
Abstract
Studies of the brains of Alzheimer’s disease (AD) patients have revealed key neuropathological features, such as the deposition of aggregates of insoluble amyloid-β (Aβ) peptides and neurofibrillary tangles (NFTs). These pathological protein deposits, including Aβ peptides (which form senile plaques) and hyperphosphorylated tau (which aggregates into NFTs), have been assumed to be ‘the cause of AD’. Aβ has been extensively targeted to develop an effective disease-modifying therapy, but with limited clinical success. Emerging therapies are also now targeting further pathological processes in AD, including neuroinflammation. This review focuses on the inflammatory and oxidative stress-related changes that occur in AD, and discusses some emerging anti-inflammatory natural products and phytomedicines. Many of the promising compounds are cytokine-suppressive anti-inflammatory drugs (CSAIDs), which target the proinflammatory AP1 and nuclear factor-κB signalling pathways and inhibit the expression of many proinflammatory cytokines, such as interleukin (IL)-1, IL-6, tumour necrosis factor-α, or nitric oxide produced by inducible nitric oxide synthase. However, many of these phytomedicines have not been tested in rigorous clinical trials in AD patients. It is not yet clear if the active compounds reach an effective concentration in the brain (due to limited bioavailability) or if they can slow down AD progression in long-term trials. The authors suggest that it is crucial for both the pharmacological and complementary medicine industries to conduct and fund those studies to significantly advance the field.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Natural phenolic and antioxidant compounds have been demonstrated to have varying mechanisms of action, relating to decreasing oxidative stress, inflammation, Aβ levels, and overall cognitive deficits. |
Despite encouraging animal data, the percentage of positive human trials in Alzheimer’s disease is surprisingly low, most likely because the therapeutics are administered too late in the disease process. |
The effectiveness of these identified compounds appears enhanced when therapy is initiated earlier in the disease. |
Future work is required to evaluate the therapeutic value of these compounds, in monotherapy as well as in combination therapy. |
1 Introduction
Alzheimer’s disease (AD) is a neurodegenerative disorder, and it is predicted that the number of people living with AD in the US alone will grow to 13.8 million in 2050 [1]. The pathology of AD is characterised by synaptic and neuronal loss, the formation of intracellular neurofibrillary tangles (NFTs) and the deposition of amyloid-β (Aβ) plaques in the brain. This is accompanied by chronic neuroinflammation, including microglial activation, as well as extensive oxidative damage [2]. Mitochondrial damage, oxidative stress, and inflammation may be initiating factors, and/or possibly the overproduction or reduced clearance of toxic Aβ peptides and aggregates. Importantly, a destructive cycle begins with this pathology as oxidative stress and inflammation promote Aβ production and hyperphosphorylation of tau, and, in turn, these pathologies also increase oxidative stress and inflammation [3]. As a result of the involvement of two proteins—Aβ and microtubule-associated protein tau (MAP-tau)—in AD pathology, therapeutic mechanisms to reduce the development of these two main disease pathologies have been studied at length over the past three decades [4]. Unfortunately, such studies have not yet led to a US FDA-approved treatment [5, 6].
Although trials have commenced recruiting ‘at risk’ presymptomatic individuals, which are highly reliant on the discovery of specific and sensitive biomarkers, the importance of investigating and targeting other contributors to the disease process cannot be understated. This review will focus on neuroinflammation as the major therapeutic target and discuss natural products and phytomedicines as potential treatment strategies for AD. The authors have concentrated on prior published works that have been central or pivotal to the various topics of discussion, using the PubMed, ISI Web of Science, EMBASE and BIOSIS electronic databases.
2 Inflammation and the Immune Response in Alzheimer’s Disease (AD)
2.1 The Role of Microglia and Astrocytes in Chronic Neuroinflammation in AD
Two types of brain cells participate in an inflammatory response—astrocytes and microglia. Microglia and astrocytes are often found to be associated with amyloid plaques, as well as markers of inflammation and oxidant stress [2, 7, 8]. However, the inflammatory response at the site of injury releases growth factors and cytokines, with trophic, mitogenic, chemotactic and angiogenic activities, in addition to reactive oxygen species (ROS) and reactive nitrogen species (RNS) [9].
The overproduction of microglial proinflammatory mediators can be neurotoxic [10, 11], which is why it has been shown that neuroinflammation (which also increases with aging) promotes neurological diseases such as AD, Parkinson’s disease and motor neuron disease. As mentioned above, Aβ, alone and in combination with other activators, is able to activate microglia, initiating or aggravating an inflammatory response [12]; in turn, inflammation can increase Aβ production.
Rodent studies have suggested there are two types of activated microglia, namely the M1 and M2 phenotype microglia, with the M1 microglia inducing a proinflammatory response, and the M2 cells having a neuroprotective role—reducing inflammation, phagocytosing Aβ and reducing Aβ-mediated toxicity [13]. The polarisation and balance of microglia in the CNS is important as it has been shown to influence learning and memory [14]. Microglia are not always harmful; there is evidence that they may reduce AD pathology by contributing to the clearance of Aβ, through phagocytosis of Aβ and releasing of enzymes responsible for Aβ degradation. Microglia (most likely M2 microglial cells) also secrete anti-inflammatory cytokines and growth factors, which can be neuroprotective. The normal microglial phagocytosis of damaged cells is also important in maintaining a healthy brain environment; however, damaged cells can become potent inflammatory stimuli, resulting in more tissue damage.
On the other hand, microglia become less efficient in their neurosupportive role as a result of aging. In addition, they can also become overactive (or primed) in response to stimulation, for example, through higher levels of Aβ or its modification, or synergistic interaction with advanced glycation end products (AGEs) [12, 15]. Furthermore, phagocytosis by microglia can also activate the respiratory burst, a rapid release in ROS, such as superoxide radicals and hydrogen peroxide (H2O2), from cells [16].
Mice models of aging and familial AD have recently provided further evidence of overstimulation and aging aspects of microglia. For example, studies of AD-model brain slice cultures have demonstrated that the addition of young microglia or their conditioned medium could induce microglial proliferation and reduce amyloid plaque size [17]. Other studies have shown that microglia isolated from aged mouse brains have a gene expression pattern aimed at proinflammatory processes, phagocytosis, and lipid homeostasis. Furthermore, studies conducted using brains from middle-aged people have demonstrated that signs of inflammation are already present, through the age-dependent increases in CD68 (associated with phagocytosis) and HLA-DR (associated with antigen presentation) seen in microglia of white matter. Increased microglial activity is also detectable in early-onset AD brain tissue, as well as in late-onset AD cases [18].
Neurofibrillary ‘ghost’ tangles initially formed from hyperphosphorylation of tau and then exposed to the extracellular environment are also a trigger for neuroinflammation. Furthermore, studies of traumatic brain injury, a well-known precursor to NFT formation in later life, have shown that the neuroinflammation that occurs after injury can persist for years post-injury and that any future injuries tend to provoke overreactive glial responses in later years, most likely increasing tau pathology [19].
Although astrocytes are important in maintaining neuronal health by reacting as part of the normal immune responses of the brain, astrogliosis can also contribute to neuronal inflammation [20]. However, some microglial products such as interleukin (IL)-1β, tumour necrosis factor (TNF)-α, IL-6, and C1q activate astrocytes, which can lead to neuronal dysfunction and death. It was recently shown that microglia can activate a subtype of astrocytes. These ‘A1’ astrocytes lose their ability to promote neuronal survival, growth, synaptogenesis, and phagocytosis, and are also highly toxic to neurons, although the exact mechanism of toxicity is still unknown [21]. These astrocytes have been detected in several neurodegenerative conditions, including AD, Huntington’s disease, Parkinson’s disease, multiple sclerosis, and amyotrophic lateral sclerosis, underscoring the central role of chronic microglial and astrocyte activation in neurodegenerative conditions. The A1 astrocytes were shown to be induced specifically by microglial secretion of IL-1α, TNFα and C1q (a first subcomponent of the C1 complex in complement activation) [21].
2.2 The Biology of Oxidative Stress and its Relevance for AD
Oxidative stress results from the imbalance between ROS production and neutralisation. Oxidative stress causes two types of cell death: moderate oxidation can trigger cell apoptosis, and more intense stress (with concurrent ATP depletion) may cause necrosis [2]. The peroxidation of polyunsaturated fatty acid (PUFA) leads to damage to cell membranes, and also generates several aldehyde byproducts, including malondialdehyde and C3–C10 straight chain aldehydes, as well as α, β-unsaturated aldehydes such as 4-hydroxy-2-nominal (HNE) and acrolein. Of these, the α, β-unsaturated aldehydes in particular are important effectors of tissue damage. They show high reactivity with cysteine, histidine, and lysine [22].
Free radicals, particularly the hydroxyl radicals, also attack nucleic acids. This leads to strand breaks, crosslinking and base modifications, which may contribute to alterations in protein production and propagate neuron dysfunction and death. Mitochondrial DNA (mtDNA) is more susceptible to free radical-mediated damage than nuclear DNA, as a result of its proximity to the site of ROS production. As Dizdaroglu et al. argue in their review, DNA attack by ROS can lead to the generation of more than 20 oxidized base adducts, with 8-hydroxydeoxyguanosine (8-OHdG) the most prominent because of the relatively low oxidation potential of guanine [23].
These adducts promote DNA–DNA and DNA–protein crosslinking that can limit transcription [24]. Mitochondria are especially vulnerable to oxidative stress as energy production, the production of antioxidant enzymes, and the maintenance of membrane potential are all decreased. This can cause a further increase in ROS, leading to cell death by apoptosis [25, 26].
2.3 Advanced Glycation End Products and Carbonyl Stress in AD
AGEs are compounds that form due to the non-enzymatic glycation of amino groups on proteins, lipids, or nucleic acids, by reducing sugars and reactive aldehydes [27, 28]. They are more often formed under hyperglycaemic conditions, and some AGEs are also derived from the diet, particularly from foods fried or roasted at very high temperatures, meat and processed meat products, and full-fat cheese. AGEs have been identified as modifications to many of the long-lived protein deposits in AD, such as plaques, tangles and Hirano bodies. AGEs exhibit toxic effects related to their ability to promote oxidative stress and inflammation [29, 30], following binding to cell surface receptors such as RAGE or crosslinking with other proteins, altering their structure and function [28, 31]. A major AGE receptor is RAGE, which is found throughout the body, including the brain, and levels of RAGE are increased in AD [32]. AGEs binding to RAGE induces the activation of various intracellular cascades, which involve the nuclear factor-κB (NF-κB) pathway and inflammatory mediators such as IL-6, and TNFα, as well as C-reactive protein, all of which are linked to increased inflammation and oxidative stress [33]. RAGE has also been implicated in cardiovascular diseases and type II diabetes, both known risk factors for AD [34]. Recent studies have found many links between AGEs, RAGE, microglial activation, and disrupted Aβ metabolism, including the excessive transfer of Aβ from the blood to the brain via RAGE located on the vascular side of the blood–brain barrier [35].
2.4 Involvement of the Complement System in AD
The complement system involves a collection of proteins, including C1–C9, which participate in an amplifying cascade of enzymes, resulting in large numbers of downstream complement factors. The complement system participates in antigen–antibody reactions when outside the brain. However, within the brain, complement protein C1q binds to Aβ fibrils, which are then phagocytosed by microglia. This may explain the 10- to 80-fold increase in C1q expression levels found in AD brain regions highly affected by AD pathology, such as the entorhinal cortex, hippocampus, and mid-temporal gyrus (compared with non-AD brains) [36]. These fibrils may then remain largely undegraded by the microglia, resulting in a ‘frustrated phagocytosis’ that could lead to increased microglia activation, if the toxic elements of the phagolysosome are released into the cell environment [37]. In vitro fibrillar amyloid activates both the classical and alternative complement cascades [38], which could lead to further degeneration as a result of this possible frustrated phagocytosis. In line with this postulation, chronic treatment with a C5a antagonist, PMX205 (hydrocinnamate-[Om-Pro-D-cyclohexylalanine-Trp-Arg]), decreased AD pathology in two mouse models [39]. C1q has also been shown to bind to NFTs, although possibly to a lesser extent [40].
Studies of cerebral spinal fluid (CSF) in AD patients have shown higher levels of C1q, C3d and C4d in the early stages of AD, and levels show a positive correlation with plaque number from mild to severe cases. Furthermore, the patterns of complement factor expression change over time, indicating a temporal pattern of complement protein expression in relation to Aβ plaque deposition or other AD pathology [41], and possibly providing biomarkers for the disease.
In contrast, the increase in components of the terminal pathway are considered pathogenic. For example, increases in the levels of C5a and its receptor occur in AD and AD-model transgenic mice, and the specific inhibition of C5a reduces amyloid build-up in transgenic AD mice. A recent cell culture study confirmed that C5a increases the damage to primary neurons caused by Aβ fibrils via the C5a receptor, with the conclusion being that C5aR1 antagonists may provide benefit as AD therapeutics [42]. Interestingly, another study has found that a deficiency in the C5a receptor leads to lower body weight, lower plasma triglyceride and non-esterified fatty acid levels in C5a receptor knockout mice fed a high-sucrose diet, indicating C5a receptors also have a role in fat storage and metabolism [43], abnormalities of which have been linked to AD.
2.5 Cytokines and Chemokines in AD
Activation of microglia by Aβ induces the production of inflammatory cytokines, including IL-1β and TNFα via NF-κB pathway activation [44]. Activated microglia also upregulate inducible nitric oxide synthases (iNOS), which cause oxidative stress by generating nitric oxide (NO), leading to peroxynitrite [33].
Increases in IL-1β further aggravate the inflammatory response induction of amyloid precursor protein (APP) expression. IL-1 may promote the phosphorylation of the tau protein, leading to the formation of NFTs and possibly contributing to neuronal death [45]. Many cytokines and chemokines, including IL-1β, IL-6, TNFα, IL-8, tumour growth factor-β (TGF-β) and macrophage inflammatory protein-1α (MIP-1α), are upregulated in AD brains [46]. Animal models of familial AD, such as the Tg2567 mouse model (overexpressing APP with the Swedish mutation), also show enhanced levels of TNFα, IL-1α, IL-1β, chemoattractant protein-1 and cyclooxygenase (COX)-2 [47]. In addition, it has also been reported that mice overexpressing the mutant human P301 tau protein have an increased immunoreactivity to IL-1 and COX-2 [48]. The production of interleukins and other cytokines and chemokines, for example by interaction of AGEs and Aβ amyloid with receptors for AGEs, may also lead to microglial activation, astrogliosis (reactive astrocytes) and further secretion of proinflammatory molecules and amyloid, perpetuating the pathological cascade of AD [49]. Interestingly, activation of microglia and macrophages also increase the production of the AGE precursor methylglyoxal, leading to the formation of proinflammatory AGEs [50, 51].
Genetic studies have also indicated the central role of inflammation in AD. For example, genome-wide association studies have identified several immune system-related genes linked to AD, such as CLU (clusterin), CR1 (complement receptor 1) and TREM2 (triggering receptor expressed on myeloid cells 2) [52]. More recent meta-analyses found other AD linkages to more immune system proteins, including HLA-DRB5-DRB1 (HLA class II histocompatibility antigen, DRB5 beta, allelic variant DRB1) and INPP5D (inositol polyphosphate-5-phosphatase D, linked to cancers of the immune system) [53], as well as MS4A4A (a cell surface protein found on monocytes) [54].
All the above data suggest that the progression of many neurodegenerative diseases, including AD [55], arise from a cycle of self-perpetuating inflammatory neurotoxicity. Stages that may occur include the following.
-
1.
A variety of inflammatory triggers lead to the initial microglial activation. Such triggers can be peripheral (e.g. peripheral chronic inflammation or sepsis) or central (e.g. degenerating and dying neurons or amyloid deposits). In one widely investigated mouse model, the Glial Fibrillary Acid Protein (GFAP)-IL6 mouse, the trigger is the cytokine IL-6 [56].
-
2.
Stimulated microglia release toxic factors such as TNFα and ROS, which cause damage to neighboring neurons. This process can be inhibited by cytokine-suppressive anti-inflammatory drugs (CSAIDs) [10].
-
3.
The release of microglia activators from damaged or dying neurons, such as damage-associated molecular pattern molecules (DAMPs), resulting in subsequent microglial activation and self-perpetuating vicious cycle.
Consequently, targeting chronic neuroinflammation is a novel disease-modifying treatment approach for many neurodegenerative diseases, including AD [57].
3 Consequences of Chronic Neuroinflammation
The amyloid hypothesis of AD suggests that the disease may start with abnormally high levels of extracellular Aβ oligomers (due to excessive production or a reduction in clearance of the peptide), triggering oxidative stress and inflammation [58], an effect that is later enhanced by tau. The inflammatory response increases over time as the disease progresses [59]. In summary, many studies indicate that inflammation plays a role in the process of amyloid plaque formation (and amyloid deposits lead to further inflammation in a vicious cycle). That inhibition of this inflammatory cascade may attenuate amyloidogenic processes and slow down the progression of AD [58].
