
Abstract
Accumulating evidence suggests that Alzheimer’s disease may manifest as a metabolic disorder with pathology and/or dysfunction in numerous tissues. Adults with Alzheimer’s disease suffer with significantly more comorbidities than demographically matched Medicare beneficiaries (Zhao et al, BMC Health Serv Res 8:108, 2008b). Reciprocally, comorbid health conditions increase the risk of developing Alzheimer’s disease (Haaksma et al, PLoS One 12(5):e0177044, 2017). Type 2 diabetes mellitus is especially notable as the disease shares many overlapping pathologies observed in patients with Alzheimer’s disease, including hyperglycemia, hyperinsulinemia, insulin resistance, glucose intolerance, dyslipidemia, inflammation, and cognitive dysfunction, as described in Chap. 8 of this book (Yoshitake et al, Neurology 45(6):1161–1168, 1995; Leibson et al, Am J Epidemiol 145(4):301–308, 1997; Ott et al, Neurology 53(9):1937–1942, 1999; Voisin et al, Rev Med Interne 24(Suppl 3):288s–291s, 2003; Janson et al. Diabetes 53(2):474–481, 2004; Ristow M, J Mol Med (Berl) 82(8):510–529, 2004; Whitmer et al, BMJ 330(7504):1360, 2005, Curr Alzheimer Res 4(2):103–109, 2007; Ohara et al, Neurology 77(12):1126–1134, 2011). Although nondiabetic older adults also experience age-related cognitive decline, diabetes is uniquely associated with a twofold increased risk of Alzheimer’s disease, as described in Chap. 2 of this book (Yoshitake et al, Neurology 45(6):1161–1168, 1995; Leibson et al, Am J Epidemiol 145(4):301–308, 1997; Ott et al. Neurology 53(9):1937–1942, 1999; Ohara et al, Neurology 77(12):1126–1134, 2011). Good glycemic control has been shown to improve cognitive status (Cukierman-et al, Diabetes Care 32(2):221–226, 2009), and the use of insulin sensitizers is correlated with a lower rate of cognitive decline in older adults (Morris JK, Burns JM, Curr Neurol Neurosci Rep 12(5):520–527, 2012). At the molecular level, the mechanistic/mammalian target of rapamycin (mTOR) plays a key role in maintaining energy homeostasis. Nutrient availability and cellular stress information, both extracellular and intracellular, are integrated and transduced through mTOR signaling pathways. Aberrant regulation of mTOR occurs in the brains of patients with Alzheimer’s disease and in numerous tissues of individuals with type 2 diabetes (Mannaa et al, J Mol Med (Berl) 91(10):1167–1175, 2013). Moreover, modulating mTOR activity with a pharmacological inhibitor, rapamycin, provides wide-ranging health benefits, including healthy life span extension in numerous model organisms (Vellai et al, Nature 426(6967):620, 2003; Jia et al, Development 131(16):3897–3906, 2004; Kapahi et al, Curr Biol 14(10):885–890, 2004; Kaeberlein et al, Science 310(5751):1193–1196, 2005; Powers et al, Genes Dev 20(2):174–184, 2006; Harrison et al, Nature 460(7253):392–395, 2009; Selman et al, Science 326(5949):140–144, 2009; Sharp ZD, Strong R, J Gerontol A Biol Sci Med Sci 65(6):580–589, 2010), which underscores its importance to overall organismal health and longevity. In this chapter, we discuss the physiological role of mTOR signaling and the consequences of mTOR dysregulation in the brain and peripheral tissues, with emphasis on its relevance to the development of Alzheimer’s disease and link to type 2 diabetes.
Access provided by Autonomous University of Puebla. Download chapter PDF
Similar content being viewed by others
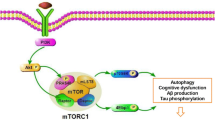
10.1 Introduction to mTOR Signaling
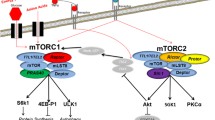
mTOR is an evolutionarily conserved serine/threonine kinase, and member of the phosphoinositide-3-kinase (PI3K)-related kinases (PIKK) family; others include ATM, ATR, DNA-PK, and hSMG1. mTOR functions as the catalytic subunit of two independent multi-protein holoenzyme complexes, mTORC1 and mTORC2 (Fig. 10.1). Common to both core complexes are mTOR, mammalian lethal with Sec13 protein 8 (mLST8, also known as GβL), and both interact with DEP domain-containing mTOR-interacting protein (Deptor) and the Tel2/Tti1 complex. Furthermore, mLST8 is critical in promoting mTOR-mediated tumor progression (Kakumoto et al. 2015), although it has no effect on physiological mTOR activity in vitro or in vivo (Guertin et al. 2006). Unique to the mTORC1 core complex is raptor and transient interactions with nutrient-sensitive proline-rich AKT substrate 40 kDa (PRAS40) (Wullschleger et al. 2006). Raptor has many functions that regulate mTORC1 assembly, substrate recruitment, subcellular localization, and amino acid sensing (Hara et al. 2002; Kim et al. 2002; Sancak et al. 2008). PRAS40 and Deptor are both substrates of mTORC1, whereby mTORC1-mediated phosphorylation weakens their association with the complex and promotes mTORC1 kinase activity (Oshiro et al. 2007; Sancak et al. 2007; Thedieck et al. 2007; Vander Haar et al. 2007; Wang et al. 2008; Peterson et al. 2009). Rictor, RPTOR-independent companion of mTOR, is the defining component of mTORC2. Rictor and raptor are mutually exclusive mTOR-binding partners and required for mTORC2 and mTORC1 activity, respectively (Jacinto et al. 2004; Sarbassov et al. 2004). The core protein complex of mTORC2 also consists of the mammalian stress-activated protein kinase-interacting protein (mSIN1); and protein observed with Rictor-1 (Protor-1, also known as PRR5). Proteins with nutrient-sensitive or transient interactions with the mTORC2 core are DEPTOR, IKK, Sestrin3, Xpln, and TSC2 (Fig. 10.1).

mTOR complex proteins. Activation of both mTORC1 and mTORC2 occurs through growth factor and mitogen PI3K-dependent signaling. Active mTORC1 increases macromolecule biogenesis and suppress autophagy. Active mTORC2 promotes cell survival, proliferation, and cytoskeletal remodeling. AKT activates mTORC1 through PI3K signaling or downstream of mTORC2 activation and thus serves as a connecting node to these independent complexes. mTORC1 and mTORC2 share in common mTOR and mLST8 as core proteins and Tel2/Tti1 and Deptor as transient and/or nutrient-sensing regulatory proteins. Unique to mTORC1 is the scaffold protein Raptor and transient and/or nutrient-sensing proteins PRAS40. Rictor, Protor, and mSIN1 are unique to the mTORC2 core complex, as are the transient and/or nutrient-sensing proteins IKK, Sestrin3, Xpln, and TSC2
Elevated cellular energy, nutrient availability, and mitogens activate mTOR signaling. Excess cellular resource availability, as in diabetes and obesity, increases mTOR signaling through the convergence of numerous signals. For example, hormones such as insulin and IGF1 activate mTOR through phosphoinositide 3-kinase (PI3K). In the presence of insulin, the transmembrane tyrosine kinase insulin receptor (IR) becomes activated and autophosphorylates tyrosine residues located in the intracellular portion of the receptor. The signal is rapidly transduced by the phosphorylation of tyrosine residues on insulin receptor substrate 1 through 4 (IRS-1 through IRS-4) (Sun et al. 1991, 1995; Lavan et al. 1997a, b). Among the distinct pathways next affected, the most prominent is mTORC2/AKT. Activated mTORC2 promotes cell survival, cytoskeletal remodeling, cell growth, and cell proliferation by activating AKT, SGK1, and PKC while inhibiting MST1 (Sarbassov et al. 2005; Cybulski and Hall 2009; Laplante and Sabatini 2012; Manning and Toker 2017). AKT also is an upstream activator of mTORC1 and an indirect substrate of mTORC1 during times of hyperactive mTOR signaling; thus, AKT serves as a signaling node between mTORC1 and mTORC2. Specifically, AKT activates mTORC1 through inhibitory phosphorylation of TSC1/2, an mTORC1 inhibitor (Figs. 10.1 and 10.2).

