
Abstract
Trans-cinnamaldehyde (TCA), an essential oil in cinnamon powder, may have beneficial effects as a treatment for stroke which is the second leading cause of death worldwide. Post-ischemic inflammation induces neuronal cell damage after stroke, and activation of microglia, in particular, has been thought as the main contributor of proinflammatory and neurotoxic factors. The purpose of this study was to investigate the neuroprotective effects of TCA in an animal model of ischemia/reperfusion (I/R)-induced brain injury and the neuroprotective mechanism was verified in LPS-induced inflammation of BV-2 microglial cells. Our results showed that TCA (10–30 mg/kg, p.o.) significantly reduced the infarction area, neurological deficit score and decreased iNOS and COX-2 protein expression level in I/R-induced injury brain tissue. It inhibited 0.5 µg/ml LPS-induced NO production in BV-2 microglial cells without affecting cell viability, reduced protein expression of iNOS and COX-2, and attenuated inhibition of p53 protein. TCA also suppressed the effects of LPS-induced nuclear translocation of NF-κB p65 and p50 and increased cytosolic IκBα. It also reduced LPS-induced mRNA expression of iNOS, COX-2, and TNFα. We concluded that TCA has a potential neuroprotective effect to against the ischemic stroke, which may be via the inhibition of neuroinflammation through attenuating iNOS, COX-2 expression and NF-κB signaling pathway.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Stroke, a global health issue, causes financial, emotional and other heavy burdens for families and societies worldwide. Ischemic stroke, occlusion of blood vessel in brain, accounts for a majority of stroke cases (about 87 % statistically) (Mozaffarian et al. 2015). Inflammation is the key risk factor to neuron damage in stroke (Tuttolomondo et al. 2009) and neurodegenerative diseases (Block et al. 2007; Block and Hong 2005). Microglia, the brain-resident macrophage, becomes activated within a few hours after brain ischemia (Block et al. 2007; Chew et al. 2006; Lee et al. 2014; Nimmerjahn et al. 2005). Although microglia activation is required for host defense and neuron survival (Kettenmann et al. 2013), the overactivation of microglia produce excess neurotoxic products such as superoxide, nitrite oxide (NO) or tumor necrosis factor α (TNF-α) (Colton and Gilbert 1987; Moss and Bates 2001; Liu et al. 2002; Lee et al. 1993) causing cell death.
Lipopolysaccharide (LPS), the polysaccharide component of the cell walls of gram-negative bacteria, is the most frequently used model to investigate the inflammatory responses of microglia (Gao et al. 2002; Zheng et al. 2008; Le et al. 2001). LPS can be recognized by the pattern recognition receptors (PRRs) of microglia and then binds to the Toll-like receptor 4 evoking a complex array of intracellular signaling pathways (Anderem and Ulevitch 2000; Jung et al. 2009). In addition, stimulation of microglia with LPS induces the release of tumor necrosis factor-α (TNF-α) and facilitates TNF-α- or NO-mediated neuron death in vitro (Hemmer et al. 2001). Inducible NO synthase (iNOS) is quickly transcribed and expressed in microglia after stimulation with bacterial LPS and cytokines (Kim et al. 2000; Choi et al. 2008). COX-2 (cyclooxygenase-2) also plays a predominant role in inflammation (Minghetti 2004). LPS induces PGE2 production in human microglia through COX-2 induction (Hsu and Wen 2002). The molecular mechanisms of inflammation induced by LPS in microglial cells have been well reported (Wang et al. 2002; Bhat et al. 1998; Woo et al. 2003; Pawate et al. 2004; Xie et al. 2004). NF-κB pathway, one of the most important mechanisms in inflammatory diseases, is the key transcription factor in the regulation of proinflammatory cytokines, iNOS and COX-2 (Baeuerle and Henkel 1994; Baldwin 2001). At the molecular level, NF-κB induces activation of cytoplasmic proteins and the nuclear translocation of p65, p50 subunit of NF-κB (Karin and Ben-Neriah 2000; Delhase et al. 2000). Previous studies have shown that LPS causes the nuclear translocation of p65 subunit of NF-κB through IκB degradation (Baeuerle and Baltimore 1988; Henkel et al. 1993).
Cinnamon powder, one of the most famous spices, is made from the inner bark of cinnamon trees and can be found in many types of recipes and foods. The extract from cinnamon bark has also been used as a traditional medicine. Some studies have found that cinnamon may have beneficial health properties, such as lowering blood sugar and cholesterol levels, and reducing inflammation (Bernardo et al. 2015; Shalaby and Saifan 2014; Pahan 2015). Cinnamaldehyde, an essential oil in cinnamon powder, is the source of the distinct smell and flavor of cinnamon (Adams et al. 2004). Trans-cinnamaldehyde displays diverse pharmacological properties including anti-inflammatory activity (Wu et al. 2012; Lee et al. 2002; Kim et al. 2007; Amalaradjou et al. 2010; Shahverdi et al. 2007). Our preliminary data showed that cinnamon powder significantly reduced ischemia/reperfusion (I/R)-induced cerebral infarction (data not shown). The purposes of the present study were to examine the neuroprotective effect of trans-cinnamaldehyde in I/R-induced brain injury in vivo and anti-inflammatory mechanisms in vitro by LPS-induced insults in BV-2 cells.
Materials and Methods
Chemicals
LPS (0111:B4), MTT, Griess reagent and trans-cinnamaldehyde were purchased from Sigma-Aldrich (St Louis, MO, USA).