Inflammation appears to influence the metabolism of APP directly, increasing Aβ production, or affecting the clearance and breakdown of Aβ. In neurons, for example, cytokines upregulate β-secretase (BACE1) expression [60]. In fact, oxidative stress, inflammation, hypoxia and ischaemia are all capable of increasing BACE1 activity [61]. These data are supported by experiments showing increased expression of BACE1 in NT2 cells under oxidative stress and in reactive astrocytes in models of gliosis [61]. The enzyme ADAM17, which is thought to be the regulated APP α-secretase enzyme that precludes Aβ production by cleaving APP within the Aβ region, is also involved in the shedding of extracellular domains of at least 40 other transmembrane proteins, including cytokines, growth factors and receptors. However, changes in ADAM17 have been shown to play a role in atherosclerosis, in adipose tissue metabolism, insulin resistance and diabetes, some of which are known risk factors for AD. Furthermore, in inflammation, the proinflammatory activity of TNFα is mostly mediated by type 1 TNFα receptors that undergo cleavage via ADAM17 [62]. ADAM17 and TNFα have been shown to be involved in stress-induced activation of NFκB, a central regulator of inflammation, and responses to free radicals, etc. All the above evidence indicates that ADAM17 may influence APP metabolism on many levels [63].
Astrocytes have been shown to clear Aβ by endosomal/lysosomal pathways, yet some enzymes involved in Aβ breakdown, such as insulin-degrading enzyme (IDE) and neprilysin, can only degrade Aβ monomers. The uptake of Aβ by astrocytes can also be carried out, by the low-density lipoprotein receptor-related protein (LRP1), scavenger receptor class B member 1 (SCARB1), and the receptor for AGEs (RAGE), yet some receptors such as LRP1 are again only capable of monomer uptake, thus an increasing amount of oligomers and fibrils will eventually reduce the effectiveness of astrocyte-mediated Aβ clearance (for a review, see Batarseh et al. [64]).
This is not an exhaustive list, just examples of the many changes to APP-regulating enzymes that are linked to inflammation, but it might give a valuable insight into the potential targets of the natural products and phytomedicines discussed in the later parts of this review.
4 Medications for the Treatment of AD
4.1 Current Shortfalls and Limitation of Symptomatic Treatment with Acetylcholinesterase Inhibitors and Memantine
Several medications are currently used to treat AD, however these do not provide a cure or halt the disease but merely provide some temporary improvement [65, 66].
One of the defining hallmarks in the pathogenesis of AD is a deficit in the neurotransmitter acetylcholine [67]. Acetylcholinesterase inhibitors (AChEIs) were therefore developed many years ago to augment acetylcholine-mediated neurotransmission. As a result, the best current treatment for mild to moderate AD is to improve cognitive function by treatment with AChEIs. Examples include donepezil, rivastigmine and galantamine, which are licensed as medications for symptomatic treatment of AD, despite the fact that the benefit of these agents is modest.
Donepezil is now widely used and is administered once daily [68, 69]. Rivastigmine may be a suitable alternative for patients who are unresponsive to donepezil or galantamine [70]. Another AChEI that has shown some promise in AD treatment research is physostigmine [71].
Another target in AD is excitotoxity. Glutamatergic overstimulation may result in neuronal cell death, a phenomenon that involves the NMDA receptor, neuronal calcium overload and has been termed excitotoxicity [72]. Based on these findings, the glutamatergic inhibitor memantine is also used to treat AD, although usually only in the moderate to severe stages of the disease. Memantine blocks NMDA receptors and is sometimes used in conjunction with AChEIs in the moderate AD stages to improve symptoms and slow progression of the disease. However, again, the improvements are temporary, lasting approximately 6–18 months [73].
5 Treatment Approaches with Anti-Inflammatory Agents
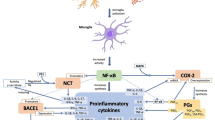
Disease-modifying treatments (treatments that influence the progress of the disease) are not yet available. The next section will introduce these potential treatments, including natural products and phytomedicines (Fig. 1).

Main effects of selected natural products and phytomedicines in Alzheimer's disease (AD) pathogenesis. A summary of compounds discussed in this review, and their main effects on various processes in AD pathogenesis and relevant pathological hallmarks in AD. TLR4 Toll-like receptor 4, iNOS inducible nitric oxide synthase, Aβ amyloid-β peptide, APP amyloid precursor protein, ADAM a disintegrin and metalloproteinase, MEK mitogen-activated protein kinase, ERK extracellular signal-regulated kinase, TNF tumour necrosis factor, IL interleukin, PGE prostaglandin, COX cyclooxygenase, RBC red blood cells, NEP neprilysin, BDNF brain-derived neurotrophic factor, BACE β-secretase, CRP C-reactive protein, IDE insulin-degrading enzyme, GFAP glial fibrillary acidic protein, SIRT1 sirtuin 1
5.1 Non-Steroidal Anti-Inflammatory Drugs
The pathology of AD involves an inflammatory pathway, and some epidemiological evidence indicated that the use of non-steroidal anti-inflammatory drugs (NSAIDs) was associated with a reduced risk of AD. However, many studies have been conducted to try to verify this potential utility of NSAIDs, and reviews and meta-analyses concluded that there was no clear evidence of any benefit of NSAIDs in preventing or treating AD [74, 75].
However, NSAIDs are designed as inhibitors of COXs and do not interfere with the expression of proinflammatory cytokines such as TNFα, or radicals such as NO. These findings illustrate the need to develop novel and safe anti-inflammatory drugs with a broader range of anti-inflammatory effects than conventional NSAIDs. Cytokine-suppressive anti-inflammatory drugs (CSAIDs) have emerged as a new class of anti-inflammatory drugs as they have a broader range of action than NSAIDs in the treatment of chronic neuroinflammation. CSAIDs target the p38 MAPK and NF-κB signalling pathways to inhibit the expression of a variety of proinflammatory cytokines [76].
5.2 Anti-Inflammatory Natural Products for the Treatment of AD
Many plant extracts and their active constituents have shown a variety of medicinal pharmacological properties as an anti-inflammatory treatment for AD (Fig. 1). A preliminary start to identifying clinically significant compounds for the treatment of AD includes testing them for inhibition of induced NO production in cell lines, such as the gold standard for these assays, the macrophage cell line RAW 264.7. Potency is a very important parameter in this respect as only potent compounds are likely to reach a therapeutically effective concentration in the brain. Table 1 shows the half maximal inhibitory concentration (IC50) value of some clinically important natural products that have been studied for their anti-inflammatory activity in macrophages, and further investigated for the treatment of AD.
5.2.1 Cinnamon/Cinnamaldehyde
Cinnamaldehyde (CA) is a flavonoid that is largely responsible for the flavor and aroma in cinnamon. Sri Lankan cinnamon (C. zeylanicum) was shown to be one of the most potent anti-inflammatory foods out of 115 Australian foods tested [77]. The most abundant compounds in cinnamon extract were E-cinnamaldehyde and o-methoxycinnamaldehyde [78].
The neuroprotective effects of CA were shown in ischaemia/reperfusion-induced brain injury, as it reduced the infarction area and neurological deficit score [79].
Cinnamon can also prevent tau aggregation in vitro and can even dissolve tangles isolated from AD brain [80]. The anti-tangle activity of CA was due to its binding to the two cysteine residues in tau. Furthermore, these compounds protected tau from oxidation caused by H2O2. Epicatechin can also react with lipid peroxidation products such as acrolein [81].
Recent studies of a mouse model of cerebral ischaemia have added further evidence of CA inhibiting inflammation, partly mediated by reduced expression of signal transduction molecules such as toll-like receptor 4 (TLR4) and tumour necrosis receptor-associated factor 6, and by reducing the nuclear translocation of NF-κB, resulting in an attenuated increase in TNFα and IL-1β, amongst others [82]. The inhibition of TLR4/NOX4 signalling is known to reduce oxidative stress/nitrative stress in neuronal damage and apoptosis pathways. A recent study of lipopolysaccharide (LPS)-induced cardiac dysfunction in rats found that CA reduced intracellular ROS production and reduced TNFα, IL-1β and IL-6 levels in LPS-stimulated rats by blocking the TLR4, NOX4, MAPK and autophagy signals [83].
CA also reduces inflammation in mice fed a high-fat diet [84], and in other mice and microglial studies involving LPS-induced inflammation, trans-cinnamaldehyde (another anti-inflammatory compound isolated from Cinnamomum cassia) was shown to improve synaptic plasticity in the mice. It also reduced NO production by accelerating the degradation of iNOS mRNA (in turn thought to occur due to trans-cinnamaldehyde inhibiting the MEK1/2–ERK1/2 signalling pathway) and reduced IL-1β release from primary microglia [85].
In summary, cinnamon efficiently inhibits oxidative stress and proinflammatory pathways, tau accumulation, Aβ aggregation, and toxicity in vivo and in vitro models; however, more clinical trials with cinnamon are needed [86].
5.2.2 (−)Epigallocatechin-3-Gallate (EGCG) and Other Green Tea Polyphenols
Green tea is derived from the leaves of the Camellia sinensis plant. Green tea polyphenols are potent antioxidants, and scavengers of ROS and RNS in vitro [87]. Many studies have shown that raising the dietary intake of polyphenols can reduce oxidative stress and reduce the risk for oxidative stress-related diseases, including AD, Parkinson’s disease, stroke and Huntington’s disease [88]. A subclass of green tea polyphenols, called catechins, has been investigated through various studies evaluating their efficacy. Of the four major catechins, (−)epigallocatechin-3-gallate (EGCG) is the major constituent (approximately 60%), followed by (−)-epigallocate-chin (EGC), (−)-epicatechin (EC), and, lastly, (−)-epicatechin-3-gallate (ECG) [87]. EGCG can prevent neuronal cell death caused by Aβ neurotoxicity in vitro [89]. Another in vitro study demonstrated that EGCG reduced Aβ generation in neuronal-like cells and primary neuronal cultures, and also promoted the non-amyloidogenic α-secretase proteolytic pathway [90]. Furthermore, when 12-month-old Tg2576 mice were administered EGCG 20 mg/kg for 60 days via intraperitoneal injections, decreased brain levels of Aβ and Aβ plaque load were detected. EGCG was also shown to promote protein kinase C (PKC)-dependent activation of the α-secretase pathway [89, 90].
Studies of green tea have shown that acetylcholinesterase can be inhibited by green (and white) tea, with EGCG being the most potent [91]. Furthermore, recent studies have indicated that polyphenols bind directly to acetylcholinesterase (and butyrylcholinesterase), collectively suggesting that green tea may function similarly to therapies currently available for AD [92]. Furthermore, primary neuronal cultures exposed to glutamate to induce neurotoxicity were found to be protected by green tea polyphenols as they inhibited the glutamate-induced ROS release. The green tea polyphenols improved mitochondrial function in cells and reduced the changes in Bax, caspase-3, and Bcl-2 that were caused by glutamate exposure [93], suggesting polyphenols were able to provide protection via antioxidative and anti-apoptotic pathways. Interestingly, these antioxidative properties are also thought to be the reason behind why green tea polyphenols, together with tai chi, have been found to reduce oxidative stress and reduce signs of osteoporosis in postmenopausal women [94].
Similar results have been obtained in studies of the double transgenic mouse AD model (APP/PS1), where mice were given green tea polyphenols in drinking water along with treadmill exercises for 4 months. The intervention reduced deficits in spatial learning and memory and lowered soluble Aβ1-40 and Aβ1-42 levels, along with oxidative stress markers in the brain. The treatment also raised brain-derived neurotrophic factor (BDNF) levels in the brain and activated Akt/GSK-3/CREB signalling [95]. Other AD-model mouse studies have shown that EGCG could prevent LPS-induced memory impairment, as well as the LPS-induced initiation of astrocytes increasing cytokine expression (TNFα, IL-1β, IL-6). Therefore, it may be possible that EGCG can reduce the inflammation associated with AD [96]. Overall, EGCG appears to be the most bioactive polyphenol and has important anti-inflammatory and anti-atherogenic properties, as well as protective effects against neuronal damage and brain edema. Other recent studies have shown that EGCG can suppress TNFα, IL-1β, IL-6, and iNOS, and restore levels of intracellular antioxidants against Aβ-induced and free radical-induced proinflammatory effects in microglia. EGCG could also restore levels of nuclear erythroid-2-related factor 2 (Nrf2; transcription factor that regulates expression of antioxidant proteins) and haem oxygenase-1 (HO-1; an enzyme that degrades haem into CO, biliverdin and free iron, all of which have anti-inflammatory and anti-apoptotic properties) [97]. Additionally, EGCG was also able to reduce Aβ-induced toxicity by reducing ROS-induced NF-κB activation and mitogen-activated protein kinase (MAPK) signalling [97, 98].
Despite the many in vitro and animal studies that have indicated many positive effects of green tea extracts, studies suggest EGCG cannot cross the blood–brain barrier in humans. Thus, the positive effects seen in cell culture, particularly in AD-model mice, may not occur in humans, or at least not to the same extent. It has been suggested that this may be due to differences in dose and time of administration, or possible differences in metabolism between mice and humans (reviewed by Cascella et al. [98]). However, many dietary and preventative studies are currently suggesting that a healthy diet high in antioxidant polyphenols (such as EGCG) combined with exercise and other physical activity can help decrease the risk of AD, at least partly by reducing oxidative stress and chronic inflammation-associated conditions such as obesity, cardiovascular disease, and insulin-resistance and type II diabetes [99]. Therefore, benefits may be obtained systemically that may translate to reduced oxidative stress and inflammation in the brain, and further research, including epidemiological studies and clinical trials of green tea extracts, should be carried out.
5.2.3 Curcumin
Curcumin is a component of the roots of the turmeric plant (Curcuma longa Linn) [100]. Turmeric is used for flavor, colour and as a food preservative, in addition to being long used to treat a variety of illnesses in traditional Indian medicine [101]. Curcumin has been demonstrated to have both antioxidant and radical scaving activities [102, 104] and is more potent than vitamin E as an antioxidant. It has also been reported to have anti-inflammatory properties [10, 103] and can inhibit the aggregation of Aβ [105]. Curcumin interferes with signalling downstream of the IL-6 receptor, through suppressing STAT3 phosphorylation [106, 107]. Furthermore, curcumin has broad cytokine-suppressive anti-inflammatory action, influencing the expression of COX-2, iNOS, TNFα, and IL-1, -2, -6, -8, and -12.
The effects of curcumin in AD mouse models have been investigated, whereby feeding a curcumin diet (500 ppm) to 17-month old Tg2576 mice for 6 months led to reduced amyloid levels and plaque burden. Curcumin injected peripherally was able to enter the brain and bind to amyloid plaques [105]. The ability of curcumin to bind to pathological amyloid fibrils is thought to be due to its structural similarity to the water-soluble Congo red dye, which also binds strongly to amyloid fibrils. Curcuminoids may also reduce Aβ levels as the components of bisdemethoxycurcumin (BDMC) can reduce BACE1 mRNA and protein levels, while demethoxycurcumin (DMC) can reduce BACE1 mRNA expression [108]. Aged female rats administered curcumin for 12 days showed improved spatial memory (in the Morris water maze test) and again showed signs of reduced oxidative damage [109].
Formulations of curcumin that can cross the blood–brain barrier have been investigated as they are not thought to reach the brain in high concentrations. As a result, highly bioavailable curcumin preparations such as ‘Longvida’ (Verdure Sciences, Noblesville, IN, USA) have been produced and these can achieve micromolar concentrations in the brain [110]. In humans, ‘Longvida’ curcumin (400 mg) was shown to significantly improve working memory and mood after 4 weeks of treatment in a randomised, double-blind, placebo-controlled human trial [111]. Additionally, the pharmacokinetic properties of curcumin (however, only when encapsulated in liposomes or micelles) are favourable for use as a therapeutic.
In a similar study, solid lipid curcumin particles (SLCP) were suggested to have greater permeability and be more neuroprotective than natural curcumin. Inhibition of Aβ aggregation, anti-amyloid, and anti-inflammatory responses of curcumin and/or SLCP were tested in mouse models of AD. 5xFAD- and age-matched wild-type mice were given injections of curcumin or SLCP (for 2 or 5 days). A larger decrease in Aβ plaque loads in the hippocampus was observed with SLPC when compared with natural curcumin. Similarly, relative to natural curcumin, SLCP produced a larger decrease of pyknotic, or tangle-like, neurons in the hippocampus [112]. Another study utilising 5xFAD mice demonstrated that intragastric administration of curcumin 150 or 300 mg/kg/day for 60 days resulted in reduced Aβ pathology through inhibition of BACE1 expression, and an improvement in both spatial learning and memory, when compared with untreated control mice [113].
