
Abstract
Diabetes mellitus is a worldwide impacting disorder and the ratio through which the number of diabetic patients had increased worldwide, puts medical professionals to serious stress for its effective management. Due to its polygenic origin and involvement of multiple genes to its pathophysiology, leads to understanding of this ailment more complex. It seems that current interventions, such as dietary changes, life style changes and drug therapy such as oral hypoglycaemics and insulin, are unable to halt the trend. There are various novel and emerging targets on which the researchers are paying attention to combat with this ailment successfully. Human glucokinase (GK) enzyme is one of these novel and emerging targets for management of diabetes. Its availability in the pancreas and liver cells makes this target more lucrative. GK’s presence in the pancreatic and hepatic cells plays a very important function for the management of glucose homoeostasis. Small molecules that activate GK allosterically provide an alternative strategy for restoring/improving glycaemic regulation, especially in type 2 diabetic patients. Although after enduring many setbacks in the development of the GK activators, interest has been renewed especially due to introduction of novel dual acting GK activator dorzagliatin, and a novel hepato-selective GK activator, TTP399. This review article has been formulated to discuss importance of GK in glucose homeostasis, recent updates on small molecules of GK activators, clinical status of GK activators and challenges in development of GK activators.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Diabetes (type 2) is distinguished by high level of glycaemic content in blood caused due to inadequate pancreatic β-cell secretion of insulin associated with insulin resistance, which is mostly evident in the liver and skeletal muscles. Diabetic complications, if left untreated, will result in vision loss, peripheral neuropathy, reduced renal function and various cardiovascular disorders like stroke and heart diseases [1, 2]. Since the condition has a polygenic basis and multiple genes (over 20 according to the most recent count) are implicated in its pathogenesis, western lifestyles marked by minimum workout and excessive intake of caloric food are essential devastating factors for the emerging outbreak of type 2 diabetes (T2D) worldwide [3, 4]. It seems that current interventions, such as dietary changes and drug therapy such as insulin formulations and oral hypoglycaemics, are unable to halt the trend. In addition, an overdose of oral hypoglycaemic drugs may possibly lead to hypoglycaemia and many other adverse drug events [5]. As a result, new approaches are needed, such as the production of new chemical entities with novel mechanisms of action [6,7,8,9,10]. Activation of enzyme glucokinase (GK) might be able to address this unmet need. Due to glucose sensing function of GK in β-cells of pancreas and its role in the clearance of hepatic glucose and glycogen biosynthesis, all of which are inhibited in type 2 diabetic patients, GK has been recognized as an excellent target for designing of novel and effective antidiabetic medications [3, 11]. This review article has been designed to discuss role of GK in glucose homeostasis, recent updates on small molecules of GK activators, clinical status of GK activators and challenges in development of GK activators.
Glucokinase (GK)
GK (hexokinase IV or D, ATP: D-glucose-6-phosphotransferase, EC 2.7.1.2) is one sort of hexokinase (out of four) having unique properties. It is a cytoplasmic enzyme which catalyses the early reactions of glucose breakdown and convert glucose to its phosphorylated product glucose-6-phosphate (G-6-P) [12, 13]. It is expressed mainly in pancreatic β-cells and liver cells due to which it plays a significant role in the glucose homeostasis [14,15,16]. At glucose concentration of 8.0 mM, GK reaches at its half-maximal activity, while the other three hexokinases become saturated at significantly lower blood glucose values (1.0 mM). Hence, GK’s glucose metabolism increases as blood glucose levels rise from fasting to postprandial levels following carbohydrate-rich meal consumption [3, 6, 17]. GK is not blocked by G-6-P and it has a non-Michaelis-Menten sigmoidal concentration curve of glucose and its inflection point lies in the range of 4–5 mmol/L that is comparable to the insulin secretion threshold. This leads to guaranteed graded response to changes in the level of glucose, and GK activity enters a plateau period when glucose levels are near to the physiological limit for secretion of insulin caused by glucose (5 mM) [18, 19]. GK regulatory protein more commonly known as GKRP binds with GK enzyme with strong affinity at threshold glucose concentrations, i.e., 5 mmol/l and inactivate GK after sequestering it in the nucleus [20]. GK control in the liver is essential for postprandial glycogen synthesis and storage, rather than utilizing glucose as its main energy source. Glucose supply to the brain is maintained during hunger, while liver glycogen stocks decline, by an increasing the degree of gluconeogenesis in the hepatocytes [11, 21,22,23,24].