Animal Cerebral Ischemia Model
Adult male C57BL/6 mice (weight 22–28 g), purchased from National Laboratory Animal Center, were used in this study. Animals were fed with standard chow and housed in standard cages at a constant room temperature of 22 ± 1 °C. Relative humidity of 55 ± 5 % was maintained with 12-h inverted light–dark cycle for 1 week at least prior to the experiment. The experimental protocol was approved by the Institutional Animal Care and Use Committee (IACUC), China Medical University (Permit Number: 101-148-N). The minimum number of animals and duration of observations required to obtain reliable data are used. For infarct volume studies, the animals were divided into four groups: the I/R group (ischemia/reperfusion induction, as a control group) and TCA treatment groups (10, 20, 30 mg/kg, orally administration 60 min before ischemia surgery). Animals were subjected to two-vessel ligation. All surgical procedures were performed by sterile/aseptic techniques. The mice were anesthetized with chloral hydrate (0.4 g/kg, ip). Ligation of the right middle cerebral artery (MCA) and right common carotid arteries (CCA) was performed by methods described previously, with slight modifications (Yang et al. 1997). The right CCA was clamped with non-traumatic arterial clip. The right MCA was ligated with 10-0 nylon suture. After 120 min of ischemia, the suture on the MCA and arterial clip on CCA were removed to allow reperfusion. Core body temperature was monitored with a thermostat probe and maintained at 37 °C with a heating pad during anesthesia. After recovery from anesthesia, body temperature was maintained at 37 °C with a heat lamp (Ding et al. 2007).
Evaluation of Neurological Deficits and Measurement of Infarction Area
Twenty-four hours following reperfusion, neurological function of mice was assessed as previously described (Hattori et al. 2000). The degree of neurological deficits were divided 0–4 (higher score is for more severe neurological deficits) as follows: 0 = no deficit; 1 = forelimb weakness; 2 = circling to affected side; 3 = partial paralysis on affected side and 4 = no spontaneous motor activity. After completion of neurological evaluation, mice were deeply anesthetized by an intraperitoneal injection of chloral hydrate (0.8 g/kg) followed by an intracardiac perfusion and decapitation. After decapitation, the brain was removed and sectioned into 2-mm-thick slices and then immersed for 15 min into 2 % TTC solution at 37 °C. Animals with extended hemorrhage in their skull base were excluded from the study. The area of the ischemic damage of the hemispheres was measured for each brain slice using an Image-Pro Plus 6.0 (Media Cybernetics, MD, USA). Complete lack of TTC staining was defined as core (Bederson et al. 1986). Purple–red brain tissue was penumbra positioned between core, and viable brain tissue was stained red (Gill et al. 1995). Total infarction area was the sum of core and penumbra areas. Results were given as the means of SEM for the different treatment groups.
iNOS and COX-2 Protein Expression Level in Ischemic Brain Tissue
After completion of neurological evaluation, mice were deeply anesthetized, decapitated, and the brains were removed for protein extraction. Tissue lysates were prepared in lysis buffer, and protein concentrations in the tissue lysates were determined by Bio-Rad protein assay kit (Richmond, CA, USA). Samples of protein (50 μg) were electrophoresed using 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membrane. The iNOS, COX-2 were assayed by specific antibodies (Santa Cruz Biotech, Santa Cruz, CA, USA, and Cell Signaling Technology, Danvers, MA, USA). Immunodetection was done using an enhanced chemiluminescence detection kit (Amersham, Piscataway, NJ, USA).
Cell Culture
Murine BV-2 microglial cells were kindly provided by Professor Jau-Shyong Hong (Neurobiology laboratory/Neuropharmacology group, NIEHS/NIH, NC, USA). BV-2 microglial cells were cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10 % FBS, 100 units/ml penicillin and 100 mg/ml streptomycin, and kept at 37 °C in a humidified incubator with 5 % CO2 and 95 % air. A scraper transferred confluent cultures every 2–3 days.
MTT Assay
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) assay determined cell viability. The BV-2 microglia cells were cultured at the density of 5 × 104 cells/well in 96-well plate. BV-2 microglia cell cultures were treated with TCA (12.5, 25, 50 μM) and LPS (0.5 μg/ml) for 24 h. MTT was added to each well, and the cells were incubated for 1 h at 37 °C. After culture media were discarded, DMSO was added to dissolve the formazan dye. The optical density was measured at 570 nm.
Nitrite Quantification
NO generation was measured by the accumulation of nitrite in the culture medium. The colorimetric assay was used to determine nitrite with Griess reagent. BV-2 cells (5 × 104 cells/well) in 96-well plates in 200 μl culture medium were treated with LPS (0.5 μg/ml) for 24 h. Hundred microliters of isolated supernatant was added with an equal volume of Griess reagent in 96-well plates for 10 min at room temperature and light avoidance. Standard solution of sodium nitrite prepared in cell-culture medium was used to determine nitrite concentrations. The absorbance at 570 nm was read using an Elisa reader (Triad LT, DYNEX Technologies Inc, VA, USA). Each experiment was duplicated three times.
Western Blot Analysis
Cell lysates were prepared in lysis buffer. Protein concentrations in the cell lysates were determined by Bio-Rad protein assay kit (Richmond, CA, USA). Samples of protein (50 μg) were electrophoresed using 10 % sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) and then transferred to nitrocellulose membrane. The iNOS, COX2, p53, phospho-IκBα, IκBα, phospho-NF-κBp65, NF-κBp65 and NF-κBp50 were assayed by specific antibodies (Santa Cruz Biotech, Santa Cruz, CA, USA Cell Signaling Technology). Immunodetection was done using an enhanced chemiluminescence detection kit (Amersham, Piscataway, NJ, USA) (Huang et al. 2011).