Interestingly, curcumin’s fluorescent activity, preferential binding to Aβ, and structural similarities with other traditional amyloid-binding dyes collectively make it a promising candidate for labelling and imaging of Aβ plaques in vivo [114].
However, one point of criticism is that even after considerable research and many clinical trials, no curcumin drug has eventuated, raising doubt regarding the clinical efficacy of curcumin. The comment by Nelson et al. that “Curcumin is best typified, therefore, as a missile that continually blows up on the launch pad, never reaching the atmosphere or its intended target(s). These results have given curcumin the label of pharmacodynamically fierce (hits many targets) yet pharmacokinetically feeble (does not get to its targets)” is characteristic of the skepticism that has developed in the field, and many scientists believe that the commercial success of curcumin is based on insufficient clinical evidence. However, a recent article in the Nature journal states that “a PubMed search under ‘curcumin double-blind placebo-controlled clinical trial’ yields 49 entries, of which 17 trials show efficacy. In addition, there are 27 other clinical trials and at least 5 animal studies of curcumin that point to therapeutic benefits” [115].
The effect of curcumin in preventing cognitive decline was investigated in a population of community-dwelling older subjects. These individuals were administered either a placebo or Biocurcumax™ (a non-liposomal preparation) 1500 mg/day, and subsequent clinical and cognitive tests were performed at baseline and at the 6- and 12-month follow-up assessments. While statistical analysis suggested a significant result in the Montreal Cognitive Assessment (MoCA), which assesses general cognitive function, this was found to be driven by a decrease in MoCA performance function by the placebo group at 6 months, which did not occur in the curcumin-treated group, contributing to the significant result observed. However, this was the only positive outcome of the study as no other differences were observed between the groups for all other clinical and cognitive measures [117].
Overall, the few clinical trials that have been conducted with curcumin have produced mixed results, with weak evidence suggesting the use of curcumin in dementia patients [118]. The majority of clinical trials demonstrated no cognitive benefits (for a review, see Goozee et al. [116]). However, these negative results may be due to two main reasons. First, AD stage at the time of curcumin administration may have been too advanced. Epidemiological data support the concept that curcumin can reduce AD risk if taken/eaten regularly at much younger ages, and clinical studies need to be carried out at preclinical stages of AD, to slow or prevent AD pathogenesis [116]. Second, the use of low-bioavailability curcumin preparations instead of high-bioavailability (liposomal) formulations may have also impacted clinical efficacy in these trials [119, 120]. In order to be clinically effective, the concentration of curcumin in the target tissue needs to be in the low micromolar range, as the IC50 values from data from the in vitro assays suggest. It would be crucial for the pharmaceutical and complementary medicine industries to prove, at least in an animal model, that their curcumin preparation reaches a therapeutically effective concentration in the brain to support any claims of CNS-related beneficial activities [76, 120,121,122].
5.2.4 Gingko and its Associated Terpenoids and Flavonol Glycosides
Leaf extracts of Ginkgo biloba are widely used for AD, vascular dementia, and other aging-related memory disorders. The principal bioactive components of ginkgo include terpenoids (e.g. ginkgolide and bilobalide) and flavonol glycosides (e.g. quercetin and kaempferol) [123].
In preclinical studies, ginkgo has been shown to possess multiple pharmacological properties, including antioxidant and anti-inflammatory effects, anti-anxiety effects via inhibiting corticosteroid production, enhanced glucose utilisation and ATP production, enhanced erythrocyte deformability to improve blood circulation, increased NO production, and decreased platelet aggregation, all of which are potentially beneficial in dementia and other neurodegenerative diseases [2, 123].
The mechanisms of action underlying the anti-neuroinflammatory effects of ginkgo have been well-documented. In LPS-activated primary microglial cells, EGb 761, a standardised ginkgo extract, has been shown to significantly inhibit the proinflammatory mediator prostaglandin E2 in a dose-dependent manner [124]. In addition, EGb 761 significantly downregulated several inflammatory cytokines, including AGE- or LPS-induced NO, TNFα, IL-6 and IL-1 [125]. EGb 761 and its key constituents, quercetin and ginkgolide B, have also been shown to elicit a concentration-dependent inhibition of Aβ(1-42)-induced decrease of cell viability, intracellular ROS, mitochondrial dysfunction and activation of caspase 3, JNK, ERK1/2 and Akt in SH-SY5Y cells [126].
Data from animal studies are consistent with that of the in vitro findings. In an APP-transgenic AD model in mice, treatment with EGb 761 for 5 months significantly reduced the travel time and distance of travel of animals in the Barnes Maze test, attenuated the loss of synaptic structure protein (including PSD-95, Munc 18-1, and SNAP25) and inhibited inflammatory activation of microglia in the brain [127]. The effectiveness of gingko treatment has been demonstrated in numerous clinical trials in both healthy and dementia populations. In healthy young adults, ginkgo has been shown to improve the speed of processing, working memory, and executive function [128].
In a randomised, double-blind, placebo-controlled trial, 216 participants with AD and/or vascular dementia were randomly allocated to receive a 24-week treatment of EGb 761 or placebo. The results demonstrated a significant improvement in attention and memory function in participants who received the Ginkgo biloba treatment [129]. In a more recent study, 24-week treatment with EGb 761 was associated with a significant improvement in cognitive function and neuropsychiatric symptoms in 404 patients diagnosed with dementia or vascular dementia [130].
Ginkgo’s beneficial therapeutic effects have been demonstrated in several systematic reviews and meta-analyses that show ginkgo treatment stabilises or slows decline in cognition, function, and behaviour in dementia patients [131,132,133].
5.2.5 Ginseng and Ginsenoside
The root of Panax ginseng (ginseng) has been traditionally used for centuries to manage a wide range of disorders, including AD, in many Asian countries. The principal bioactive components of ginseng are ginsenosides, a type of terpenoid dammarane glycoside, and more than 60 different ginsenosides, such as Rb1, Rb2, Rb3, Rc Rd, Re, Rg1, Rg2 and Rg3 have been identified in ginseng root [134]. The mechanisms underlying the anti-AD effects of ginseng are complex, including inhibition of Aβ formation, stimulation of soluble APP-α formation, anti-inflammation, anti-apoptosis, a decrease in oxidation stress, and enhancement of CNS cholinergic function [135,136,137,138].
Anti-inflammatory effects of ginsenosides have been shown in numerous in vitro and in vivo studies. Ginsenoside Re and Rg18 reduced LPS-induced proinflammatory mediators such as iNOS, COX2, TNFα, and iNOS through inhibiting phosphorylation of p38MAPK and STAT1 in BV2 microglial cells [139]. Similarly, in a primary mixed culture of astrocytes from postnatal Sprague–Dawley rats, total ginseng saponins significantly inhibited TNFα, IL-1β, iNOS and COX2 [140]. These in vitro results have received support from in vivo studies in animals. Ginsenoside Rb1 significantly reduced LPS-induced upregulation of microglia cell activation characterised by Iba1 protein expression and mRNA expression of TNFα, IL-1β and IL6 in mice [141]. Similar results were observed with ginsenoside Rg1 in an LPS-induced neuroinflammation model in mice [142].
Early evidence exists to support the clinical use of ginseng in AD. In healthy human subjects, ginseng has been shown to modestly improve thinking and working memory [143, 144]. In two small, open-label trials, 12 and 24 weeks of ginseng treatment significantly improved Alzheimer’s Disease Assessment Scale-cognitive subscale (ADAS-cog) and Mini-Mental State Examination (MMSE) scores in patients with AD [145, 146]. Combined treatment of ginseng and ginkgo has also been demonstrated to improve cognitive function in humans [147,148,149].
5.2.6 Polygala Tenuifolia (Yuan Zhi)
Polygala tenuifolia Willd (PT) has long been used in traditional Chinese herbal medicine for treating various conditions. It is also frequently used as a component of traditional herbal formulas in other Asian countries such as Korea and Japan.
Several crude and purified extracts of PT, including tenuigenin, tenuifolin, BT-11 and PGS32, have been tested for their anti-AD activities. For example, tenuifolin was shown to inhibit Aβ secretion from transfected cells in vitro [150], most likely through inhibition of BACE1 (β-secretase) [151]. BT-11 was shown to protect against Aβ 25-35 peptide-induced neurotoxicity in vitro and in vivo [150]. The PT extract has shown anti-inflammatory activities, including inhibition of the production of inflammatory mediators such as NO, PGE2, IL-1β and TNFα, as well as iNOS and COX-2 expressions in LPS-stimulated murine BV2 microglia; the effect is likely to be mediated by inhibiting the NF-κB and TLR signalling pathways [152, 153].
A recent study on tenuifolin also found that it inhibited cellular production of NO and PGE2, downregulated the MAPK and NF-κB pathways, but upregulated the Nrf2/HO-1 pathways [151]. Another PT extract, PGS32, was shown to enhance neurotransmission and induce long-term potentiation (LTP) in the hippocampus, possibly involving MAP kinase activation and an increase in BDNF concentrations [154]. Studies on active components of PT were also reported, including senegenin [155], which was shown to protect against Aβ peptide-induced neurotoxicity in PC-12 cells [155]. Clinical studies showed PT formulations have memory-enhancing effects in elderly humans [156, 157]. Furthermore, a Japanese herbal formula, Kami-untan-to (KUT) improved cognition and brain perfusion of AD patients in a 12-week trial in combination therapy with donepezil [158].
5.2.7 Cannabis/Cannabidiol
Cannabidiol (CBD) is one of the major phytocannabinoids expressed in cannabis sativa and was first isolated in 1940. CBD has very low toxicity, is rapidly distributed, and is lipophilic, allowing it to easily pass through the blood–brain barrier [159]. The pharmacological profile of CBD includes anticonvulsive, anti-anxiety, and antipsychotic effects, as well as anti-nausea and anti-inflammatory properties [160].
The function of CBD in brain circuits and the endocannabinoid system, in particular, appears complex. It shows very low affinity to the cannabinoid receptors 1 and 2 (CB1 and CB2) and seems to be an inverse agonist at CB2. CBD also acts at glutamate receptors, serotonergic receptors (e.g. the human 5-HT1a receptors, where it inhibits 5-HT reuptake) and peroxisome proliferator-activated receptors (PPAR)-γ, and has allosteric properties at μ- and δ-opioid receptors at very high concentrations [161].
In the context of AD, it is of interest that CBD possesses antioxidant, anti-inflammatory, and neuroprotective properties, and decreases Aβ production and tau hyperphosphorylation in vitro [161]. In a first in vivo study where mice were inoculated with Aβ42 in the hippocampus, CBD dose-dependently suppressed Aβ-induced iNOS and IL-1β protein expression, as well as the related NO and IL-1β release. CBD also decreased Aβ-induced glial fibrillary acidic protein (GFAP) mRNA and protein expression (i.e. a marker of activated astrocytes) [162].
In line with this early finding, Martin-Moreno and co-workers found that mice injected intravenously with fibrillar Aβ exhibited deficits in Morris water maze learning, which were reversed post chronic CBD treatment. This beneficial effect of chronic CBD was accompanied by decreased levels of IL-6 [163]. However, most preclinical in vivo research into AD utilises transgenic mouse models for the disease. Indeed, the therapeutic potential of CBD has more recently been evaluated in AD transgenic mice [164].
Karl and colleagues conducted the first two studies evaluating the remedial and preventative properties of CBD in AD transgenic mice. For the remedial study, CBD was administered chronically to male double-transgenic APPswe/PS1∆E9 (APPxPS1) mice after the onset of AD-like symptoms (i.e. cognitive deficits and Aβ pathology). CBD treatment reversed deficits in social recognition memory and object recognition deficits in these AD transgenic mice [165]. In the preventative study, APPxPS1 male mice were treated with vehicle or CBD gel pellets on a daily basis using a voluntary oral administration scheme, starting at an age when AD-like pathophysiology is still sparse in these mice (i.e. for 8 months starting at 2.5 months of age). CBD prevented the development of a social recognition deficit in APPxPS1 males. The therapeutic-like effect of CBD was not associated with changes in Aβ levels or oxidation; however, the findings suggested that CBD might modulate cytokine levels, in particular TNFα [166].
The therapeutic potential of CBD for AD in combination treatments with Δ9-tetrahydrocannabinol (THC) has also been assessed. In the first experiment, Sativex® (a mixture of a THC and CBD at a ratio of 1: 1) treatment of parkin-null, human tau overexpressing (PK−/−/TauVLW) mice decreased neuroinflammation (e.g. GFAP levels) and iNOS levels in the cerebral cortex and reduced stress-related behaviours typical for PK−/−/TauVLW mice [167]. Most relevant to AD, Sativex® also reduced cortical and hippocampal Aβ, plaques and phosphorylated tau, and stimulated autophagy. In a second study, APPxPS1 transgenic mice at the early symptomatic phase were treated with botanical extracts high in THC content (and low CBD concentration, i.e. < 0.5%), or high in CBD content (and low THC concentration, i.e. < 2.5%). Another group of control and AD transgenic mice was treated with a combination of CBD plus THC [168]. All treatments preserved object recognition memory of AD transgenic mice. The CBD/THC combination AD transgenic group also exhibited improvements in fear-associated learning, reductions in cortical soluble Aβ42 (but not Aβ40) levels, as well as changed plaque composition. The combination treatment was also most effective in lowering inflammation and changing neuroinflammatory processes in APPxPS1 mice [168].
Few clinical trials have investigated the effect of cannabis/CBD in dementia patients. Medical cannabis oil (MCO) containing THC was used as an add-on to pharmacotherapy and was evaluated for relieving behavioural and psychological symptoms of dementia (BPSD) in a 4-week trial. Significant reductions in Clinical Global Impression (CGI) severity score and delusions, agitation/aggression, irritability, apathy, sleep, and caregiver distress were recorded [169].
5.2.8 Resveratrol
Resveratrol (3,5,4ʹ-trihydroxystilbene) is found in many plant species, including grapes and berries, as well as some medicinal herbs such as Polygonum cuspidatum. It is best known as a key ingredient for the health benefits of wine, derived from observations that wine consumption is related to a reduced risk of dementia, which links to wine polyphenols [170,171,172].
Numerous studies have demonstrated various bioactivities of resveratrol, including antioxidant, anti-inflammatory, improved vascular function, neuroprotective, modulations of insulin sensitivity, and glucose and lipid metabolisms, indicating its potential application in cardiovascular conditions and AD [121, 173,174,175,176,177]. Corpas et al. [178] reported that supplementation of resveratrol 100 mg/kg for 10 months in 3xTg-AD mice resulted in neuroprotection against amyloid and tau pathology, measured through increased levels of neprilysin, reduction of BACE1, increased proteasome protein levels and upregulation of the sirtuin 1 (SIRT1) pathways. Corpas et al. also demonstrated improved cognitive performance in spatial learning and memory tests.
The action of resveratrol against pathological effects in AD involves multiple mechanisms. Resveratrol changes Aβ synthesis, oxidative stress, inflammation, apoptosis and neurodegeneration as follows:
-
1.
Modulation of APP metabolism, decreased APP cleavage, enhanced Aβ peptide clearance and reduced Aβ synthesis and aggregation [179,180,181].
-
2.
Modulation of metabolic homeostasis, such as inhibition of the production of AGEs and regulation of apoptosis proteins and cell signalling pathways [182]. The actions of resveratrol on glucose metabolism and insulin sensitivity may help to correct the relevant abnormalities or reduce the risk factors involved in AD development [183]. Sirtuin signalling plays an important role in the regulation of cell growth and apoptosis and is also linked to AD development [182]. SIRT1 level was lower in patients with AD and mild cognitive impairment, and has also been suggested as an early marker for the detection of AD [184].
-
3.
Inhibition of inflammatory mediators and ROS. Resveratrol was reported to inhibit proinflammatory cytokines and inflammatory mediators, such as TNFα, IL-1β, prostacyclin, and prostaglandins [177].
The anti-inflammatory effect of resveratrol has been confirmed in clinical studies. A recent analysis of 17 randomised controlled clinical trials involving 736 subjects revealed that resveratrol significantly reduced TNFα and hs-CRP levels [185]. It has been suggested that resveratrol may improve select measures of cognitive performance and mood in adults, although the current literature is still limited [186]. In summary, these findings support using resveratrol as an adjunct therapy for AD management.
5.2.9 Apigenin
Apigenin (4ʹ,5,7-trihydroxyflavone) is a natural flavonoid found in many vegetables and fruits, such as chamomile, parsley, celery, apples, peppermint, oregano, rosemary, and grapefruit. Among these, dried fresh parsley (Petroselinum crispum) is extremely rich in apigenin, with a content of 4.5 g per 100 g [187]. The systemic bioavailability of apigenin is poor for two main reasons. First, a large portion of ingested apigenin is excreted unabsorbed [188], and, second, its poor availability is also due to its extensive metabolism in the liver (especially phase II), including conjugation reactions that lead to the formation of sulfated and glucuronidated apigenin [189]. Nevertheless, new nanoscience techniques can be used in order to improve its oral bioavailability. For example, the use of a formulation made of carbon nanopowder and apigenin has improved its availability by 275%, compared with unmodified apigenin [190].