Biological functions of GK
GK is responsible for glucose phosphorylation and convert it to G-6-P after its entry into the cell. Low levels of insulin and high glucose also cause stimulation of GK, a glucose-specific enzyme that is unaffected by the phosphorylated component, G-6-P. GK is found in many organs, including the pancreas and liver, which are also essential for glucose metabolism [16]. GK stimulates the synthesis of glycogen in the hepatic cells, and regulate the synthesis of insulin in pancreatic β-cells, hence proved as a key player in blood glucose regulation [25]. Mutations (nonsense, missense and many other mutations) in the genes of GK enzyme may lead to development of many types of diabetes. The maturity onset diabetes (MODY) group with the name MODY2 includes GK mutation-related diabetes. Patients with MODY2 had lower hepatic glycogen production after three meals compared to the general population, and their hyperglycaemic state had a weak inhibitory effect on liver glucose performance. GK contributes an important function in overall blood glucose homeostasis as it is a primary enzyme expressed in the hepatic cells which has a strong controlling effect on hepatic glucose clearance ultimately reducing hyperglycaemia, and is responsible for release of glucose stimulated insulin secretion [3, 6]. Inactivating mutations in the gene of GK enzyme decreased the enzyme’s affinity for its substrate (i.e., glucose) or compromised expression of GK which leads to development of maturity onset diabetes of the young type 2 (MODY2), while opposite to this, i.e., activating mutations in the GK gene, decreased glucose level in the blood [26].
Role of GK in glucose homeostasis
Although glucose homeostasis is a complicated process and majorly can be clarified on the basis of its two important hormones glucagon and insulin. Former is secreted by α-cells of pancreatic islet and maintains energy by preserving euglycemia during fasting. This is done by increasing hepatic glucose supply by encouraging gluconeogenesis (de novo glucose generation from non-carbohydrate sources) and glycogenolysis (glycogen catabolism and glucose liberation from the liver). In contrast to this, insulin is generated by pancreatic β-cells, reduces blood glucose in postprandial state by facilitating glucose utilization in various peripheral tissues and increased glucose absorption by hepatic cells after switching it to glycogen synthesis mode [27, 28]. The location of GK gene on chromosome is 7p15.3-p15.1, and it is made up of 12 exon that extend 45,168 base pairs and encodes for a 465-amino-acids with molecular weight of 52,191 Da that is expressed predominantly in various organs like pancreas, liver, brain and many more [26]. GK induce secretion of insulin from pancreatic β-cells depending upon the glucose concentration hence referred as “glucose sensor” and as a “gate-keeper” in liver cells as facilitating glucose absorption in hepatocytes as well as synthesis and accumulation of glycogen. As GK enzyme is activated, its desired substrate glucose catabolised to its phosphorylated form G-6-P which again act as a substrate for the synthesis of glycogen in the liver [29, 30]. When concentration of glucose is <10 mM, hepatic GK enzyme remain in inactive state due to its association with GKRP, which confers much lower affinity for its substrate (glucose) then pancreatic β-cells, which is stimulated only during the well feed condition to fulfil its role of increasing hepatic glucose intake [11, 31, 32]. In the hepatic cell, GKRP functions as a competitive regulator of glucose, sequestering GK at low blood glucose level and detaching from enzyme GK while glycaemic content of the blood increases. Apart from the pancreas and hepatic cells, GK is also present in different parts of body’s like entero-endocrine, anterior pituitary and nerve cells. Hence, GK has significant role in overall glucose homeostasis by inducing secretion of insulin, facilitating glucose absorption and switching the liver cells to glycogen synthesis mode.33 Blood glucose levels are lowered in both direct and indirect forms as a result of the above GK-mediated pathways [3, 20].
GK as a potential therapeutic target for T2D
Role in pancreas
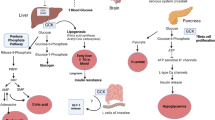
In pancreatic β-cell and liver parenchymal cells, glucose transporter 2 (GLUT2) aids glucose transport across the plasma membrane. The rate of conversion of glucose to G-6-P, which is catalysed by GK enzyme, limits the amount and speed of glucose transport. Phosphorylation of glucose to G-6-P causes glycogen synthesis in hepatocytes (L-Type GK) and glucose metabolism to pyruvate in pancreatic β-cells (B-Type GK). This results in an increase in the Kreb’s cycle and electron transport, resulting in a rise in the ATP/ADP ratio, which blocks ATP-sensitive K+ ion channels, producing membrane depolarization and as a consequence, Ca2+ influx. This causes the pancreatic β-cells to secrete insulin into the bloodstream (Fig. 1) [3, 6, 17, 33, 34].

Role of Glucokinase enzyme in pancreatic β cell. GK: Glucokinase enzyme; G-6-P: Glucose-6-phosphate; F-6-P: Fructose-6-phosphate; F-1,6-BP: Fructose 1,6 bisphosphate; PEP: Phospho-enol pyruvate; GLUT2: Glucose transporter 2
Role in liver
The GK enzyme converts glucose to G-6-P, which triggers glucose absorption, storage in the form of glycogen, and inhibition of gluconeogenesis (synthesis of glucose from non-carbohydrate sources) in liver hepatocytes, resulting in lower blood glucose levels (Fig. 2). GK can be activated by small molecules attaching to the GK enzyme allosterically or disrupting the GK-GKRP complex. Compounds that have any of the aforementioned effects might be useful in the treatment of T2D [3, 17, 33,34,35,36,37].