Hyperactive mTORC1 signaling contributes to insulin resistance. Insulin signaling activates mTORC1 to promote macromolecule biogenesis and decrease autophagy. Activated mTORC1/S6K1 feeds back on the pathway to block insulin signaling through inhibitory phosphorylation of insulin receptor substrate 1 IRS1 (Ser307 and Ser636/639) and Grb10. Persistent nutrient availability induces insulin resistance through the sustained mTORC1/S6K1 inhibitory phosphorylation on IRS1 and Grb10 activation. Acute rapamycin releases the autoinhibitory signaling and prevents insulin resistance
mTORC1 also responds to cellular energy, i.e., ATP/ADP ratio through AMPK (Hardie and Ashford 2014). During low cellular energy, AMPK inhibits mTORC1 through both direct and indirect mechanisms: direct phosphorylation of raptor disrupts the mTORC1 complex, and phosphorylation of the mTOR inhibitor TSC2 dampens mTORC1 activity. Increasing cellular amino acid (Wang and Proud 2009) or glucose concentration activates mTORC1 through the Rag family of GTPases. In response, mTORC1 localizes to the lysosome and stimulates macromolecule biosynthesis. In contrast, during starvation or low nutrient availability, mTORC1 negatively regulates autophagy by inhibiting autophagy initiation and lysosomal biogenesis through phosphorylation of the ULK complex and TFEB, respectively (Roczniak-Ferguson et al. 2012; Settembre et al. 2012). During high cellular energy resources, mTORC1 also promotes ribosome biogenesis and mRNA translation through phosphorylation and activation of downstream substrates: ribosomal S6 kinases (S6Ks) and the inhibitory eIF4E-binding proteins (4E-BPs). mTORC1-activated S6K phosphorylates substrates to promote translation and elongation (i.e., 40S ribosomal subunit proteins, eiF4B and eEF2K), while mTORC1-phosphorylated 4E-BPs promote cap-dependent translation through their participation in the eIF4F complex (Laplante and Sabatini 2012; Saxton and Sabatini 2017a, b). Also, mTORC1 regulates lipid biosynthesis through activation of SREBP1, a major lipogenic transcription factor that controls genes involved in fatty acid and cholesterol synthesis, (Lamming and Sabatini 2013). In summary, mTOR is at the nexus of cellular metabolism, growth, and survival.
10.2 Physiological mTOR Signaling in the Brain
As in other cell types where mTOR regulates cell growth and proliferation, mTOR has an essential role in neuronal progenitor proliferation and in the temporal control of neuronal differentiation through insulin/IGF1-mediated PI3K/mTOR activation (Han et al. 2008; Fishwick et al. 2010; Malagelada et al. 2011). The principle growth factors that activate mTORC1 in neurons are brain-derived neurotrophic factor (BDNF), insulin, insulin-like growth factor I (IGF-I), and vascular endothelial growth factor (VEGF) (Kim et al. 2008; Takei and Nawa 2014). The use of transgenic animal models and pharmacological approaches has provided strong evidence for the necessary role of mTOR signaling in the brain throughout life stages. Nestin-driven ablation of mTORC1 (raptor deletion) and mTORC2 (rictor deletion) activity was performed to determine the contribution of each complex on brain development (Cloetta et al. 2013; Thomanetz et al. 2013). Both studies found microcephaly, as well as reduced neuron cell number, size, morphology, and function indicating a necessary role of mTOR signaling during neuronal development. Interestingly, genetic upregulation of mTOR activity during forebrain development also resulted in microcephaly due to neural progenitor apoptosis in transgenic mice; in contrast upregulating mTOR in the adult brain resulted in cortical hypertrophy (Kassai et al. 2014). mTOR signaling is critical for axonal and dendritic sprouting, growth and regeneration during development (Inoki et al. 2006; Jossin and Goffinet 2007; Li et al. 2008; Nie et al. 2010), and in the mature nervous system. For example, in the adult central nervous system (CNS), neurons fail to regenerate; in contrast axonal sprouting and axonal growth can occur in response to injury in peripheral nervous system (PNS). This differential response to neuronal injury in the CNS and PNS is partially mediated through mTORC1 signaling. In the PNS, neuronal injury upregulates mTORC1; conversely, injury to CNS neurons causes mTORC1 downregulation (Park et al. 2008; Liu et al. 2010). In CNS neuron injury models, upregulation of mTORC1 by deleting negative regulators PTEN or TSC1 both enhanced axonal sprouting and regeneration retinal ganglion cells (Park et al. 2008); and PTEN deletion had the same benefits on corticospinal tract neuronal injury (Liu et al. 2010). Expression of constitutively active forms of mTOR pathway members, AKT and Rheb, also induce axonal regrowth. These studies indicate that mTORC1 activation is sufficient to promote axonal growth in both CNS and PNS neurons following injury and may have therapeutic importance to neurodegeneration and peripheral neuropathy in patients diagnosed with Alzheimer’s disease and diabetes.
Proper mTOR regulation also is necessary for proper brain function. The use of rapamycin has elegantly demonstrated the requirement of mTORC1 activity for long-term potentiation (LTP) (Tang et al. 2002; Cammalleri et al. 2003; Banko et al. 2005; Ehninger et al. 2008; Stoica et al. 2011) and memory consolidation via mTORC1-mediated translational control (Stoica et al. 2011). mTORC1-dependent translational control also mediates changes in dendritic spine morphology that is required for memory storage and recall (Henry et al. 2017). Consolidation of memories also requires mTORC1 (Dash et al. 2006; Parsons et al. 2006; Schicknick et al. 2008; Stoica et al. 2011; Gafford et al. 2013) and mTORC2 activation for cytoskeletal remodeling (Costa-Mattioli and Monteggia 2013; Huang et al. 2013). While these studies collectively indicate the importance of mTOR in overall neuronal function, other experimental results contradict the necessity of mTOR in LTP (Antion et al. 2008), specifically in the dentate gyrus (Panja et al. 2009). Along these lines, mTORC1 hyperactivity induces seizures and epilepsy in rodent models (Sharma et al. 2010; Kassai et al. 2014; Rotschafer and Razak 2014; Russo et al. 2014), and genetic mutations resulting in hyperactive mTORC1 have been reported in familial focal epilepsy (Dibbens et al. 2013). In models with hyperactive mTOR, such as aging and Alzheimer’s disease, decreasing mTOR activity with rapamycin results in enhanced learning and memory (Zhou et al. 2009; Spilman et al. 2010; Majumder et al. 2011; Ehninger 2013; Neff et al. 2013; Ozcelik et al. 2013). Collectively these studies underscore that mTOR requires precise temporal regulation and level of activation for proper neuronal and brain function.
10.3 mTOR and Insulin Resistance in the Brain
In the late 1970s, the brain was deemed an insulin-insensitive organ; however, insulin now is well appreciated for its major physiological roles in the brain, as described in Chap. 1 of this book. While insulin is not a major regulator of glucose metabolism or transport of glucose in the brain (Marfaing et al. 1990; Hasselbalch et al. 1999; Seaquist et al. 2001), insulin and insulin-like growth factor type 1 (IGF-1) are important modulators of neurogenesis, neuronal growth, survival and differentiation, synaptic plasticity, lipid metabolism, and protein homeostasis (D’Ercole et al. 1996; Popken et al. 2004; Bedse et al. 2015). These processes are mediated through mTOR via the PI3K/mTOR axis of insulin signaling. Several studies report overactivation of the PI3K/mTOR pathway as an early feature of Alzheimer’s disease (Pei et al. 2008; Caccamo et al. 2010; Tramutola et al. 2015) that can lead to aberrant glucose metabolism, energy production, and regulation of protein synthesis and degradation (Oddo 2012; O’Neill 2013; Tramutola et al. 2015). In Alzheimer’s disease, neurons become increasingly resistant to insulin and IGF-1 (Moloney et al. 2010). Neuronal insulin resistance is maintained by overactivation of the PI3K/mTOR axis, which inhibits IRS1 activity via a negative feedback mechanism (Tramutola et al. 2015) (Fig. 10.2). In this way, insulin is both upstream and downstream of mTOR. The contribution of insulin-independent (e.g., oxidative stress, inflammation, protein accumulation, etc.,) chronic activation of mTORC1 signaling may be more responsible for the brain insulin resistance found in Alzheimer’s disease and is discussed in the following sections.
