
Abstract
The mechanistic target of rapamycin (mTOR) is an important molecule that connects aging, lifespan, energy balance, glucose and lipid metabolism, and neurodegeneration. Rapamycin exerts effects in numerous biological activities via its target protein, playing a key role in energy balance, regulation of autophagy, extension of lifespan, immunosuppression, and protection against neurodegeneration. There are many similar pathophysiological processes and molecular pathways between Alzheimer’s disease (AD) and type 2 diabetes mellitus (T2DM), and pharmacologic agents used to treat T2DM, including glucagon-like peptide-1 (GLP-1) analogs, seem to be beneficial for AD. mTOR mediates the effects of GLP-1 analogs in the treatment of T2DM; hence, I hypothesize that mTOR is a key molecule for mediating the protective effects of GLP-1 for AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Evidence suggests that there may be a common molecular pathway connecting Alzheimer’s disease (AD), type 2 diabetes mellitus (T2DM) and longevity, potentially involving the mechanistic target of rapamycin (mTOR). |
mTOR is a member of the phosphatidylinositol 3-kinase-related kinase family of protein kinases and is known to regulate key biological functions such as cellular growth, proliferation and survival, protein synthesis, apoptosis, and autophagy. Dysregulation of mTOR has been implicated in T2DM, cancer, and neurodegeneration. |
Drugs used to treat T2DM, such as the glucagon-like peptide-1 (GLP-1) analogs, are currently being investigated as potential drugs for the treatment of AD. We hypothesize that mTOR mediates the protective effect of GLP-1 in AD. |
1 Background
A bioactive molecule that plays a positive role in one pathophysiologic process or disease may act in a similar way upon another. By the same token, a bioactive molecule may worsen a pathophysiologic process or disease and have the same detrimental effect in another. For example, the apolipoprotein E (APOE) ε2 isoform is beneficial for Alzheimer’s disease (AD), type 2 diabetes mellitus (T2DM), and cardiovascular disease, as well as extending one’s lifespan. On the other hand, the APOE ε4 allele is a risk factor for AD and cardiovascular disease and may shorten a patient’s lifespan [1]. Nitric oxide is beneficial for cardiovascular disease and also reduces the probability of contracting T2DM and AD [2]. This phenomenon can be understood if we treat the body as a whole. Just as APOE ε2, ε3, and ε4 affect the brain, they also affect the cardiovascular system; hence, we hypothesize that there might be a common molecular pathway connecting AD, T2DM, and longevity.
The mechanistic target of rapamycin (mTOR) is a bioactive molecule known to connect lifespan, AD, T2DM, and aging. Based on common pathophysiologic processes between T2DM and AD, as well as the publication of positive clinical data, a new therapeutic strategy is being developed that utilizes glucagon-like peptide-1 (GLP-1), a pharmacologic agent for T2DM to protect against AD. A complex linkage between T2DM, AD, mTOR, aging, and lifespan offers a clue as to why GLP-1 protects against AD.
2 Rapamycin and Mechanistic Target of Rapamycin (mTOR)
Rapamycin was first discovered in 1970 in a soil sample from Rapa Nui, and was initially studied for its potent antifungal properties. Rapamycin was subsequently identified to have a number of important biological functions, which has led to US FDA approval for several indications over the last 20 years [3].
Based on its immunosuppressant properties, rapamycin was approved by the FDA for use as a transplant medicine, particularly kidney transplantation [4]. Due to its antiproliferative effects on vascular smooth muscle cells, rapamycin was also approved by the FDA to be used in coronary-artery eluting stents [4]. Rapamycin’s beneficial effect on lifespan was shown in budding yeast in 2006, in mice in 2009, fruitfly in 2010, and nematodes in 2012. Subsequently, rapamycin was approved by the FDA to treat cardiac hypertrophy [5, 6], various cancers, including renal cell carcinoma, mantle cell lymphoma and pancreatic cancer [7], as well as tuberous sclerosis. Recently, neuroscientists have been focusing on the protective effects of rapamycin in neurodegenerative disease, particularly AD [8].
2.1 The mTOR Molecular Pathway
Rapamycin exerts its biological effects by binding to its target protein. The TOR was first identified as a physical target for rapamycin in the budding yeast and, soon afterwards, in mammals [9].
mTOR is a serine/threonine protein kinase that controls cellular growth as well as cellular homeostasis [10], and was isolated in Saccharomyces cerevisiae with the generation of rapamycin-resistant TOR mutants [11]. The TOR is divided into two subtypes: TOR1 and TOR2.
Basically, mTOR is a 289 kDa protein with a C-terminal containing 600 amino acid residues in which 110 amino acid residues are homologous with mammalian phosphatidylinositol 3-kinase (PI3K) [12, 13]. mTOR is a key protein on the PI3K pathway and regulates the protein synthesis needed for cellular growth and metabolism, as well as nutrient uptake [14, 15].
mTOR functions as part of two distinct signaling complexes with different regulatory mechanisms and functions: mTOR complex 1 (mTORC1) and mTOR complex 2 (mTORC2). mTORC1, in particular, is considered critical in the regulation of cellular functions and is activated by growth factors, including growth hormone, insulin and insulin-like growth factor-1 (IGF-1) via PI3K, phosphoinositide-dependent kinase-1 (PDK-1), and Akt [16, 17].
3 A Link between mTOR, Aging, Lifespan, Alzheimer’s Disease (AD) and Type 2 Diabetes Mellitus (T2DM)
Aging is the most significant risk factor for developing AD; however, more studies are needed to determine the pathophysiologic processes by which aging contributes to AD. mTOR is likely to play an important role.
In the last decades, scientists have been focusing on the mTORC1 pathway for its effects on lifespan and its potential as a pharmacologic target for aged-related disease. It is assumed that mTOR signaling impacts lifespan and aged-related disease by regulating autophagy-mediated clearance of damaged cellular constituents. Moreover, mTORC1 signaling contributes to the regulation of synaptic plasticity and memory formation. Hence, we speculate that pharmacologic agents inhibiting the mTOR pathway are potential interventions for delaying age-related neurodegeneration.
Santos et al. [8] reviewed the molecular interplay between mTOR and AD pathophysiological processes. Aging is a progressive process, with the accumulation of deleterious changes occurring in cells and tissue, leading to increased odds of disease and death [17].
Calorie restriction (CR) is a key age-delaying intervention. Reducing food intake by 30–40% without malnutrition can extend lifespan by 15–40% and delay the onset of age-related diseases [18].
4 Evidence for a Link between AD and T2DM
4.1 Epidemiological Evidence
Epidemiological studies have shown that T2DM almost doubles the risk of AD [19]. Individuals with T2DM are at an approximately 60% greater risk of developing dementia than those without T2DM [20]. Furthermore, elevation of glucose levels correlate with an increased risk of AD [21].
In the AD brain, some bioactive factors related to T2DM are damaged. Levels of IGF-1 binding protein-2 (IGFBP-2), a major IGF-1 binding protein in the AD temporal cortex, are decreased [22].
Utilizing positron emission tomography (PET) with the tracer 18F-fluorodeoxyglucose (FDG), Mosconi et al. [23] found a consistent reduction in energy utilization and glucose uptake, as well as progressive cerebral glucose metabolism reductions, in patients with AD. Other molecular factors associated with AD and T2DM include the presence of APOE ε4, chronic inflammation, and upregulation of apoptosis [24, 25].
4.2 Insulin and Insulin-Like Growth Factor in the Brain
Generally speaking, AD is a neurodegenerative disease that is characterized by amyloid-β (Aβ) accumulation, phosphorylated tau (τ) aggregation, and neuroinflammation. AD can also be regarded as a metabolic disease, with evidence based on epidemiologic and experimental studies showing that insulin, as well as IGF resistance, lead to the inability of the brain to utilize glucose efficiently and respond to critical trophic factor signals [26].
Glucose possesses special significance for the brain’s energy supply as its main energy source. Insulin signaling impairments may damage neuronal survival, energy production, gene expression, plasticity, and white matter integrity, etc. The relationship between disturbance of glucose metabolism, damage of insulin/IGF signaling and AD pathophysiology include (1) accumulation of the Aβ precursor protein (AβPP) and Aβ; (2) activation of kinases involved in phosphorylation of τ; (3) generation of reactive oxygen species; (4) oxidative damage and endoplasmic reticulum stress; (5) mitochondrial dysfunction; and (6) proinflammatory and proapoptosis cascades [27, 28].
Extensive expression of insulin and the IGF polypeptide and its receptor is seen in the brain, especially in AD-targeted structures. In the brain, the insulin/IGF pathway is essential for cell survival, growth, differentiation, migration, gene expression, protein synthesis, cytoskeletal assembly, energy metabolism, synapse formation, neurotransmitter function, and plasticity. Hence, impairing insulin/IGF signaling results in damage to the structural and functional integrity of the brain [28].
Deficits in cerebral glucose utilization happen at a very early stage in patients with AD, and impairments in glucose metabolism and insulin signaling may even occur ahead of the initial stages of cognitive dysfunction [29]. In other words, disturbance of glucose metabolism and insulin resistance could occur prior to an AD pathophysiologic event.
The significance of insulin signaling in the brain is not only to stimulate its glucose uptake and utilization but also to promote memory and cognition. Resistance to or deficiency of insulin in the brain damages glucose metabolism and aggravates brain energy imbalance, as well as leading to increased reactive oxygen species production, mitochondrial dysfunction, and DNA damage, all of which, as mentioned above, drive proapoptosis, proinflammatory, and pro-AβPP/Aβ cascades [30].