Role of GK enzyme in liver. GK: Glucokinase; GKRP: Glucokinase regulatory protein; G-6-P: Glucose-6phosphate; F-6-P: Fructose-6-phosphate; F-1,6-BP: Fructose-1,6 bisphosphate; PEP: Phospho-enol pyruvate; GLUT2: Glucose transporter 2; GS: Glycogen synthase
Overview of GK activators
Hoffman La Roche’s invention of allosteric GK activator drug candidates in the beginning of twenty-first century was a landmark moment. It offered pharmacological guidance for glucose sensor model based on GK enzyme and potential experimental methods and optimism for the treatment of diabetes. From that time GK activator’s ability has been extensively studied by both academia as well as industry [3, 6]. Since the first GK activator was launched in 2003 [38], several others have been developed and evaluated as potential type 2 antidiabetic agents [39,40,41,42,43,44,45,46,47,48,49,50]. They are all small molecules that can bind to enzyme GK at its allosteric site, stabilizing a high-affinity conformation to facilitate GK activation. The chemical composition of these small compound GK activators can be used to categorize them (carbon-, urea-, 1,2,4 and 1,3,5- substituted aryl- centred amides and others) [37, 51, 52]. Second categorization option is based on the site of action site: hepato-selective GK activators that perform their function in the hepatic cells with or without causing disruption in the inhibitory complex, i.e., GK-GKRP interaction and pancreatic and liver dual GK activators [53, 54]. By adding a carboxyl group to the composition of GK activators, the compound is unable to reach the pancreas, resulting in liver specificity. Hypoglycaemia is a typical adverse effect that varies based on the type of compound and dosage. The glucose lowering effects has not been maintained in clinical trials that have lasted months rather than hours. Insulin is released too often when GK is activated at lower glucose concentrations, exacerbating peripheral insulin resistance. GK activators regulate blood glycaemic content by improving the potential of β-cells towards detection of glucose and its proportional insulin secretion in a glucose-dependent manner. Furthermore, these activators increase the disposal of glucose and reduce hepatic glucose production. Every GK activator has the ability to cause a distinct conformation at allosteric site of GK (active form), and the resultant complexes can have distinct kinetic profiles [55]. Many small molecule GK activators were shown to facilitate insulin secretion through a Ca2+ dependent pathway in previous studies [56]. Another way GK activators promote insulin secretion is by fixing defects in pancreatic cell oxygen intake and intracellular Ca2+ reaction in T2D patients [57]. In liver, GK activators can activate GK directly, as well as facilitate the dissociation of the GK/GKRP complex, which activates GK and stimulates glycolysis and glycogen synthesis [37]. Various types of natural extracts (such Allium hirtifolium and Sapiumellipticum) or phytoconstituents (such as ganoderan B, glucolipsins A and B, eupatilin, coagulanolide, mangiferin, and kaempferol) were reported with potent GK stimulating property [58,59,60,61,62,63,64,65,66,67,68]. Several hundreds of GK activators have been developed by various pharmaceutical companies over the last 20 years. Just a limited number of GK activators have advanced to various phases of clinical investigations, but the majority of these have been halted owing to hypoglycaemia and liver side effects [17, 37]. In clinical trials, drugs like TMG-123, PF 04937319, R1511 or GK3HMS5552, TTP3999 and Dorzagliatin were shown to efficiently regulate blood glucose levels. R1440 GKA2, GKA 50, YH-GKA, PSN 010, MK-0941, ZYGK1, and Ro-28-1675 are among the other agents undergoing preclinical testing. Some GK activator agents, such as Piragliatin, ARRY-403, etc., were dragged out from the clinical investigations due their toxic effects and a loss of efficacy over prolonged use [37, 69,70,71,72,73]. Some of prominent GK activators (which advanced in clinical investigations) are presented in Table 1.