10.4 mTOR in Alzheimer’s Disease Pathogenesis
The human brain consumes ~20% of the body’s glucose-derived energy to maintain its metabolic demands; compared to its modest 2% body weight, the brain’s requirement is much higher than that of other tissues (Erbsloh et al. 1958). Neurons are the most energy-demanding cell type in the adult human brain and require continuous delivery of glucose from the blood (Howarth et al. 2012). Numerous clinical studies have shown that hypergylcemia causes cognitive impairment (Umegaki et al. 2008; Cukierman-Yaffe et al. 2009; Crane et al. 2013). Early stages of Alzheimer’s disease are characterized by deficits in cerebral glucose utilization (Caselli et al. 2008; Mosconi et al. 2008, 2009), brain insulin resistance, and insulin deficiency (Frolich et al. 1998) that continue to worsen as the disease progresses (Hoyer et al. 1991; Rivera et al. 2005). Furthermore, the impairment in insulin and IGF signaling is directly associated with neuropathological markers of Alzheimer’s disease, including Aβ and phosphorylated tau (Orr et al. 2014). Aβ and tau pathologies are associated with mTOR hyperactivity in brains of patients with Alzheimer’s disease and corresponding mouse models (An et al. 2003; Pei and Hugon 2008; Caccamo et al. 2010, 2011, 2013; Oddo 2012). Several groups have shown that Alzheimer’s disease-associated pathology and cognitive deficits can be mitigated by using the mTOR inhibitor rapamycin to restore mTOR activity (Caccamo et al. 2010; Spilman et al. 2010; Majumder et al. 2011, 2012; Ozcelik et al. 2013), even in the presence of an insulin resistance-inducing diet (Orr et al. 2014). Since Alzheimer’s disease is associated with increased mTOR activity, overactivation of both mTOR and insulin can contribute to brain insulin resistance in Alzheimer’s disease through overlapping mechanisms.
Experimentally inducing insulin resistance in Alzheimer’s disease models greatly exacerbates pathogenesis, cognitive decline, and insulin signaling (Cao et al. 2007; Ke et al. 2009; Jolivalt et al. 2010; Plaschke et al. 2010; Takeda et al. 2010; Bitel et al. 2012; Orr et al. 2014). Studies investigating the cellular mechanisms have found mTOR is a critical signaling molecule exacerbating cognitive deficits and pathogenic protein accumulation in diet (Ma et al. 2013; Orr et al. 2014) and pharmacological (i.e., use of a single streptozotocin injection to impair pancreatic β cells along with high-fat and/or high-carbohydrate diet) models of type 1/2 diabetes pathologies (Ma et al. 2015). Alzheimer’s disease and type 2 diabetes rat models exhibit brain mTOR hyperactivity through oxidative stress, inflammation, protein accumulation, etc. (discussed below). When these diseases are combined, behavioral deficits are worsened coincident with increased mTOR and tau gene and protein expression levels (Ma et al. 2013, 2015). Administering 20% sucrose to drinking water of the 3xTgAD transgenic mouse model of Alzheimer’s disease caused significant weight gain and insulin resistance (Orr et al. 2014). The peripheral phenotypes were accompanied by an increase in pathogenic Aβ, phosphorylated tau deposition, and mTORC1 pathway upregulation in the brain. The restoration of mTOR activity via rapamycin prevented the sucrose-induced exacerbation of Aβ and tau pathology, to strongly implicate disregulated mTORC1 as a nexus of peripheral insulin resistance and brain pathologies (Orr et al. 2014).
Neuronal autophagy is highly efficient and necessary for neuronal health. Loss of autophagy is neurotoxic even in the absence of disease and/or toxic protein accumulation (Hara et al. 2006; Komatsu et al. 2006). Disregulated autophagy (either hyper- or hypoautophagy) has been reported in neurodegenerative disease (Nixon 2013). In Alzheimer’s disease mouse models with hyperactive mTOR, autophagic dysfunction is often cited as a contributing cellular mechanism responsible for the deleterious effects of protein accumulation (Caccamo et al. 2010, 2014; Majumder et al. 2011). Mechanistically, hyperactive mTOR decreases autophagy through ULK and TFEB inhibition (please refer to Fig. 10.1). In the rTg(tauP301L)4510 mouse model of Alzheimer’s disease-associated tau accumulation (Santacruz et al. 2005), TFEB is significantly downregulated (Polito et al. 2014). Experimentally increasing TFEB resulted in decreased pathogenic neurofibrillary tangle formation and brain atrophy concomitant with improved synaptic function and cognitive behavior (Polito et al. 2014). Maintaining proper autophagy in the Alzheimer’s disease brain is especially critical in a diabetes-like condition. Experimentally inducing insulin resistance in 3xTgAD mice caused an upregulation of autophagy-associated proteins in the brain (Fig. 10.3). The upregulation of Beclin 1, Atg7, Atg5, and Atg3 occurred in the absence of increased LC3 II/I ratio, which suggests an accumulation of autophagasomes. A decrease in autophagic flux occurs when either autophagasome formation or lysosomal clearance mechanisms are impaired; the resulting accumulation of defective autophagasomes and autolysosomes is capable of exacerbating Alzheimer’s disease pathogenesis (Orr and Oddo 2013). Notably, treatment with rapamycin restored autophagic flux deficits in these 3xTgAD insulin-resistant mice indicating that restoring brain mTOR activity can overcome the negative effective effects of peripheral obesity and insulin resistance.

Diet-induced insulin resistance caused autophagic impariment in the 3xTgAD Alzheimer’s disease mouse model. (a) Protein expression analyses of brain tissue from 3xTgAD Alzheimer’s disease mice indicate that dietary-induced insulin resistance increased autophagy-associated proteins. (b) The absence of concomitant LC3-II upregulation is indicative of autophagasome constipation. Treatment with rapamycin prevented the autophagasome accumulation and restored autophagic flux to control levels. (Open bar, control mice; solid bar, sucrose; gray bar, sucrose + rapamycin). N = 8 mice/group. One-way ANOVA Tukey’s post hoc *, p < 0.05; **, p < 0.005; ***, p < 0.0001
On a cellular level, the impaired energy metabolism maintained by mTOR-mediated inhibition of insulin and IGF-1 responses leads to oxidative stress and inflammation, which contributes to the pathogenesis of Alzheimer’s disease, as described in Chap. 3 of this book (Craft and Watson 2004). Chronic overactivation of mTOR has been linked to systemic inflammation (Liu et al. 2016b; Paschoal et al. 2017), and there is evidence that mTOR also regulates neuroinflammation in several different disease models. For instance, high-fat diet-induced obesity is associated with increased neuroinflammation and overactivation of mTOR; and when neuroinflammation is reduced, mTOR activity also decreases (Dasuri et al. 2016).