Talbot et al. [26] revealed molecular and biochemical evidence of insulin/IGF resistance and deficiencies in the postmortem AD brain, which included reduced levels of insulin/IGF receptor binding and responsiveness to insulin/IGF stimulation. Damage to insulin signaling is involved in the pathogenesis of AD. Suppression of the brain’s insulin receptor causes cognitive impairment, as well as molecular and biochemical abnormalities [31].
4.3 Common Animal Model for T2DM and AD
Because of shared pathophysiological processes, common animal models have been developed for T2DM and AD studies. The new AD animal models that display insulin desensitization in the brain have been previously reviewed [32].
The ob/ob transgenic mouse is a mutant mouse that is fed excessively and becomes profoundly obese. Characteristic pathological changes of AD are observed in the ob/ob mice brain, which is also used as an animal model for T2DM. Long-term potentiation (LTP) induced in area CA1 is completely abolished in ob/ob mice [33], and these animals displayed anxious behavior in an elevated plus-maze [34]. Levels of τ cleavage and hyperphosphorylation at several residues, including Ser199/202, Ser262 and Thr231 in the cortex of ob/ob mice, were increased compared with wild-type (WT) animals [35].
APP (+)-ob/ob mice (APP23 transgenic mice crossed with ob/ob mice) present Alzheimer’s-like cognitive dysfunction and show notable cerebrovascular inflammation and severe amyloid angiopathy correlated with cognitive deficits [36].
The db/db mouse is a model of obesity, T2DM, and dyslipidemia, and also exhibits profound deficits in spatial learning and memory [37,38,39]. APOE-dependent processing of the AβPP plays a big part in the development of AD. A significant upregulation of brain APOE messenger RNA (mRNA) was observed in the db/db mouse [40], while increases in phosphorylated τ were also found in db/db mice [41].
High-fat (HF) diet-induced T2DM is a classic and widely used diabetic animal model. An HF diet can also induce AD-related pathophysiology. Furthermore, it has been shown that animals receiving HF/low carbohydrate (LC) food could increase the level of soluble Aβ in the brain [42]. Rats receiving an HF diet for 10 weeks developed both insulin resistance and cognitive deficits.
In a transgenic animal model of AD, impaired learning and memory of mice overexpressing mutant AβPP and presenilin-1 (PS1dE9) is exacerbated by HF diet treatment. The HF diet alters τ isoform expression and increases phosphorylation of τ at the Ser202 site in female mice regardless of genotypes [43]. Another study has shown that a HF diet increases the Aβ burden and level of τ hyperphosphorylation. Furthermore, this administration also induces the reduction of cortical levels of the postsynaptic marker drebrin [44].
Streptozotocin, which is used to induce T2DM, is also used to induce AD in animal models. T2DM induced by streptozotocin, with administration via intraperitoneal injection, is a classic animal model with extensive utilization. Meanwhile, the administration of streptozotocin to induce AD-related pathophysiologic processes has already been widely used as a model. Insulin signaling in the brain is interrupted by intracerebroventricular (ICV) administration of streptozotocin. A range of AD pathophysiologies, including cognitive impairment, brain cholinergic signaling deficits, oxidative stress, Aβ burden, and τ hyperphosphorylation, have been induced by streptozotocin ICV administration [45,46,47]. Rats with brain insulin desensitization induced by streptozotocin ICV administration have also presented τ protein hyperphosphorylation and Aβ-like aggregations in meningeal vessels [48].
Chen et al. [49] studied 84 AD-related genes that are involved in APP processing, τ/cytoskeleton, synapse function, autophagy, apoptosis, AD-related protein kinases, insulin signaling, glucose metabolism, and the mTOR pathway. They compared the expressions of these genes in the hippocampus and the cerebral cortex of streptozotocin-treated mice with that of 3×Tg-AD mice in quantitative polymerase chain reaction (q-PCR) arrays. By comparison, more genes related to insulin signaling and glucose metabolism are downregulated in streptozotocin mice [49].
4.4 Impairment of Insulin Signaling and Amyloid-β Neurotoxicity
Studies focusing on the relationship between T2DM and AD have shown that insulin/IGF is a key factor for normal AβPP/Aβ metabolism, and impairment of insulin/IGF signaling leads to Aβ pathology; insulin boosts trafficking of AβPP/Aβ from the trans-Golgi network to the plasma membrane for extracellular secretion [50]. AβPP/Aβ oligomers inhibit the neuronal transmission of insulin-stimulated signals by desensitizing and reducing the surface expression of insulin receptors. Furthermore, AβPP/Aβ interferes with insulin’s binding affinity to its own receptor, damaging insulin signaling and worsening insulin resistance [51]. Impairments of insulin signaling wreck AβPP processing, as well as AβPP/Aβ clearance [52], while insulin also prevents the intracellular accumulation and degradation of AβPP-Aβ [53]. Intracellular AβPP/Aβ directly interferes with PI3 kinase activation of Akt, impairs neuronal survival, and increases glycogen synthase kinase (GSK)-3β, as well as hyperphosphorylation of τ.
4.5 Impairment of Insulin Signaling and Tau Phosphorylation
Insulin/IGF signaling regulates τ expression and hyperphosphorylation. PI3K connects mTOR, T2DM, and AD as an upstream factor of mTOR. In the AD brain, insulin/IGF resistance promotes τ hyperphosphorylation by regulating PI3K, Akt, and Wnt/β-catenin. GSK is a rate-limited enzyme in the synthesis process of glycogen, and also promotes τ hyperphosphorylation. Insulin/IGF resistance also promotes τ hyperphosphorylation by activating GSK-3β [54]. Inhibiting insulin and IGF signaling impairs τ gene expression and further induces AD τ pathology.
4.6 Insulin Resistance and Oxidative Stress in AD
Considering that glucose is a key energy source for the brain, impairment of insulin signaling inevitably leads to deficiencies in the brain’s energy metabolism. Consequently, the brain is damaged by oxidative stress, mitochondrial dysfunction, and proinflammatory cytokine activation [55]. Oxidative stress attacks bioactive substances and damages molecules such as DNA, RNA, lipids, and proteins. As a result, the structural and functional integrity of the brain’s neurons is impaired due to the loss of membrane functions, disruption of the cytoskeleton, dystrophy of synaptic terminals, deficits in neurotransmitter functions and plasticity, and the perturbation of signaling pathways for energy metabolism, homeostasis, and cell survival [30].
4.7 Further Evidence of a Relationship between AD and T2DM
Besides the relationship between T2DM and AD reviewed above, there is more evidence connecting T2DM to AD [32, 56, 57]. Such evidence includes a study that found decreases in IGFBP-2, a major IGF-1 binding protein, in the AD temporal cortex [22]. This indicates that insulin and IGF signaling is impaired in patients who are not diabetic, resulting in the impairment of glucose utilization, cell metabolism, growth factor gene expression, and cell repair.
Compared with the age-matched control group, AD patients have shown regional glucose metabolism impairment in the parietal-temporal lobe, posterior cingulate cortex, and frontal areas [58, 59]. The reduction in energy utilization and glucose uptake has been measured using PET with the tracer FDG, and a clear reduction in the PET signal has been found in the cortex of AD patients. Consistent and progressive cerebral glucose metabolism reduction has been demonstrated in the AD brain utilizing FDG-PET studies [23]. Other molecular factors associated with AD and T2DM include the presence of APOE ε4, chronic inflammation, and the upregulation of apoptosis [60, 61].
Table 1 summarizes some of the key commonalities between T2DM and AD.
5 Impact of T2DM Therapies on AD
Numerous studies have demonstrated that pharmacologic agents that are used to treat T2DM also benefit patients with AD [24], which implies a common molecular pathway to explain the protective effect of GLP-1 for T2DM and AD.
mTOR function, interlinking aging, T2DM and AD, offers a possible molecular explanation for why pharmacological agents for the treatment of T2DM can also inhibit AD pathophysiological processes.
5.1 Neuroprotective Effects of Antidiabetic Drugs for AD
Some of the neuroprotective effects of pharmacologic agents for T2DM have previously been demonstrated (Table 2). It has been reported that insulin injection successfully alleviated streptozotocin-induced cognitive decline, as well as improved learning and memory [62]. In addition, insulin injection has been shown to reduce Aβ, as well as increase hippocampal levels of insulin-degrading enzymes, insulin receptor expression, Akt, and somatostatin [62]. Moreover, insulin and IGF have been shown to prevent brain atrophy and cognitive impairment induced by streptozotocin ICV treatment in rats [63].
Other kinds of drugs have been developed to increase insulin sensitivity, and these may also induce the desired neuroprotective effects. In obese, leptin-resistant (db/db) mice, metformin was shown to attenuate the rise of total τ and phosphorylated τ, attenuate the loss of synaptophysin, and cause a drop in Aβ levels [32].
Glimepiride, a sulphonylurea used for the treatment of T2DM, protected neurons against synapse damage induced by Aβ. A possible molecular mechanism of treatment is associated with the loss of specific glycosylphosphatidylinositol-anchored proteins, including the cellular prion protein that acts as a receptor for Aβ42, increased synaptic gangliosides, and altered cell signaling [64]. In addition, glimepiride attenuates Aβ-induced cholesterol increase and activation of cytoplasmic phospholipase A2 in synapses. Furthermore, soluble cellular prion protein released from neurons treated by glimepiride neutralised Aβ-induced synapse damage [48].