Challenges with GK activators
The use of older generation GK activators posed significant questions about efficacy and safety. Hypoglycaemia, initiation of fatty liver, hepatic cells lipidosis, and decreased efficacy over the period of time proved to be the most significant side effects observed with GK activators. In reality, during the early stages of GK activators growth, the incidences of hypoglycemia and dyslipidemia as a result of over-stimulating effect of pancreatic and hepatic GK, respectively, were seen as possible intimidations [18, 82]. Acute insulin release (disproportionate to the stimulus) as a result of an exaggerated glucose reaction could normally occur as a result of activation of GK and was always a possibility. Piragliatin and MK-0941 were shown to cause hypoglycemic episodes more frequently. To counter this risk, partial activators with a higher dependence on glucose levels were designed to reduce the risk of over activation at low glucose concentrations [51]. Agents that are hepato-selective were also developed and tested [54]. With the partial GK activator, PF-04937319, the chances of hypoglycaemia were reduced. According to the pathophysiological processes associated with activation of GK that may contribute to dyslipidemia is due to excessive G-6-P accumulation, arising due to hyper stimulation of hepatic GK enzyme stimulates glycolysis via intermediate product fructose-2,6-bisphosphate, this indicates correspond to G-6-P rise and acts through feed forward allosteric activation mechanisms. As a result of this effect (glycolysis activation), acetyl CoA levels rises, this causes increased inflow to fatty acids and triglycerides, as well as increased hepatic de-novo lipogenesis. According to the report, this corresponds to the initial stage of fatty liver disease (non-alcoholic), which may vary from simple steatosis to its complicated form, i.e., steato-hepatitis [83]. Furthermore, long-term exposure may be required for the latter. Compound MK-0941 was shown to cause acute hyperlipidosis. While rise in level of fatty acid by less than 20% is not as significant as that caused by a high-glycemic low-fat diet, it is also undesirable in T2D patients who are also prediagnosed to dyslipidemia, NAFLD and high blood pressure [80]. Histological studies in mouse models indicating double-strand breaks in DNA, presumably accounting for activation of the p53 tumour suppressor and consequent β -cell death, have also been used to propose toxicity of GK activators on the β-cells [19, 84, 85].
Conclusion and future perspective
Diabetes is a metabolic condition that affects people all over the world, and its prevalence is increasing on a daily basis in both developed and developing nations. Reduced physical activity and sedentary lifestyles have a higher chance of exacerbating the disease’s consequences. Despite the fact that there are several medicines that operate through various pathways, no one can claim that any of the currently available medications can completely reverse disease development. Small molecules that activate GK provide an alternative strategy for restoring/improving glycaemic regulation in patients with T2D. GK activators reduce hepatic glucose production by increasing insulin secretion and glycogen synthesis. Despite a number of setbacks in their growth, the GK activators class has reawakened interest, especially after the release of dorzagliatin, a dual-acting and novel GK activator and TTP399, a hepato-selective novel GK activators. With the use of older generation GK activators, both efficacy (diminished long-term efficacy) and safety concerns (hypoglycemia, fatty liver, and dyslipidaemia) were raised. Clinically relevant and prolonged glycaemic effectiveness, as well as a minimal risk of adverse effects, such as hypoglycemia, hepato-steatosis, and hypertriglyceridemia, are the basic conditions for new GK activators to be considered as a serious alternative to prior GK activator therapies. A new compound’s desirable properties would be the ability to treat long-term consequences of persistent hyperglycemia and/or change the disease’s natural course.
References
Defronzo RA. Banting Lecture. From the triumvirate to the ominous octet: a new paradigm for the treatment of type 2 diabetes mellitus. Diabetes. 2009;58(4):773–795.
Grewal AS, Beniwal M, Pandita D, Sekhon BS, Lather V. Recent updates on peroxisome proliferator-activated receptor δ agonists for the treatment of metabolic syndrome. Med Chem. 2016;12:3–21.
Grewal AS, Sekhon BS, Lather V. Recent updates on glucokinase activators for the treatment of type 2 diabetes mellitus. Mini Rev Med Chem. 2014;14(7):585–602.
Lin X, Xu Y, Pan X, Xu J, Ding Y, Sun X, Song X, Ren T, Shan P. Global, regional, and national burden and trend of diabetes in 195 countries and territories: an analysis from 1990 to 2025. Sci Rep. 2020;10:14790.
Alhadramy MS. Diabetes and oral therapies: a review of oral therapies for diabetes mellitus. J Taibah Univ Med Sci. 2016;11(4):317–29.
Pal M. Recent advances in glucokinase activators for the treatment of type 2 diabetes. Drug Discov Today. 2009;14(15–16):784–92.
Rochester CD, Akiyode O. Novel and emerging diabetes mellitus drug therapies for the type 2 diabetes patient. World J Diabetes. 2014;5(3):305–15.
Marín-Peñalver JJ, Martín-Timón I, Sevillano-Collantes C, Del Cañizo-Gómez FJ. Update on the treatment of type 2 diabetes mellitus. World J Diabetes. 2016;7(17):354–95.
Belay E, Abera A, Mehari A, Gebremeskel G, Endrias A, Endriset K. Achievements of diabetes goals and their determinants in type 2 diabetic patients attending outpatient diabetic clinic in northern Ethiopia. Int J Chronic Dis. 2017;2017:5713187.
Verma AK, Goyal Y, Bhatt D, Dev K, Alsahli MA, Rahmani AH, Almatroudi A. A compendium of perspectives on diabetes: a challenge for sustainable health in the modern era. Diabetes Metab Syndr Obes. 2021;2021(14):2775–87.
Sharma P, Singh S, Thakur V, Sharma N, Grewal AS. Novel and emerging therapeutic drug targets for management of type 2 diabetes mellitus. Obes Med. 2021;23:100329.
Matschinsky F. Glucokinase as glucose sensor and metabolic signal generator in pancreatic beta-cells and hepatocytes. Diabetes. 1990;39(6):647–52.