However, much of the work showing mTOR-dependent regulation of neuroinflammation originates in models characterized by acute neuroinflammation. For instance, inhibiting mTOR after intracranial hemorrhage reduces neuronal death, decreases expression of the proinflammatory cytokines TNF-α, IL1β, IL-6, and caspase-3, and reduces microglial activation (Li et al. 2016a; Wang and Zhang 2017). In fact, mTOR has been shown to specifically regulate cytokine-dependent microglial activation, such that pharmacological inhibition of mTOR reduces microglial, but not astrocytic, responses to inflammatory cytokine application (Dello Russo et al. 2009). Additionally, inhibition of mTOR with rapamycin also prevents proinflammatory M1 macrophage polarization and can shift macrophage polarization toward an anti-inflammatory M2 type (Xie et al. 2014b; Li et al. 2016b). In contrast, studies performed in cultured microglia indicate that rapamycin generally enhances proinflammatory activity (Xie et al. 2014b). This discrepancy suggests that mTOR regulates proinflammatory activity indirectly. Specifically, rapamycin likely acts on other cell types which then decrease proinflammatory markers in macrophages and microglia. Overall, these data indicate that mTOR regulates neuroinflammation through the activation of microglia and proinflammatory cytokines. However, since much of this work has been performed in disorders characterized by acute neuroinflammation, these results should be confirmed in disease states with chronic neuroinflammation, such as that found in neurodegenerative diseases.
Specific to Alzheimer’s disease, neuronal insulin resistance is linked to neuroinflammation through activation of the TNF-α/JNK pathway, which inhibits IRS-1. Aβ oligomers (Bomfim et al. 2012; Lourenco et al. 2013) and misfolded tau (Kovac et al. 2011) activate TNF-α, which alters IRS-1/mTOR signaling. As insulin negatively regulates Aβ deposition and tau phosphorylation, dysregulation of insulin signaling increases these pathologies (de la Monte 2014). Additionally, Aβ oligomers inhibit the expression of neuronal surface insulin receptors (Zhao et al. 2008a) and IRS1 phosphorylation at Ser307 (Bomfim et al. 2012), which uncouples the interaction of IRS1 and IR (Harrington et al. 2004; Werner et al. 2004). Moreover, tau directly induces abnormal IRS-1 phosphorylation (Yarchoan et al. 2014), suggesting that Alzheimer’s disease-associated pathologies can further contribute to aberrant insulin signaling in Alzheimer’s disease. Therefore, Aβ and tau can influence abnormal insulin signaling both directly and indirectly through activation of the TNF-α/JNK pathway. Furthermore, dysregulation of brain insulin signaling in Alzheimer’s disease is directly associated with increased Aβ accumulation, tau phosphorylation, and proinflammatory mediators (Di Domenico et al. 2017), as well as mTOR overactivation (Oddo 2012). Therefore, aberrant mTOR signaling is positioned at the crossroad of brain insulin resistance and neuroinflammation in Alzheimer’s disease and acts as a vicious positive feed forward loop to accelerate disease progression.
Brain insulin resistance has been reported in nondiabetic Alzheimer’s disease patients (Talbot et al. 2012), which is exacerbated by diabetes (Yoshitake et al. 1995; Leibson et al. 1997; Ott et al. 1999; Ohara et al. 2011). These reports indicate that brain insulin resistance in Alzheimer’s disease can be a local disease process, as described in Chap. 3 of this book, but also is influenced by peripheral insulin resistance. For example, using rapamycin to reduce mTOR activity in Alzheimer’s disease alleviates brain insulin resistance but simultaneously exacerbates peripheral insulin resistance and obesity (Orr et al. 2014). Similarly, others have reported peripheral insulin resistance induced by rapamycin and rapalogs in wild-type mice, as well as an increased incidence of hyperglycemia and diabetes associated with pharmaceutical inhibition of the mTOR pathway in human clinical trials, indicating that physiological mTOR pathway activity levels are tissue specific (Johnston et al. 2008; Lamming et al. 2012; Geuna et al. 2015). Due to the promising therapeutic benefits of decreasing mTOR activity on various health conditions (Dazert and Hall 2011), recent work is now focused on intermittent dosing regimens to reduce the negative effects on peripheral insulin resistance and obesity while maintaining the benefits of decreasing mTOR activity in brain (Arriola Apelo et al. 2016a, b).
10.4.1 Peripheral Insulin Resistance
While the cause of elevated mTOR in Alzheimer’s disease brain remains inconclusive, excess nutrient intake as seen in type 2 diabetes and obesity leads to the upregulation of mTORC1/S6K1 signaling in insulin-sensitive tissues (Um et al. 2004; Khamzina et al. 2005; Shigeyama et al. 2008). In response, the primary function of mTORC1 is to shift catabolism to growth-promoting anabolism through the synthesis of the major macromolecules (i.e., proteins, lipids, and nucleotides). In times of high nutrient availability, mTOR directly phosphorylates the insulin receptor leading to its internalization; this, in turn, results in a decrease of mTOR signaling (Wullschleger et al. 2006). However, chronic mTOR hyperactivity leads to pathogenic conditions including insulin resistance, a key feature of type 2 diabetes (Saha et al. 2011). Chronic mTORC1 activation by nutrient excess contributes to triacylglycerol or lipid deposition in white adipose tissue, liver, and muscle cells (Bentzinger et al. 2008; Cota et al. 2008; Polak et al. 2008; Sengupta et al. 2010). The induction of an S6K1-dependent negative feedback loop is one consequence from chronic mTORC1 hyper-activation and would lead to attenuation of AKT signaling in multiple tissues and insulin resistance (Harrington et al. 2004; Shah et al. 2004; Um et al. 2004; Khamzina et al. 2005). Therefore, it is conceivable that mTORC1 activation is required for an initial physiological adaptation of nutrient excess and obesity, but a negative feedback loop on IRS signaling caused by chronic and persistent hyper-activation could lead to development of insulin resistance.
10.4.1.1 Adipose Tissue
Obesity, defined as having a body mass index (BMI) of greater than 30 kg per m2, greatly increases the risk of type 2 diabetes and has dichotomous effects on Alzheimer’s disease risk depending on the individual’s life stage. Specifically, obesity in midlife increases the risk of developing late-life Alzheimer’s disease, whereas an increased BMI in older ages is protective (Whitmer et al. 2005, 2007; Emmerzaal et al. 2015; Rodriguez-Casado et al. 2017). As an exocrine organ, adipose influences the activity and function of many tissues, including the brain. Through these neural connections, adipose tissue is well integrated in controlling whole-body glucose homeostasis in both normal and disease states. Mechanistically, mTOR signaling is hyperactive in numerous tissues derived from obese human and animal models due to the convergence of nutrients and growth factor stimuli (Um et al. 2004; Khamzina et al. 2005; Tremblay et al. 2007; Woods et al. 2008; Cota 2009; Kucejova et al. 2016; Tsai et al. 2016). Here we discuss the role of adipose tissue in type 2 diabetes and Alzheimer’s disease, with particular focus on adipose-derived hormone signaling and how mTOR activity influences the interaction between obesity and cognition in Alzheimer’s disease.
There are two main types of adipose tissue, white and brown. White adipose stores excess energy derived from food intake. In general, chronic nutrient stress signals resulting in uncontrolled white adipose tissue expansion promotes heart disease, diabetes, and metabolic syndrome. In contrast, brown adipose tissue is important for energy expenditure and non-shivering thermoregulation because it contains high mitochondrial content in the uncoupled state. This makes brown adipose tissue an attractive target for the treatment of obesity and type 2 diabetes. Interestingly, 3xTgAD Alzheimer’s disease mice have impaired thermoregulation, which is exacerbated with age (Tournissac et al. 2017) and may suggest potential interactions between Alzheimer’s associated protein aggregation and brown adipose tissue dysfunction. In line with this notion, cold exposure in these mice increases tau phosphorylation, full length APP, and soluble APP fragments; placing the animals in a thermoneutral environment reverses the effects on APP (Tournissac et al. 2017). In mice, white adipose tissue browning can alter whole-body energy expenditure, resulting in weight loss and increased insulin sensitivity. mTOR activity has a role in this cold stress-induced transition, resulting in an intermediate “beige” phenotype (Liu et al. 2016a). Furthermore, genetic activation of mTOR in white adipose tissue results in enhanced mitochondrial function and protection from high-fat diet-induced obesity and insulin resistance (Magdalon et al. 2016). In brown adipose tissue, genetically upregulating mTOR induces a transition to white adipose tissue, which can be reversed by pharmacologically decreasing mTOR with rapamycin treatment (Xiang et al. 2015). These dichotomous effects of mTOR in each adipose tissue type underscore its complexity.