Based on the hypothesis that a copper-overload state is central to the pathogenesis of diabetic and AD damage, Cooper [65] proposed an important new target for therapeutic intervention, suggesting that triethylenetetramine (TETA), a highly selective divalent copper [Cu(II)] chelator, prevents or reverses diabetic and AD copper overload, thereby suppressing oxidative stress.
Alagiakrishnan et al. [66] reviewed the protective effects of antidiabetic drugs for patients with early AD and mild cognitive impairment. Metformin has shown a neuroprotective effect and prevented neuronal apoptosis induced by etoposide or oxygen-glucose deprivation. One possible molecular mechanism to explain this effect is that metformin reverses insulin receptor phosphorylation and hence improves neuronal survival [67]. Gupta et al. [68] showed that metformin enhanced insulin action and prevented the molecular and pathological changes observed in AD in a cell culture line of insulin resistance.
In an epidemiologic study involving subjects aged 50 years and older, Hsu et al. [69] found that metformin and sulfonylureas significantly decreased the risk of dementia. Glyburide and glipizide have been demonstrated to have properties of mTOR antagonists but their efficacy in preventing AD needs to be determined [70].
Intranasal administration of insulin is used to improve cognition due to rapid delivery of insulin to the brain via olfactory and trigeminal perivascular channels. Intranasal insulin has been shown to protect against neuronal insulin resistance induced by Aβ oligomers [71]. An epidemiologic survey showed that intranasal insulin administration of 20 IU over a period of 21 days resulted in subjects with early AD able to retain more verbal information and show superior attention and functional status [72].
A protective effect of rosiglitazone has been shown against neuronal insulin resistance induced by Aβ oligomers [73]. Furthermore, pioglitazone improved cognitive performance in streptozotocin-treated rats [74]. In a 6-month trial, Watson et al. [75] showed that rosiglitazone, at a dose of 4 mg, prevented memory loss in patients with early AD and amnestic mild cognitive impairment (MCI). Similarly, Risner et al. [76] also showed that rosiglitazone ameliorated impairment of brain glucose metabolism.
5.2 Protective Effects of Glucagon-Like Peptide-1 (GLP-1) for AD
GLP-1, a new pharmacologic agent for T2DM, has also shown neuroprotective effects. Our studies indicated that [Val(8)] GLP-1 might reverse behavioral and pathological damage induced by streptozotocin, including damage to learning and memory ability, reduction of phosphorylated τ levels, and pathologic changes to the nucleus and nucleolus [77].
It has been reported that treatment with exedin-4, a GLP-1 mimetic, improves impaired learning and memory performance, and also ameliorates τ hyperphosphorylation induced by streptozotocin ICV [49].
Liraglutide, another analog of GLP-1, was shown to rescue the hippocampal LTP that is severely impaired in ob/ob mice [33]. Treatment with liraglutide was also shown to improve impaired spatial learning and memory, reduce brain levels and aggregation of Aβ, increase cellular proliferation and differentiation into neurons, reduce synapse loss, and attenuate inflammation response in the brain [78].
Drugs that act as incretin receptor agonists or inhibit the proteolytic degradation of incretins [dipeptidyl peptidase 4 (DPP4) inhibitors] have been approved for use in T2DM treatment since 2005. The neuroprotective properties of these drugs have recently been shown in cell cultures and animal models [79].
Geniposide, a monomer extracted from the fruits of G. Jasminoides Ellis, is a GLP-1 receptor agonist that appears to possess pharmacologic functions in protecting against T2DM and AD [80]. Our study showed that the central administration of geniposide reversed streptozotocin-induced spatial learning deficits by 40%, reduced τ phosphorylation by 30%, and averted streptozotocin-induced neural pathology, including paired helical filament (PHF)-like structures, accumulation of vesicles in the synaptic terminal, abnormalities of endoplasmic reticulum, and early stages of apoptosis. In molecular terms, we showed an elevated expression of GSK3β(pS-9) but suppressed GSK3β(pY-216), indicating that geniposide reduced streptozotocin-induced GSK3β hyperactivity [81].
6 mTOR: A Key Molecule in AD Protection
The mTOR protein is expressed throughout the body and is present in the brain, cardiopulmonary system, gastrointestinal system, immune system, skeletal system, and reproductive system [82], and has extensive biologic effects in the brain.
6.1 Autophagy: A Key Way for mTOR to Protect Against AD
Autophagy is essential for cell survival, proliferation, differentiation, and homeostasis via a catabolic pathway for the degradation of cytoplasmic material in the lysosomal system.
Recent studies have shown that autophagy may be a key pathway to connect mTOR and AD. Inhibition of mTOR promotes autophagy and in turn enhances cell survival in the event of decreased nutrient availability via the breakdown of cell constituents into amino acids and other small molecules [83]. Autophagy is also expected to pay an important role in prolonging lifespan induced by CR and sirtuin 1 (SIRT1) activators [84].
mTOR is involved in cellular events via several pathways: regulating translation and ribosome biogenesis in the biosynthetic process, eliminating damaged cellular constituents by autophagy, and regulating metabolism. Hence, mTOR is regarded as a key molecule in the control of the aging process.
Based on the evidence outlined in this article, we hypothesize that mTOR inhibitors are a promising candidate to slow the aging process and the onset of age-related disorders such as cancer, cardiovascular disease, T2DM, and neurodegenerative disease. Rapamycin enhances the autophagic clearance of different proteins with long polyglutamines, and reduces their toxicity [85]. Caccamo et al. [86] explored the effect of mTOR in the cognitive deficits associated with AD and found that suppression of mTOR signaling rescued memory deficits and reduced Aβ deposits. Mechanistically, the reduction in mTOR signaling may promote the autophagy and restoration of hippocampal gene expression.
6.2 mTOR and Neurodegeneration
Maiese [87] thoroughly reviewed the protective effect of mTOR in neural regeneration. mTOR might also protect against neurodegenerative diseases by promoting neural regeneration. Using multiple animal models and complementary genetic and pharmacological approaches, Caccamo et al. [88] explored the action of mTOR in τ phosphorylation and neurodegeneration and its molecular mechanism. They showed that enhancing mTOR activity elevates total τ and phosphorylated τ, and inhibiting mTOR signaling ameliorates τ pathology and the associated behavioral deficits. Mechanistically, they evidenced that GSK3β and autophagy are involved in the association between mTOR and τ. Furthermore, Caccamo et al. hypothesized that hyperactive mTOR signaling may present a molecular pathway by which aging contributes to the development of AD. Autophagy is also attributed to Aβ peptide generation from AβPP during autophagic turnover of APP-rich organelles.
Given that aging is the most important risk factor for most neurodegenerative diseases, the effects of mTOR on neuronal stem cell should be investigated further. As a key molecule regulating lifespan, understanding the role of mTOR signaling in neuronal stem cell development and migration might contribute to the understanding of neurodegenerative diseases. Activating the mTOR pathway is a key step in neuronal stem cell premature differentiation and impaired maturation [89], and 4EBP is a key target for neural stem cell self-renewal [90]. mTOR pathways control neuronal migration and cortical patterning [91], as well as insulin-induced neuronal differentiation of neuronal progenitor cells [92]. Utilizing mTOR(+/−) mice, Gangloff et al. [93] showed that in the absence of mTOR signaling, embryonic stem cell proliferation is inhibited. mTOR is also required for neural stem cell differentiation and neural stem cell self-renewal.
Extensive neuroprotective protective pathways originating from mTOR involve not only PI3K and protein kinase B (PKB)/Akt but also growth factors, transcription factors, wingless pathways, sirtuins, neurotransmitter modulation, and lipid metabolism [94]. As a result, neuronal protection and regeneration may require mTOR modulation in conjunction with multiple signaling components of mTOR, such as phosphorylated p70S6K, proline-rich Akt substrate of 40 kDa, BAD, and Bcl-xL [95].
The degree of mTOR activation plays a critical role in neuronal protection and regeneration. mTOR activation protects dopamine neurons from oxidative stress-mediated autophagy [96]. Conversely, prolonged activation of mTOR leads to dyskinesia in AD patients [97]. Inhibition of mTOR in models of AD induces the reduction of β-site APP cleaving enzyme-1 [98], inhibiting senile plaque formation and preventing Aβ generation [99]. In contrast, mTOR activation protects neuronal networks controlling memory [100]. Thus, it seems that the balance of mTOR activity is critical for neuroprotection and prevention against neurodegenerative disease.
6.3 Linking mTOR and AD
Aging is the most important risk factor for AD. In fact, the normal aging brain also undergoes processes that resemble those occurring in the AD brain. mTOR is involved in the basic molecular mechanisms of the aging brain, neuronal development, and plasticity [101]. AD is pathologically characterized by the formation of extracellular senile plaques composed of Aβ and intracellular neurofibrillary tangles (NFTs) containing hyperphosphorylated τ protein, and clinically characterized by the decline in learning and memory, change of personality, and decrease in judgment, vision, and association sensory-motor function.
Wu et al. [102] showed that CR significantly prolongs lifespan via gene regulation. The expression levels of 490 genes are regulated by CR in an age-dependent manner, with the most obvious impact at middle age. These genes, especially those related to the Wingless (Wnt) and Notch signaling pathways, are mainly involved in development and neurogenesis. The numerous genes involved in stress and inflammatory responses, as well as apoptosis, are also regulated by CR. Furthermore, CR altered the expression of genes associated with mTOR. The inhibition of mTOR by rapamycin could significantly prevent neuronal apoptosis induced by paraquat [102]. All of these revealed a close linkage between CR and mTOR signaling in neuroprotection.