Matschinsky F, Porte D. Glucokinase activators (GKAs) promise a new pharmacotherapy for diabetics. F1000 Med Rep. 2010;2:43.
Meglasson MD, Matschinsky F. Pancreatic islet glucose metabolism and regulation of insulin secretion. Diabetes Metab Rev. 1986;2(3–4):163–214.
Matschinsky F, Liang Y, Kesavan P, Wang L, Froguel P, Velho G, Cohen D, Permutt MA, Tanizawa Y, Jetton TL, Niswender K, Magnuson MA. Glucokinase as pancreatic beta cell glucose sensor and diabetes gene. J Clin Invest. 1993;92(5):2092–8.
Iynedjian PB. Molecular physiology of mammalian glucokinase. Cell Mol Life Sci. 2009;66(1):27–42.
Li W, Zhang X, Sun Y, Liu Z. Recent clinical advances of glucokinase activators in the treatment of diabetes mellitus type 2. Pharmazie. 2020;75(6):230–5.
Agius L. Lessons from glucokinase activators: the problem of declining efficacy. Expert Opin Ther Pat. 2014;24(11):1155–9.
Nakamura A, Terauchi Y. Present status of clinical deployment of glucokinase activators. J Diabetes Investig. 2015;6(2):124–32.
Agius L. Targeting hepatic glucokinase in type 2 diabetes: weighing the benefits and risks. Diabetes. 2008;58(1):18–20.
Van Schaftingen E, Vandercammen A, Detheux M, Davies DR. The regulatory protein of liver glucokinase. Adv Enzym Regul. 1992;32:133–48.
Van Schaftingen E. Short-term regulation of glucokinase. Diabetologia. 1994;37:S43–7.
Baltrusch S, Francini F, Lenzen S, Tiedge M. Interaction of glucokinase with the liver regulatory protein is conferred by leucine-asparagine motifs of the enzyme. Diabetes. 2005;54(10):2829–37.
Agius L. Targeting hepatic glucokinase in type 2 diabetes: weighing the benefits and risks. Diabetes. 2009;58(1):18–20.
Gomis RR, Favre C, García-Rocha M, Fernández-Novell JM, Ferrer JC, Guinovart JJ. Glucose 6-phosphate produced by gluconeogenesis and by glucokinase is equally effective in activating hepatic glycogen synthase. J Biol Chem. 2003;278(11):9740–6.
Osbak KK, Colclough K, Saint-Martin C, Beer NL, Bellanné-Chantelot C, Ellard S, Gloyn AL. Update on mutations in glucokinase (GCK), which cause maturity-onset diabetes of the young, permanent neonatal diabetes, and hyperinsulinemic hypoglycemia. Hum Mutat. 2009;30(11):1512–26.
Rines AK, Sharabi K, Tavares CD, Puigserver P. Targeting hepatic glucose metabolism in the treatment of type 2 diabetes. Nat Rev Drug Discov. 2016;15(11):786–804.
Röder PV, Wu B, Liu Y, Han W. Pancreatic regulation of glucose homeostasis. Exp Mol Med. 2016;48(3):e219.
Perseghin G. Exploring the in vivo mechanisms of action of glucokinase activators in type 2 diabetes. J Clin Endocrinol Metab. 2010;95:4871–3.
Petersen MC, Vatner DF, Shulman GI. Regulation of hepatic glucose metabolism in health and disease. Nat Rev Endocrinol. 2017;13(10):572–87.
Choi JM, Seo MH, Kyeong HH, Kim E, Kim HS. Molecular basis for the role of glucokinase regulatory protein as the allosteric switch for glucokinase. Proc Natl Acad Sci U S A. 2013;110(25):10171–6.
Raimondo A, Rees MG, Gloyn AL. Glucokinase regulatory protein: complexity at the crossroads of triglyceride and glucose metabolism. Curr Opin Lipidol. 2015;26(2):88–95.
Matschinsky F, Wilson DF. The central role of glucokinase in glucose homeostasis: a perspective 50 years after demonstrating the presence of the enzyme in islets of langerhans. Front Physiol. 2019;10:148.
Toulis KA, Nirantharakumar K, Pourzitaki C, Barnett AH, Tahrani AA. Glucokinase activators for type 2 diabetes: challenges and future developments. Drugs. 2020;80:467–75.
Walker DG, Rao S. The role of glucokinase in the phosphorylation of glucose by rat liver. Biochem J. 1964;90(2):360–8.
Al-Hasani H, Tschöp MH, Cushman SW. Two birds with one stone: novel glucokinase activator stimulates glucose-induced pancreatic insulin secretion and augments hepatic glucose metabolism. Mol Interv. 2003;3(7):367–70.
Grewal AS, Lather V, Charaya N, Sharma N, Singh S, Kairys V. Recent developments in medicinal chemistry of allosteric activators of human glucokinase for type 2 diabetes mellitus therapeutics. Curr Pharm Des. 2020;26(21):2510–52.