Nuclear Protein Extraction and mRNA Analysis by Quantitative Real-Time PCR
Cytoplasmic proteins and nuclear protein was isolated by the ProteoJET™ Cytoplasmic and Nuclear Protein Extraction Kit (Fermentas). Total RNA was isolated and purified from BV-2 microglial cells using the TRIzol method and ChargeSwitch® Total RNA cell kits (Invitrogen, CA, USA). Using Thermo Scientific Verso cDNA Synthesis Kit reverse-transcribed RNA to generate cDNA in a Bio-Rad’s iQ5 multicolor real-time PCR according to the manufacturer’s instructions. The generated cDNA was used to quantify real-time PCR through real-time analysis using Invitrogen’s SYBR® GreenER™ qPCR SuperMix. The primer sets used were purchased from Dharmacon and were
-
GAPDH, forward primer: 5′-GGCTGGCATTGCTCTCAA-3′ and reverse primer: 5′-GCTGTAGCCGTATTCATTGTC-3′;
-
TNF, forward primer: 5′-AACTCCAGGCGGTGCCTAT-3′ and reverse primer: 5′-GGAGGCCATTTGGGAAC-3′;
-
COX2, forward primer: 5′-TAGCAGATGACTGCCCAACT-3′ and reverse primer: 5′-GAATCAGGAAGCTCCTTA-3′;
-
iNOS, forward primer: 5′-CGTGAAGAAAACCCCTTGTG-3′ and reverse primer: 5′-GATTCTGGAACATTCTGTGCTG-3′.
Thermal cycles were programed according to the manufacturer’s instructions for each primer. To normalize the data for each experiment, GAPDH was used as a reference gene. Using ΔΔC T calculations determined fold expression of gene.
Statistics
Statistical analysis was performed using the software SPSS version 10.0. The values given were mean ± SE from 3 to 4 experiments of each group. Statistical analysis between two samples was performed using Student’s t test. Statistical comparisons of more than two groups were performed using one-way analysis of variance (ANOVA) with Duncan’s test. A P value <0.05 was considered statistically significant.
Results
TCA Ameliorated Ischemia/Reperfusion Brain Injury and Reduced iNOS and COX-2 Protein Expression Level in I/R-Induced Brain Injury of Mice
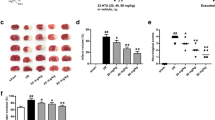
TCA (10, 20 and 30 mg/kg, p.o.) significantly reduced the infarction area in I/R-induced brain injury (Fig. 1a, b) and neurological deficit score (Fig. 1c) in mice in a dose-dependent manner (P < 0.001). The reduction rates of TCA (10, 20, 30 mg/kg, p.o.) on infarction area are 60, 68 and 80 %, respectively. TCA (10, 20, 30 mg/kg, p.o.) demonstrated a 13, 40 and 50 % reduction of neurological deficit score, respectively. Moreover, TCA also significantly reduced iNOS and COX-2 protein expression levels in ischemic mice brain (Fig. 2a–c) in a dose-dependent manner (P < 0.001). The reduction rates of TCA (10, 20, 30 mg/kg, p.o.) on iNOS protein expression level are 70, 88 and 95 %, respectively. TCA (10, 20, 30 mg/kg, p.o.) illustrated a 37, 55 and 73 % reduction of COX-2 protein expression level.

Effects of trans-cinnamaldehyde (TCA) on ischemia/reperfusion (I/R)-induced brain injury in mice. Coronal section of brain after ischemia for 120 min and followed by reperfusion for 24 h. Then, staining with TTC, the infarction areas appeared white and non-infarction areas appeared red–purple in color. S1 → S4 slices from frontal lobe. a TTC of control mice and mice treated with TCA (10, 20, 30 mg/kg, p.o.) 60 min before I/R. b Statistical bars of cerebral infarction area. c Neurological deficit score of mice 24 h after I/R. Scale bar 1 cm. Data are mean ± SE from more than three independent experiments. ***P < 0.001 compared with the control (I/R) group (Color figure online)

Effects of trans-cinnamaldehyde (TCA) on protein expression level of iNOS and COX-2 in I/R-induced brain injury mice. a Western blot of iNOS and COX-2 in control group and mice treated with TCA (10, 20, 30 mg/kg, p.o.) 60 min before I/R injury. b Statistical bars of protein expression level of iNOS. c Statistical bars of protein expression level of COX-2. Data are mean ± SE from more than three independent experiments. ***P < 0.001 compared with the control (I/R) group
TCA Inhibited NO Production, Reduced Protein Expression Level of iNOS, COX-2, But Increased Expression of p53 in LPS-Challenged BV-2 Microglial Cells
Results of BV-2 cells treated with TCA (12.5, 25, 50 μM) plus LPS (0.5 μg/ml) or LPS (0.5 μg/ml) alone for 24 h was showed in Fig. 3a. TCA did not induce cytotoxicity. It significantly inhibited NO production at concentration of 50 μM after 24 h treatment compared with the LPS-only group (P < 0.001) (Fig. 3b). TCA (12.5, 25, 50 μM) displayed a 10, 17 and 53 % reduction of NO production, respectively. TCA also reduced cytosolic protein levels of iNOS, COX-2 (Fig. 4a, b) and attenuated inhibition of p53 elicited by LPS (Fig. 4c). The reduction rates of TCA (12.5, 25, 50 μM) on iNOS protein expression level are 9, 26 and 35 %, respectively, and 50 μM of TCA showed a 13 % reduction of COX-2 protein expression level in LPS-induced BV-2 microglia cells.

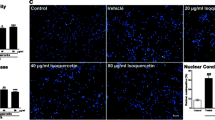
Effects of trans-cinnamaldehyde (TCA) on NO production in LPS-challenged BV-2 microglial cells. Trans-cinnamaldehyde significantly reduced NO production (b) without affecting the BV-2 cell viability (a) induced by LPS (0.5 μg/ml). Data are mean ± SE from more than three independent experiments. ### P < 0.001 compared with untreated group. *P < 0.05, **P < 0.01, ***P < 0.001, compared with LPS alone

Effects of trans-cinnamaldehyde (TCA) on expression of iNOS, COX-2 and p53 in LPS-challenged BV-2 microglial cells. Trans-cinnamaldehyde attenuated expression of iNOS (a), COX-2 (b) proteins, while increased expression of p53 (c) protein BV-2 cells, which were simultaneously treated different concentrations of trans-cinnamaldehyde (12.5, 25, 50 μM) and LPS (0.5 μg/ml) for 24 h. Protein level of iNOS, COX-2 and p53 was analyzed by Western blotting. β-actin was used as a control of protein loading. Data are mean ± SE from more than three independent experiments. ### P < 0.001 compared with untreated group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with LPS alone
TCA Reduced NF-κB Activation and IκB Degradation Induced by LPS
In Fig. 5a, b, it showed that treatment with LPS for 15 min stimulated nuclear translocation of NF-κB, while TCA at 12.5–50 μM significantly suppressed the nuclear translocation of p65 and p50. The effect of TCA on the expression of cytosol IκBα by Western blot analysis showed that it increased the level of IκBα (Fig. 5c), but had no obvious significance on total IκB (Fig. 4d) compared to LPS-challenged-only group.