Recently, apigenin has raised interest for its strong antioxidant and anti-inflammatory effects. Several in vivo and in vitro studies underline its antioxidant effects through radical scavenging action and its role in the upregulation of antioxidant defenses [191]. For example, apigenin was found to be a potent neuroprotective compound against microglial insult (at low micromolar concentrations) by inhibiting the transcriptional activation of iNOS (producing NO) and TFNα, IL-6 and COX-2 (IC50 = 5 µM) [10]. In addition, apigenin significantly decreased the presence of malondialdehyde and several other inflammatory markers, and increased the activity of superoxide dismutase and glutathione (GSH) peroxidase activity [192, 193]. Finally, apigenin has an important role in the upregulation of apoptosis genes. Zhang et al. analysed the expressions of several apoptosis genes in injured rats and found that the rats fed apigenin showed a significant decrease in the expression of some related apoptotic genes compared with the control group [193].
Apigenin is able to enter the brain by passing through the blood–brain barrier, reaching a concentration of 1.2 µM after daily intraperitoneal administration of apigenin 20 mg/kg for 1 week [191]. Furthermore, a variety of studies indicate the CNS effects of apigenin when delivered intraperitoneally or orally [194]. For example, 3 months’ treatment of apigenin (40 mg/kg) improved memory deficits in an amyloid-based transgenic mouse model of AD, the APP/PS1 mouse, when tested in the Morris water maze test [194]. Another study, using the Kunming mouse model for age-related decline, demonstrated a role of apigenin in preserving the function and integrity of the blood–brain barrier, which is usually destroyed under pathological conditions [195]. Furthermore, Chen et al. tested the effect of apigenin on aged rats that were administered isoflurane to induce memory impairment. Apigenin was administered at a single dose via intraperitoneal injection and, after treatment, Chen et al. examined the acetylation of histone H3 lysine 9 and H4 lysine 12 in the hippocampus and found that apigenin suppressed isoflurane-induced histone deacetylation [196]. In addition, they upregulated some proinflammatory cytokines, such as IL-2, IL-4 and IL-10, and all were found to be suppressed after apigenin treatment, restoring them to control conditions [196]. On the other hand, a preclinical study conducted on SH-SY5Y human neuroblastoma cells confirmed that apigenin has a central role in the protection of neuron loss, but, surprisingly, no antioxidant effects were detected. Therefore, this effect is likely due to a direct interaction of this compound with the caspase pathway [197]. Caspase-3 has a key role in apoptosis, and Kang et al. found that apigenin blocked caspase-3, which led the inhibition of apoptosis [197].
Only a few clinical studies on apigenin have been performed in humans in the area of inflammation, oxidative stress and neuroprotection. One clinical study, conducted in a randomised group of volunteers fed a diet high in parsley for 2 weeks, demonstrated an increase in the antioxidant enzymes superoxide dismutase and GSH reductase [198].
5.2.10 Other Polyphenolic Antioxidants
Many other food-derived polyphenolic antioxidants have been investigated. For example, the polyphenols from Oriental plums have been shown to improve cognitive function, lower cholesterol levels, and lower the diet-induced overexpression of BACE1, Aβ and 24-hydroxycholesterol in mice fed a high cholesterol diet [199]. Furthermore, polyphenol stilbenes, which can be found in grapes and berries, have demonstrated positive effects on Nrf2 in oxidative stress [200]. Certain flavonoids, such as scutellarin, daidzein, genistein, and fisetin, are thought to increase neurotrophic factor expression, whereas apigenin and ferulic acid increase CREB phosphorylation (for a review, see Moosavi et al. [201]). Cinnamon extracts and derivatives have also been shown to provide anti-inflammatory and cardioprotective actions [202].
The importance of olive oil in the traditional Mediterranean diet, a diet that epidemiological studies have indicated can lead to good health and longevity, has led to studies of the polyphenols in olive oil and its byproducts. For example, recent studies have found that oleuropein aglycone, or a mix of polyphenols obtained from olive mill waste water, can improve cognitive function in transgenic AD-model mice, as well as reduce Aβ1-42 levels and deposition in certain brain regions [203]. Other studies have shown similar neuroprotective effects, such as reduced oxidative stress, improved cell signalling and reduced Aβ1-42 aggregation, through diets high in olive oil or diets that include olive oil polyphenol supplements (for a review, see Casamenti and Stefani [204]). Further studies of olive oil and olive extracts are needed to show how valuable these products may be in preventing neurodegeneration.
5.2.11 Omega-3 (n-3) Essential Fatty Acids
Docosahexaenoic acid (DHA) is an essential omega-3 (n-3) PUFA. DHA is known to be an important n-3 PUFA in the brain, where it accounts for approximately 15% of fatty acids in grey matter [205]. There has been great interest in dietary DHA supplementation in AD, with the view of helping to protect from neuronal degeneration.
Epidemiological data suggest that low dietary intake of DHA is a candidate risk factor for AD [206], and DHA levels have been shown to be decreased in the AD brain [207]. Florent et al. showed that DHA provided cortical neurons with a higher level of resistance to the toxic effects of soluble Aβ oligomers in vitro [208]. The study by Calon et al. showed that reducing dietary n-3 PUFA in Tg2576 transgenic mice resulted in a loss of postsynaptic proteins and behavioural deficits, while a DHA-enriched diet prevented these effects [209]. Arsenault et al. [210] observed that a high intake of DHA for 8–10 months in 3xTg-AD mice resulted in neuroprotective effects and enhanced cognition compared with control 3xTg-AD mice consuming a diet containing no n-3 essential fatty acids. These researchers also demonstrated that acute exposure to DHA could not elicit the same results that were observed after chronic DHA treatment.
Additional studies have shown that DHA protects neurons from Aβ accumulation and toxicity, and decreases cognitive impairment in AD models [211]. Cole and Frautschy showed that DHA supplementation in 17-month-old Tg2576 transgenic mice markedly decreased Aβ accumulation, oxidative damage and cognitive deficits [212]. Feeding 3xTg-AD mice a high-fat diet with DHA for 4 months reduced phosphorylated JNK, insulin receptor substrate-1 (IRS-1) and tau phosphorylation in the brain, and was also accompanied by cognitive improvement in a Y-maze test [213]. Another study demonstrated that chronic treatment of a derivative of DHA (2-hydroxy DHA) in 5xFAD mice showed improved performance in a radial arm maze test, along with restoration of cell proliferation in the dentate gyrus, but no changes in the presence of Aβ plaques [214].
Several recent studies regarding the influence of apolipoprotein E (APOE) alleles on omega-3 fatty acids have been carried out. For example, APOE ε4 mice, compared with mice carrying other APOE alleles, suffered greater cognitive impairments and anxiety, as well as a greater omega-3 fatty acid depletion in organs and tissues, when fed a diet deficient in omega-3 fatty acids, yet these levels could be restored by switching to a diet rich in omega-3 fatty acids [215]. This suggests that long-term omega-3 fatty acid supplementation in middle-aged to elderly people should be encouraged, especially in APOE ε4 carriers. However, a study of macrophages from patients with mild cognitive impairment who have taken fish-derived omega-3 fatty acid supplements found that the expression of cytoprotective genes increased, whereas proapoptotic gene expression decreased. Aβ clearance by macrophage phagocytosis was improved, and the MMSE scores of the patients also improved, although these positive changes were only found in APOE ε3/ε3 patients. The other subgroup, APOE ε3/ε4 patients, had a high variability in responses [216]. This may reflect other vulnerabilities to AD pathogenesis caused by ε4 alleles that cannot be overcome with n-3 fatty acid supplements. In support of this, other differences seen in APOE ε4 carriers have been detected. For example, ε4 carriers who developed mild cognitive impairment/AD had high arachidonic acid/DHA ratios in blood phospholipids, compared with cognitively normal ε4 carriers and non-ε4 carriers [217]. There are also many studies that have shown APOE ε4 is less efficient than other APOE forms in other aspects of lipid metabolism, receptor binding and Aβ clearance [218], and these differences may also influence the potential benefits of DHA supplementation.
A recent placebo-controlled 3-year clinical trial of n-3 fatty acid supplementation (± a multidomain lifestyle intervention) did not identify any benefits from the treatments [219]; however, the cohort included people over 70 years of age who already had memory complaints, and it may be that intervention at this age (and at this stage of developing AD) is too late. A review of DHA supplementation with respect to AD pathogenesis stage supports this concept, indicating that high-dose DHA supplementation in APOE ε4 carriers before the onset of AD dementia may decrease the incidence of AD, but does not appear to have any benefits once dementia is established [220].
Other studies on the distribution of unsaturated fatty acids in AD brain have shown alterations in the metabolism of unsaturated fatty acids, indicating both global metabolic perturbations in AD and changes related to specific features of AD pathology [221]. Another mechanism whereby n-3 fatty acids may influence AD pathogenesis is by affecting IDE, a major Aβ-degrading enzyme in the brain. N-3 eicosapentaenoic acid (EPA) has been shown to increase IDE enzyme activity and gene expression [222]. DHA has also been shown to directly stimulate IDE enzyme activity, resulting in enhanced Aβ degradation in the extracellular space [222].
In summary, many studies indicate that dietary supplementation with DHA, or improving the diet to increase the n-3: n-6 fatty acid ratio, a ratio that would ideally be around 1: 4 (but Western diets typically cause this ratio to be from 1:8 to 1:20), may decrease Aβ accumulation, inflammation, and oxidative stress, and consequently reduce the risk of AD or slow its pathogenesis. However, studies suggest that such supplementation or dietary change needs to be lifelong, or at least started around mid-life, to be effective in reducing AD risk.
5.2.12 α-Lipoic Acid
Studies suggest that lipoic acid (LA) has a multitude of diverse pharmacologic and antioxidant properties [223]. The R-form (RLA) of LA is the naturally occurring stereoisomer, but commercially available preparations of LA often contain racemic mixtures of RLA and S-LA (SLA).
LA targets a variety of pathophysiological mechanisms relevant for AD.
-
1.
Increases neuronal acetylcholine levels (the neurotransmitter most deficient in AD brains) by activation of neuronal choline acetyltransferase (ChAT) [224].
-
2.
Chelates copper and iron and other transition metals, and subsequently inhibits the Fenton reaction, in which H2O2 is converted to hydroxyl radicals [225].
-
3.
Scavenges ROS (most importantly the highly toxic hydroxyl radical) [226].
-
4.
Neutralises and ameliorates the pathophysiological effects of reactive carbonyl compounds and AGEs, as well as lipid peroxidation products [227].
-
5.
Induces enzymes involved in GSH synthesis and other antioxidant protective enzymes, possibly through activation of the transcription factor Nrf-2 [175, 228].
-
6.
Leads to a higher activity of glyoxalase I (by increasing GSH, a cofactor of glyoxalase I), thereby decreasing the concentration of methylglyoxal. This is important for AD as glyoxalase I activity is reduced in AD brains [229, 230].
-
7.
Normalises levels of reduced GSH in AGE-challenged neuronal cells [231].
-
8.
Downregulates redox-sensitive inflammatory signals.
-
9.
Increases glucose uptake and utilisation [232].
The metal chelating abilities of LA are particularly important as Aβ binds to metal ions in the brain (copper, iron, and zinc), which induces the Aβ peptide to precipitate and form plaques. Since amyloid aggregates are non-covalently crosslinked by AGEs, as well as by metals (iron and copper) [233, 234], it was hypothesised that LA blocks aggregate formation or dissolves already-formed amyloid deposits. Indeed, Fonte et al. dissolved Aβ aggregates with metal ion chelators, and also demonstrated that LA enhanced the extraction of Aβ from mouse brains in a transgenic mouse model of familial AD [235].
Furthermore, the combination of Aβ with redox-active copper or iron ions might be involved in the production of H2O2 from molecular oxygen [236], which subsequently produces neurotoxic hydroxyl radicals by the Fenton or Haber–Weiss reactions. To test the effect of LA on metal levels in vitro, the effect of an RLA diet on brain iron levels and antioxidant status was investigated in aged rats [237]. Indeed, iron levels in the brain of LA-fed older animals were lower when compared with controls, and were similar to levels seen in younger rats [237].
As mentioned earlier, AD is characterised by chronic activation of microglia and astrocytes, particularly around senile plaques, producing free radicals and secreting proinflammatory cytokines [15, 27, 238]. ROS are involved in these proinflammatory signalling processes through a process known as ‘redox-sensitive signal transduction’, and are able to regulate the level of effector kinases that can influence the expression of various proinflammatory molecules such as IL-1β, IL-6, and iNOS [239]. LA targets these pathways through eliminating free radicals (which act as second messengers in these pathways), and thereby inhibits proinflammatory signal transduction pathways, overall attenuating the creation of more free radicals and more toxic cytokines [125].
Cell membranes contain unsaturated fatty acids such as linoleic acid and arachidonic acid, which can, through chemical and cell-mediated oxidation, be converted into lipid peroxidation products such as 4-hydroxynonenal (HNE) and acrolein [227]. These lipid peroxidation products not only crosslink Aβ and tau [240, 241] but also inhibit the activity of the mitochondrial enzymes pyruvate dehydrogenase (PDH) and α-ketoglutarate dehydrogenase (KGDH) through binding LA, a component of these two enzyme complexes. Increased levels of these lipid peroxidation products (found in AD brains) may therefore be partially responsible for the mitochondrial dysfunction and decreased ATP production observed in AD brains, which contribute to further neurodegeneration [242]. In several rodent models, dietary supplementation with LA resulted in a decrease in lipid peroxidation, and also prevented decay in LA-dependent mitochondria enzymes, reducing both the extent of oxidative damage and the proinflammatory state in the brain [243,244,245].
Recent studies have suggested new mechanisms whereby LA influences AD pathogenesis. For example, cell culture studies have found that an increase in BACE1 activity can diminish glucose oxidation by inhibiting key mitochondrial decarboxylation reactions, which in turn diminishes substrate delivery to the mitochondria. ALA (or β-hydroxybutyrate) was found to alleviate this effect of BACE1 [246]. Furthermore, studies of high-fat diet-fed rats showed that LA could reduce the diet-induced damage to neuronal insulin signalling and cognitive deficits in the rats [247]. Hippocampus expression levels of the vesicular glutamate transporter (VGlut1), required for the release of glutamate, were also reduced by the high-fat diet. LA was found to reverse this change, leading the authors to suggest LA might reduce the glutamatergic deficit seen in AD. Similarly, mice fed a high-fat diet developed insulin resistance, lower brain glucose uptake, lower levels of glucose transporters, changes to glucose metabolism, and, ultimately, synaptic loss. LA treatment was again found to prevent many of these metabolic changes and preserve synaptic plasticity [248]. This was attributed to the insulin-like effect of LA [248], which the researchers had previously demonstrated in AD-model transgenic mice, where LA was found to prevent the transgene-associated decrease in glucose uptake, to reduce the decrease in IRS activation and mediate greater downstream Akt signalling [249]. All of these positive effects of LA suggest LA supplements or foods high in LA (for example, broccoli, spinach, red meat, tomatoes) should be encouraged. Innovative drugs based on the effects of LA are being developed [250].
No randomised, placebo-controlled trials have tested LA for the prevention or treatment of AD, although two open-label studies reported that LA did benefit patients with AD [251, 252].
6 Future Directions
Future work is still required to further evaluate the value of these compounds and foods in clinical trials, as well as the efficacy of using combinations of these compounds for the prevention and treatment of AD. It might also be prudent to investigate plant-derived extracts in search of more potent and safer compounds, including those with traditional or ethnobotanical evidence [253,254,255,256]. Combination treatments may reduce cognitive deficits (memory and learning), oxidative stress, neuroinflammation, Aβ levels, Aβ plaque load, and tau hyperphosphorylation and aggregation, thereby providing an important insight into possible complementary medicine preventative treatment strategies for AD.
7 Conclusions
Due to the critical roles of amyloid, tau, inflammation, and oxidative stress in AD pathogenesis and progression, identifying therapeutic agents that target multiple pathological features is a rational approach that requires further exploration. Natural compounds and phytomedicines may represent such agents, as discussed in this review, as they have antioxidant, anti-inflammatory, anti-amyloidogenic and neuroprotective effects. A summary of their comparative in vitro potency in a standard anti-inflammatory test is shown in Table 1. Phytonutrients in spices, fruits and vegetables may represent the ideal candidates as they attenuate plaque and tangle formation; decrease neuroinflammation, oxidative and carbonyl stress; and are likely to be safe for long-term treatments at the presymptomatic/clinical stages of the disease (Fig. 1).