Grimsby J, Sarabu R, Corbett WL, Haynes NE, Bizzarro FT, Coffey JW, Guertin KR, Hilliard DW, Kester RF, Mahaney PE, Marcus L, Qi L, Spence CL, Tengi J, Magnuson MA, Chu CA, Dvorozniak MT, Matschinsky F, Grippo JF. Allosteric activators of glucokinase: potential role in diabetes therapy. Science. 2003;301(5631):370–3.
Behera P, Behera D, Satpati S, Agnihotri G, Nayak S, Padhi P, Dixit A. Molecular modeling and identification of novel glucokinase activators through stepwise virtual screening. J Mol Graph Model. 2015;57:122–30.
Park K, Lee BM, Hyun KH, Han T, Lee DH, Choi HH. Design and synthesis of acetylenyl benzamide derivatives as novel glucokinase activators for the treatment of T2DM. ACS Med Chem Lett. 2015;6:296–301.
Paczal A, Bálint B, Wéber C, Szabó ZB, Ondi L, Theret I, De Ceuninck F, Bernard C, Ktorza A, Perron-Sierra F, Kotschy A. Structure-activity relationship of azaindole-based glucokinase activators. J Med Chem. 2016;59:687–706.
Cheruvallath Z, Gwaltney SL, Sabat M, Tang M, Wang H, Jennings A, Hosfield D, Lee B, Wu Y, Halkowycz P, Grimshaw CE. Discovery of potent and orally active 1,4-disubstituted indazoles as novel allosteric glucokinase activators. Bioorg Med Chem Lett. 2017;27(12):2678–82.
Zhang L, Hu S, Lei L, Zhang Y, Zhang L, Song H, Shen Z, Feng Z. Design, synthesis and evaluation of novel derivatives of orotic acid amide as potent glucokinase activators. Lett Drug Des Discov. 2017;14(3):252–61.
Singh R, Lather V, Pandita D, Judge V, Arumugam KN, Grewal AS. Synthesis, docking and antidiabetic activity of some newer benzamide derivatives as potential glucokinase activators. Lett Drug Des Discov. 2017;14(5):540–53.
Charaya N, Pandita D, Grewal AS, Lather V. Design, synthesis and biological evaluation of novel thiazol-2-yl benzamide derivatives as glucokinase activators. Comput Biol Chem. 2018;73:221–9.
Bano S, Khan AU, Asghar F, Usman M, Badshah A, Ali S. Computational and pharmacological evaluation of ferrocene-based acyl ureas and homoleptic cadmium carboxylate derivatives for anti-diabetic potential. Front Pharmacol. 2018;8:1001.
Grewal AS, Kharb R, Prasad DN, Dua JS, Lather V. N-pyridin-2-yl benzamide analogues as allosteric activators of glucokinase: design, synthesis, in vitro, in silico and in vivo evaluation. Chem Biol Drug Des. 2019;93(3):364–72.
Grewal AS, Kharb R, Prasad DN, Dua JS, Lather V. Design, synthesis and evaluation of novel 3,5-disubstituted benzamide derivatives as allosteric glucokinase activators. BMC Chem. 2019;13(1):2.
Singh S, Arora S, Dhalio E, Sharma S, Arora K, Grewal AS. Design and synthesis of newer N-benzimidazol-2yl benzamide analogues as allosteric activators of human glucokinase. Med Chem Res. 2021;30:760–70.
Arora S, Grewal AS, Sharma N, Arora K, Dhalio E, Singh S. Design, synthesis, and evaluation of some novel N-benzothiazol-2-yl benzamide derivatives as allosteric activators of human glucokinase. J Appl Pharm Sci. 2021;11(Suppl 1):038–47.
Filipski KJ, Pfefferkorn JA. A patent review of glucokinase activators and disruptors of the glucokinase–glucokinase regulatory protein interaction: 2011–2014. Expert Opin Ther Pat. 2014;24(8):875–91.
Sarabu R, Berthel SJ, Kester RF, Tilley JW. Novel glucokinase activators: a patent review (2008–2010). Expert Opin Ther Pat. 2011;21(1):13–33.
Pfeferkorn JA. Strategies for the design of hepatoselective glucokinase activators to treat type 2 diabetes. Expert Opin Drug Discov. 2013;8(3):319–30.
Egan A, Vella A. TTP399: an investigational liver-selective glucokinase (GK) activator as a potential treatment for type 2 diabetes. Expert Opin Investig Drugs. 2019;28(9):741–7.
Dransfield PJ, Pattaropong V, Lai S, Fu Z, Kohn TJ, Du X, Cheng A, Xiong Y, Komorowski R, Jin L, Conn M, Tien E, DeWolf WE Jr, Hinklin RJ, Aicher TD, Kraser CF, Boyd SA, Voegtli WC, Condroski KR, et al. Novel series of potent glucokinase activators leading to the discovery of AM-2394. ACS Med Chem Lett. 2016;7(7):714–8.