Effects of trans-cinnamaldehyde (TCA) on NF-κB and IκB induced by LPS. Trans-cinnamaldehyde curbed NF-κBp65 (a) and NF-κBp50 activation (b) and IκBα degradation (c, d) induced by LPS. BV-2 cells were simultaneously treated different concentrations of trans-cinnamaldehyde (12.5, 25, 50 μM) and LPS 0.5 μg/ml for 15 min. Protein levels of NF-κB (nuclear protein) and IκBα (cytosol and total proteins) were analyzed by Western blotting. Actin and H3 were used as the control of protein loading. ### P < 0.001 compared with untreated group. *P < 0.05, **P < 0.01, ***P < 0.001 compared with LPS alone
TCA Reduced mRNA Expression Levels of iNOS, COX-2 and TNFα Induced by LPS
Data in Fig. 6a–c showed that TCA reduced mRNA expression of iNOS, COX-2, and TNFα in BV-2 cells treated with LPS. Significant effects on mRNA levels were observed after 4 and 12 h of TCA incubation.

Effects of trans-cinnamaldehyde (TCA) on mRNA expression levels of iNOS, COX-2 and TNFα induced by LPS. Trans-cinnamaldehyde reduced mRNA expression of iNOS (a), COX-2 (b) and TNFα (c) in cells treated with LPS. BV-2 cells were treated with LPS (0.5 µg/mL) or trans-cinnamaldehyde (50 μM) for 12 h or 4 h, and total RNA was subjected to quantitative RT-PCR. The RT products were labeled with SYBR Green dye. Relative iNOS, COX-2 and TNFα mRNA expressions (2 – ΔC t ) were determined by real-time PCR and calculated by subtracting the C t value for iNOS, COX-2 and TNFα from GAPDH mRNA, respectively. ΔC t = C t COX-2 − C t GAPDH. Each value represents the mean ± SE of three independent experiments. ## P < 0.01, ### P < 0.001 compared with untreated group. **P < 0.01, ***P < 0.001 compared with LPS alone
Discussion
Inflammation plays an important role in the pathogenesis of ischemic stroke, and the activation of microglia is the key factor contributing to neuroinflammation (Tuttolomondo et al. 2009). LPS activates NF-κB resulting in the induction of several inflammation-related early genes (Baeuerle 1991; Yamamoto and Gaynor 2001) particularly gene expression of iNOS (Griscavage et al. 1996) and COX-2 (Umezawa et al. 2000). LPS activates NF-κB resulting in the induction of several inflammation-related early genes (Baeuerle 1991; Yamamoto and Gaynor 2001) particularly gene expression of iNOS and COX-2 (Umezawa et al. 2000). LPS induces cellular inflammation by up-regulating pro-inflammatory factors such as iNOS, COX-2 and TNFα (Block et al. 2007). Those genes are regulated by NF-κB (Mattson and Camandola 2001). Trans-cinnamaldehyde, an essential oil in cinnamon powder, has anti-inflammatory effects (Kim et al. 2007). In our data, it showed that trans-cinnamaldehyde significantly ameliorated the infarct volume in mice brain subjected to ischemia and reduced the neurological deficit score in ischemia mice revealed that trans-cinnamaldehyde had neuroprotection against cerebral ischemia in vivo (Fig. 1). In addition, trans-cinnamaldehyde markedly attenuated protein expression level of iNOS and COX-2 in ischemia brain tissue that revealed its anti-inflammatory activity in vivo. The effect of trans-cinnamaldehyde has not only showed neuroprotection in a mouse ischemic stroke model, but also revealed anti-inflammatory activity in activated BV-2 microglial cells elicited by an LPS challenge. The anti-inflammatory mechanisms of trans-cinnamaldehyde is via the inhibition of inflammatory molecules including production of NO and expression of iNOS, COX-2, NF-κB, IκB, mRNA of iNOS, TNFα and the increasing p53 levels.
Cerebral ischemia involves both innate and adaptive immunity (Famakin 2014). Microglia originates from the hematopoietic system and has been regarded as the innate immune cells taking responsibility for immune surveillance in the CNS (Ginhoux et al. 2010). However, once activated by inflammatory stimuli, such as LPS, the cell wall component of gram-negative bacteria, microglia can become overactivated and cause neurotoxicity (Block et al. 2007). LPS can trigger an immune response, and it has been a technique to activate microglia (Gao et al. 2002; Gibbons and Dragunow 2006). BV-2 cells derived from raf/myc-immortalized murine neonatal microglia are a recognized model of primary microglia (Henn et al. 2009; Laurenzi et al. 2001). In the present study, we determined whether trans-cinnamaldehyde would be neuroprotective in BV-2 cells incubated with LPS and in a mouse model of cerebral ischemia.
Trans-cinnamaldehyde reduced production of nitric oxide and expression of iNOS mRNA and iNOS protein stimulated by LPS-induced inflammation in BV-2 cells. These results are similar to earlier studies showing that trans-cinnamaldehyde reduced the expression of iNOS and COX-2 in different models, such as 6-OHD (Pyo et al. 2013), carrageenan-induced mouse paw edema (Liao et al. 2012) and LPS-stimulated mouse macrophage (RAW264.7) (Liao et al. 2012; Lee et al. 2005).