Utilizing an animal model expressing heterozygous mutations in the TSC1 or TSC2 genes, Ehninger et al. [103] showed that cognitive dysfunction was reversed by inhibiting mTOR. Rapamycin rescued not only synaptic plasticity but also behavioral decline. Similarly, Puighermanal et al. [104] also showed that delta-9-tetrahydrocannabinol-induced memory deficits are rescued by rapamycin. Ramírez et al. [105] showed that rapamycin increases the expression of SV2, a presynaptic vesicular protein linked to neurotransmitter release, resulting in increasing frequency of miniature excitatory postsynaptic currents and calcium transients of rat hippocampal primary neurons. As a result, rapamycin-treated hippocampal neurons are resistant to the synaptotoxic effect induced by Aβ oligomers.
The relationship between Aβ, phosphorylated τ, and mTOR arouses a medical hypothesis that mTOR is a key molecular pathway in the AD pathophysiologic process. Lafay-Chebassier et al. [106] showed an impairment of the mTOR signaling pathway and inhibition of the phosphorylation of S6K in vivo and vitro, as well as a positive correlation of the ratio of phospho-S6K/S6K with Mini-Mental State Examination scores in AD patients. Aβ-induced deregulation of the mTOR pathway led to the impairment of synaptic plasticity in the Tg2576 transgenic mouse model, which was rescued by mTOR upregulation [107]. Conversely, other researchers have demonstrated that rapamycin exacerbates Aβ production [108] and promotes Aβ production via inhibition of an enzyme involved in the non-amyloidogenic processing pathway [109]. Some studies suggest that rapamycin protects against AD, while others found opposite results. Studies have demonstrated that Aβ oligomers, but not fibrils or monomers, lead to an elevation in the levels of activated mTOR and Akt. Inhibitors of mTOR, Akt, or PI3K blocked Aβ oligomer-induced neuronal cell cycle events [110].
Rapamycin also inhibited expression of τ in wild type and transgenic mice [85]. Similarly, rapamycin reversed behavioral deficits and autophagy-dependent decrease in Aβ and hyperphosphorylated τ in PDAPP transgenic mice [99], as well as in 3×TgAD mice, and might regulate translation of τ mRNA in the AD brain [111].
Li et al. [112] investigated the relationship between levels of translation control elements, including total and phosphorylated forms of mTOR, 4E-BP1, eEF2, and τ, in the medial temporal cortex from 20 AD and 10 control brains. They showed that levels of phosphorylated mTOR (Ser2481) and phosphorylated 4E-BP1 (Thr70 and Ser65) dramatically increase in the AD brain, and are positively significantly correlated with total τ and phosphorylated τ. Levels of phosphorylated eEF2K significantly increased and total eEF2 significantly decreased in AD compared with controls. This suggests that there are obvious abnormalities of elements related to translation control in the AD brain, and their aberrant changes may upregulate the translation of τ mRNA, contributing to hyperphosphorylated τ accumulation in NFT-bearing neurons.
Overexpression of SIRT1 improves cognitive decline and reverses the AD pathophysiologic processes. Julien et al. [113] showed that reduction of SIRT1 correlates with the accumulation of Aβ and τ in the cerebral cortex of AD patients. SIRT1 modulates Aβ production, while mTOR regulates autophagy. This interplay between SIRT1, mTOR, Aβ, and autophagy implies a promising therapeutic intervention.
It is widely accepted that mTOR is a key molecule involved in AD; however, the effect of rapamycin in AD is more controversial since it alleviates the neuropathological burden in some studies but exacerbates AD pathologic events in others. More studies must be conducted to clarify the role of mTOR, as well as the potential therapeutic effects of rapamycin, in AD.
mTOR is involved in Aβ metabolism. The build-up of Aβ improves mTOR signaling, whereas a decrease in mTOR signaling debases Aβ levels. The mTOR is a key molecule for controlling protein homeostasis and hence neuronal functions. Furthermore, mTOR signaling regulates learning and memory, and proteins, related to neurodegeneration. Caccamo et al. [88, 111] showed that pharmacologically repairing the mTOR signaling pathway with rapamycin reverses cognitive decline and ameliorates Aβ and τ pathology by increasing autophagy. Indeed, autophagy induction is involved in the inhibition of Aβ mediated by rapamycin. Such results provide evidence that rapamycin can improve learning and memory and reduce Aβ and τ pathology.
Wang et al. [114] reported that curcumin, an inhibitor of Aβ, not only attenuated cognitive impairment but also inhibited the generation of Aβ in APP/PS1 double transgenic AD mice. Furthermore, Wang et al. [114] also showed that curcumin significantly inhibited the expression of PI3K, phosphorylated Akt, and mTOR. Therefore, the authors hypothesized that curcumin inhibits Aβ generation and induces autophagy by downregulating the PI3K/Akt/mTOR signaling pathway. Tramutola et al. [115] analyzed the PI3K/Akt/mTOR pathway in post-mortem tissue of the inferior parietal lobule at three different stages of AD: late AD, amnestic MCI and preclinical AD. They found that the alteration of mTOR signaling and autophagy occurs at the early stage of AD. This study signified that hyperactivation of the PI3K/Akt/mTOR pathway was synchronous with autophagy impairment in the brain of MCI and AD subjects. Further increases of two mTOR downstream targets (p70S6K and 4EBP1) was also found in AD and MCI subjects. The study by Tramutola et al. [115] revealed the link between Aβ and the PI3K/Akt/mTOR axis, and provided further insights into the relationship between AD pathology and insulin resistance.
The μ-opioid receptor (OPRM1) performs extensive biologic functions, including antinociception, neuroprotection, hippocampal plasticity, and activation of the downstream effectors of mTOR. Wang et al. [116] assessed the effects of OPRM1 activation on Aβ oligomer-induced neurotoxicity and the activities of mTOR, Akt, and p70 S6k in primary cultured cortical neurons, with the results showing that morphine, an agonist of OPRM1, dose-dependently weakened Aβ oligomer-induced neurotoxicity. Morphine significantly rescued mTOR signaling by reversing changes to mTOR and its upstream signaling molecule Akt, as well as its downstream molecule p70 S6k, that were induced by Aβ oligomers. The neuroprotective effect of morphine is reversed by an OPRM1 selective antagonist and PI3K, Akt, and mTOR inhibitors. Wang et al. [116] hypothesized that OPRM1 activation attenuated Aβ oligomer-induced neurotoxicity via an mTOR signaling pathway.
It is well known that leptin inhibits appetite; however, a recent study also showed that leptin protects against AD. To explore whether the PI3K/Akt/mTOR pathway is involved in the AD protective effect of leptin, Yamamoto et al. [117] treated neurons with leptin and inhibitors of PI3K, Akt, and rapamycin, and measured Aβ expression. The results showed that Aβ assembled on the cell surface of neurons was greatly inhibited after treatment with leptin. This reduction was markedly inhibited after pretreatment with LY294002, triciribine, and rapamycin. The results also suggested that leptin significantly inhibits Aβ assembly in the neuronal surface through the PI3k/Akt/mTOR pathway.
It is also well known that suppression of the ribosomal protein S6K1 increases both healthspan and lifespan. Therefore, Caccamo et al. [118] explored the relationship between S6K1 expression and AD pathophysiologic processes. Using a mouse model of AD, they found that genetic reduction of S6K1 improved synaptic plasticity and spatial memory deficits, and reduced the accumulation of Aβ and τ. Mechanistically, S6K1 dysregulation is involved in the decline of synaptic and memory function, as well as the accumulation of Aβ and phosphorylation of τ. Hence, S6K1 may represent a molecular link between aging and AD.
7 mTOR, Insulin Signaling and T2DM
The mTOR pathway is important in insulin signaling, and abnormity in this pathway is involved in hypoinsulinemia, glucose intolerance, insulin insensitivity to glucose secretion, and a decrease in β-cell size [119]. An increase of phosphorylation of p70S6K and 4EBP1 improves insulin secretion and resists β-cell damage induced by streptozotocin [120]. In contrast, mTOR inhibition leads to insulin resistance, reduces β-cell function and mass, limits insulin secretion, and results in T2DM [121]. Although inhibition of mTOR with rapamycin reduces food intake and prevents HF diet-induced obesity, rapamycin administration attenuates glucose uptake and metabolism in skeletal muscle through prevention of insulin-generated Akt activation and alteration in the translocation of glucose transporters to the plasma membrane [122].
Although insulin is a potent activator of mTOR through Akt regulatory pathways, mTOR may have a negative feedback loop, and led to glucose intolerance through inhibition of the insulin receptor substrate 1 (IRS-1). For example, mTOR signaling through the tuberous sclerosis complex (TSC1, hamartin/TSC2, tuberin) can inactivate IRS and phosphorylate p70S6K to block IRS-1 activity by direct phosphorylation [123]. As a result, activation of the mTOR pathway may lead to poor insulin signaling and insulin resistance.
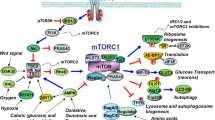
mTOR is activated by insulin and in turn accelerates the AD pathologic processes (Fig. 1).