The authors believe that after having determined the effective brain concentration of the active compounds in a suitable rodent model, the pharmaceutical and complementary medicine industry should initiate sufficiently powered, long-term, double-blind, placebo-controlled clinical trials with an effective dose (using high bioavailability preparations) to further determine the therapeutic value of these agents in AD.
References
Association Alzheimer’s. 2016 Alzheimer’s disease facts and figures. Alzheimers Dement. 2016;12(4):459–509.
Retz W, Gsell W, Münch G, Rosler M, Riederer P. Free radicals in Alzheimer’s disease. J Neural Transm Suppl. 1998;54:221–36.
Butterfield DA, Griffin S, Münch G, Pasinetti GM. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J Alzheimers Dis. 2002;4(3):193–201.
Gotz J, Xia D, Leinenga G, Chew YL, Nicholas H. What renders TAU toxic. Front Neurol. 2013;4:72.
Münch G, Robinson SR. Potential neurotoxic inflammatory responses to Abeta vaccination in humans. J Neural Trans. 2002;109(7–8):1081–7.
Münch G, Robinson SR. Alzheimer’s vaccine: a cure as dangerous as the disease? J Neural Trans. 2002;109(4):537–9.
Durany N, Münch G, Michel T, Riederer P. Investigations on oxidative stress and therapeutical implications in dementia. Eur Arch Psychiatry Clin Neurosci. 1999;249(Suppl 3):68–73.
Lüth HJ, Münch G, Arendt T. Aberrant expression of NOS isoforms in Alzheimer’s disease is structurally related to nitrotyrosine formation. Brain Res. 2002;953(1–2):135–43.
von Bernhardi R, Ramirez G. Microglia-astrocyte interaction in Alzheimer’s disease: friends or foes for the nervous system? Biol Res. 2001;34(2):123–8.
Hansen E, Krautwald M, Maczurek AE, Stuchbury G, Fromm P, Steele M, et al. A versatile high throughput screening system for the simultaneous identification of anti-inflammatory and neuroprotective compounds. J Alzheimer’s Dis. 2010;19(2):451–64.
Münch G, Gasic-Milenkovic J, Dukic-Stefanovic S, Kuhla B, Heinrich K, Riederer P, et al. Microglial activation induces cell death, inhibits neurite outgrowth and causes neurite retraction of differentiated neuroblastoma cells. Exp Brain Res. 2003;150(1):1–8.
Gasic-Milenkovic J, Dukic-Stefanovic S, Deuther-Conrad W, Gartner U, Münch G. beta-Amyloid peptide potentiates inflammatory responses induced by lipopolysaccharide, interferon -gamma and ‘advanced glycation endproducts’ in a murine microglia cell line. Eur J Neurosci. 2003;17(4):813–21.
Francos-Quijorna I, Amo-Aparicio J, Martinez-Muriana A, Lopez-Vales R. IL-4 drives microglia and macrophages toward a phenotype conducive for tissue repair and functional recovery after spinal cord injury. Glia. 2016;64(12):2079–92.
Tremblay ME, Lecours C, Samson L, Sanchez-Zafra V, Sierra A. From the Cajal alumni Achucarro and Rio-Hortega to the rediscovery of never-resting microglia. Front Neuroanat. 2015;9:45.
Münch G, Thome J, Foley P, Schinzel R, Riederer P. Advanced glycation endproducts in ageing and Alzheimer’s disease. Brain Res Brain Res Rev. 1997;23(1–2):134–43.
Sierra A, Abiega O, Shahraz A, Neumann H. Janus-faced microglia: beneficial and detrimental consequences of microglial phagocytosis. Front Cell Neurosci. 2013;7:6.
Daria A, Colombo A, Llovera G, Hampel H, Willem M, Liesz A, et al. Young microglia restore amyloid plaque clearance of aged microglia. EMBO J. 2017;36(5):583–603.
Raj D, Yin Z, Breur M, Doorduin J, Holtman IR, Olah M, et al. Increased white matter inflammation in aging- and Alzheimer’s disease brain. Front Mol Neurosci. 2017;10:206.
Leyns CEG, Holtzman DM. Glial contributions to neurodegeneration in tauopathies. Mol Neurodegener. 2017;12(1):50.
Fuller S, Münch G, Steele M. Activated astrocytes: a therapeutic target in Alzheimer’s disease? Expert Rev Neurother. 2009;9(11):1585–94.
Liddelow SA, Guttenplan KA, Clarke LE, Bennett FC, Bohlen CJ, Schirmer L, et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature. 2017;541(7638):481–7.
Picklo MJ, Montine TJ, Amarnath V, Neely MD. Carbonyl toxicology and Alzheimer’s disease. Toxicol Appl Pharmacol. 2002;184(3):187–97.
Dizdaroglu M, Jaruga P, Birincioglu M, Rodriguez H. Free radical-induced damage to DNA: mechanisms and measurement. Free Radical Biol Med. 2002;32(11):1102–15.
Requena JR, Levine RL, Stadtman ER. Recent advances in the analysis of oxidized proteins. Amino Acids. 2003;25(3):221–6.
Guo L, Tian J, Du H. Mitochondrial dysfunction and synaptic transmission failure in Alzheimer’s disease. J Alzheimers Dis. 2017;57(4):1071–86.
Zhu X, Perry G, Moreira PI, Aliev G, Cash AD, Hirai K, et al. Mitochondrial abnormalities and oxidative imbalance in Alzheimer disease. J Alzheimers Dis. 2006;9(2):147–53.
Münch G, Schinzel R, Loske C, Wong A, Durany N, Li JJ, et al. Alzheimer’s disease: synergistic effects of glucose deficit, oxidative stress and advanced glycation endproducts. J Neural Trans. 1998;105(4–5):439–61.
Münch G, Schicktanz D, Behme A, Gerlach M, Riederer P, Palm D, et al. Amino acid specificity of glycation and protein-AGE crosslinking reactivities determined with a dipeptide SPOT library. Nat Biotechnol. 1999;17(10):1006–10.
Loske C, Neumann A, Cunningham AM, Nichol K, Schinzel R, Riederer P, et al. Cytotoxicity of advanced glycation endproducts is mediated by oxidative stress. J Neural Trans. 1998;105(8–9):1005–15.
Gasic-Milenkovic J, Loske C, Deuther-Conrad W, Münch G. Protein, “AGEing”–cytotoxicity of a glycated protein increases with its degree of AGE-modification. Z Gerontol Geriatr. 2001;34(6):457–60.
Abate G, Marziano M, Rungratanawanich W, Memo M, Uberti D. Nutrition and AGE-ing: focusing on Alzheimer’s disease. Oxidative Med Cell Longevity. 2017;2017:7039816.
Srikanth V, Maczurek A, Phan T, Steele M, Westcott B, Juskiw D, et al. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging. 2011;32(5):763–77.
Dukic-Stefanovic S, Gasic-Milenkovic J, Deuther-Conrad W, Münch G. Signal transduction pathways in mouse microglia N-11 cells activated by advanced glycation endproducts (AGEs). J Neurochem. 2003;87(1):2609–15.
Matrone C, Djelloul M, Taglialatela G, Perrone L. Inflammatory risk factors and pathologies promoting Alzheimer’s disease progression: is RAGE the key? Histol Histopathol. 2015;30(2):125–39.
Yan SS, Chen D, Yan S, Guo L, Du H, Chen JX. RAGE is a key cellular target for Abeta-induced perturbation in Alzheimer’s disease. Front Biosci. 2012;4:240–50.
Yasojima K, Schwab C, McGeer EG, McGeer PL. Up-regulated production and activation of the complement system in Alzheimer’s disease brain. Am J Pathol. 1999;154(3):927–36.
Rogers J, Lue L-F. Microglial chemotaxis, activation, and phagocytosis of amyloid β-peptide as linked phenomena in Alzheimer’s disease. Neurochem Int. 2001;39(5–6):333–40.
Fonseca MI, Ager RR, Woodruff TM, Chu S-H, Yazan O, Sanderson S, et al. Chronic treatment with C5a antagonist decreases pathology in two mouse models of Alzheimer’s disease. Alzheimer’s Dementia. 2008;4(4 Suppl 1):T188–T.
Fonseca MI, Ager RR, Chu SH, Yazan O, Sanderson SD, LaFerla FM, et al. Treatment with a C5aR antagonist decreases pathology and enhances behavioral performance in murine models of Alzheimer’s disease. J Immunol. 2009;183(2):1375–83.
Shen Y, Lue L, Yang L, Roher A, Kuo Y, Strohmeyer R, et al. Complement activation by neurofibrillary tangles in Alzheimer’s disease. Neurosci Lett. 2001;305(3):165–8.
Daborg J, Andreasson U, Pekna M, Lautner R, Hanse E, Minthon L, et al. Cerebrospinal fluid levels of complement proteins C3, C4 and CR1 in Alzheimer’s disease. J Neural Transm. 2012;119(7):789–97.
Hernandez MX, Namiranian P, Nguyen E, Fonseca MI, Tenner AJ. C5a increases the injury to primary neurons elicited by fibrillar amyloid beta. ASN Neuro. 2017;9(1):1759091416687871.
Roy C, Gupta A, Fisette A, Lapointe M, Poursharifi P, Richard D, et al. C5a receptor deficiency alters energy utilization and fat storage. PLoS One. 2013;8(5):e62531.
Snow WM, Albensi BC. Neuronal gene targets of NF-kappaB and their dysregulation in Alzheimer’s disease. Front Mol Neurosci. 2016;9:118.
Tanji K, Mori F, Imaizumi T, Yoshida H, Satoh K, Wakabayashi K. Interleukin-1 induces tau phosphorylation and morphological changes in cultured human astrocytes. NeuroReport. 2003;14(3):413–7.
Akiyama H, Arai T, Kondo H, Tanno E, Haga C, Ikeda K. Cell mediators of inflammation in the Alzheimer disease brain. Alzheimer Dis Assoc Disord. 2000;14(Suppl 1):S47–53.
Münch G, Apelt J, Rosemarie Kientsch E, Stahl P, Lüth HJ, Schliebs R. Advanced glycation endproducts and pro-inflammatory cytokines in transgenic Tg2576 mice with amyloid plaque pathology. J Neurochem. 2003;86(2):283–9.
Khandelwal PJ, Dumanis SB, Herman AM, Rebeck GW, Moussa CE. Wild type and P301L mutant Tau promote neuro-inflammation and alpha-Synuclein accumulation in lentiviral gene delivery models. Mol Cell Neurosci. 2012;49(1):44–53.
Srikanth V, Maczurek A, Phan T, Steele M et al. Advanced glycation endproducts and their receptor RAGE in Alzheimer’s disease. Neurobiol Aging. 2011;32(5):763–77. https://doi.org/10.1016/j.neurobiolaging.2009.04.016.
Dhananjayan K, Gunawardena D, Hearn N, Sonntag T, Moran C, Gyengesi E, et al. Activation of macrophages and microglia by interferon-gamma and lipopolysaccharide increases methylglyoxal production: a new mechanism in the development of vascular complications and cognitive decline in Type 2 diabetes mellitus? J Alzheimers Dis. 2017;59(2):467–79.
Gyengesi E, Liang H, Millington C, Sonego S, Sirijovski D, Gunawardena D, et al. investigation into the effects of tenilsetam on markers of neuroinflammation in GFAP-IL6 mice. Pharm Res. 2018;35(1):22.
Patel A, Rees SD, Kelly MA, Bain SC, Barnett AH, Prasher A, et al. Genetic variants conferring susceptibility to Alzheimer’s disease in the general population; do they also predispose to dementia in Down’s syndrome. BMC Res Notes. 2014;7(1):42.
Lambert JC, Ibrahim-Verbaas CA, Harold D, Naj AC, Sims R, Bellenguez C, et al. Meta-analysis of 74,046 individuals identifies 11 new susceptibility loci for Alzheimer’s disease. Nat Genet. 2013;45(12):1452–8.
Naj AC, Jun G, Beecham GW, Wang LS, Vardarajan BN, Buros J, et al. Common variants at MS4A4/MS4A6E, CD2AP, CD33 and EPHA1 are associated with late-onset Alzheimer’s disease. Nat Genet. 2011;43(5):436–41.
Block ML, Zecca L, Hong JS. Microglia-mediated neurotoxicity: uncovering the molecular mechanisms. Nat Rev Neurosci. 2007;8(1):57–69.
Millington C, Sonego S, Karunaweera N, Rangel A, Aldrich-Wright JR, Campbell IL, et al. Chronic neuroinflammation in Alzheimer’s disease: new perspectives on animal models and promising candidate drugs. Biomed Res Int. 2014;2014:309129.
Heneka MT, Golenbock DT, Latz E. Innate immunity in Alzheimer’s disease. Nat Immunol. 2015;16(3):229–36.
Butterfield DA, Griffin S, Münch G, Pasinetti GM. Amyloid beta-peptide and amyloid pathology are central to the oxidative stress and inflammatory cascades under which Alzheimer’s disease brain exists. J Alzheimer’s Dis. 2002;4(3):193–201.
McGeer PL, McGeer EG. The amyloid cascade-inflammatory hypothesis of Alzheimer disease: implications for therapy. Acta Neuropathol. 2013;126(4):479–97.
Sastre M, Klockgether T, Heneka MT. Contribution of inflammatory processes to Alzheimer’s disease: molecular mechanisms. Int J Dev Neurosci. 2006;24(2–3):167–76.
Chami L, Checler F. BACE1 is at the crossroad of a toxic vicious cycle involving cellular stress and beta-amyloid production in Alzheimer’s disease. Mol Neurodegener. 2012;5(7):52.
Chanthaphavong RS, Loughran PA, Lee TY, Scott MJ, Billiar TR. A role for cGMP in inducible nitric-oxide synthase (iNOS)-induced tumor necrosis factor (TNF) alpha-converting enzyme (TACE/ADAM17) activation, translocation, and TNF receptor 1 (TNFR1) shedding in hepatocytes. J Biol Chem. 2012;287(43):35887–98.
Xu J, Mukerjee S, Silva-Alves CR, Carvalho-Galvao A, Cruz JC, Balarini CM, et al. A disintegrin and metalloprotease 17 in the cardiovascular and central nervous systems. Front Physiol. 2016;7:469.
Batarseh YS, Duong QV, Mousa YM, Al Rihani SB, Elfakhri K, Kaddoumi A. Amyloid-beta and astrocytes interplay in amyloid-beta related disorders. Int J Mol Sci. 2016;17(3):338.
Calhoun A, King C, Khoury R, Grossberg GT. An evaluation of memantine ER + donepezil for the treatment of Alzheimer’s disease. Expert Opin Pharmacother. 2018;19(15):1711–7.
Birks JS, Harvey RJ. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2018;6:CD001190.
Davis BM, Mohs RC, Greenwald BS, Mathe AA, Johns CA, Horvath TB, et al. Clinical studies of the cholinergic deficit in Alzheimer’s disease. I. Neurochemical and neuroendocrine studies. J Am Geriatr Soc. 1985;33(11):741–8.
Birks JS, Harvey R. Donepezil for dementia due to Alzheimer’s disease. Cochrane Database Syst Rev. 2003(3):CD001190.
Doody RS, Cummings JL, Farlow MR. Reviewing the role of donepezil in the treatment of Alzheimer’s disease. Curr Alzheimer Res. 2012;9(7):773–81.
Birks J, Grimley Evans J, Iakovidou V, Tsolaki M, Holt FE. Rivastigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2009(2):CD001191.
Coelho F, Birks J. Physostigmine for Alzheimer’s disease. Cochrane Database Syst Rev. 2001(2):CD001499.
Danysz W, Parsons CG. The NMDA receptor antagonist memantine as a symptomatological and neuroprotective treatment for Alzheimer’s disease: preclinical evidence. Int J Geriatr Psychiatry. 2003;18(Suppl 1):S23–32.
Wang CH, Wang LS, Zhu N. Cholinesterase inhibitors and non-steroidal anti-inflammatory drugs as Alzheimer’s disease therapies: an updated umbrella review of systematic reviews and meta-analyses. Eur Rev Med Pharmacol Sci. 2016;20(22):4801–17.
Miguel-Alvarez M, Santos-Lozano A, Sanchis-Gomar F, Fiuza-Luces C, Pareja-Galeano H, Garatachea N, et al. Non-steroidal anti-inflammatory drugs as a treatment for Alzheimer’s disease: a systematic review and meta-analysis of treatment effect. Drugs Aging. 2015;32(2):139–47.
Jaturapatporn D, Isaac MG, McCleery J, Tabet N. Aspirin, steroidal and non-steroidal anti-inflammatory drugs for the treatment of Alzheimer’s disease. Cochrane Database Syst Rev. 2012;15(2):Cd006378.