The anti-inflammatory effect of trans-cinnamaldehyde is attributed to its pharmacological action and not due to cytotoxicity. Cell viability was similar in LPS (0.5 μg/ml) treatment alone or co-treatment with LPS (0.5 μg/ml) and trans-cinnamaldehyde (12.5–50 μM). Previous studies have reported that activated microglia increase levels of COX-2 and TNF-α (Mozaffarian et al. 2015; Zhao et al. 2007). COX-2, a rate-limiting enzyme for the synthesis of prostaglandins, is a characteristic of neuroinflammation (Hoozemans et al. 2002). TNF-α, the proinflammatory cytokine, is known to up-regulate the transcription of COX-2 and iNOS genes (Hoozemans et al. 2002). Trans-cinnamaldehyde at 50 μM inhibited the LPS-induced increase in COX-2 protein levels and the expression of COX-2 mRNA and TNF-α mRNA. These data indicate that trans-cinnamaldehyde diminishes production of pro-inflammatory molecules induced by LPS. LPS stimulates inflammation via the activation of the NF-κB signaling pathway (Anderson 2000; Jung et al. 2009). NF-κB is an important transcription factor in responding pro-inflammatory stimuli, such as LPS. NF-κB regulates transcription of several genes such as iNOS, COX-2 and TNF-α, whose proteins products involve in inflammatory pathways (Baldwin 1996). Our present data showed that trans-cinnamaldehyde reduced p65 and p50 activation and IκBα. The anti-inflammatory effect of trans-cinnamaldehyde is consistent with previous reports (Tung et al. 2008, 2010; Ballabeni et al. 2010). Trans-cinnamaldehyde suppressed age-related NF-κB activation and its targeting inflammatory genes iNOS and COX-2 in rat kidney (Kim et al. 2007).
The p53 and NF-κB pathways are the two main pathways that respond to cell stress. p53, the major tumor suppressor, is activated by intrinsic stress signals (e.g., DNA damage, oncogene expression, spindle poisoning), while NF-κB is responding to extrinsic stress signals (e.g., the presence of infectious agents) (Gudkov et al. 2011). Several reports suggest that cells expressing NF-κB inhibit p53 activity and p53 responses, and, inversely, cells with up-regulated p53 suppress NF-κB activity (O’Prey et al. 2010; Hudson et al. 1999; Yonish-Rouach et al. 1991). Therefore, agents that inhibit NF-κB can activate p53 (Gurova et al. 2005). There is an abundance of evidence showing that p53 is an inhibitor of inflammation due to its antagonism of NF-κB (Gudkov et al. 2011; Komarova et al. 2005; Yamanishi et al. 2002; Okuda et al. 2003). We found that trans-cinnamaldehyde significantly inhibited both p53 reduction (Fig. 4c) and NF-κB activation (Fig. 5a, b) in LPS-stimulated BV-2 cells, and those findings are in agreement with those earlier studies (Youn et al. 2008).
In conclusion, we demonstrated that trans-cinnamaldehyde, an essential oil in cinnamon powder, significantly reduced infarct area in cerebral ischemia mouse model. It inhibited nitric oxide production and inflammation which were by down-regulating gene expression of iNOS, COX-2 and TNF-α, and suppressing the NF-κB and p53 pathways (showed as Fig. 7). These in vivo and in vitro results are strong evidence that trans-cinnamaldehyde can be a potential therapeutic agent to reduce inflammation associated with ischemic stroke.

Proposed action mechanism of trans-cinnamaldehyde (TCA) on neuroinflammation and neuroprotection. Trans-cinnamaldehyde inhibited nitric oxide production and inflammation which were by down-regulating gene expression of iNOS, COX-2 and TNF-α, and suppressing the NF-κB pathway
References
Adams, T. B., Cohen, S. M., Doull, J., Feron, V. J., Goodman, J. I., Marnett, L. J., et al. (2004). The FEMA GRAS assessment of cinnamyl derivatives used as flavor ingredients. Food and Chemical Toxicology, 42, 157–185.
Amalaradjou, M. A., Narayanan, A., Baskaran, S. A., & Venkitanarayanan, K. (2010). Antibiofilm effect of trans-cinnamaldehyde on uropathogenic Escherichia coli. The Journal of Urology, 184(1), 358–363.
Anderem, A., & Ulevitch, R. J. (2000). Toll-like receptors in the induction of the innate immune response. Nature, 406(6797), 782–787.
Anderson, K. M. (2000). Toll signaling pathway in the innate immune response. Current Opinion in Immunology, 12, 13–19.
Baeuerle, P. (1991). The inducible transcription activator NF-kB: Regulation by distinct protein subunits. Biochimica et Biophysica Acta, 1072, 63–80.
Baeuerle, P. A., & Baltimore, D. (1988). I kappa B: A specific inhibitor of the NF-kappa B transcription factor. Science, 242(4878), 540–546.
Baeuerle, P. A., & Henkel, T. (1994). Function and activation of NF-kappa B in the immune system. Annual Review of Immunology, 12, 141–179.
Baldwin, A. S, Jr. (1996). The NF-kappa B and I kappa B proteins: New discoveries and insights. Annals Review of Immunology, 14, 649–683.
Baldwin, A. S. (2001). Control of oncogenesis and cancer therapy resistance by the transcription factor NF-kappaB. Journal of Clinical Investigation, 107, 241–246.
Ballabeni, V., Tognolini, M., Giorgio, C., Bertoni, S., Bruni, R., & Barocelli, E. (2010). Ocotea quixos Lam. Essential oil: In vitro and in vivo investigation on its anti-inflammatory propertie. Fitoterapia, 81(4), 289–295.
Bederson, J. B., Pitts, L. H., Germano, S. M., Nishimura, M. C., Davis, R. L., & Bartkowski, H. M. (1986). Evaluation of 2, 3, 5-triphenyltetrazolium chloride as a stain for detection and quantification of experimental cerebral infarction in rats. Stroke, 17, 1304–1308.
Bernardo, M. A., Silva, M. L., Santos, E., Moncada, M. M., Brito, J., Proenca, L., et al. (2015). Effect of cinnamon tea on postprandial glucose concentration. Journal of Diabetes Research. Article ID 913651.