Adipose tissue secretes numerous peptide hormones and cytokines (referred to as adipokines) including leptin, adiponectin, resistin, and lipoprotein lipase LPL allowing it to communicate with distant organs such as the brain (Ailhaud 2006). The effects of adipose-to-neuronal signaling have been demonstrated in vitro (Wan et al. 2015) and in vivo (Yamazaki et al. 2015). Human neuronal cells cultured with conditioned media from healthy female (males were not included in this study) adipocytes exhibit neuroprotection against toxic oxidative stress while increasing the activation of inflammatory proteins like JNK and ERK (Wan et al. 2015). In vivo, the systemic administration of adipocyte-conditioned media improved latency times in tail suspension and forced swim tasks in the 5xAD transgenic Alzheimer’s disease mouse line (Yamazaki et al. 2015). In line with these findings, leptin treatment alone enhanced neural progenitor cell proliferation in transgenic APP/PS1 mice, which is thought to confer some benefit to brain function in these mice (Perez-Gonzalez et al. 2011). Notably, leptin plasma concentration was positively associated with reduced incidence of Alzheimer’s disease and whole brain and hippocampal weight in the Framingham Heart study, a large cohort study, to provide translational relevance of these in vitro and preclinical findings (Lieb et al. 2009; Paz-Filho et al. 2010).
The level of leptin in the CNS is dependent on the amount of body fat (Klein et al. 1996) and is correlated with BMI (Schwartz et al. 1996). Activation of mTOR is required for leptin biogenesis and is an important signaling event for healthy adipose tissue expansion (Roh et al. 2003). Like in the Alzheimer’s disease brain, mTOR activity is elevated in adipose tissue from obese and insulin-resistant human subjects (Catalan et al. 2015). mTOR activation induces vascularization of white adipose tissue especially under nutrient replete conditions (Soumya et al. 2013). This process is critical for healthy adipose tissue expansion as evidenced by adipose tissue atrophy and insulin resistance in adipose-specific mTOR knockout mice (Shan et al. 2016). Though elevated mTOR is associated with obesity, it reduces adipose-derived inflammation, especially during high-fat diet feeding (Liu et al. 2016b; Paschoal et al. 2017).
Leptin receptors are highly expressed in several brain regions, highlighting the diversity and importance of leptin signaling in the brain. For example, leptin is involved in regulation of appetite and energy expenditure through its activity in the hypothalamus, satiety through signaling on neurons in the arcuate nucleus (Klein et al. 1996; Schwartz et al. 1996; Elmquist et al. 1998), and long-term potentiation, learning and memory in the hippocampus (Harvey et al. 2006). In Alzheimer’s disease transgenic mice, leptin receptor expression is downregulated (Pedros et al. 2015), which lends support to the idea that patients with Alzheimer’s disease have significant leptin resistance similar to obese and high-fat diet states. While leptin exerts many benefits to brain and neuronal health, a negative association between plasma leptin levels and brain region-specific volumes has been reported (Bentzinger et al. 2008). Obese individuals show greater brain atrophy in old age (Debette et al. 2010; Ho et al. 2010, 2011), which may result from alterations to leptin signaling (i.e., leptin resistance) as well as other adipose-derived molecules. For example, plasma and cerebral spinal fluid (CSF) levels of adipose-derived adiponectin are negatively correlated with cognitive acuity; specifically adiponectin levels increase in a stepwise fashion from control to mild cognitive impairment (MCI) and are even higher in patients with Alzheimer’s disease (Une et al. 2011). However, like the inconsistent findings with leptin, other studies reported lower circulating adiponectin levels in patients with Alzheimer’s disease than controls and concluded adiponectin levels provide no predictive value for disease progression (Teixeira et al. 2013). In vitro work further highlights the complexity of adiponectin signaling. For instance, globular adiponectin can increase the secretion of inflammatory cytokines in human astrocyte-like cells (Wan et al. 2014), but in cultured microglia, it can suppress Aβ-induced inflammatory signaling (Song et al. 2017). These studies have not been confirmed in vivo but underscore the diverse effects of adiponectin signaling dependent on cell type.
Adipose tissue also secretes non-peptide biologically active molecules such as activated lipids. One of the hallmarks of type 2 diabetes is elevated systemic free fatty acid (FFA) levels, which arises from resistance to insulin-induced suppression of lipolysis in adipose tissue (DeFronzo 2004). Furthermore, high levels of peripheral norepinephrine (NE) can also induce triacylglyceride (TAG) breakdown and release of FFA into circulation (Raclot and Groscolas 1993). In turn high levels of FFA can increase inflammation and induce insulin resistance (Liang et al. 2013) in a vicious cycle. Interestingly, one of the first brain regions to degenerate in Alzheimer’s disease is the locus coeruleus, the primary producer of the NE to the brain (Grinberg et al. 2011). This decrease in central NE has been linked to elevated levels of NE in plasma and CSF of patients with Alzheimer’s disease (Raskind et al. 1984). Increased peripheral NE may contribute to suppression of mTOR-mediated glucose uptake into fat cells and increased lipolysis (Mullins et al. 2014). Furthermore, NE mediates beta-adrenergic signaling-induced glucose uptake and mTOR activity in brown adipose tissue (Mullins et al. 2014). These findings suggest a potential link between Alzheimer’s disease and increased susceptibility to insulin resistance. While elevated FFA levels have been shown to induce insulin resistance, FFAs have also been shown to accelerate the formation of Aβ and tau oligomers (Wilson and Binder 1997). Furthermore, Alzheimer’s disease patients have increased CSF levels of the FFA shuttles (albumins) (Elovaara et al. 1985). CSF albuminuria is suggestive of blood-brain barrier leakage and elevated FFAs in the brain, which can facilitate toxic protein aggregation. Moreover, excess circulating FFAs cause accumulation of triglycerides and activated lipids in the form of long-chain fatty acyl-CoA esters in other tissues such as skeletal muscle, liver, and β-cells which disrupts the normal metabolic and secretory functions of these tissues. Thus, adipose tissue is the primary organ responsible for controlling systemic lipid levels, which greatly impacts the modulation of both glucose and lipid homeostasis and underscores the importance of maintaining proper BMI in preventing systemic metabolic dysfunction as in type 2 diabetes and Alzheimer’s disease. It is generally accepted that two features are critical for obesity to elicit type 2 diabetes: skeletal muscle insulin resistance and the failure of pancreatic β cells to secrete the required levels of insulin needed to maintain euglycemia, which will be discussed below.
10.4.1.2 Skeletal Muscle
Skeletal muscle is the largest organ in the human body and comprises 30–50% of an individual’s total body weight. It is classically known for its roles in the generation of power, locomotion, metabolism, and thermogenesis. Skeletal muscle is critically important in the regulation of metabolic homeostasis; it is the major site of glucose disposal (~80%), and muscle insulin resistance is considered one of the major hallmarks of type 2 diabetes (DeFronzo and Tripathy 2009). The risk for sarcopenia, the progressive decline in skeletal muscle mass resulting in decreased muscle strength and physical performance (Cruz-Jentoft et al. 2010), and physical disability increases in older patients with type 2 diabetes mellitus (Kalyani et al. 2010). Sarcopenia negatively impacts cognition (Canon and Crimmins 2011), overall health and quality of life. Motor, sensory, and coordination deficits are not common early in Alzheimer’s disease; however, abnormal weight and muscle loss are clinical features of Alzheimer’s disease progression (McKhann et al. 1984; Burns et al. 2010; Takagi et al. 2017). Moreover, acquired resistance to insulin signaling and action in skeletal muscle is associated with obesity and promotes the development of type 2 diabetes mellitus. In this section, we will discuss the role of skeletal muscle in metabolic homeostasis, dysfunctions in type 2 diabetes and Alzheimer’s disease, and the role of mTOR signaling in these processes.