Venigalla M, Gyengesi E, Münch G. Curcumin and Apigenin - novel and promising therapeutics against chronic neuroinflammation in Alzheimer’s disease. Neural Regen Res. 2015;10(8):1181–5.
Gunawardena D, Shanmugam K, Low M, Bennett L, Govindaraghavan S, Head R, et al. Determination of anti-inflammatory activities of standardised preparations of plant- and mushroom-based foods. Eur J Nutr. 2014;53(1):335–43. https://doi.org/10.1007/s00394-013-0531-9.
Gunawardena D, Karunaweera N, Lee S, van Der Kooy F, Harman DG, Raju R, et al. Anti-inflammatory activity of cinnamon (C. zeylanicum and C. cassia) extracts—identification of E-cinnamaldehyde and o-methoxy cinnamaldehyde as the most potent bioactive compounds. Food & function. 2015;6(3):910–9.
Chen YF, Wang YW, Huang WS, Lee MM, Wood WG, Leung YM, et al. Trans-Cinnamaldehyde, an essential oil in cinnamon powder, ameliorates cerebral ischemia-induced brain injury via inhibition of neuroinflammation through attenuation of iNOS, COX-2 expression and NFkappa-B signaling pathway. Neuromolecular Med. 2016;18(3):322–33. https://doi.org/10.1007/s12017-016-8395-9.
Peterson DW, George RC, Scaramozzino F, LaPointe NE, Anderson RA, Graves DJ, et al. Cinnamon extract inhibits tau aggregation associated with Alzheimer’s disease in vitro. J Alzheimers Dis. 2009;17(3):585–97.
George RC, Lew J, Graves DJ. Interaction of cinnamaldehyde and epicatechin with tau: implications of beneficial effects in modulating Alzheimer’s disease pathogenesis. J Alzheimers Dis. 2013;36(1):21–40.
Zhao J, Zhang X, Dong L, Wen Y, Zheng X, Zhang C, et al. Cinnamaldehyde inhibits inflammation and brain damage in a mouse model of permanent cerebral ischaemia. Br J Pharmacol. 2015;172(20):5009–23.
Zhao H, Zhang M, Zhou F, Cao W, Bi L, Xie Y, et al. Cinnamaldehyde ameliorates LPS-induced cardiac dysfunction via TLR4-NOX4 pathway: the regulation of autophagy and ROS production. J Mol Cell Cardiol. 2016;101:11–24.
Khare P, Jagtap S, Jain Y, Baboota RK, Mangal P, Boparai RK, et al. Cinnamaldehyde supplementation prevents fasting-induced hyperphagia, lipid accumulation, and inflammation in high-fat diet-fed mice. BioFactors (Oxford, England). 2016;42(2):201–11.
Zhang L, Zhang Z, Fu Y, Yang P, Qin Z, Chen Y, et al. Trans-cinnamaldehyde improves memory impairment by blocking microglial activation through the destabilization of iNOS mRNA in mice challenged with lipopolysaccharide. Neuropharmacology. 2016;110(Pt A):503–18.
Momtaz S, Hassani S, Khan F, Ziaee M, Abdollahi M. Cinnamon, a promising prospect towards Alzheimer’s disease. Pharmacol Res. 2018;130:241–58.
Mandel S, Weinreb O, Amit T, Youdim MB. Cell signaling pathways in the neuroprotective actions of the green tea polyphenol (-)-epigallocatechin-3-gallate: implications for neurodegenerative diseases. J Neurochem. 2004;88(6):1555–69.
Bhullar KS, Rupasinghe HP. Polyphenols: multipotent therapeutic agents in neurodegenerative diseases. Oxid Med Cell Longevity. 2013;2013:891748.
Levites Y, Amit T, Mandel S, Youdim MB. Neuroprotection and neurorescue against Abeta toxicity and PKC-dependent release of nonamyloidogenic soluble precursor protein by green tea polyphenol (-)-epigallocatechin-3-gallate. Faseb J. 2003;17(8):952–4.
Rezai-Zadeh K, Shytle D, Sun N, Mori T, Hou H, Jeanniton D, et al. Green tea epigallocatechin-3-gallate (EGCG) modulates amyloid precursor protein cleavage and reduces cerebral amyloidosis in Alzheimer transgenic mice. J Neurosci. 2005;25(38):8807–14.
Okello EJ, Leylabi R, McDougall GJ. Inhibition of acetylcholinesterase by green and white tea and their simulated intestinal metabolites. Food Funct. 2012;3(6):651–61.
Ali B, Jamal QM, Shams S, Al-Wabel NA, Siddiqui MU, Alzohairy MA, et al. In silico analysis of green tea polyphenols as inhibitors of AChE and BChE enzymes in Alzheimer’s disease treatment. CNS Neurol Disord: Drug Targets. 2016;15(5):624–8.
Cong L, Cao C, Cheng Y, Qin XY. Green tea polyphenols attenuated glutamate excitotoxicity via antioxidative and antiapoptotic pathway in the primary cultured cortical neurons. Oxidative Med Cell Longevity. 2016;2016:2050435.
Qian G, Xue K, Tang L, Wang F, Song X, Chyu MC, et al. Mitigation of oxidative damage by green tea polyphenols and Tai Chi exercise in postmenopausal women with osteopenia. PLoS One. 2012;7(10):e48090.
Zhang Z, Wu H, Huang H. Epicatechin plus treadmill exercise are neuroprotective against moderate-stage amyloid precursor protein/presenilin 1 mice. Pharm Magaz. 2016;12(Suppl 2):S139–46.
Lee YJ, Choi DY, Yun YP, Han SB, Oh KW, Hong JT. Epigallocatechin-3-gallate prevents systemic inflammation-induced memory deficiency and amyloidogenesis via its anti-neuroinflammatory properties. J Nutr Biochem. 2013;24(1):298–310.
Cheng-Chung Wei J, Huang HC, Chen WJ, Huang CN, Peng CH, Lin CL. Epigallocatechin gallate attenuates amyloid beta-induced inflammation and neurotoxicity in EOC 13.31 microglia. Eur J Pharmacol. 2016;770:16–24.
Cascella M, Bimonte S, Muzio MR, Schiavone V, Cuomo A. The efficacy of Epigallocatechin-3-gallate (green tea) in the treatment of Alzheimer’s disease: an overview of pre-clinical studies and translational perspectives in clinical practice. Infect Agents Cancer. 2017;12:36.
Rege SD, Geetha T, Broderick TL, Babu JR. Can Diet and Physical Activity Limit Alzheimer’s Disease Risk? Curr Alzheimer Res. 2017;14(1):76–93.
Ringman JM, Frautschy SA, Cole GM, Masterman DL, Cummings JL. A potential role of the curry spice curcumin in Alzheimer’s disease. Curr Alzheimer Res. 2005;2(2):131–6.
Kirby L, Lehmann P, Majeed A. Dementia in people aged 65 years and older: a growing problem? Population trends. 1998;(92):23–8.
Sreejayan N, Rao MN. Free radical scavenging activity of curcuminoids. Arzneimittelforschung. 1996;46(2):169–71.
Pan MH, Lin-Shiau SY, Lin JK. Comparative studies on the suppression of nitric oxide synthase by curcumin and its hydrogenated metabolites through down-regulation of IkappaB kinase and NFkappaB activation in macrophages. Biochem Pharmacol. 2000;60(11):1665–76.
Zhao BL, Li XJ, He RG, Cheng SJ, Xin WJ. Scavenging effect of extracts of green tea and natural antioxidants on active oxygen radicals. Cell Biophys. 1989;14(2):175–85.
Yang F, Lim GP, Begum AN, Ubeda OJ, Simmons MR, Ambegaokar SS, et al. Curcumin inhibits formation of amyloid beta oligomers and fibrils, binds plaques, and reduces amyloid in vivo. J Biol Chem. 2005;280(7):5892–901.
Bharti AC, Donato N, Aggarwal BB. Curcumin (diferuloylmethane) inhibits constitutive and IL-6-inducible STAT3 phosphorylation in human multiple myeloma cells. J Immunol. 2003;171(7):3863–71.
Ray B, Lahiri DK. Neuroinflammation in Alzheimer’s disease: different molecular targets and potential therapeutic agents including curcumin. Curr Opin Pharmacol. 2009;9(4):434–44.
Liu H, Li Z, Qiu D, Gu Q, Lei Q, Mao L. The inhibitory effects of different curcuminoids on beta-amyloid protein, beta-amyloid precursor protein and beta-site amyloid precursor protein cleaving enzyme 1 in swAPP HEK293 cells. Neurosci Lett. 2010;485(2):83–8.
Belviranli M, Okudan N, Atalik KE, Oz M. Curcumin improves spatial memory and decreases oxidative damage in aged female rats. Biogerontology. 2013;14(2):187–96.
Begum AN, Jones MR, Lim GP, Morihara T, Kim P, Heath DD, et al. Curcumin structure-function, bioavailability, and efficacy in models of neuroinflammation and Alzheimer’s disease. J Pharmacol Exp Ther. 2008;326(1):196–208.
Cox KH, Pipingas A, Scholey AB. Investigation of the effects of solid lipid curcumin on cognition and mood in a healthy older population. J Psychopharmacol. 2015;29(5):642–51.
Maiti P, Paladugu L, Dunbar GL. Solid lipid curcumin particles provide greater anti-amyloid, anti-inflammatory and neuroprotective effects than curcumin in the 5xFAD mouse model of Alzheimer’s disease. BMC Neurosci. 2018;19(1):7.
Zheng K, Dai X, Xiao N, Wu X, Wei Z, Fang W, et al. Curcumin ameliorates memory decline via inhibiting BACE1 expression and beta-amyloid pathology in 5xFAD transgenic mice. Mol Neurobiol. 2017;54(3):1967–77.
Maiti P, Hall TC, Paladugu L, Kolli N, Learman C, Rossignol J, et al. A comparative study of dietary curcumin, nanocurcumin, and other classical amyloid-binding dyes for labeling and imaging of amyloid plaques in brain tissue of 5x-familial Alzheimer’s disease mice. Histochem Cell Biol. 2016;146(5):609–25.
Heger M. Drug screening: don’t discount all curcumin trial data. Nature. 2017;543(7643):40.
Goozee KG, Shah TM, Sohrabi HR, Rainey-Smith SR, Brown B, Verdile G, et al. Examining the potential clinical value of curcumin in the prevention and diagnosis of Alzheimer’s disease. Br J Nutr. 2016;115(3):449–65.
Rainey-Smith SR, Brown BM, Sohrabi HR, Shah T, Goozee KG, Gupta VB, et al. Curcumin and cognition: a randomised, placebo-controlled, double-blind study of community-dwelling older adults. Br J Nutr. 2016;115(12):2106–13.
Brondino N, Re S, Boldrini A, Cuccomarino A, Lanati N, Barale F, et al. Curcumin as a therapeutic agent in dementia: a mini systematic review of human studies. Sci World J. 2014;2014:174282.
Purpura M, Lowery RP, Wilson JM, Mannan H, Münch G, Razmovski-Naumovski V. Analysis of different innovative formulations of curcumin for improved relative oral bioavailability in human subjects. Eur J Nutr. 2018;57(3):929–938. https://doi.org/10.1007/s00394-016-1376-9.
Ullah F, Liang A, Rangel A, Gyengesi E, Niedermayer G, Münch G. High bioavailability curcumin: an anti-inflammatory and neurosupportive bioactive nutrient for neurodegenerative diseases characterized by chronic neuroinflammation. Arch Toxicol. 2017;91(4):1623–34.
Venigalla M, Sonego S, Gyengesi E, Sharman MJ, Münch G. Novel promising therapeutics against chronic neuroinflammation and neurodegeneration in Alzheimer’s disease. Neurochem Int. 2016;95:63–74.
Purpura M, Lowery RP, Wilson JM, Mannan H, Münch G, Razmovski-Naumovski V. Analysis of different innovative formulations of curcumin for improved relative oral bioavailability in human subjects. Eur J Nutr. 2018;57(3):929–38.
Chan P-C, Xia Q, Fu PP. Ginkgo biloba leave extract: biological, medicinal, and toxicological effects. J Environ Sci Health Part C Environ Carcinogen Ecotoxicol Rev. 2007;25(3):211–44.
Gargouri B, Carstensen J, Bhatia HS, Huell M, Dietz GPH, Fiebich BL. Anti-neuroinflammatory effects of Ginkgo biloba extract EGb761 in LPS-activated primary microglial cells. Phytomedicine. 2018;15(44):45–55.
Wong A, Dukic-Stefanovic S, Gasic-Milenkovic J, Schinzel R, Wiesinger H, Riederer P, et al. Anti-inflammatory antioxidants attenuate the expression of inducible nitric oxide synthase mediated by advanced glycation endproducts in murine microglia. Eur J Neurosci. 2001;14(12):1961–7.
Shi C, Zhao L, Zhu B, Li Q, Yew DT, Yao Z, et al. Protective effects of Ginkgo biloba extract (EGb761) and its constituents quercetin and ginkgolide B against beta-amyloid peptide-induced toxicity in SH-SY5Y cells. Chem Biol Interact. 2009;181(1):115–23.
Liu X, Hao W, Qin Y, Decker Y, Wang X, Burkart M, et al. Long-term treatment with Ginkgo biloba extract EGb 761 improves symptoms and pathology in a transgenic mouse model of Alzheimer’s disease. Brain Behav Immun. 2015;46:121–31. https://doi.org/10.1016/j.bbi.2015.01.011.
Kennedy DO, Jackson PA, Haskell CF, Scholey AB. Modulation of cognitive performance following single doses of 120 mg Ginkgo biloba extract administered to healthy young volunteers. Hum Psychopharmacol Clin Exp. 2007;22(8):559–66.
Kanowski S, Herrmann WM, Stephan K, Wierich W, Horr R. Proof of efficacy of the ginkgo biloba special extract EGb 761 in outpatients suffering from mild to moderate primary degenerative dementia of the Alzheimer type or multi-infarct dementia. Pharmacopsychiatry. 1996;29(2):47–56.
Ihl R, Tribanek M, Bachinskaya N, Group GS. Efficacy and tolerability of a once daily formulation of Ginkgo biloba extract EGb 761(R) in Alzheimer’s disease and vascular dementia: results from a randomised controlled trial. Pharmacopsychiatry. 2012;45(2):41–6.
Gauthier S, Schlaefke S. Efficacy and tolerability of Ginkgo biloba extract EGb 761(R) in dementia: a systematic review and meta-analysis of randomized placebo-controlled trials. Clin Interv Aging. 2014;9:2065–77.
Tan MS, Yu JT, Tan CC, Wang HF, Meng XF, Wang C, et al. Efficacy and adverse effects of ginkgo biloba for cognitive impairment and dementia: a systematic review and meta-analysis. J Alzheimers Dis. 2015;43(2):589–603.
Yang G, Wang Y, Sun J, Zhang K, Liu J. Ginkgo biloba for mild cognitive impairment and alzheimer’s disease: a systematic review and meta-analysis of randomized controlled trials. Curr Top Med Chem. 2016;16(5):520–8.
Ong WY, Farooqui T, Koh HL, Farooqui AA, Ling EA. Protective effects of ginseng on neurological disorders. Front Aging Neurosci. 2015;7:129.
Yang L, Hao J, Zhang J, Xia W, Dong X, Hu X, et al. Ginsenoside Rg3 promotes beta-amyloid peptide degradation by enhancing gene expression of neprilysin. J Pharm Pharmacol. 2009;61(3):375–80.
Wang Y, Feng Y, Fu Q, Li L. Panax notoginsenoside Rb1 ameliorates Alzheimer’s disease by upregulating brain-derived neurotrophic factor and downregulating Tau protein expression. Exp Ther Med. 2013;6(3):826–30.
Radad K, Gille G, Liu L, Rausch W-D. Use of ginseng in medicine with emphasis on neurodegenerative disorders. J Pharmacol Sci. 2006;100(3):175–86.
Kim HJ, Jung SW, Kim SY, Cho IH, Kim HC, Rhim H, et al. Panax ginseng as an adjuvant treatment for Alzheimer’s disease. J Ginseng Res. 2018;42(4):401–11.
Kim M, Choi SY, Kim KT, Rhee YK, Hur JY. Ginsenoside Rg18 suppresses lipopolysaccharide-induced neuroinflammation in BV2 microglia and amyloid-β-induced oxidative stress in SH-SY5Y neurons via nuclear factor erythroid 2-related factor 2/heme oxygenase-1 induction. J Funct Foods. 2017;31:71–8.
Kim SH, Shim SH, Choi DS, Kim JH, Kwon YB, Kwon JK. Modulation of LPS-stimulated astroglial activation by ginseng total saponins. J Ginseng Res. 2011;35(1):80–5.
Lee JS, Song JH, Sohn NW, Shin JW. Inhibitory effects of ginsenoside Rb1 on neuroinflammation following systemic lipopolysaccharide treatment in mice. Phytother Res. 2013;27(9):1270–6.
