabetes Obes Metab. 2015;17:751–9.
Bonadonna RC, Heise T, Arbet-Engels C, Kapitza C, Avogaro A, Grimsby J, et al. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95:5028–36.
Drabkin M, Yogev Y, Zeller L, Zarivach R, Zalk R, Halperin D, et al. Hyperuricemia and gout caused by missense mutation in d-lactate dehydrogenase. J Clin Invest. 2019;129:5163–8.
Köttgen A, Albrecht E, Teumer A, Vitart V, Krumsiek J, Hundertmark C, et al. Genome-wide association analyses identify 18 new loci associated with serum urate concentrations. Nat Genet. 2013;45:145–54.
Litwack G. Chapter 6—Insulin and sugars. In: Litwack G, editor. Hum Biochem. Boston: Academic Press; 2018. pp. 131–60. https://www.sciencedirect.com/science/article/pii/B9780123838643000065. Accessed 19 Aug 2023.
Wang Y, Viollet B, Terkeltaub R, Liu-Bryan R. AMP-activated protein kinase suppresses urate crystal-induced inflammation and transduces colchicine effects in macrophages. Ann Rheum Dis. 2016;75:286–94.
Yang W, Zhu D, Gan S, Dong X, Su J, Li W, et al. Dorzagliatin add-on therapy to metformin in patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022;28:974–81.
Zhu D, Li X, Ma J, Zeng J, Gan S, Dong X, et al. Dorzagliatin in drug-naïve patients with type 2 diabetes: a randomized, double-blind, placebo-controlled phase 3 trial. Nat Med. 2022;28:965–73.
Wang K, Shi M, Yang A, Fan B, Tam CHT, Lau E, et al. GCKR and GCK polymorphisms are associated with increased risk of end-stage kidney disease in Chinese patients with type 2 diabetes: the Hong Kong Diabetes Register (1995–2019). Diabetes Res Clin Pract. 2022;193: 110118.
Lian J, Guo J, Chen Z, Jiang Q, Ye H, Huang X, et al. Positive association between GCKR rs780093 polymorphism and coronary heart disease in the aged Han Chinese. Dis Markers. 2013;35:863–8.
Zahedi AS, Akbarzadeh M, Sedaghati-Khayat B, Seyedhamzehzadeh A, Daneshpour MS. GCKR common functional polymorphisms are associated with metabolic syndrome and its components: a 10-year retrospective cohort study in Iranian adults. Diabetol Metab Syndr. 2021;13:20.
Zhu D, Gan S, Liu Y, Ma J, Dong X, Song W, et al. Dorzagliatin monotherapy in Chinese patients with type 2 diabetes: a dose-ranging, randomised, double-blind, placebo-controlled, phase 2 study. Lancet Diabetes Endocrinol. 2018;6:627–36.
Zhu X-X, Zhu D-L, Li X-Y, Li Y-L, Jin X-W, Hu T-X, et al. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycemic control and pancreatic β-cell function in patients with type 2 diabetes: a 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018;20:2113–20.
Chow E, Wang K, Lim CKP, Tsoi STF, Fan B, Poon E, et al. Dorzagliatin, a dual-acting glucokinase activator, increases insulin secretion and glucose sensitivity in glucokinase maturity-onset diabetes of the young and recent-onset type 2 diabetes. Diabetes. 2023;72:299–308.
Chen L, Zhang J, Sun Y, Zhao Y, Liu X, Fang Z, et al. A phase I open-label clinical trial to study drug-drug interactions of dorzagliatin and sitagliptin in patients with type 2 diabetes and obesity. Nat Commun. 2023;14:1405.
Rosengren A, Jing X, Eliasson L, Renström E. Why treatment fails in type 2 diabetes. PLoS Med. 2008;5: e215.
Tysoe O. Sulfonylurea secondary failure mechanism identified. Nat Rev Endocrinol. 2023;19:189–189.
Xu H, Sheng L, Chen W, Yuan F, Yang M, Li H, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of novel glucokinase activator HMS5552: results from a first-in-human single ascending dose study. Drug Des Dev Ther. 2016;10:1619–26.
Miao J, Fu P, Ren S, Hu C, Wang Y, Jiao C, et al. Effect of renal impairment on the pharmacokinetics and safety of dorzagliatin, a novel dual-acting glucokinase activator. Clin Transl Sci. 2022;15:548–57.
Kiyosue A, Hayashi N, Komori H, Leonsson-Zachrisson M, Johnsson E. Dose-ranging study with the glucokinase activator AZD1656 as monotherapy in Japanese patients with type 2 diabetes mellitus. Diabetes Obes Metab. 2013;15:923–30.
Ericsson H, Sjöberg F, Heijer M, Dorani H, Johansson P, Wollbratt M, et al. The glucokinase activator AZD6370 decreases fasting and postprandial glucose in type 2 diabetes mellitus patients with effects influenced by dosing regimen and food. Diabetes Res Clin Pract. 2012;98:436–44.