Skeletal muscle is a major determinant of the basal metabolic rate (Zurlo et al. 1990, 1994; Janssen et al. 2000), which is the amount of energy that an individual requires for the body to maintain function while at rest. These basic processes include breathing, blood circulation, and controlling body temperature. The basal metabolic rate accounts for about 60–75% of the daily calorie expenditure by individuals and determines whether an individual maintains, gains, or loses weight. As in most tissues of the adult human body, mTOR signaling in skeletal muscle is critical for protein synthesis. Since muscle mass is regulated by the balance between protein synthesis and degradation, mTOR signaling in skeletal muscle is paramount for maintaining muscle mass. Furthermore, skeletal muscle mTOR activity is key to integrating nutrient, growth factor, and stress signaling, as well as promoting muscle mass accrual after exercise (Saxton and Sabatini 2017a, b). Both mTORC1 and mTORC2 are highly expressed in skeletal muscle. mTORC1 signaling is essential for maintaining muscle mass and has been associated with muscle hypertrophy (Bodine et al. 2001). Moreover, muscle-specific raptor knockout mice with diminished mTORC1 signaling exhibited severe muscle atrophy leading to early death (Bentzinger et al. 2008). Paradoxically, sustained activation of mTORC1 in muscle-specific TSC1 knockout mice also led to myopathy and reduced survival due to an inability of the body to remove damaged cells as a result of impaired autophagy (Castets et al. 2013). Similar to brain mTOR signaling, these data suggest that proper mTORC1 activity is important for maintaining optimal tissue health, and subsequently an individual’s overall metabolic rate.
In contrast to mTORC1, the major function of mTORC2 in the muscle is the regulation of glucose uptake through PI3K/AKT signaling. Consistent with this notion, muscle-specific rictor knockout mice showed impaired insulin-stimulated glucose transport (Kumar et al. 2008). Injecting mice with the mTOR inhibitor, AZD8055, suppressed insulin-stimulated glucose disposal in the muscle (Kleinert et al. 2014). However, in muscle-specific Rictor knockout mice, AZD8055 did not cause further defects in glucose regulation, suggesting that AZD8055 impairs glucose metabolism in the muscle, in part by blocking mTORC2. These findings support the notion that mTORC2 signaling critically regulates glucose metabolism in muscle and subsequently whole-body metabolism.
Similar to brain and adipose tissue, muscle mTORC1 signaling is elevated in obesity and diabetes (Khamzina et al. 2005). It has been demonstrated that mTORC1 activation of S6K1 results in serine phosphorylation of IRS-1, leading to suppressed insulin signaling (Tzatsos and Kandror 2006) and subsequently insulin resistance (Khamzina et al. 2005). Furthermore, rodent studies showed that mice with constitutive 4E-BP1 activity in the muscle, i.e., reduced downstream mTORC1 signaling, were resistant to age and diet-induced insulin resistance (Tsai et al. 2015). These studies suggest that downstream targets of mTORC1 signaling in the muscle play important roles in glucose metabolism.
In the brain during Alzheimer’s disease pathogenesis, elevated mTOR activity is associated with dysregulation of insulin signaling and the accumulation of pathogenic proteins (Caccamo et al. 2014; Orr et al. 2014). Experimental evidence suggests that skeletal muscle of obese and/or Alzheimer’s disease animal models may develop similar pathogenic protein accumulation and lead to disruptions in glucose regulation. Specifically, a high-cholesterol diet given to rabbits induced Aβ and phosphorylated tau deposition in skeletal muscle (Sparks et al. 1994). In transgenic Alzheimer’s disease mice, mutant APP was found within skeletal muscle as well (Monteiro-Cardoso et al. 2015). These findings are also relevant to human patients: amyloid beta 42 (Aβ42) expression is detectable in skeletal muscle from cognitively normal older adults and is elevated in the skeletal muscle from patients with Alzheimer’s disease as assessed during autopsy (Arai et al. 1991; Kuo et al. 2000). Collectively these findings suggest that hyperactive mTOR may be common to numerous tissues in patients with Alzheimer’s disease and even produce similar protein accumulation pathologies, which could contribute to the muscular dysfunction that is present in Alzheimer’s disease.
Skeletal mass and function have critical roles in the overall health of an organism. Gait speed, primarily governed by muscle function, is one of the closest predictors of mortality in older individuals (Studenski et al. 2011; Toots et al. 2013). Muscle atrophy and dysfunction in patients with Alzheimer’s disease has been confirmed in several independent studies (McKhann et al. 1984; Burns et al. 2010; Takagi et al. 2017). Reduced motor function and grip strength have been associated with an increased risk of mild cognitive impairment (MCI), a diagnosis which often progresses to Alzheimer’s disease (Boyle et al. 2009). Moreover, poor swallowing function in patients with Alzheimer’s disease has been directly attributed to skeletal muscle dysfunction (Takagi et al. 2017). A loss of the homeostatic control of energy metabolism has been implicated as the common cause underling both brain and skeletal muscle dysfunctions detected in Alzheimer’s disease subjects and is attributed to mitochondrial dysfunction (Hauptmann et al. 2009; Du et al. 2010; Boncompagni et al. 2012; Ding et al. 2013). Transgenic Alzheimer’s disease mice recapitulate defects in skeletal muscle function similar to patients with Alzheimer’s disease (Schuh et al. 2014). Specifically decreased skeletal muscle maximal respiratory capacity (Schuh et al. 2014), acetylcholinesterase and catalase activity, and altered skeletal muscle mitochondrial membrane composition (Monteiro-Cardoso et al. 2015) have all been reported in mouse models of Alzheimer’s disease. Of note, acetylcholinesterase inhibitors are used as a standard treatment for Alzheimer’s disease, although they provide little benefit (Kaduszkiewicz et al. 2005). Given that acetylcholinesterase activity in skeletal muscle is impaired in Alzheimer’s disease (Monteiro-Cardoso et al. 2015), using acetylcholinesterase inhibitors may also affect skeletal muscle function, which could impact systemic metabolism.
Muscle is increasingly recognized as an endocrine organ that communicates with other tissues, including the brain, through the secretion of myokines, soluble factors that regulate metabolism and other biological processes in a systemic manner (Henningsen et al. 2010; Pedersen and Hojman 2012; Schnyder and Handschin 2015). Myokines include cytokines, peptides, and other metabolites, which are released by muscle primarily in response to exercise (Schnyder and Handschin 2015). BDNF is predominantly expressed in the brain (Matthews et al. 2009) and is a recently identified myokine (Matthews et al. 2009). BDNF is a member of the neurotrophin family and is essential in regulating the survival, growth, and maintenance of neurons (Hofer and Barde 1988). BDNF plays a key role in learning and memory (Tyler et al. 2002) and in the regulation of body mass and energy homeostasis (Wisse and Schwartz 2003). Patients with Alzheimer’s disease showed lower levels of BDNF in the brain and circulation (Connor et al. 1997; Laske et al. 2006). Interestingly, reduced plasma BDNF levels were also found in obesity and type 2 diabetes (Krabbe et al. 2007). Muscle contractions during exercise produce BDNF mRNA and protein in skeletal muscle, where it seems to play a role in enhancing glucose metabolism. Whether BDNF acts as a myokine that can improve Alzheimer’s disease, however, is currently under debate. Muscle-derived BDNF does not appear to be released into the circulation suggesting that BDNF primarily acts locally in the muscle (Miura et al. 2012). Other neurotrophin family members such as neurotrophin 3 (NT-3) and NT-4/5 have also been found in the skeletal muscle (Omura et al. 2005). While the systemic effects of these aforementioned myokines is not yet understood, evidence for improved memory in Rhesus monkeys and humans due to the upregulation of muscle-derived cathepsin B has been elegantly demonstrated (Moon et al. 2016). Upregulated cathepsin B also improves learning and memory in Alzheimer’s disease mouse models (Embury et al. 2017). Because skeletal muscle release of cathepsin B is increased with exercise, myokines may be directly contributing to the exercise-associated cognitive benefits reported in Alzheimer’s disease (Brandt and Pedersen 2010).