Shin JW, Ma SH, Lee JW, Kim DK, Do K, Sohn NW. Ginsenoside Rg1 attenuates neuroinflammation following systemic lipopolysaccharide treatment in mice. Korea J Herbol. 2013;28(6):145–53.
Kennedy DO, Scholey AB, Drewery L, Marsh VR, Moore B, Ashton H. Electroencephalograph effects of single doses of Ginkgo biloba and Panax ginseng in healthy young volunteers. Pharmacol Biochem Behav. 2003;75(3):701–9.
Reay JL, Kennedy DO, Scholey AB. Effects of Panax ginseng, consumed with and without glucose, on blood glucose levels and cognitive performance during sustained ‘mentally demanding’ tasks. J Psychopharmacol (Oxford, England). 2006;20(6):771–81.
Heo JC, Woo SU, Kweon MA, Park JY, Lee HK, Son M, et al. Aqueous extract of the Helianthus annuus seed alleviates asthmatic symptoms in vivo. Int J Mol Med. 2008;21(1):57–61.
Heo JH, Lee ST, Oh MJ, Park HJ, Shim JY, Chu K, et al. Improvement of cognitive deficit in Alzheimer’s disease patients by long term treatment with korean red ginseng. J Ginseng Res. 2011;35(4):457–61.
Kennedy DO, Scholey AB, Wesnes KA. Differential, dose dependent changes in cognitive performance following acute administration of a Ginkgo biloba/Panax ginseng combination to healthy young volunteers. Nutr Neurosci. 2001;4(5):399–412.
Wesnes KA, Ward T, McGinty A, Petrini O. The memory enhancing effects of a Ginkgo biloba/Panax ginseng combination in healthy middle-aged volunteers. Psychopharmacology. 2000;152(4):353–61.
Yakoot M, Salem A, Helmy S. Effect of Memo®, a natural formula combination, on Mini-Mental State Examination scores in patients with mild cognitive impairment. Clin Interv Aging. 2013;8:975–81.
Liu YM, Li ZY, Hu H, Xu SP, Chang Q, Liao YH, et al. Tenuifolin, a secondary saponin from hydrolysates of polygalasaponins, counteracts the neurotoxicity induced by Abeta25-35 peptides in vitro and in vivo. Pharmacol Biochem Behav. 2015;128:14–22.
Lv J, Jia H, Jiang Y, Ruan Y, Liu Z, Yue W, et al. Tenuifolin, an extract derived from tenuigenin, inhibits amyloid-beta secretion in vitro. Acta Physiol (Oxf). 2009;196(4):419–25.
Kim HM, Lee EH, Na HJ, Lee SB, Shin TY, Lyu YS, et al. Effect of Polygala tenuifolia root extract on the tumor necrosis factor-alpha secretion from mouse astrocytes. J Ethnopharmacol. 1998;61(3):201–8.
Cheong MH, Lee SR, Yoo HS, Jeong JW, Kim GY, Kim WJ, et al. Anti-inflammatory effects of Polygala tenuifolia root through inhibition of NF-kappaB activation in lipopolysaccharide-induced BV2 microglial cells. J Ethnopharmacol. 2011;137(3):1402–8.
Xue W, Hu JF, Yuan YH, Sun JD, Li BY, Zhang DM, et al. Polygalasaponin XXXII from Polygala tenuifolia root improves hippocampal-dependent learning and memory. Acta Pharmacol Sin. 2009;30(9):1211–9.
Jesky R, Chen H. The neuritogenic and neuroprotective potential of senegenin against Abeta-induced neurotoxicity in PC 12 cells. BMC Complement Altern Med. 2016;23(16):26.
Shin KY, Lee JY, Won BY, Jung HY, Chang KA, Koppula S, et al. BT-11 is effective for enhancing cognitive functions in the elderly humans. Neurosci Lett. 2009;465(2):157–9.
Lee JY, Kim KY, Shin KY, Won BY, Jung HY, Suh YH. Effects of BT-11 on memory in healthy humans. Neurosci Lett. 2009;454(2):111–4.
Maruyama M, Tomita N, Iwasaki K, Ootsuki M, Matsui T, Nemoto M, et al. Benefits of combining donepezil plus traditional Japanese herbal medicine on cognition and brain perfusion in Alzheimer’s disease: a 12-week observer-blind, donepezil monotherapy controlled trial. J Am Geriatr Soc. 2006;54(5):869–71.
Grotenhermen F. Pharmacokinetics and pharmacodynamics of cannabinoids. Clin Pharmacokinet. 2003;42(4):327–60.
Pertwee RG. The diverse CB1 and CB2 receptor pharmacology of three plant cannabinoids: delta9-tetrahydrocannabinol, cannabidiol and delta9-tetrahydrocannabivarin. Br J Pharmacol. 2008;153(2):199–215.
Karl T, Garner B, Cheng D. The therapeutic potential of the phytocannabinoid cannabidiol for Alzheimer’s disease. Behav Pharmacol. 2017;28(2 and 3-Spec Issue):142–60.
Esposito G, Scuderi C, Savani C, Steardo L Jr, De Filippis D, Cottone P, et al. Cannabidiol in vivo blunts beta-amyloid induced neuroinflammation by suppressing IL-1beta and iNOS expression. Br J Pharmacol. 2007;151(8):1272–9.
Martin-Moreno AM, Reigada D, Ramirez BG, Mechoulam R, Innamorato N, Cuadrado A, et al. Cannabidiol and other cannabinoids reduce microglial activation in vitro and in vivo: relevance to Alzheimer’s disease. Mol Pharmacol. 2011;79(6):964–73.
Watt G, Karl T. In vivo Evidence for Therapeutic Properties of Cannabidiol (CBD) for Alzheimer’s Disease. Front Pharmacol. 2017;8:20.
Cheng D, Low JK, Logge W, Garner B, Karl T. Chronic cannabidiol treatment improves social and object recognition in double transgenic APPswe/PS1E9 mice. Psychopharmacology. 2014;231(15):3009–17.
Cheng D, Spiro AS, Jenner AM, Garner B, Karl T. Long-term cannabidiol treatment prevents the development of social recognition memory deficits in Alzheimer’s disease transgenic mice. J Alzheimer’s Dis. 2014;42(4):1383–96.
Casarejos MJ, Perucho J, Gomez A, Munoz MP, Fernandez-Estevez M, Sagredo O, et al. Natural cannabinoids improve dopamine neurotransmission and tau and amyloid pathology in a mouse model of tauopathy. J Alzheimers Dis. 2013;35(3):525–39.
Aso E, Sanchez-Pla A, Vegas-Lozano E, Maldonado R, Ferrer I. Cannabis-based medicine reduces multiple pathological processes in AbetaPP/PS1 mice. J Alzheimers Dis. 2015;43(3):977–91.
Shelef A, Barak Y, Berger U, Paleacu D, Tadger S, Plopsky I, et al. Safety and efficacy of medical cannabis oil for behavioral and psychological symptoms of dementia: an-open label, add-on. Pilot Study. J Alzheimers Dis. 2016;51(1):15–9.
Arntzen KA, Schirmer H, Wilsgaard T, Mathiesen EB. Moderate wine consumption is associated with better cognitive test results: a 7 year follow up of 5033 subjects in the Tromsø Study. Acta Neurol Scand Suppl. 2010;190:23–9.
Mendes D, Oliveira MM, Moreira PI, Coutinho J, Nunes FM, Pereira DM, et al. Beneficial effects of white wine polyphenols-enriched diet on Alzheimer’s disease-like pathology. J Nutr Biochem. 2018;55:165–77.
Truelsen T, Thudium D, Grønbaek M, Copenhagen City Heart S. Amount and type of alcohol and risk of dementia: the Copenhagen City Heart Study. Neurology. 2002;59(9):1313–9.
Kulkarni SS, Canto C. The molecular targets of resveratrol. Biochim Biophys Acta. 2015;1852(6):1114–23.
Nguyen NT, Ooi L, Piller SC, Münch G. Proenergetic effects of resveratrol in the murine neuronal cell line Neuro2a. Mol Nutr Food Res. 2013;57(11):1901–7.
Steele ML, Fuller S, Patel M, Kersaitis C, Ooi L, Münch G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol. 2013;1(1):441–5.
Steele ML, Fuller S, Patel M, Kersaitis C, Ooi L, Münch G. Effect of Nrf2 activators on release of glutathione, cysteinylglycine and homocysteine by human U373 astroglial cells. Redox Biol. 2013;1:441–5.
Steiner N, Balez R, Karunaweera N, Lind JM, Münch G, Ooi L. Neuroprotection of Neuro2a cells and the cytokine suppressive and anti-inflammatory mode of action of resveratrol in activated RAW264.7 macrophages and C8-B4 microglia. Neurochem Int. 2016;95:46–54. https://doi.org/10.1016/j.neuint.2015.10.013.
Corpas R, Grinan-Ferre C, Rodriguez-Farre E, Pallas M, Sanfeliu C. Resveratrol Induces Brain Resilience Against Alzheimer Neurodegeneration Through Proteostasis Enhancement. Mol Neurobiol. 2018.
Jia Y, Wang N, Liu X. Resveratrol and Amyloid-Beta: Mechanistic Insights. Nutrients. 2017. https://doi.org/10.3390/nu9101122.
Ge JF, Qiao JP, Qi CC, Wang CW, Zhou JN. The binding of resveratrol to monomer and fibril amyloid beta. Neurochem Int. 2012;61(7):1192–201.
Granzotto A, Zatta P. Resveratrol acts not through anti-aggregative pathways but mainly via its scavenging properties against Abeta and Abeta-metal complexes toxicity. PLoS One. 2011;6(6):e21565.
Drygalski K, Fereniec E, Korycinski K, Chomentowski A, Kielczewska A, Odrzygozdz C, et al. Resveratrol and Alzheimer’s disease. From molecular pathophysiology to clinical trials. Exp Gerontol. 2018;113:36–47.
Jayasena T, Poljak A, Smythe G, Braidy N, Münch G, Sachdev P. The role of polyphenols in the modulation of sirtuins and other pathways involved in Alzheimer’s disease. Ageing Res Rev. 2013;12(4):867–83. https://doi.org/10.1016/j.arr.2013.06.003.
Kumar R, Chaterjee P, Sharma PK, Singh AK, Gupta A, Gill K, et al. Sirtuin1: a promising serum protein marker for early detection of Alzheimer’s disease. PLoS One. 2013;8(4):e61560.
Koushki M, Dashatan NA, Meshkani R. Effect of resveratrol supplementation on inflammatory markers: a systematic review and meta-analysis of randomized controlled trials. Clin Ther. 2018;40(7):1180-92.e5.
Marx W, Kelly JT, Marshall S, Cutajar J, Annois B, Pipingas A, et al. Effect of resveratrol supplementation on cognitive performance and mood in adults: a systematic literature review and meta-analysis of randomized controlled trials. Nutr Rev. 2018;76(6):432–43.
Seema Bhagwat DBH, Joanne MH. USDA Database for the Flavonoid Content Content of Selected Foods. US Department of Agriculture Agricultural Research Service. 2013;15(12):1348–62.
Tang D, Chen K, Huang L, Li J. Pharmacokinetic properties and drug interactions of apigenin, a natural flavone. Expert Opin Drug Metabol Toxicol. 2017;13(3):323–30.
Gradolatto A, Basly J-P, Berges R, Teyssier C, Chagnon M-C, Siess M-H, et al. Pharmacokinetics and metabolism of apigenin in female and male rats after a single oral administration. Drug Metab Dispos. 2005;33(1):49–54.
Ding SM, Zhang ZH, Song J, Cheng XD, Jiang J, Jia XB. Enhanced bioavailability of apigenin via preparation of a carbon nanopowder solid dispersion. Int J Nanomed. 2014;9(1):2327–33.
Miroljub Popovic’ MC-BOB-G, Julián C. Short Report The flavonoid apigenin delays forgetting of passive avoidance conditioning in rats. J Biol Chem. 2014;264(11):6009–12.
Liang YC, Huang YT, Tsai SH, Lin-Shiau SY, Chen CF, Lin JK. Suppression of inducible cyclooxygenase and inducible nitric oxide synthase by apigenin and related flavonoids in mouse macrophages. Carcinogenesis. 1999;20(10):1945–52.
Zhang F, Li F, Chen G. Neuroprotective effect of apigenin in rats after contusive spinal cord injury. Neurol Sci. 2014;35(4):583–8.
Zhao L, Wang JL, Liu R, Li XX, Li JF, Zhang L. Neuroprotective, anti-amyloidogenic and neurotrophic effects of apigenin in an Alzheimer’s disease mouse model. Molecules. 2013;18(8):9949–65.
Liu R, Zhang T, Yang H, Lan X, Ying J, Du G. The flavonoid apigenin protects brain neurovascular coupling against amyloid-β25-35-induced toxicity in mice. J Alzheimer’s Dis. 2011;24(1):85–100.
Chen L, Xie W, Xie W, Zhuang W, Jiang C, Liu N. Apigenin attenuates isoflurane-induced cognitive dysfunction via epigenetic regulation and neuroinflammation in aged rats. Arch Gerontol Geriatr. 2017;73:29–36. https://doi.org/10.1016/j.archger.2017.07.004.
Kang SS, Lee JY, Choi YK, Kim GS, Han BH. Neuroprotective effects of flavones on hydrogen peroxide-induced apoptosis in SH-SY5Y neuroblostoma cells. Bioorg Med Chem Lett. 2004;14(9):2261–4.
Nielsen SE, Young JF, Daneshvar B, Lauridsen ST, Knuthsen P, Sandström B, et al. Effect of parsley (Petroselinum crispum) intake on urinary apigenin excretion, blood antioxidant enzymes and biomarkers for oxidative stress in human subjects. Br J Nutr. 1999;81(6):447–55.
Kuo PH, Lin CI, Chen YH, Chiu WC, Lin SH. A high-cholesterol diet enriched with polyphenols from Oriental plums (Prunus salicina) improves cognitive function and lowers brain cholesterol levels and neurodegenerative-related protein expression in mice. Br J Nutr. 2015;113(10):1550–7.
Reinisalo M, Karlund A, Koskela A, Kaarniranta K, Karjalainen RO. Polyphenol stilbenes: molecular mechanisms of defence against oxidative stress and aging-related diseases. Oxidative Med Cell Longevity. 2015;2015:340520.
Moosavi F, Hosseini R, Saso L, Firuzi O. Modulation of neurotrophic signaling pathways by polyphenols. Drug Design Dev Therapy. 2016;10:23–42.
Hariri M, Ghiasvand R. Cinnamon and chronic diseases. Adv Exp Med Biol. 2016;929:1–24.
Pantano D, Luccarini I, Nardiello P, Servili M, Stefani M, Casamenti F. Oleuropein aglycone and polyphenols from olive mill waste water ameliorate cognitive deficits and neuropathology. Br J Clin Pharmacol. 2017;83(1):54–62.
Casamenti F, Stefani M. Olive polyphenols: new promising agents to combat aging-associated neurodegeneration. Expert Rev Neurother. 2017;17(4):345–58.
Salem N Jr, Litman B, Kim HY, Gawrisch K. Mechanisms of action of docosahexaenoic acid in the nervous system. Lipids. 2001;36(9):945–59.
Calon F, Lim GP, Morihara T, Yang F, Ubeda O, Salem N Jr, et al. Dietary n-3 polyunsaturated fatty acid depletion activates caspases and decreases NMDA receptors in the brain of a transgenic mouse model of Alzheimer’s disease. Eur J Neurosci. 2005;22(3):617–26.
Prasad MR, Lovell MA, Yatin M, Dhillon H, Markesbery WR. Regional membrane phospholipid alterations in Alzheimer’s disease. Neurochem Res. 1998;23(1):81–8.
Florent S, Malaplate-Armand C, Youssef I, Kriem B, Koziel V, Escanye MC, et al. Docosahexaenoic acid prevents neuronal apoptosis induced by soluble amyloid-beta oligomers. J Neurochem. 2006;96(2):385–95.
Calon F, Lim GP, Yang F, Morihara T, Teter B, Ubeda O, et al. Docosahexaenoic acid protects from dendritic pathology in an Alzheimer’s disease mouse model. Neuron. 2004;43(5):633–45.
Arsenault D, Julien C, Tremblay C, Calon F. DHA improves cognition and prevents dysfunction of entorhinal cortex neurons in 3xTg-AD mice. PLoS ONE. 2011;6(2):e17397.
Hashimoto M, Tanabe Y, Fujii Y, Kikuta T, Shibata H, Shido O. Chronic administration of docosahexaenoic acid ameliorates the impairment of spatial cognition learning ability in amyloid beta-infused rats. J Nutr. 2005;135(3):549–55.