Insulin activates mTOR through PI3K/Akt-mediated pathways. PI3K is activated by insulin and subsequent activation of Akt. Once active, Akt can result in the activation of mTOR through a series of signaling pathways. Akt can also directly phosphorylate PRAS40 and reduce its binding to Raptor, and release mTORC1 from its suppression by PRAS40. Upon activation, mTOR phosphorylates its two major downstream targets, p70S6K and eukaryotic initiation factor 4E-BP1, and mediates pathologic processes that relate to AD. 4E-BP1 4E-binding protein 1, AD Alzheimer’s disease, Akt protein kinase B, mLST8 mammalian lethal with SEC 13 protein 8, mTOR mechanistic target of rapamycin, mTORC1 mechanistic target of rapamycin complex 1, p70S6K 70 kDa ribosomal protein S6 kinase, PI3K phosphoinositide 3 kinase, PRAS 40 proline-rich AKT substrate 40 kDa
8 mTOR as a Regulator of GLP-1 in T2DM and AD
Recent research suggests that mTOR might be a key molecule for mediating the protective effect of GLP-1 in both T2DM and AD. GLP-1 enhances glucose-dependent proinsulin biosynthesis and secretion, and stimulates cellular growth and proliferation. Kwon et al. [124] demonstrated that GLP-1 dose-dependently enhances phosphorylation of S6K1 in a rapamycin-sensitive manner. GLP-1-derived cyclic adenosine monophosphate (cAMP) is involved in the metabolic activation of mTOR by mobilizing intracellular calcium stores, to upregulate mitochondrial dehydrogenases and enhance ATP production. ATP is then involved in modulating KATP channels as a substrate for adenylyl cyclase, and possibly directly regulating mTOR activation.
Based on the investigation of the coordinated effects of glucose and GLP-1 on the expression of adenosine 5′-monophosphate-activated protein kinase (AMPK) and p70S6K, Hurtado-Carneiro et al. [125] showed that GLP-1 treatment attenuated the activities of AMPK and p70S6K. Furthermore, they also showed that GLP-1 modulated AMPK and p70S6K activities in both fasted and refed obese Zucker and lean control rats. Xie et al. [126] showed that GLP-1 stimulates β-cell proliferation via the phosphorylated PKB-dependent stimulation of mTORC1/S6K1, whose activation is mediated through the autocrine/paracrine activation of the IGF-1 receptor (IGF-1R). They further hypothesized that the mTOR/S6K1 molecular pathway is key to islet cell DNA replication induced by GLP-1 in the presence of glucose, and that this pathway is activated by stimulating IGF-1R, as small interfering RNA (siRNA)-mediated knockdown of the IGF-1R effectively blocked exendin-4-stimulated PKB and mTORC1 activation [126].
Based on the effect of autophagy in Aβ clearance, Jiang et al. [127] explored whether temsirolimus, an mTOR inhibitor, promotes autophagic clearance of Aβ and thus provides protective effects in cellular and animal models of AD. They showed that temsirolimus enhanced Aβ clearance in HEK293-APP695 cells and the brain of APP/PS1 mice in an autophagy-dependent manner. Meanwhile, temsirolimus attenuated cellular apoptosis in the hippocampus of APP/PS1 mice, which was accompanied by an improvement in spatial learning and memory abilities.
In order to explore the effect of the mTOR molecular pathway in which GLP-1 treats T2DM and protects against AD pathologic changes, Kimura et al. [128] measured the expression and phosphorylation of PI3K/Akt/mTOR/GCLc/redox balance in PC12 cells treated with methylglyoxal and/or GLP-1. The result showed that enhancement of PI3K/Akt/mTOR/GCLc/redox signaling is key for the neuroprotective effect of GLP-1.
Methylglyoxal is a highly reactive α-oxoaldehyde with strong oxidant and glycation properties that induces severe disorders, e.g. obesity, metabolic syndrome, T2DM, aging, cardiovascular atherosclerosis, hypertension, psoriasis, and AD. GLP-1 protects against methylglyoxal-induced apoptosis, which corresponded to the physiopathology of PI3K, Akt, and mTOR, as well as the following upregulation of GCLc and restoration of the redox imbalance. Inhibition of PI3K, Akt, and mTOR, induced by their inhibitors LY294002, Akt-1, and rapamycin, weakens the protective effect of GLP-1, e.g. GCLc upregulation and inhibition of methylglyoxal-induced PC12 apoptosis. Similarly, the GLP-1-induced redox restoration was also attenuated by rapamycin. Furthermore, Chang and Tseng [129] showed that GLP-1, but not metformin, suppressed prolonged AMPK activation, and reversed apoptosis and mitochondrial dysfunction induced by methylglyoxal. They hypothesized that mechanistically, PKA- and PI3K-dependent pathways can be attributed to the protective effect of GLP-1.
The AMPK-related family of kinases contains 13 members. These kinases regulate glucose and energy homeostasis activated in response to an increase in AMP/ATP ratio induced by low-nutrient conditions. AMPK is an inhibitor of mTOR signaling, which coordinates nutritional status with protein synthesis in pancreatic β cells. Synapses of amphids defective (SAD-A) kinase is one of AMPK-related kinases [130]. Studies have revealed an extensive role of SAD-A kinase, from stimulating glucose-stimulated insulin secretion (GSIS), islet β-cell size and mass, to regulation of GLP-1 response as the first tissue-specific effector of mTORC1 signaling. SAD-A is extensively expressed in the pancreas and the brain [130]. What is interesting is that SAD-A is exclusively expressed in the primary target tissue of GLP-1 and hence causes weight loss by suppressing appetite [131]. SAD-A is a pancreas-specific regulator of the GLP-1 effect in islet β cells. GLP-1 activates islet β cells, and overexpression of SAD-A greatly enhances the GLP-1 effect on GSIS. In the brain, SAD-A controls neuronal polarity and axon specification [132].
Cumulative evidence suggests a critical role of mTORC1 in regulating pancreatic β-cell mass and function. Repressors of mTORC1 lead to increased islet β-cell mass and enhanced GSIS [133]. Targeted deletion of S6K1 or ablation of the S6K1 phosphorylation site leads to hypoinsulinemia, defective GSIS, and a reduction in islet β-cell size [134]. Furthermore, inhibition of mTORC1 with rapamycin also reduces islet mass and insulin content, and exacerbates T2DM [135]. Nie et al. [136] demonstrated that SAD-A kinase plays a key role in regulating β-cell function and size as a specific mediator of mTORC1 signaling in islet β cells. Global SAD-A deletion damages islet β-cell functions, which include growth retardation, hypoinsulinemia, insulin deficiency, petite islets, and diminished β-cell mass [119]. Conversely, overexpression of SAD-A obviously increases MIN6 β cells. Activation of SAD-A is induced by GLP-1 and forskolin in islet cells, and overexpression of SAD-A significantly enhances the GLP-1 effect on GSIS from isolated mouse islets [137].
Recent studies provide more evidence for the hypothesis that mTOR exerts a key effect in the protection of GLP-1. Xu et al. [138] showed that GLP-1 activated AMPK, and hence inhibited cell proliferation and fibronectin secretion in rats, which may partly explain the benefits of GLP-1 on the kidney. Briaud et al. [139] demonstrated that glucose-induced Erk-1/2 phosphorylation was inhibited by blocking calcium influx with verapamil, or inhibiting protein kinase A (PKA) with H89. Increasing cAMP levels by GLP-1 potentiated glucose-induced Erk-1/2 phosphorylation via PKA activation. Glucose markedly activates mTOR and promotes DNA synthesis in an amino acid-dependent manner in islets.
In conclusion, the neuroprotective effect of GLP-1 is due to an enhancement of PI3K/Akt/mTOR/GCLc/redox signaling. To explore whether GLP-1 protects β cells via AMPK/mTOR signaling, Miao et al. [140] evaluated the effect of GLP-1 on cellular ATP levels and mTOR pathway protein expression. Their results suggested that the enhancement of β-cell proliferation induced by GLP-1 is mediated, at least in part, by AMPK/mTOR signaling. GLP-1 also prevents β-cell glucolipotoxicity by activating mTOR. Zhang et al. [141] showed that GLP-1 decreased islet apoptosis, enhanced islet function and survival, and promoted expression of the IGF receptor, phosphorylated Akt and phosphorylated mTOR in islets of T2DM monkeys. The protective effects of GLP1 on streptozotocin-induced islet injury were inhibited by blocking the IGFR/Akt/mTOR signaling pathways utilizing NVP-AEW541 or Wortmannin in vitro. Hence, activation of the IGFR/Akt/mTOR signaling pathways is important for the protective effects of GLP-1 on streptozotocin-induced islet injury.
9 Conclusions
Common pathophysiologic processes between AD and T2DM expedite a strategy for AD treatment utilizing GLP-1, a pharmacologic agent currently used to treat T2DM. Rapamycin exerts extensive biological activities via mTOR, with roles in energy balance, regulation of autophagy, extension of lifespan, immunosuppression, and protection against neurodegeneration. Accumulated evidence shows that mTOR might be involved in the molecular pathway by which GLP-1 protects against T2DM. Based on a review of the relationship between mTOR, AD, aging, T2DM, and GLP-1, we put forward a hypothesis that GLP-1 shows a protective effect against AD via the mTOR molecular pathway.
References
Martins IJ, Hone E, Foster JK, Sünram-Lea SI, Gnjec A, Fuller SJ, et al. Apolipoprotein E, cholesterol metabolism, T2DM, and the convergence of risk factors for Alzheimer’s disease and cardiovascular disease. Mol Psychiatry. 2006;11:721–36.
Wang F, Guo X, Shen X, Kream RM, Mantione KJ, Stefano GB. Vascular dysfunction associated with type 2 T2DM and Alzheimer’s disease: a potential etiological linkage. Med Sci Monit Basic Res. 2014;20:118–29.