Bhat, N. R., Zhang, P., Lee, J. C., & Hogan, E. L. (1998). Extracellular signal-regulated kinase and p38 subgroups of mitogen-activated protein kinases regulate inducible nitric oxide synthase and tumor necrosis factor-alpha gene expression endotoxin- stimulated primary glial cultures. Journal of Neuroscience, 18, 1633–1641.
Block, M. L., & Hong, J. S. (2005). Microglia and inflammation-mediated neurodegeneration: Multiple triggers with a common mechanism. Progress in Neurobiology, 76, 77–98.
Block, M. L., Zecca, L., & Hong, J. S. (2007). Microglia-mediated neurotoxicity: Uncovering the molecular mechanisms. Nature Reviews Neuroscience, 8, 57–69.
Chew, L. J., Takanohashi, A., & Bell, M. (2006). Microglia and imflammation: Impact on developmental brain injuries. Mental Retardation and Developmental Disabilities Research, 12, 105–112.
Choi, S. K., Park, Y. S., Choi, D. K., & Chang, H. I. (2008). Effects of astaxanthin on the production of NO and the expression of COX-2 and iNOS in LPS-stimulated BV2 microglial cells. Journal of Microbiology and Biotechnology, 18(12), 1990–1996.
Colton, C. A., & Gilbert, D. L. (1987). Production of superoxide anions by a CNS macrophage, the microglia. FEBS Letters, 223, 284–288.
Delhase, M., Li, N., & Karin, M. (2000). Signalling pathways: Kinase regulation in inflammatory response. Nature, 406, 367–368.
Ding, D. C., Shyu, W. C., Chiang, M. F., Lin, S. Z., Chang, Y. C., Wang, H. J., et al. (2007). Enhancement of neuroplasticity through upregulation of β1-integrin in human umbilical cord-derived stromal cell implanted stroke model. Neurobiology of Disease, 27, 339–353.
Famakin, B. M. (2014). The immune response to acute focal cerebral ischemia and associated post-stroke immunodepression: A focused review. Aging and Disease, 5(5), 307–326.
Gao, H. M., Jiang, J., Wilson, B., Zhang, W., Hong, J. S., & Liu, B. (2002). Microglial activation-mediated delayed and progressive degeneration of rat nigral dopaminergic neurons: Relevance to Parkinson’s disease. Journal of Neurochemistry, 81, 1285–1297.
Gibbons, H. M., & Dragunow, M. (2006). Microglia induce neural cell death via a proximity-dependent mechanism involving nitric oxide. Brain Research, 1084(1), 1–15.
Gill, R., Sibson, N. R., Hatfield, R. H., Burdett, N. G., Carpenter, T. A., Hall, L. D., et al. (1995). A comparison of the early development of ischemic damage following permanent middle cerebral artery occlusion in rats as assessed using magnetic resonance imaging and histology. Journal of Cerebrocascular Blood Flow and Metabolism, 15, 1–11.
Ginhoux, F., Greter, M., Leboeuf, M., Nandi, S., See, P., Gokhan, S., et al. (2010). Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science, 330(6005), 841–845.
Griscavage, J. M., Wilk, S., & Ignarro, L. J. (1996). Inhibitors of the proteasome pathway interfere with induction of nitric oxide synthase in macrophages by blocking activation of transcription factor NF-kB. Proceedings of National Academy of Science USA, 93, 3308–3312.
Gudkov, A. V., Gurova, K. V., & Komarova, E. A. (2011). Inflammation and p53: A tale of two stresses. Genes and Cancer, 2(4), 503–516.
Gurova, K. V., Hill, J. E., Guo, C., et al. (2005). Small molecules that reactivate p53 in renal cell carcinoma reveal a NF-kappaB-dependent mechanism of p53 suppression in tumors. Proceedings of National Academy of Science USA, 102(48), 17448–17453.
Hattori, K., Lee, H., Hurn, P. D., Crain, B. J., Traystman, R. J., & DeVries, A. C. (2000). Cognitive deficits after focal cerebral ischemia in mice. Stroke, 31, 1939–1944.
Hemmer, K., Fransen, L., Vanderstichele, H., Vanmechelen, E., & Heuschling, P. (2001). An in vitro model for the study of microglia-induced neurodegeneration: Involvement of nitric oxide and tumor necrosis factor-a. Neurochemistry International Journal, 38, 557–565.
Henkel, T., Machleidt, T., Alkalay, I., Krönke, M., Ben-Neriah, Y., & Baeuerle, P. A. (1993). Rapid proteolysis of I kappa B-alpha is necessary for activation of transcription factor NF-kappa B. Nature, 365(6442), 182–185.
Henn, A., Lund, S., Hedtjärn, M., Schrattenholz, A., Pörzgen, P., & Leist, M. (2009). The suitability of BV2 cells as alternative model system for primary microglia cultures or for animal experiments examining brain inflammation. ALTEX, 26(2), 83–94.
Hoozemans, J. J., Veerhuis, R., Janssen, I., van Elk, E. J., Rozemuller, A. J., & Eikelenboom, P. (2002). The role of cyclo-oxygenase 1 and 2 activity in prostaglandin E(2) secretion by cultured human adult microglia: Implications for Alzheimer’s disease. Brain Research, 951, 218–226.
Hsu, H. Y., & Wen, M. H. (2002). Lipopolysaccharide-mediated reactive oxygen species and signal transduction in the regulation of interleukin-1 gene expression. Journal of Biological Chemistry, 277, 22131–22139.
Huang, W. W., Ko, S. W., Tsai, H. Y., Chung, J. G., Chiang, J. H., Chen, K. T., et al. (2011). Cantharidin induces G2/M phase arrest and apoptosis in human colorectal cancer colo 205 cells through inhibition of CDK1 activity and caspase-dependent signaling pathways. International Journal of Oncology, 38(4), 1067–1073.