Cole GM, Frautschy SA. Docosahexaenoic acid protects from amyloid and dendritic pathology in an Alzheimer’s disease mouse model. Nutr Health. 2006;18(3):249–59.
Ma QL, Yang F, Rosario ER, Ubeda OJ, Beech W, Gant DJ, et al. Beta-amyloid oligomers induce phosphorylation of tau and inactivation of insulin receptor substrate via c-Jun N-terminal kinase signaling: suppression by omega-3 fatty acids and curcumin. J Neurosci. 2009;29(28):9078–89.
Fiol-deRoque MA, Gutierrez-Lanza R, Terés S, Torres M, Barceló P, Rial RV, et al. Cognitive recovery and restoration of cell proliferation in the dentate gyrus in the 5XFAD transgenic mice model of Alzheimer’s disease following 2-hydroxy-DHA treatment. Biogerontology. 2013;14(6):763–75 13p.
Nock TG, Chouinard-Watkins R, Plourde M. Carriers of an apolipoprotein E epsilon 4 allele are more vulnerable to a dietary deficiency in omega-3 fatty acids and cognitive decline. Biochim Biophys Acta. 2017 Oct;1862(10 Pt A):1068–78.
Olivera-Perez HM, Lam L, Dang J, Jiang W, Rodriguez F, Rigali E, et al. Omega-3 fatty acids increase the unfolded protein response and improve amyloid-beta phagocytosis by macrophages of patients with mild cognitive impairment. Faseb J. 2017;31(10):4359–69.
Abdullah L, Evans JE, Emmerich T, Crynen G, Shackleton B, Keegan AP, et al. APOE epsilon4 specific imbalance of arachidonic acid and docosahexaenoic acid in serum phospholipids identifies individuals with preclinical Mild Cognitive Impairment/Alzheimer’s Disease. Aging. 2017;9(3):964–85.
Hauser PS, Ryan RO. Impact of apolipoprotein E on Alzheimer’s disease. Curr Alzheimer Res. 2013;10(8):809–17.
Andrieu S, Guyonnet S, Coley N, Cantet C, Bonnefoy M, Bordes S, et al. Effect of long-term omega 3 polyunsaturated fatty acid supplementation with or without multidomain intervention on cognitive function in elderly adults with memory complaints (MAPT): a randomised, placebo-controlled trial. Lancet Neurol. 2017;16(5):377–89.
Yassine HN, Braskie MN, Mack WJ, Castor KJ, Fonteh AN, Schneider LS, et al. Association of docosahexaenoic acid supplementation with alzheimer disease stage in apolipoprotein E epsilon4 carriers: a review. JAMA Neurol. 2017;74(3):339–47.
Snowden SG, Ebshiana AA, Hye A, An Y, Pletnikova O, O’Brien R, et al. Association between fatty acid metabolism in the brain and Alzheimer disease neuropathology and cognitive performance: a nontargeted metabolomic study. PLoS Med. 2017;14(3):e1002266.
Grimm MO, Mett J, Stahlmann CP, Haupenthal VJ, Blumel T, Stotzel H, et al. Eicosapentaenoic acid and docosahexaenoic acid increase the degradation of amyloid-beta by affecting insulin-degrading enzyme. Biochemistry and cell biology = Biochimie et biologie cellulaire. 2016;94(6):534–42.
Packer L, Witt EH, Tritschler HJ. alpha-Lipoic acid as a biological antioxidant. Free Radic Biol Med. 1995;19(2):227–50.
Münch G, Maczurek A, Hager K, Kenklies M, Sharman M, Martins R, et al. Lipoic acid as an anti-inflammatory and neuroprotective treatment for Alzheimer’s disease. Adv Drug Deliver Rev. 2008;60(13–14):1463–70.
Holmquist L, Stuchbury G, Berbaum K, Muscat S, Young S, Hager K, et al. Lipoic acid as a novel treatment for Alzheimer’s disease and related dementias. Pharmacol Ther. 2007;113(1):154–64.
Matsugo S, Yan LJ, Han D, Trischler HJ, Packer L. Elucidation of antioxidant activity of alpha-lipoic acid toward hydroxyl radical. Biochem Biophys Res Commun. 1995;208(1):161–7.
Gasic-Milenkovic J, Loske C, Münch G. Advanced glycation endproducts cause lipid peroxidation in the human neuronal cell line SH-SY5Y. J Alzheimers Dis. 2003;5(1):25–30.
Steele ML, Fuller S, Maczurek AE, Kersaitis C, Ooi L, Münch G. Chronic inflammation alters production and release of glutathione and related thiols in human U373 astroglial cells. Cell Mol Neurobiol. 2013;33(1):19–30.
Kuhla B, Boeck K, Schmidt A, Ogunlade V, Arendt T, Münch G, et al. Age- and stage-dependent glyoxalase I expression and its activity in normal and Alzheimer’s disease brains. Neurobiol Aging. 2007;28(1):29–41.
Kuhla B, Luth HJ, Haferburg D, Boeck K, Arendt T, Münch G. Methylglyoxal, glyoxal, and their detoxification in Alzheimer’s disease. Ann N Y Acad Sci. 2005;1043:211–6.
Deuther-Conrad W, Loske C, Schinzel R, Dringen R, Riederer P, Münch G. Advanced glycation endproducts change glutathione redox status in SH-SY5Y human neuroblastoma cells by a hydrogen peroxide dependent mechanism. Neurosci Lett. 2001;312(1):29–32.
de Arriba SG, Loske C, Meiners I, Fleischer G, Lobisch M, Wessel K, et al. Advanced glycation endproducts induce changes in glucose consumption, lactate production, and ATP levels in SH-SY5Y neuroblastoma cells by a redox-sensitive mechanism. J Cereb Blood Flow Metab. 2003;23(11):1307–13.
Loske C, Gerdemann A, Schepl W, Wycislo M, Schinzel R, Palm D, et al. Transition metal-mediated glycoxidation accelerates cross-linking of beta-amyloid peptide. Eur J Biochem/FEBS. 2000;267(13):4171–8.
Münch G, Mayer S, Michaelis J, Hipkiss AR, Riederer P, Muller R, et al. Influence of advanced glycation end-products and AGE-inhibitors on nucleation-dependent polymerization of beta-amyloid peptide. Biochem Biophys Acta. 1997;1360(1):17–29.
Fonte J, Miklossy J, Atwood C, Martins R. The severity of cortical Alzheimer’s type changes is positively correlated with increased amyloid-beta Levels: resolubilization of amyloid-beta with transition metal ion chelators. J Alzheimers Dis. 2001;3(2):209–19.
Huang X, Atwood CS, Hartshorn MA, Multhaup G, Goldstein LE, Scarpa RC, et al. The A beta peptide of Alzheimer’s disease directly produces hydrogen peroxide through metal ion reduction. Biochemistry. 1999;38(24):7609–16.
Suh JH, Moreau R, Heath SH, Hagen TM. Dietary supplementation with (R)-alpha-lipoic acid reverses the age-related accumulation of iron and depletion of antioxidants in the rat cerebral cortex. Redox Rep. 2005;10(1):52–60.
Griffin WS, Sheng JG, Roberts GW, Mrak RE. Interleukin-1 expression in different plaque types in Alzheimer’s disease: significance in plaque evolution. J Neuropathol Exp Neurol. 1995;54(2):276–81.
Lander HM, Ogiste JS, Teng KK, Novogrodsky A. p21ras as a common signaling target of reactive free radicals and cellular redox stress. J Biol Chem. 1995;270(36):21195–8.
Richter T, Münch G, Luth HJ, Arendt T, Kientsch-Engel R, Stahl P, et al. Immunochemical crossreactivity of antibodies specific for “advanced glycation endproducts” with “advanced lipoxidation endproducts”. Neurobiol Aging. 2005;26(4):465–74.
Kuhla B, Haase C, Flach K, Lüth HJ, Arendt T, Münch G. Effect of pseudophosphorylation and cross-linking by lipid peroxidation and advanced glycation end product precursors on tau aggregation and filament formation. J Biol Chem. 2007;282(10):6984–91.
Pocernich CB, Butterfield DA. Acrolein inhibits NADH-linked mitochondrial enzyme activity: implications for Alzheimer’s disease. Neurotox Res. 2003;5(7):515–20.
Thakurta IG, Chattopadhyay M, Ghosh A, Chakrabarti S. Dietary supplementation with N-acetyl cysteine, alpha-tocopherol and alpha-lipoic acid reduces the extent of oxidative stress and proinflammatory state in aged rat brain. Biogerontology. 2012;13(5):479–88.
Monette JS, Gomez LA, Moreau RF, Dunn KC, Butler JA, Finlay LA, et al. (R)-alpha-Lipoic acid treatment restores ceramide balance in aging rat cardiac mitochondria. Pharmacol Res. 2011;63(1):23–9.
Aliev G, Liu J, Shenk JC, Fischbach K, Pacheco GJ, Chen SG, et al. Neuronal mitochondrial amelioration by feeding acetyl-L-carnitine and lipoic acid to aged rats. J Cell Mol Med. 2008 Mar 28.
Findlay JA, Hamilton DL, Ashford ML. BACE1 activity impairs neuronal glucose oxidation: rescue by beta-hydroxybutyrate and lipoic acid. Front Cell Neurosci. 2015;9:382.
Rodriguez-Perdigon M, Solas M, Moreno-Aliaga MJ, Ramirez MJ. Lipoic acid improves neuronal insulin signalling and rescues cognitive function regulating VGlut1 expression in high-fat-fed rats: implications for Alzheimer’s disease. Biochim Biophys Acta. 2016;1862(4):511–7.
Liu Z, Patil I, Sancheti H, Yin F, Cadenas E. Effects of lipoic acid on high-fat diet-induced alteration of synaptic plasticity and brain glucose metabolism: a PET/CT and (13)C-NMR study. Sci Rep. 2017;7(1):5391.
Sancheti H, Kanamori K, Patil I, Diaz Brinton R, Ross BD, Cadenas E. Reversal of metabolic deficits by lipoic acid in a triple transgenic mouse model of Alzheimer’s disease: a C NMR study. J Cereb Blood Flow Metab. 2014;34(2):288–96.
Estrada M, Perez C, Soriano E, Laurini E, Romano M, Pricl S, et al. New neurogenic lipoic-based hybrids as innovative Alzheimer’s drugs with sigma-1 agonism and beta-secretase inhibition. Future Med Chem. 2016;8(11):1191–207.
Hager K, Marahrens A, Kenklies M, Riederer P, Münch G. Alpha-lipoic acid as a new treatment option for Alzheimer [corrected] type dementia. Arch Gerontol Geriatr. 2001;32(3):275–82.
Hager K, Kenklies M, McAfoose J, Engel J, Münch G. Alpha-lipoic acid as a new treatment option for Alzheimer’s disease–a 48 months follow-up analysis. J Neural Transm Suppl. 2007;72:189–93.
Singh A, Münch G, Reddell P, Radzieta M, Jensen S, Raju R. A new anti-inflammatory phenolic monosaccharide from the Australian native rainforest plant Elaeocarpus eumundi. Nat Prod Commun. 2018;13(6):731–3.
Raju R, Singh A, Reddell P, Münch G. Anti-inflammatory activity of prenyl and geranyloxy furanocoumarins from Citrus garrawayi (Rutaceae). Phytochem Lett. 2018;27:197–202.
Akthar MA, Münch G, Bodkin F, Raju R. A New Anti-inflammatory Chromone from the leaves of Eucalyptus viminalis. Nat Prod Commun. 2018;13(10):1297–300.
Akhtar MA, Raju R, Beattie KD, Bodkin F, Münch G. Medicinal plants of the Australian aboriginal dharawal people exhibiting anti-inflammatory activity. Evid Based Complement Alternat Med. 2016;2016:2935403.
Xagorari A, Papapetropoulos A, Mauromatis A, Economou M, Fotsis T, Roussos C. Luteolin inhibits an endotoxin-stimulated phosphorylation cascade and proinflammatory cytokine production in macrophages. J Pharmacol Exp Ther. 2001;296(1):181–7.
Lee S-J, Woo Kang H, Yuan Lee S, Jin Hur S. Green tea polyphenol epigallocatechin-3-o-gallate attenuates lipopolysaccharide-induced nitric oxide production in RAW264.7 cells. J Food Nutr Res. 2014;2:425–8.
Du Z, Liu Z, Ning Z, Liu Y, Song Z, Wang C, et al. Prospects of boswellic acids as potential pharmaceutics. Planta Med. 2015;81:259–71.
Eräsalo H, Hämäläinen M, Leppänen T, Mäki-Opas I, Laavola M, Haavikko R, et al. Natural stilbenoids have anti-inflammatory properties in vivo and down-regulate the production of inflammatory mediators NO, IL6, and MCP1 possibly in a PI3K/Akt-dependent manner. J Nat Prod. 2018;81:1131–42.
Brouet I, Ohshima H. Curcumin, an anti-tumour promoter and anti-inflammatory agent, inhibits induction of nitric oxide synthase in activated macrophages. Biochem Biophys Res Commun. 1995;206(2):533–40.
Lee SH, Lee SY, Lee HJ, Hong JT, Son DJ, Yoo HS, et al. Inhibitory effect of 2′-hydroxycinnamaldehyde on nitric oxide production through inhibition of NF-κB activation in RAW 264.7 cells. Biochem Pharmacol. 2005;69(5):791–9.
Chen YC, Shen SC, Chen LG, Lee TJF, Yang LL. Wogonin, baicalin, and baicalein inhibition of inducible nitric oxide synthase and cyclooxygenase-2 gene expressions induced by nitric oxide synthase inhibitors and lipopolysaccharide. Biochem Pharmacol. 2001;61:1417–27.
Chiou WF, Lin JJ, Chen CF. Andrographolide suppresses the expression of inducible nitric oxide synthase in macrophage and restores the vasoconstriction in rat aorta treated with lipopolysaccharide. Br J Pharmacol. 1998;125:327–34.
Shanmugam K, Holmquist L, Steele M, Stuchbury G, Berbaum K, Schulz O, et al. Plant-derived polyphenols attenuate lipopolysaccharide-induced nitric oxide and tumour necrosis factor production in murine microglia and macrophages. Mol Nutr Food Res. 2008;52:427–38.
Park YC, Lee CH, Kang HS, Kim KW, Chung HT, Kim HD. Ginsenoside-Rh1 and Rh2 inhibit the induction of nitric oxide synthesis in murine peritoneal macrophages. Biochem Mol Biol Int. 1996;40:823–31.
Lee HJ, Hyun EA, Yoon WJ, Kim BH, Rhee MH, Kang HK, et al. In vitro anti-inflammatory and anti-oxidative effects of Cinnamomum camphora extracts. J Ethnopharmacol. 2006;103(2):208–16.
Ippoushi K, Azuma K, Ito H, Horie H, Higashio H. [6]-Gingerol inhibits nitric oxide synthesis in activated J774.1 mouse macrophages and prevents peroxynitrite-induced oxidation and nitration reactions. Life Sci. 2003;73:3427–37.
Kim HK, Cheon BS, Kim YH, Kim SY, Kim HP. Effects of naturally occurring flavonoids on nitric oxide production in macrophage cell line RAW 264.7 and their structure – activity relationships. Biochem Pharmacol. 1999;58:759–65.
Hämäläinen M, Nieminen R, Vuorela P, Heinonen M, Moilanen E. Anti-inflammatory effects of flavonoids: Genistein, kaempferol, quercetin, and daidzein inhibit STAT-1 and NF-$κ$B activations, whereas flavone, isorhamnetin, naringenin, and pelargonidin inhibit only NF-$κ$B activation along with their inhibitory effect Mediators Inflamm. 2007;2007:1–10.
Kotanidou A, Xagorari A, Bagli E, Kitsanta P, Fotsis T, Papapetropoulos A, et al. Luteolin reduces lipopolysaccharide-induced lethal toxicity and expression of proinflammatory molecules in mice. Am J Respir Crit Care Med. 2002;165(6):818–23.
Acknowledgements
The authors thank Prof. Annemarie Hennessy for critical reading of this article.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No sources of funding were used to assist in the preparation of this manuscript.
Conflicts of interest
Gerald Münch has received funding in this subject matter from the Australian Health and Medical Research Council, and also from Verdure Sciences, Indena and Nutrafur for research projects. Matthew J. Sharman, Giuseppe Verdile, Shanmugam Kirubakaran, Christina Parenti, Ahilya Singh, Georgina Watt, Chun Guang Li, Tim Karl and Dennis Chang have no conflicts of interest to declare that are directly relevant to the content of this manuscript.
Rights and permissions
About this article

Cite this article
Sharman, M.J., Verdile, G., Kirubakaran, S. et al. Targeting Inflammatory Pathways in Alzheimer’s Disease: A Focus on Natural Products and Phytomedicines. CNS Drugs 33, 457–480 (2019). https://doi.org/10.1007/s40263-019-00619-1
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-019-00619-1