Efanov AM, Barrett DG, Brenner MB, Briggs SL, Delaunois A, Durbin JD, Giese U, Guo H, Radloff M, Gil GS, Sewing S, Wang Y, Weichert A, Zaliani A, Gromada J. A novel glucokinase activator modulates pancreatic islet and hepatocyte function. Endocrinology. 2005;146:3696–701.
Doliba NM, Fenner D, Zelent B, Bass J, Sarabu R, Matschinsky F. Repair of diverse diabetic defects of β-cells in man and mouse by pharmacological glucokinase activation. Diabetes Obes Metab. 2012;14(Suppl 3):109–19.
Mahmoodi M, Zarei S, Rezaeian M, Arababadi MK, Ghasemi H, Khoramdelazad H, Rezayati N, Hasanshahi G, Hosseini-Zijoud S-M. Persian shallot (Allium hirtifolium Boiss) extract elevates glucokinase (GCK) activity and gene expression in diabetic rats. Am J Plant Sci. 2013;4(7):1393–9.
Ighodaro OM, Akinloye OA, Ugbaja RN, Omotainse SO. Sapium ellipticum (Hochst) Pax ethanol leaf extract modulates glucokinase and glucose-6-phosphatase activities in streptozotocin induced diabetic rats. Asian Pac J Trop Biomed. 2017;7(6):544–8.
Angadi K, Gundampati R, Jagannadham M, Kandru A. Molecular docking studies of guggultetrol from Nymphaea pubescens with target glucokinase (GK) related to type-II diabetes. J Appl Pharm Sci. 2012;3(2):127–31.
Hikino H, Ishiyama M, Suzuki Y, Konno C. Mechanisms of hypoglycemic activity of ganoderan B: a glycan of Ganoderma lucidum fruit bodies. Planta Med. 1989;55(5):423–8.
Kang YJ, Jung UJ, Lee MK, Kim HJ, Jeon SM, Park YB, Chung HG, Baek NI, Lee KT, Jeong TS, Choi MS. Eupatilin, isolated from Artemisia princeps Pampanini, enhances hepatic glucose metabolism and pancreatic beta-cell function in type 2 diabetic mice. Diabetes Res Clin Pract. 2008;82(1):25–32.
Qian-Cutrone J, Ueki T, Huang S, Mookhtiar KA, Ezekiel R, Kalinowski SS, Brown KS, Golik J, Lowe S, Pirnik DM, Hugill R, Veitch JA, Klohr SE, Whitney JL, Manly SP. Glucolipsin a and B, two new glucokinase activators produced by Streptomyces purpurogeniscleroticus and Nocardia vaccinii. J Antibiot. 1999;52(3):245–55.
Grewal AS, Sharma N, Singh S, Arora S. Molecular docking studies of phenolic compounds from Syzygium cumini with multiple targets of type 2 diabetes. J Pharm Technol Res Manag. 2018;6(2):125–33.
Singh AB, Singh N, Akanksha J, Maurya R, Srivastava AK. Coagulanolide modulates hepatic glucose metabolism in C57BL/KsJ-db/db mice. Hum Exp Toxicol. 2012;31(10):1056–65.
Min Q, Cai X, Sun W, Gao F, Li Z, Zhang Q, Wan LS, Li H, Chen J. Identification of mangiferin as a potential glucokinase activator by structure-based virtual ligand screening. Sci Rep. 2017;7:44681.
Jeyabaskar S, Viswanathan T, Mahendran R, Nishandhini M. In silico molecular docking studies to investigate interactions of natural camptothecin molecule with diabetic enzymes. Res J Pharm Technol. 2017;10(9):2917–22.
Grewal AS, Sharma N, Singh S. Molecular docking investigation of compounds from Sapium ellipticum (Hochst) Pax as allosteric activators of human glucokinase. Int J Pharm Qual Assur. 2019;10(4):588–96.
Scheen AJ. Investigational insulin secretagogues for type 2 diabetes. Expert Opin Investig Drugs. 2016;25(4):405–22.
Kamimura H, Ito S, Chijiwa H, Okuzono T, Ishiguro T, Yamamoto Y, Nishinoaki S, Ninomiya SI, Mitsui M, Kalgutkar AS, Yamazaki H, Suemizu H. Simulation of human plasma concentration-time profiles of the partial glucokinase activator PF-04937319 and its disproportionate N-demethylated metabolite using humanized chimeric mice and semi-physiological pharmacokinetic modeling. Xenobiotica. 2017;47(5):382–93.
Tsumura Y, Tsushima Y, Tamura A, Hasebe M, Kanou M, Kato H Kobayashi T. TMG-123, a novel glucokinase activator, exerts durable effects on hyperglycemia without increasing triglyceride in diabetic animal models. PLoS One 2017;12(2):e0172252.