Denney WS, Denham DS, Riggs MR, Amin NB. Glycemic effect and safety of a systemic, partial glucokinase activator, PF-04937319, in patients with type 2 diabetes mellitus inadequately controlled on metformin: a randomized, crossover, active-controlled study. Clin Pharmacol Drug Dev. 2016;5:517–27.
Liu D, Du Y, Yao X, Wei Y, Zhu J, Cui C, et al. Safety, tolerability, pharmacokinetics, and pharmacodynamics of the glucokinase activator PB-201 and its effects on the glucose excursion profile in drug-naïve Chinese patients with type 2 diabetes: a randomised controlled, crossover, single-centre phase 1 trial. EClinicalMedicine. 2021;42: 101185.
Zhi J, Zhai S. Effects of piragliatin, a glucokinase activator, on fasting and postprandial plasma glucose in patients with type 2 diabetes mellitus. J Clin Pharmacol. 2016;56:231–8.
Vella A, Freeman JLR, Dunn I, Keller K, Buse JB, Valcarce C. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Sci Transl Med. 2019;11:eaau3441.
Gao Q, Zhang W, Li T, Yang G, Zhu W, Chen N, et al. The efficacy and safety of glucokinase activators for the treatment of type-2 diabetes mellitus. Medicine (Baltim). 2021;100: e27476.
A 12-week, phase 2, randomized, double-blind, placebo controlled, dose-ranging, parallel group study to evaluate the efficacy and safety of once daily Pf-04991532 and sitagliptin in adult patients with type 2 diabetes mellitus inadequately controlled on metformin. ClinicalTrials.gov; 2013 Jun. Report No.: NCT01336738. Pfizer. https://clinicaltrials.gov/study/NCT01336738. Accessed 21 Feb 2024.
Safety and tolerability of multiple ascending doses of LY2608204 in patients with type 2 diabetes mellitus. ClinicalTrials.gov; 2018 Oct. Report No.: NCT01247363. Eli Lilly and Company. https://clinicaltrials.gov/study/NCT01247363. Accessed 21 Feb 2024.
Tsumura Y, Tsushima Y, Tamura A, Hasebe M, Kanou M, Kato H, et al. TMG-123, a novel glucokinase activator, exerts durable effects on hyperglycemia without increasing triglyceride in diabetic animal models. PLoS ONE. 2017;12: e0172252.
Klein KR, Freeman JLR, Dunn I, Dvergsten C, Kirkman MS, Buse JB, et al. The SimpliciT1 study: a randomized, double-blind, placebo-controlled phase 1b/2 adaptive study of TTP399, a hepatoselective glucokinase activator, for adjunctive treatment of type 1 diabetes. Diabetes Care. 2021;44:960–8.
A study of LY2599506 in patients with type 2 diabetes. https://clinicaltrials.gov/study/NCT01024244. Accessed 7 Mar 2023.
A study of LY2599506 (Oral Agent Medication: Glucokinase Activator 1) in type 2 diabetes mellitus. https://clinicaltrials.gov/study/NCT01029795. Accessed 7 Mar 2023.
A study to compare two forms of LY2608204 in healthy people. https://clinicaltrials.gov/study/NCT01313286. Accessed 7 Mar 2023.
Acknowledgements
Upinder Kaur and Sankha Shubhra Chakrabarti thank the Institutions of Eminence Scheme at the Banaras Hindu University for research support.
Author information
Authors and Affiliations
Corresponding authors
Ethics declarations
Funding
No funding was received to write this review.
Conflict of Interest
Upinder Kaur, Bhairav Kumar Pathak, Tharik Jalal Meerashahib, Dondapati Venkata Vamshi Krishna, and Sankha Shubhra Chakrabarti have no conflicts of interest that are directly relevant to the content of this article.
Ethics Approval
Not applicable as the review did not involve any human or animal experiments.
Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Availability of Data and Material
Not applicable. The article reviews data from other published papers, which are available in the public domain.
Code Availability
Not applicable.
Authors’ Contributions
UK: planned the study, supervised the data extraction, verified the extracted data, performed the literature search, and wrote the first draft of the paper. BKP: performed the literature search, assisted in writing the paper and the presentation of data, and verified the extracted data. TJM: assisted in writing the paper and the tabular presentation of data, and verified the extracted data. DVVK: contributed to the tabular presentation of data and verification of the extracted data. SSC: edited the final draft of the paper and performed the literature search.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Kaur, U., Pathak, B.K., Meerashahib, T.J. et al. Should Glucokinase be Given a Chance in Diabetes Therapeutics? A Clinical-Pharmacological Review of Dorzagliatin and Lessons Learned So Far. Clin Drug Investig 44, 223–250 (2024). https://doi.org/10.1007/s40261-024-01351-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40261-024-01351-5