Hudson, J. D., Shoaibi, M. A., Maestro, R., Carnero, A., Hannon, G. J., & Beach, D. H. (1999). A proinflammatory cytokine inhibits p53 tumor suppressor activity. Journal of Experimental Medicine, 190(10), 1375–1382.
Jung, W. K., Ahn, Y. W., Lee, S. H., Choi, Y. H., Kim, S. K., Yea, S. S., et al. (2009). Ecklonia cava ethanolic extracts inhibit lioppolysaccaharide-induced cyclooxygenase-2 and inducible nitric oxide synthase expression in BV2 microglia via the MAPK kinase and NF-kB pathways. Food and Chemical Toxicology, 47, 410–417.
Karin, M., & Ben-Neriah, Y. (2000). Phosphorylation meets ubiquitination: The control of NF-[kappa] B activity. Annual Review of Immunology, 18, 621–658.
Kettenmann, H., Kirchhoff, F., & Verkhratsky, A. (2013). Microglia: New roles for the synaptic stripper. Neuron, 77(1), 10–18.
Kim, D. H., Kim, C. H., Kim, M. S., Kim, J. Y., Jung, K. J., Chung, J. H., et al. (2007). Suppression of age-related inflammatory NF-κB activation by cinnamaldehyde. Biogerontology, 8, 545–554.
Kim, W. G., Mohney, R. P., Wilson, B., Jeohn, G. H., Liu, B., & Hong, J. S. (2000). Regional difference in susceptibility to lipopolysaccharide-induced neurotoxicity in the rat brain. Role of microglia. Journal of Neuroscience, 20(16), 6309–6316.
Komarova, E. A., Krivokrysenko, V., Wang, K., et al. (2005). p53 is a suppressor of inflammatory response in mice. FASEB Journal, 19(8), 1030–1032.
Laurenzi, M. A., Arcuri, C., Rossi, R., Marconi, P., & Bocchini, V. (2001). Effects of microenvironment on morphology and function of the microglial cell line BV-2. Neurochemical Research, 26(11), 1209–1216.
Le, W., Rowe, D., Xie, W., Ortiz, I., He, Y., & Appel, S. H. (2001). Microglial activation and dopaminergic cell injury: An in vitro model relevant to Parkinson’s disease. Journal of Neuroscience, 21, 8447–8455.
Lee, H. S., Kim, B. S., & Kim, M. K. (2002). Suppression effect of Cinnamomum cassia bark-derived component on nitric oxide synthase. Journal of Agriculture and Food Chemistry, 50, 7700–7703.
Lee, Y. J., Lee, S. R., Choi, S. S., Yeo, H. G., Chang, K. T., & Lee, H. J. (2014). Therapeutically targeting neuroinflammation and microglia after acute ischemic stroke. Biomed Research International. Article ID 297241.
Lee, S. H., Lee, S. Y., Son, D. J., Lee, H., Yoo, H. S., Song, S., et al. (2005). Inhibitory effect of 2-hydroxycinnamaldehyde on nitric oxide production through inhibition of NF-kB activation in RAW 264.7 cells. Biochemical Pharmacology, 69, 791–799.
Lee, S. C., Liu, W., Dickson, D. W., Brosnan, C. F., & Berman, J. W. (1993). Cytokine production by human fetal microglia and astrocytes. Differential induction by lipopolysaccharide and IL-1 β. Journal of Immunology, 150, 2659–2667.
Liao, J. C., Deng, J. S., Chiu, C. S., Hou, W. C., Huang, S. S., Shie, P. H., et al. (2012). Anti-inflammatory activities of cinnamomum cassia constituents in vitro and in vivo. Evidence-Based Complementary and Alternative Medicine. Article ID 429320.
Liu, B., Gao, H. M., Wang, J. Y., Jeohn, G. H., Cooper, C. L., & Hong, J. S. (2002). Role of nitric oxide in inflammation mediated neurodegeneration. Annals of New York Academy of Sciences, 962, 318–331.
Mattson, M. P., & Camandola, S. (2001). NF-κB in neuronal plasticity and neurodegenerative disorders. Journal of Clinical Investigation, 107(3), 247–254.
Minghetti, L. (2004). Cyclooxygenase-2 (COX-2) in inflammatory and degenerative brain diseases. Journal of Neuropathology and Experimental Neurology, 63, 901–910.
Moss, D. W., & Bates, T. E. (2001). Activation of murine microglial cell lines by lipopolysaccharide and interferon-γ causes NO-mediated decreases in mitochondrial and cellular function. European Journal of Neuroscience, 13, 529–538.
Mozaffarian, D., Benjamin, E. J., Go, A. S., Arnett, D. K., Blaha, M. J., Cushman, M., et al. (2015). Heart disease and stroke statistics—2015 update: A report from the American Heart Association. Circulation, 131(4), e29–e322.
Nimmerjahn, A., Kirchhoff, F., & Helmchen, F. (2005). Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science, 308, 1314–1318.
O’Prey, J., Crighton, D., Martin, A. G., Vousden, K. H., Fearnhead, H. O., & Ryan, K. M. (2010). p53-mediated induction of Noxa and p53AIP1 requires NFkappaB. Cell Cycle, 9(5), 947–952.
Okuda, Y., Okuda, M., & Bernard, C. C. (2003). Regulatory role of p53 in experimental autoimmune encephalomyelitis. Journal of Neuroimmunology, 135(1–2), 29–37.
Pahan, K. (2015). Prospects of cinnamon in multiple sclerosis. Journal of Multiple Sclerosis, 2(3), 1000149. doi:10.4172/2376-0389.1000149.
Pawate, S., Shen, Q., Fan, F., & Bhat, N. R. (2004). Redox regulation of glial inflammatory response to lipopolysaccharide and interferon gamma. Journal of Neuroscience Research, 77(4), 540–551.
Pyo, J. H., Jeong, Y. K., Yeo, S., Lee, J. H., Jeong, M. Y., Kim, S. H., et al. (2013). Neuroprotective effect of trans-cinnamaldehyde on the 6-hydroxydopamine-induced dopaminergic injury. Biological and Pharmaceutical Bulletin, 36(12), 1928–1935.