Sarcopenia is associated with a proinflammatory state and lower cognitive function in older adults (Mazure and Swendsen 2016). Systemic inflammation was found to mediate the negative relationship between sarcopenia and cognitive function, especially in females (Mazure and Swendsen 2016). Interestingly, exercise training has been shown to lower all-cause mortality partially because it exerts an anti-inflammatory effect. Chronic “sterile” low-grade systemic inflammation has been implicated in the pathogenesis of a number of aging-related chronic illnesses including both Alzheimer’s disease and type 2 diabetes. The etiology of sterile inflammation is still not clear and likely is a result of several factors, including physical inactivity, altered immune cell function, and somatic cell damage, among others, that can be largely abrogated by exercise. Exercise training reduces visceral fat mass, which is a major source of inflammation (please refer to the section on adipose). The production of anti-inflammatory cytokines, such as IL-1ra, IL-10, and sTNFR, increases after exercise training (Ostrowski et al. 1999). Also, contracting muscle releases myokines that may create a systemic anti-inflammatory environment to improve the function of distant organs (Brandt and Pedersen 2010). Therefore, regular exercise offers protection against Alzheimer’s disease and type 2 diabetes, as well as other chronic diseases, through decreasing adiposity, improving insulin sensitivity, and releasing myokines, many of which are regulated through the mTOR signaling axis.
10.4.1.3 Pancreas
Regulation of glucose homeostasis by insulin depends on pancreatic β cell growth and function. A primary characteristic of type 2 diabetes pathogenesis is the slowly progressive pancreatic β cell failure which results in impaired insulin secretion, decreased β cell mass, decreased insulin action on peripheral tissues, and ultimately β cell death. In general, the pancreas serves two main roles: an exocrine function that aids in digestion and an endocrine function that controls blood glucose level. The endocrine component of pancreatic regulation is controlled by the islet cells (islets of Langerhans), which create and release important metabolic hormones directly into the bloodstream. The two main pancreatic hormones are insulin, which is secreted from islet β cells to suppress catabolic processes and stimulate glucose uptake. Conversely, glucagon is secreted from islet α cells and stimulates hepatic gluconeogenesis. Each β cell contains over 10,000 secretory vesicles filled with insulin (Dean 1973; Olofsson et al. 2002), which are under close regulation to ensure tight control of blood glucose levels. During type 2 diabetes pathogenesis, individuals exhibit a progressive decline in β cell function, with gradual β cell degeneration and worsening of glycaemia over time (1995; Kahn et al. 2006); eventually pancreatic islets fail to compensate for insulin resistance in peripheral tissues. Great effort has focused on understanding mechanisms responsible for pancreatic dysfunction; mTOR signaling has emerged as a key molecular regulator of interest. In this section, we will highlight cellular processes that overlap between type 2 diabetes, β cell pathology, and brain pathogenesis in Alzheimer’s disease with emphasis on hyperactive mTOR and protein accumulation.
Numerous studies highlight a positive role of mTORC1 activation in regulating β cell health in physiological conditions (Tremblay and Marette 2001; Khamzina et al. 2005; Teutonico et al. 2005; Ueno et al. 2005; Di Paolo et al. 2006; Sarbassov et al. 2006; Krebs et al. 2007; Fraenkel et al. 2008; Rachdi et al. 2008; Elghazi et al. 2010; Gu et al. 2011; Yang et al. 2011; Blandino-Rosano et al. 2012). Carbohydrate- and fat-rich Western diets provide chronic nutrient overload (Fig. 10.4). The Western diet results in chronic exposure of β cells to persistent glucose and FFAs resulting in obesity, β cell failure, and type 2 diabetes. This nutrient excess in type 2 diabetes and obesity drives elevated blood glucose, amino acids, proinflammatory cytokines, and insulin, all of which activate mTORC1 (Wellen and Thompson 2010; Zoncu et al. 2011; Efeyan et al. 2015; Kennedy and Lamming 2016). Indeed, pancreatic islets isolated from patients with type 2 diabetes and various type 2 diabetes mouse models (i.e., hyperglycemic obese high-fat diet-fed mice and leptin receptor-deficient db/db mice) display elevated mTORC1 activity (Shigeyama et al. 2008; Bartolome et al. 2014; Hatanaka et al. 2014; Yuan et al. 2017) but reduced mTORC2/AKT signaling (Wang et al. 2010; Ardestani et al. 2014; Yuan et al. 2017). Genetically and pharmacologically modulating mTOR signaling have provided compelling evidence for a critical role of both mTORC1 and mTORC2 complexes in the regulation of glucose homeostasis by modulating β cell mass and function (Tremblay and Marette 2001; Khamzina et al. 2005; Teutonico et al. 2005; Ueno et al. 2005; Di Paolo et al. 2006; Sarbassov et al. 2006; Krebs et al. 2007; Fraenkel et al. 2008; Rachdi et al. 2008; Elghazi et al. 2010; Gu et al. 2011; Yang et al. 2011; Blandino-Rosano et al. 2012).

Causes and consequences of mTOR hyperactivity in mid- and older ages. Carbohydrate- and fat-rich Western diets increase blood glucose and mTOR activity in several tissues, leading to obesity, insulin resistance, and type 2 diabetes in middle ages. With advanced age, the persistent mTOR hyperactivity contributes to tissue dysfunction and Alzheimer’s disease. Brown, brain; red, liver; orange, pancreas; beige, adipose; purple, muscle
mTORC1 regulates several pancreatic islet functions including β cell size, growth, and proliferation, insulin secretion, and protein translation by modulating mRNA translation through phosphorylation of 4E-BPs and S6K1 (McDaniel et al. 2002; Briaud et al. 2003; Kwon et al. 2004; Zahr et al. 2007; Rachdi et al. 2008; Shigeyama et al. 2008; Mori et al. 2009; Barlow et al. 2013; Rhodes et al. 2013; Xie et al. 2014a; Stamateris et al. 2016). Whole-body mTOR loss-of-function/deletion was found to be lethal (Gangloff et al. 2004; Murakami et al. 2004; Guertin et al. 2006; Jacinto et al. 2006; Shiota et al. 2006; Yang et al. 2006); however, whole-body depletion of S6K1 caused hypoinsulinemia and reduced β cell size but improved insulin sensitivity (Pende et al. 2000; Ruvinsky et al. 2005). To further gain insight into pancreatic mTOR function, transgenic mice have been created with tissue-specific mTOR modulation. Overexpressing S6K1 in β cells increases insulin secretion without affecting β cell mass (Elghazi et al. 2010). Activation of mTORC1 by targeted deletion of mTORC1 repressors, tuberous sclerosis 1 (TSC1), or tuberous sclerosis 2 (TSC2) (Rachdi et al. 2008; Shigeyama et al. 2008; Mori et al. 2009; Blandino-Rosano et al. 2012) or overexpressing Rheb (Hamada et al. 2009) significantly increases islet β cell function (Saxton and Sabatini 2017a, b). In contrast, transgenic mice with decreased mTOR function exclusively in β cells displayed reduced mTORC1 and mTORC2 signaling and developed β cell dysfunction and glucose intolerance (Alejandro et al. 2017; Blandino-Rosano et al. 2017; Chau et al. 2017; Elghazi et al. 2017; Ni et al. 2017; Sinagoga et al. 2017). While one model displayed normal β cell mass (Alejandro et al. 2017), others display reduction in β cell mass, smaller islets, and/or pancreas size (Blandino-Rosano et al. 2017; Chau et al. 2017; Elghazi et al. 2017; Ni et al. 2017; Sinagoga et al. 2017). Similarly, mTORC2 loss of function causes hyperglycemica, glucose intolerance, reduced β cell mass and proliferation, as well as reduced pancreatic insulin level and secretion (Gu et al. 2011). These studies underscore that proper regulation of mTOR signaling in β cells is critical for pancreatic function and whole-body glycemic control.