Zhu XX, Zhu DL, Li XY, Li YL, Jin XW, Hu TX, Zhao Y, Li YG, Zhao GY, Ren S, Zhang Y, Ding YH, Chen L. Dorzagliatin (HMS5552), a novel dual-acting glucokinase activator, improves glycaemic control and pancreatic β-cell function in patients with type 2 diabetes: a 28-day treatment study using biomarker-guided patient selection. Diabetes Obes Metab. 2018;20(9):2113–20.
Vella A, Freeman JL, Dunn I, Keller K, Buse JB, Valcarce C. Targeting hepatic glucokinase to treat diabetes with TTP399, a hepatoselective glucokinase activator. Sci Transl Med. 2019;11:475.
Bonadonna RC, Heise T, Arbet-Engels C, Kapitza C, Avogaro A, Grimsby J, Zhi J, Grippo JF, Balena R. Piragliatin (RO4389620), a novel glucokinase activator, lowers plasma glucose both in the postabsorptive state and after a glucose challenge in patients with type 2 diabetes mellitus: a mechanistic study. J Clin Endocrinol Metab. 2010;95(11):5028–36.
Waring MJ, Clarke DS, Fenwick MD, Godfrey L, Groombridge SD, Johnstone J, McKerrecher D, Pike KG, Rayner JW, Robb GR, Wilson I. Property based optimisation of glucokinase activators – discovery of the phase IIb clinical candidate AZD1656. Med Chem Commun. 2012;3:1077–81.
Denney WS, Denham DS, Riggs MR, Amin NB. Glycemic effect and safety of a systemic, partial glucokinase activator, PF-04937319, in patients with type 2 diabetes mellitus inadequately controlled on metformin-a randomized, crossover, active-controlled study. Clin Pharmacol Drug Dev. 2016;5(6):517–27.
Meininger GE, Scott R, Alba M, Shentu Y, Luo E, Amin H, Davies MJ, Kaufman KD, Goldstein BJ. Effects of MK-0941, a novel glucokinase activator, on glycemic control in insulin-treated patients with type 2 diabetes. Diabetes Care. 2011;34(12):2560–6.
Hinklin RJ, Baer BR, Boyd SA, Chicarelli MD, Condroski KR, DeWolf WE Jr, Fischer J, Frank M, Hingorani GP, Lee PA, Neitzel NA, Pratt SA, Singh A, Sullivan FX, Turner T, Voegtli WC, Wallace EM, Williams L, Aicher TD. Discovery and preclinical development of AR453588 as an anti-diabetic glucokinase activator. Bioorg Med Chem. 2020;28(1):115232.
Zhang X, Schneck K, Bue-Valleskey J, Yeo KP, Heathman M, Sinha V. Dose selection using a semi-mechanistic integrated glucose-insulin-glucagon model: designing phase 2 trials for a novel oral glucokinase activator. J Pharmacokinet Pharmacodyn. 2013;40(1):53–65.
Scheen AJ. New hope for glucokinase activators in type 2 diabetes? Lancet Diabetes Endocrinol. 2018;6(8):591–3.
Zheng S, Shao F, Ding Y, Fu Z, Fu Q, Ding S, Xie L, Chen J, Zhou S, Zhang H, Zhou H, Chen Y, Sun C, Zhu J, Zheng X, Yang T. Safety, pharmacokinetics, and pharmacodynamics of globalagliatin, a glucokinase activator, in Chinese patients with type 2 diabetes mellitus: a randomized, phase Ib, 28-day ascending dose study. Clin Drug Investig. 2020;40(12):1155–66.
Matschinsky F. GKAs for diabetes therapy: why no clinically useful drug after two decades of trying? Trends Pharmacol Sci. 2013;34(2):90–9.
Brouwers M, Jacobs C, Bast A, Stehouwer CDA, Schaper NC. Modulation of glucokinase regulatory protein: a double-edged sword? Trends Mol Med. 2015;21(10):583–94.
Tornovsky-Babeay S, Dadon D, Ziv O, Tzipilevich E, Kadosh T, Schyr-Ben Haroush R, Hija A, Stolovich-Rain M, Furth-Lavi J, Granot Z, Porat S, Philipson LH, Herold KC, Bhatti TR, Stanley C, Ashcroft FM, In't Veld P, Saada A, Magnuson MA, et al. Type 2 diabetes and congenital hyperinsulinism cause DNA double-strand breaks and p53 activity in beta cells. Cell Metab. 2014;19(1):109–21.
Gao Q, Zhang W, Li T, Yang G, Zhu W, Chen N, Jin H. The efficacy and safety of glucokinase activators for the treatment of type-2 diabetes mellitus: a protocol for systematic review and meta-analysis. Medicine. 2021;100(7):e24873.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare no conflict of interest.
Additional information
Publisher’s note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Sharma, P., Singh, S., Sharma, N. et al. Targeting human Glucokinase for the treatment of type 2 diabetes: an overview of allosteric Glucokinase activators. J Diabetes Metab Disord 21, 1129–1137 (2022). https://doi.org/10.1007/s40200-022-01019-x
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40200-022-01019-x