Johnson SC, Rabinovitch PS, Kaeberlein M. mTOR is a key modulator of ageing and age-related disease. Nature. 2013;493:338–45.
Tsang CK, Qi H, Liu LF, Zheng XF. Targeting mammalian target of rapamycin (mTOR) for health and diseases. Drug Discov Today. 2007;12:112–24.
Ha T, Li Y, Gao X, McMullen JR, Shioi T, Izumo S, et al. Attenuation of cardiac hypertrophy by inhibiting both mTOR and NFkappaB activation in vivo. Free Radic Biol Med. 2005;39:1570–80.
Ding Y, Sun X, Redfield M, Kushwaha S, Xu X. Target of rapamcyin (TOR)-based therapeutics for cardiomyopathy: insights from zebrafish genetics. Cell Cycle. 2012;11:428–9.
Garber K. Targeting mTOR: something old, something new. J Natl Cancer Inst. 2009;101:288–90.
Santos RX, Correia SC, Cardoso S, Carvalho C, Santos MS, Moreira PI. Effects of rapamycin and TOR on aging and memory: implications for Alzheimer’s disease. J Neurochem. 2011;117:927–36.
Laokabte M, Sabatini DM. mTOR signaling in growth control and disease. Cell. 2012;149:274–93.
Chong ZZ, Shang YC, Zhang L, Wang S, Maiese K. Mammalian target of rapamycin: hitting the bull’s-eye for neurological disorders. Oxid Med Cell Longev. 2010;3:374–91.
Chong ZZ, Maiese K. Mammalian target of rapamycin signaling in diabetic cardiovascular disease. Cardiovasc Diabetol. 2012;11:45.
Kunz J, Henriquez R, Schneider U, Deuter-Reinhard M, Movva NR, Hall MN. Target of rapamycin in yeast, TOR2, is an essential phosphatidylinositol kinase homolog required for G1 progression. Cell. 1993;73:585–96.
Sabatini DM, Pierchala BA, Barrow RK, Schell MJ, Snyder SH. The rapamycin and FKBP12 target (RAFT) displays phosphatidylinositol 4-kinase activity. J Biol Chem. 1995;270:20875–8.
Sonenberg N, Hinnebusch AG. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 2009;136:731–45.
Avruch J, Long X, Ortiz-Vega S, Rapley J, Papageorgiou A, Dai N. Amino acid regulation of TOR complex 1. Am J Physiol Endocrinol Metab. 2009;296:E592–602.
Stanfel MN, Shamieh LS, Kaeberlein M, Kennedy BK. The TOR pathway comes of age. Biochim Biophys Acta. 2009;1790:1067–74.
Harman D. The free radical theory of aging. Antioxid Redox Signal. 2003;5:557–61.
Masoro EJ. Subfield history: caloric restriction, slowing aging, and extending life. Sci Aging Knowl Environ. 2003;2003(8):RE2.
Schrijvers EM, Witteman JC, Sijbrands EJ, Hofman A, Koudstaal PJ, Breteler MM. Insulin metabolism and the risk of Alzheimer disease: the Rotterdam Study. Neurology. 2010;75:1982–7.
Chatterjee S, Peters SA, Woodward M, Mejia Arango S, Batty GD, Beckett N, et al. Type 2 T2DM as a risk factor for dementia in women compared with men: a pooled analysis of 2.3 million people comprising more than 100,000 cases of dementia. Diabetes Care. 2016;39:300–7.
Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, et al. Glucose levels and risk of dementia. N Engl J Med. 2013;369:540–8.
Moloney AM, Griffin RJ, Timmons S, O’Connor R, Ravid R, O’Neill C. Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging. 2010;31:224–43.
Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, et al. FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging. 2009;36:811–22.
Hölscher C. Drugs developed for treatment of T2DM show protective effects in Alzheimer’s and Parkinson’s diseases. Sheng Li Xue Bao. 2015;66:497–510.
Hölscher C. Potential role of glucagon-like peptide-1 (GLP-1) in neuroprotection. CNS Drugs. 2012;26:871–82.
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, et al. Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest. 2012;122:1316–38.
de la Monte SM, Re E, Longato L, Tong M. Dysfunctional pro-ceramide, ER stress, and insulin/IGF signaling networks with progression of Alzheimer’s disease. J Alzheimers Dis. 2012;2:S217–29.
de la Monte SM, Longato L, Tong M, Wands JR. Insulin resistance and neurodegeneration: roles of obesity, type 2 T2DM mellitus and non-alcoholic steatohepatitis. Curr Opin Investig Drugs. 2009;10:1049–60.
Hoyer S. Causes and consequences of disturbances of cerebral glucose metabolism in sporadic Alzheimer disease: therapeutic implications. Adv Exp Med Biol. 2004;541:135–52.
de la Monte SM, Tong M. Mechanisms of nitrosamine mediated neurodegeneration: potential relevance to sporadic Alzheimer’s disease. J Alzheimers Dis. 2009;17:817–25.
Labak M, Foniok T, Kirk D, Rushforth D, Tomanek B, Jasiński A, et al. Metabolic changes in rat brain following intracerebroventricular injections of streptozotocin: a model of sporadic Alzheimer’s disease. Acta Neurochir Suppl. 2010;106:177–81.
Gao C, Liu Y, Li L, Hölscher C. New animal models of Alzheimer’s disease that display insulin desensitization in the brain. Rev Neurosci. 2013;24:607–15.
Porter WD, Flatt PR, Hölscher C, Gault VA. Liraglutide improves hippocampal synaptic plasticity associated with increased expression of Mash1 in ob/ob mice. Int J Obes Lond. 2013;37:678–84.
Asakawa A, Inui A, Inui T, Katsuura G, Fujino MA, Kasuga M. Leptin treatment ameliorates anxiety in ob/ob obese mice. J Diabetes Complicat. 2003;17:105–7.
Kim B, Backus C, Oh S, Feldman EL. Hyperglycemia-induced Tau cleavage in vitro and in vivo: a possible link between T2DM and Alzheimer’s disease. J Alzheimers Dis. 2013;34:727–39.
Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, et al. T2DM-accelerated memory dysfunction viacerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with T2DM. Proc Natl Acad Sci USA. 2010;107:7036–41.
Farr SA, Banks WA, Morley JE. Effects of leptin on memory processing. Peptides. 2006;27:1420–5.
Li XL, Aou S, Oomura Y, Hori N, Fukunaga K, Hori T. Impairment of long-term potentiation and spatial memory in leptin receptor-deficient rodents. Neuroscience. 2002;113:607–15.
Oomura Y, Hori N, Shiraishi T, Fukunaga K, Takeda H, Tsuji M, et al. Leptin facilitates learning and memory performance and enhances hippocampal CA1 long-term potentiation and CaMK II phosphorylation in rats. Peptides. 2006;27:2738–49.
Knight DS, Mahajan DK, Qiao X. Dietary fat up-regulates the apolipoprotein E mRNA level in the Zucker lean rat brain. NeuroReport. 2001;12:3111–5.
Doherty GH, Beccano-Kelly D, Yan SD, Gunn-Moore FJ, Harvey J. Leptin prevents hippocampal synaptic disruption and neuronal cell death induced by amyloid beta. Neurobiol Aging. 2013;34:226–37.
Pedrini S, Thomas C, Brautigam H, Schmeidler J, Ho L, Fraser P, et al. Dietary composition modulates brain mass and solubilizable Abeta levels in a mouse model of aggressive Alzheimer’s amyloid pathology. Mol Neurodegener. 2009;4:40.
Hiltunen M, Khandelwal VK, Yaluri N, Tiilikainen T, Tusa M, Koivisto H, et al. Contribution of genetic and dietary insulin resistance to Alzheimer phenotype in APP/PS1 transgenic mice. J Cell Mol Med. 2012;16:1206–22.
Julien C, Tremblay C, Phivilay A, Berthiaume L, Emond V, Julien P, et al. High-fat diet aggravates amyloid-beta and tau pathologies in the 3xTg-AD mouse model. Neurobiol Aging. 2010;31:1516–31.
Blokland A, Jolles J. Behavioral and biochemical effects of an ICV injection of streptozotocin in old Lewis rats. Pharmacol Biochem Behav. 1994;47:833–7.
Lannert H, Hoyer S. Intracerebroventricular administration of streptozotocin causes long-term diminutions in learning and memory abilities and in cerebral energy metabolism in adult rats. Behav Neurosci. 1998;112:1199–208.
Saxena G, Patro IK, Nath C. ICV STZ induced impairment in memory and neuronal mitochondrial function: a protective role of nicotinic receptor. Behav Brain Res. 2011;224:50–7.
Salkovic-Petrisic M, Hoyer S. Central insulin resistance as a trigger for sporadic Alzheimer-like pathology: an experimental approach. J Neural Transm Suppl. 2007;72:217–33.
Chen Y, Tian Z, Liang Z, Sun S, Dai CL, Lee MH, et al. Brain gene expression of a sporadic (icv-STZ Mouse) and a familial mouse model (3xTg-AD mouse) of Alzheimer’s disease. PLoS One. 2012;7:e51432.
Watson GS, Peskind ER, Asthana S, Purganan K, Wait C, Chapman D, et al. Insulin increases CSF Abeta 42 levels in normal older adults. Neurology. 2003;60:1899–903.
Xie L, Helmerhorst E, Taddei K, Plewright B, Van Bronswijk W, Martins R. Alzheimer’s beta-amyloid peptides compete for insulin binding to the insulin receptor. J Neurosci. 2002;22:rc221.