Shahverdi, A. R., Monsef-Esfahani, H. R., Tavasoli, F., Zaheri, A., & Mirjani, R. (2007). Trans-cinnamaldehyde from Cinnamomum zeylanicum bark essential oil reduces the clindamycin resistance of Clostridium difficile in vitro. Journal of Food Science, 72(1), S055–S058.
Shalaby, M. A., & Saifan, H. M. (2014). Some pharmacological effects of cinnamon and ginger herbs in obese diabetic rats. Journal of Intercultural Ethnopharmacology, 3(4), 144–149.
Tung, Y. T., Chua, M. T., Wang, S. Y., & Chang, S. T. (2008). Anti-inflammation activities of essential oil and its constituents from indigenous cinnamon (Cinnamomum osmophloeum) twigs. Bioresource Technology, 99, 3908–3913.
Tung, Y. T., Yen, P. L., Lin, C. Y., & Chang, S. T. (2010). Anti-inflammation activities of essential oil and its constituents from indigenous cinnamon (Cinnamomum osmophloeum) leaves. Pharmaceutical Biology, 48(10), 1130–1136.
Tuttolomondo, A., Di Sciacca, R., Di Raimondo, D., Renda, C., Pinto, A., & Licata, G. (2009). Inflammation as a therapeutic target in acute ischemic stroke treatment. Current Topics in Medicinal Chemistry, 9(14), 1240–1260.
Umezawa, K., Ariga, A., & Matsumoto, N. (2000). Naturally occurring and synthetic inhibitors of NF-kappa B function. Anticancer Drug Design, 15, 239–244.
Wang, M. J., Lin, W. W., Chen, H. L., Chang, Y. H., Ou, H. C., Kuo, J. S., et al. (2002). Silymarin protects dopaminergic neurons against lipopolysaccharide-induced neurotoxicity by inhibiting microglia activation. European Journal of Neuroscience, 16, 2103–2112.
Woo, M. S., Jang, P. G., Park, J. S., Kim, W. K., Joh, T. H., & Kim, H. S. (2003). Selective modulation of lipopolysaccharide-stimulated cytokine expression and mitogen-activated protein kinase pathways by dibutyryl-cAMP in BV2 microglial cells. Brain Research Molecular Brain Research, 113, 86–96.
Wu, K. J., Chen, Y. F., Tsai, H. Y., Wu, C. R., & Wood, W. G. (2012). Guizhi-Fuling-Wan, a traditional Chinese herbal medicine, ameliorates memory deficits and neuronal apoptosis in the streptozotocin-induced hyperglycemic rodents via the decrease of Bax/Bcl2 ratio and caspase-3 expression. Evidence-Based Complementary and Alternative Medicine. Article ID 656150.
Xie, Z., Smith, C. J., & Van Eldik, L. J. (2004). Activated glia induce neuron death via MAP kinase signaling pathways involving JNK and p38. Glia, 45, 170–179.
Yamamoto, Y., & Gaynor, R. B. (2001). Therapeutic potential of inhibition of the NF-kB pathway in the treatment of inflammation and cancer. Journal of Clinical Investigation, 107, 135–142.
Yamanishi, Y., Boyle, D. L., Pinkoski, M. J., et al. (2002). Regulation of joint destruction and inflammation by p53 in collagen—Induced arthritis. American Journal of Pathology, 160(1), 123–130.
Yang, G., Kitagawa, K., Matsushita, K., Mabuchi, T., Yagita, Y., Yanagihara, T., et al. (1997). C57BL/6 is most susceptible to cerebral ischemia following bilateral common carotid occlusion among seven mouse strains: Selective neuronal death in the murine transient forebrain ischemia. Brain Research, 752, 209–218.
Yonish-Rouach, E., Resnitzky, D., Lotem, J., Sachs, L., Kimchi, A., & Oren, M. (1991). Wild-type p53 induces apoptosis of myeloid leukaemic cells that is inhibited by interleukin-6. Nature, 352(6333), 345–347.
Youn, H. S., Lee, J. K., Choi, Y. J., Saitoh, S. I., Miyake, K., Hwang, D. H., et al. (2008). Cinnamaldehyde suppresses toll-like receptor 4 activation mediated through the inhibition of receptor oligomerization. Bichemical Pharmacology, 75, 494–502.
Zhao, X., Zhang, Y., Strong, R., Zhang, J., Grotta, J. C., et al. (2007). Distinct patterns of intracerebral hemorrhage-induced alterations in NF-kappaB subunit, iNOS, and COX-2 expression. Journal of Neurochemistry, 101, 652–663.
Zheng, L. T., Ock, J., Kwon, B. M., & Suk, K. (2008). Suppressive effects of flavonoid fisetin on lipopolysaccharide-induced microglial activation and neurotoxicity. International Immunopharmacology, 8, 484–494.
Acknowledgments
Authors would like to thank Dr Igbavboa (Department of Pharmacology, University of Minnesota School of Medicine) for guiding the QRT-PCR technique. This work was supported by Grants from the Ministry of Science and Technology (NSC95-2320-B-039-037), China Medical University (CMU-101-ASIA-06 and CMU102-ASIA-14) and the National Institutes of Health, AG-23524 and AG-18357.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Authors have no conflict of interest to declare.
Additional information
Yu-Wen Wang and Wei-Shih Huang have equally contributed as first author.
Yuk-Man Leung: Equal contribution to corresponding author.
Rights and permissions
About this article

Cite this article
Chen, YF., Wang, YW., Huang, WS. et al. Trans-Cinnamaldehyde, An Essential Oil in Cinnamon Powder, Ameliorates Cerebral Ischemia-Induced Brain Injury via Inhibition of Neuroinflammation Through Attenuation of iNOS, COX-2 Expression and NFκ-B Signaling Pathway. Neuromol Med 18, 322–333 (2016). https://doi.org/10.1007/s12017-016-8395-9
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12017-016-8395-9