Messier C, Teutenberg K. The role of insulin, insulin growth factor, and insulin-degrading enzyme in brain aging and Alzheimer’s disease. Neural Plast. 2005;12:311–28.
Gasparini L, Netzer WJ, Greengard P, Xu H. Does insulin dysfunction play a role in Alzheimer’s disease? Trends Pharmacol Sci. 2002;23:288–93.
Tiwari SK, Seth B, Agarwal S, Yadav A, Karmakar M, Gupta SK, et al. Ethosuximide induces hippocampal neurogenesis and reverses cognitive deficits in an amyloid-β toxin-induced alzheimer rat model via the phosphatidylinositol 3-kinase (PI3K)/Akt/Wnt/β-Catenin pathway. J Biol Chem. 2015;290:28540–58.
Hoyer S, Lannert H, Nöldner M, Chatterjee SS. Damaged neuronal energy metabolism and behavior are improved by Ginkgo biloba extract (EGb761). J Neural Transm. 1999;106:1171–88.
de la Monte SM, Wands JR. Review of insulin and insulin-like growth factor expression, signaling, and malfunction in the central nervous system: relevance to Alzheimer’s disease. J Alzheimers Dis. 2005;7:45–61.
de la Monte SM. Type 3 T2DM is sporadic Alzheimer’s disease: mini-review. Eur Neuropsychopharmacol. 2014;24:1954–60.
Friedland RP, Jagust WJ, Huesman RH, Koss E, Knittel B, Mathis CA, et al. Regional cerebral glucose transport and utilization in Alzheimer’s disease. Neurology. 1989;39:1427–34.
Baker LD, Cross DJ, Minoshima S, Belongia D, Watson GS, Craft S. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with pre T2DM or early type 2 T2DM. Arch Neurol. 2011;68:51–7.
Hölscher C. T2DM as a risk factor for Alzheimer’s disease: insulin signalling impairment in the brain as an alternative model of Alzheimer’s disease. Biochem Soc Trans. 2011;39:891–7.
Sandhir R, Gupta S. Molecular and biochemical trajectories from T2DM to Alzheimer’s disease: a critical appraisal. World J Diabetes. 2015;6(12):1223–42.
Shingo AS, Kanabayashi T, Kito S, Murase T. Intracerebroventricular administration of an insulin analogue recovers STZ-induced cognitive decline in rats. Behav Brain Res. 2013;241:105–11.
Serbedžija P, Ishii DN. Insulin and insulin-like growth factor prevent brain atrophy and cognitive impairment in diabetic rats. Indian J Endocrinol Metab. 2012;16:s601–10.
Osborne C, West E, Nolan W, McHale-Owen H, Williams A, Bate C. Glimepiride protects neurons against amyloid-β-induced synapse damage. Neuropharmacology. 2016;101:225–36.
Cooper GJ. Therapeutic potential of copper chelation with triethylenetetramine in managing T2DM mellitus and Alzheimer’s disease. Drugs. 2011;71:1281–320.
Alagiakrishnan K, Sankaralingam S, Ghosh M, Mereu L, Senior P. Antidiabetic drugs and their potential role in treating mild cognitive impairment and Alzheimer’s disease. Discov Med. 2013;16:277–86.
Mielke JG, Taghibiglou C, Wang YT. Endogenous insulin signaling protects cultured neurons from oxygen-glucose deprivation-induced cell death. Neuroscience. 2006;143:165–73.
Gupta A, Bisht B, Dey CS. Peripheral insulin-sensitizer drug metformin ameliorates neuronal insulin resistance and Alzheimer’s like changes. Neuropharmacology. 2011;60:910–20.
Hsu CC, Wahlqvist ML, Lee MS, Tsai HN. Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimer Dis. 2011;24:485–93.
Khanfar MA, AbuLhader MM, Alqtaishat S, Taha MO. Pharmacophore modeling, homology modeling, and in silico screening reveal mammalian garget of rapamycin inhibitory activities for sotalol, glyburide, metipranolol, sulfamethizole, glipizide, and pioglitazone. J Mol Graph Model. 2013;42:39–49.
Pandini G, Pace V, Copani A, Squatrito S, Milardi D, Vigneri R. Insulin has multiple antiamyloidogenic effect on human neuronal cells. Endocrinology. 2013;154:375–87.
Reger MA, Watson GS, Frey WH 2nd, Baker LD, Cholerton B, Keeling ML, et al. Effects of intranasal insulin on coginition in memory-impaired older adults: modulation by APOE genotype. Neurobil Aging. 2006;27:451–8.
De Felice FG, Vieira MN, Bomfim TR, Decker H, Velasco PT, Lambert MP, et al. Protection of synapses against Alzeimer’s-liked toxins: insulin signaling prevents the pathogenic binding of A beta oligomers. Proc Natl Acad Sci USA. 2009;106:1971–6.
Pathan AR, Viswanad B, Sonkusare SK, Ramarao P. Chronic administration of pioglitazone attenuates intracerebroventricularstreptozotocin induced-memory impaiment in rat. Life Sci. 2006;79:2209–16.
Watson GS, Cholerton BA, Reger MA, Baker LD, Plymate S, Asthana S, et al. Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosglitazone: a preliminary study. Am J Geriatr Psychiatry. 2005;13:950–8.
Risner ME, Saunders AM, Altman JF, Ormandy GC, Craft S, Foley IM, et al. Rosiglitazome in Alzheimer’s disease study group. Efficacy of rosiglitazome in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J. 2006;6:246–54.
Li L, Zhang ZF, Holscher C, Gao C, Jiang YH, Liu YZ. (Val8) glucagon-like peptide-1 prevents tau hyperphosphorylation, impairment of spatial learning and ultra-structural cellular damage induced by streptozotocin in rat brains. Eur J Pharmacol. 2012;674:280–6.
McClean PL, Jalewa J, Hölscher C. Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res. 2015;293:96–106.
Matteucci E, Giampietro O. Mechanisms of neurodegeration in type 2 T2DM and the neuroprotective potential of dipeptidyl peptidase 4 inhibitors. Curr Med Chem. 2015;22:1573–81.
Liu W, Li G, Hölscher C, Li L. Neuroprotective effects of geniposide on Alzheimer’s disease pathology. Rev Neurosci. 2015;26:371–83.
Gao C, Liu Y, Jiang Y, Ding J, Li L. Geniposide ameliorates learning memory deficits, reduces tau phosphorylation and decreases apoptosis via GSK3β pathway in streptozotocin-induced alzheimer rat model. Brain Pathol. 2014;24:261–9.
Hwang SK, Kim HH. The functions of mTOR in ischemic diseases. BMB Rep. 2011;44:506–11.
Chang YY, Juhasz G, Goraksha-Hicks P, Arsham AM, Mallin DR, Muller LK, et al. Nutrient-dependent regulation of autophagy through the target of rapamycin pathway. Biochem Soc Trans. 2009;37:232–6.
Morselli E, Maiuri MC, Markaki M, Megalou E, Pasparaki A, Palikaras K, et al. Caloric restriction and resveratrol promote longevity through the Sirtuin-1-dependent induction of autophagy. Cell Death Disease. 2010;1:e10.
Berger Z, Ravikumar B, Menzies FM, Oroz LG, Underwood BR, Pangalos MN, et al. Rapamycin alleviates toxicity of different aggregate-prone proteins. Hum Mol Genet. 2006;15:433–42.
Caccamo A, De Pinto V, Messina A, Branca C, Oddo S. Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci. 2014;34:7988–98.
Maiese K. Driving neural regeneration through the mammalian target of rapamycin. Neural Regen Res. 2014;9:1413–7.
Caccamo A, Magrì A, Medina DX, Wisely EV, López-Aranda MF, Silva AJ, et al. mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell. 2013;12:370–80.
Magri L, Cambiaghi M, Cominelli M, Alfaro-Cervello C, Cursi M, Pala M, et al. Sustained activation of mTOR pathway in embryonic neural stem cells leads to development of tuberous sclerosis complex-associated lesions. Cell Stem Cell. 2011;9:447–62.
Hartman NW, Lin TV, Zhang L, Paquelet GE, Feliciano DM, Bordey A. mTORC1 targets the translational repressor 4E-BP2, but not S6 kinase 1/2, to regulate neural stem cell self-renewal in vivo. Cell Rep. 2013;5:433–44.
Malagelada C, López-Toledano MA, Willett RT, Jin ZH, Shelanski ML, Greene LA. RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J Neurosci. 2011;31:3186–96.
Han J, Wang B, Xiao Z, Gao Y, Zhao Y, Zhang J, et al. Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol Cell Neurosci. 2008;39:118–24.
Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, et al. Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol. 2004;24:9508–16.
Maiese K. Cutting through the complexities of mTOR for the treatment of stroke. Curr Neurovasc Res. 2014;11:177–86.
Shang YC, Chong ZZ, Wang S, Maiese K. Tuberous sclerosis protein 2 (TSC2) modulates CCN4 cytoprotection during apoptotic amyloid toxicity in microglia. Curr Neurovasc Res. 2013;10:29–38.
Choi KC, Kim SH, Ha JY, Kim ST, Son JH. A novel mTOR activating protein protects dopamine neurons against oxidative stress by repressing autophagy related cell death. J Neurochem. 2010;112:366–76.
Santini E, Heiman M, Greengard P, Valjent E, Fisone G, et al. Inhibition of mTOR signaling in Parkinson’s disease prevents L-DOPA-induced dyskinesia. Sci Signal. 2009;2:ra36.
Zhu Y, Wang J. Wogonin increases β-amyloid clearance and inhibits tau phosphorylation via inhibition of mammalian target of rapamycin: potential drug to treat Alzheimer’s disease. Neurol Sci. 2015;36:1181–8.
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, et al. Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e9979.
Zare Mehrjerdi F, Aboutaleb N, Habibey R, Ajami M, Soleimani M, Arabian M, et al. Increased phosphorylation of mTOR is involved in remote ischemic preconditioning of hippocampus in mice. Brain Res. 2013;1526:94–101.
Bishop NA, Lu T, Yankner BA. Neural mechanisms of ageing and cognitive decline. Nature. 2010;464:529–35.
Wu P, Jiang C, Shen Q, Hu Y. Systematic gene expression profile of hypothalamus in calorie-restricted mice implicates the involvement of mTOR signaling in neuroprotective activity. Mech Ageing Dev. 2009;130:602–10.
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, et al. Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med. 2008;14:843–8.
Puighermanal E, Marsicano G, Busquets-Garcia A, Lutz B, Maldonado R, Ozaita A. Cannabinoid modulation of hippocampal long-term memory is mediated by mTOR signaling. Nat Neurosci. 2009;12:1152–8.
Ramírez AE, Pacheco CR, Aguayo LG, Opazo CM. Rapamycin protects against Aβ-induced synaptotoxicity by increasing presynaptic activity in hippocampal neurons. Biochim Biophys Acta. 2014;1842:1495–501.
Lafay-Chebassier C, Pérault-Pochat MC, Page G, RiouxBilan A, Damjanac M, Pain S, et al. The immunosuppressant rapamycin exacerbates neurotoxicity of A beta peptide. J Neurosci Res. 2006;84:1323–34.
Ma T, Hoeffer CA, Capetillo-Zarate E, Yu F, Wong H, Lin MT, et al. Dysregulation of the mTOR pathway mediates impairment of synaptic plasticity in a mouse model of Alzheimer’s disease. PLoS One. 2010;5:e12845.
Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, et al. Macroautophagy: a novel beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol. 2005;171:87–98.
Zhang S, Salemi J, Hou H, Zhu Y, Mori T, Giunta B, et al. Rapamycin promotes beta-amyloid production via ADAM-10 inhibition. Biochem Biophys Res Commun. 2010;398:337–41.
Bhaskar K, Miller M, Chludzinski A, Herrup K, Zagorski M, Lamb BT. The PI3K-Akt-mTOR pathway regulates A beta oligomer induced neuronal cell cycle events. Mol Neurodegener. 2009;4:14.
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S. Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem. 2010;285:13107–20.
Li X, Alafuzoff I, Soininen H, Winblad B, Pei JJ. Levels of mTOR and its downstream targets 4E-BP1, eEF2, and eEF2 kinase in relationships with tau in Alzheimer’s disease brain. FEBS J. 2005;272:4211–20.
Julien C, Tremblay C, Emond V, Lebbadi M, Salem N Jr, Bennett DA, et al. Sirtuin 1 reduction parallels the accumulation of tau in Alzheimer disease. J Neuropathol Exp Neurol. 2009;68:48–58.
Wang C, Zhang X, Teng Z, Zhang T, Li Y. Downregulation of PI3K/Akt/mTOR signaling pathway in curcumin-induced autophagy in APP/PS1 double transgenic mice. Eur J Pharmacol. 2014;740:312–20.
Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, et al. Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem. 2015;133:739–49.
Wang Y, Wang YX, Liu T, Law PY, Loh HH, Qiu Y, et al. μ-Opioid receptor attenuates Aβ oligomers-induced neurotoxicity through mTOR signaling. CNS Neurosci Ther. 2015;21:8–14.
Yamamoto N, Tanida M, Kasahara R, Sobue K, Suzuki K. Leptin inhibits amyloid β-protein fibrillogenesis by decreasing GM1 gangliosides on the neuronal cell surface through PI3K/Akt/mTOR pathway. J Neurochem. 2014;131:323–32.
Caccamo A, Branca C, Talboom JS, Shaw DM, Turner D, Ma L, et al. reducing ribosomal protein S6 kinase 1 expression improves spatial memory and synaptic plasticity in a mouse model of Alzheimer’s disease. J Neurosci. 2015;35:14042–56.
Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, et al. Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature. 2000;408:994–7.
Hamada S, Hara K, Hamada T, Yasuda H, Moriyama H, Nakayama R, et al. Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes. 2009;58:1321–32.
Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, et al. mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 T2DM. Diabetes. 2008;57:945–57.
Deblon N, Bourgoin L, Veyrat-Durebex C, Peyrou M, Vinciguerra M, Caillon A, et al. Chronic mTOR inhibition by rapamycin induces muscle insulin resistance despite weight loss in rats. Br J Pharmacol. 2012;165(7):2325–40.
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, et al. The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol. 2004;166:213–23.
Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML. Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes. 2004;3:S225–32.
Hurtado-Carneiro V, Sanz C, Roncero I, Vazquez P, Blazquez E, Alvarez E. Glucagon-like peptide 1 (GLP-1) can reverse AMP-activated protein kinase (AMPK) and S6 kinase (P70S6K) activities induced by fluctuations in glucose levels in hypothalamic areas involved in feeding behaviour. Mol. Neurobiol. 2012;45:348–561.
Xie J, El Sayed NM, Qi C, Zhao X, Moore CE, Herbert TP. Exendin-4 stimulates islet cell replication via the IGF1 receptor activation of mTORC1/S6K1. J. Mol. Endocrinol. 2014;53:105–15.
Jiang T, Yu JT, Zhu XC, Tan MS, Wang HF, Cao L, et al. Temsirolimus promotes autophagic clearance of amyloid-β and provides protective effects in cellular and animal models of Alzheimer’s disease. Pharmacol Res. 2014;81:54–63.
Kimura R, Okouchi M, Fujioka H, Ichiyanagi A, Ryuge F, Mizuno T, et al. Glucagon-like peptide-1 (GLP-1) protects against methylglyoxal-induced PC12 cell apoptosis through the PI3K/Akt/mTOR/GCLc/redox signaling pathway. Neuroscience. 2009;162:1212–9.
Chang TJ, Tseng HC. Corrigendum: glucagon-like peptide-1 prevents methylglyoxal-induced apoptosis of beta cells through improving mitochondrial function and suppressing prolonged AMPK activation. Sci Rep. 2016;31(6):26917.
Nie J, Han X. Shi Y.SAD-A and AMPK kinases: the “yin and yang” regulators of mTORC1 signaling in pancreatic β cells. Cell Cycle. 2013;12:3366–9.
Burmeister MA, Ayala J, Drucker DJ, Ayala JE. Central glucagon-like peptide 1 receptor-induced anorexia requires glucose metabolism-mediated suppression of AMPK and is impaired by central fructose. Am J Physiol Endocrinol Metab. 2013;304:E677–85.
Lilley BN, Pan YA, Sanes JR. SAD kinases sculpt axonal arbors of sensory neurons through long- and short-term responses to neurotrophin signals. Neuron. 2013;79:39–53.
Mori H, Inoki K, Opland D, Münzberg H, Villanueva EC, Faouzi M, et al. Critical roles for the TSC-mTOR pathway in β-cell function. Am J Physiol Endocrinol Metab. 2009;297:e1013–22.
Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, et al. Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev. 2005;19:2199–211.
Yang SB, Lee HY, Young DM, Tien AC, Rowson-Baldwin A, et al. Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J Mol Med (Berl). 2012;90:575–85.
Nie J, Liu X, Lilley BN, Zhang H, Pan YA, Kimball SR, et al. SAD-A kinase controls islet β-cell size and function as a mediator of mTORC1 signaling. Proc Natl Acad Sci USA. 2013;110:13857–62.
Nie J, Lilley BN, Pan YA, Faruque O, Liu X, Zhang W, et al. SAD-A potentiates glucose-stimulated insulin secretion as a mediator of glucagon-like peptide 1 response in pancreatic β cells. Mol Cell Biol. 2013;33:2527–34.
Xu WW, Guan MP, Zheng ZJ, Gao F, Zeng YM, Qin Y, et al. Exendin-4 alleviates high glucose-induced rat mesangial cell dysfunction through the AMPK pathway. Cell Physiol Biochem. 2014;33:423–32.
Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ. Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes. 2003;52:974–83.
Miao XY, Gu ZY, Liu P, Hu Y, Li L, Gong YP, et al. The human glucagon-like peptide-1 analogue liraglutide regulates pancreatic beta-cell proliferation and apoptosis via an AMPK/mTOR/P70S6K signaling pathway. Peptides. 2013;39:71–9.
Zhang Y, Chen Y, Cheng J, Guo Z, Lu Y, Tian B. DPP IV inhibitor suppresses STZ-induced islets injury dependent on activation of the IGFR/Akt/mTOR signaling pathways by GLP-1 in monkeys. Biochem Biophys Res Commun. 2015;456:139–44.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This work was funded by the Ministry of Human Resources and Social Security, Shanxi province [(2010) 255]
Conflict of interest
Professor Lin Li has no conflicts of interest to report, financial or otherwise.
Rights and permissions
About this article

Cite this article
Li, L. The Molecular Mechanism of Glucagon-Like Peptide-1 Therapy in Alzheimer’s Disease, Based on a Mechanistic Target of Rapamycin Pathway. CNS Drugs 31, 535–549 (2017). https://doi.org/10.1007/s40263-017-0431-2
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40263-017-0431-2