The chronic exposure of human and rodent β cells to persistent glucose and FFAs, as occurs in obesity and type 2 diabetes, causes β cell failure (Robertson et al. 2004; Prentki and Nolan 2006; Muoio and Newgard 2008; Leibowitz et al. 2010; Alejandro et al. 2015) and apoptosis through hyperactive mTORC1 signaling and mTORC1-dependent lipid drop accumulation (Bachar et al. 2009; Vernier et al. 2012; Mir et al. 2015; Yang et al. 2015; Varshney et al. 2017; Yuan et al. 2017). Additional mTORC1-mediated cytotoxicity occurs through inhibition of autophagy (Jung et al. 2008; Masini et al. 2009; Stienstra et al. 2014; Riahi et al. 2016) and compensatory downregulation of cytoprotective mTORC2 signaling (Gu et al. 2011; Blandino-Rosano et al. 2012; Ardestani et al. 2014; Ardestani and Maedler 2016). Rapamycin treatment significantly decreased triglyceride accumulation and β cell toxicity (Vernier et al. 2012), and mTORC1-S6K1 inhibition restored insulin secretion in human islets derived from type 2 diabetic patients providing further evidence that upregulated mTORC1 activity is responsible for these pathologies (Yuan et al. 2017).
Acute rapamycin treatment improves insulin sensitivity in humans and rodents through inhibition of mTORC1 (Tremblay and Marette 2001; Khamzina et al. 2005; Ueno et al. 2005; Krebs et al. 2007). In contrast, chronic rapamycin exposure exacerbates insulin resistance in wild type (Sarbassov et al. 2006) and Alzheimer’s disease mouse models (Orr et al. 2014). The negative effects of long-term treatment occur, in part, through inhibition of mTORC2. While mTORC1 is sensitive to low-dose and short-term exposure to rapamycin, chronic rapamycin exposure also inhibits mTORC2; thus through the mTORC2/AKT signaling pathway, β cell proliferation and survival are negatively affected (Sarbassov et al. 2006). The relative contributions of long-term rapamycin-induced β cell dysfunction and insulin resistance from mTORC1 and mTORC2 signaling are not fully understood, but both complexes influence these phenotypes (Teutonico et al. 2005; Di Paolo et al. 2006). While rapamycin treatment has been proposed as a therapeutic strategy for metabolic diseases, more research is required to unravel the tissue-specific effects after acute versus chronic treatment in different metabolically active tissues. For example, negative effects of chronic mTOR inhibition have been reported in several studies using mTOR inhibitors; therefore, it is conceivable that rapamycin therapy could impair the adaptation of β cells to insulin resistance or have yet unrecognized off-target effects, resulting in diabetes (Fraenkel et al. 2008; Yang et al. 2011).
In the brain, chronic mTOR activation contributes to Aβ and tau protein aggregation; evidence continues to suggest a role of protein accumulation contributing to pancreatic dysfunction in type 2 diabetes as well. Amylin, also known as islet amyloid polypeptide (IAPP), is a 37 amino acid peptide hormone that is derived by proteolytic cleavage of a precursor protein, called islet amyloid precursor protein, and is co-secreted with insulin from pancreatic β cells (Opie 1901; Westermark et al. 1986, 1987; Cooper et al. 1987a, b). Amylin plays a role in glycemic regulation by inhibiting insulin and glucagon secretion, slowing gastric emptying and promoting satiety. Amylin accumulates in pancreatic islets forming amyloid deposits that are present in ~95% of type 2 diabetes patients (Lopes et al. 2004). Aggregated amylin is cytotoxic and is positively correlated with the clinical severity of type 2 diabetes, β cell loss, and negatively correlated with insulin secretion and glucose metabolism (Cooper et al. 1987a, b; Hull et al. 2004) suggesting a causal role in the islet dysfunction. The proteolytic processing of amylin is akin to Aβ production by amyloid precursor protein in Alzheimer’s disease brain tissue. Moreover, Aβ and amylin deregulate similar proteins in their respective tissues, of which about 25% are associated with mitochondrial dysfunction (Lim et al. 2010). Collectively, these studies suggest that pathophysiology of protein aggregation is similar between these diseases. Moreover, in 2010, autopsy performed on patients with Alzheimer’s disease found hyperphosphorylated tau, as well as amyloid protein, in pancreatic tissue (Miklossy et al. 2010).
A common feature of Alzheimer’s disease and type 2 diabetes is APP accumulation and tau hyperphosphorylation co-localizing with components of the JNK pathway in neurons and islet cells, respectively (Thedieck et al. 2007; Tramutola et al. 2015). Hyperphosphorylated tau accumulation, along with Aβ, is hallmark of Alzheimer’s disease pathogenesis (Orr et al. 2017). Studies have since characterized the biochemistry of tau protein in β cell lines and revealed six unique tau isoforms with a balanced 1:1 ratio of 3R and 4R tau isoforms (Maj et al. 2016), but with higher molecular weight than that of cerebral cortex tau isoforms (Maj et al. 2010). Human insulinomas, pancreatic tumors comprised of β cells that overproduce insulin, display elevated tau expression with an imbalance in isoforms, specifically increased 3R isoforms (Maj et al. 2016), which is hallmark of many neurodegenerative tauopathies (Orr et al. 2017). Because tau protein stabilizes microtubules, elevated tau expression in pancreatic β cells would presumably decrease insulin secretion. Experimental evidence indeed supports this notion: exposing pancreatic cell lines to sera derived from diabetic patients modestly increased tau expression (Maj et al. 2010), and overexpressing tau in vitro significantly increases β cell proliferation and decreases insulin secretion (Maj et al. 2016). In neurons, tau is primarily expressed in axons where it stabilizes microtubules to allow for proper delivery of cargo throughout the cell. The microtubules are nucleated at the centrosome-based microtubule organizing center and extend to the cell periphery. However, in pancreatic β cells, microtubules form a dense meshwork with the Golgi acting as the microtubule organizing center. This microtubule configuration limits granule density at the cell periphery and subsequently glucose-stimulated insulin secretion (Zhu et al. 2015). High glucose destabilizes microtubules and decreases microtubule density allowing for glucose-stimulated insulin secretion (Zhu et al. 2015). In diabetic mice, dysfunctional β cells display elevated microtubule density, which decreases insulin secretion. These data indicate that tau expression and microtubule regulation in β cells plays an important role in pancreatic dysfunction and type 2 diabetes pathogenesis.
10.5 Concluding Remarks
Dr. Alois Alzheimer initially described Alzheimer’s disease as the histological accumulation of Aβ-containing plaques and tau-containing neurofibrillary tangles concomitant with neurodegeneration and dementia. Since this seminal report in 1906, basic scientists and physicians have primarily approached Alzheimer’s disease as a brain-specific disease. However, Alzheimer’s disease increasingly is being recognized as a complex systemic disorder involving numerous tissues that manifest extensive comorbid health conditions. The finding that type 2 diabetes doubles the risk for developing Alzheimer’s disease is one such example that has prompted scientists to investigate pathological changes in peripheral tissues and systems. This slowly emerging change in dogma is accompanied by a new vision for therapeutic treatment. Instead of targeting a single aberrant gene or protein, biomedical researchers are beginning to witness benefits of modulating molecules that are highly integrated into numerous cellular pathways across tissues. Pathologically hyperactive mTOR activity is a common mechanism linking Alzheimer’s disease and type 2 diabetes across multiple tissue types. Therefore, targeting mTOR epitomizes this new strategy: therapeutic modulation improves Alzheimer’s disease pathogenesis (Caccamo et al. 2010, 2013) while simultaneously delivering numerous health benefits, most notably extended life span (Vellai et al. 2003; Jia et al. 2004; Kapahi et al. 2004; Kaeberlein et al. 2005; Powers et al. 2006; Harrison et al. 2009; Selman et al. 2009; Sharp and Strong 2010).
Embarking on this new treatment era does not come without challenges. Therapeutic approaches to precisely alter only pathological, but not physiological, mTOR signaling have not yet been developed. Since mTOR is a highly integrated signaling pathway that regulates cellular metabolism and physiology across multiple tissues, maintaining a physiological level of mTOR signaling will be critical to the success of mTOR inhibitors as a pharmacotherapeutic intervention in Alzheimer’s disease. While rapamycin has celebrated numerous successes, a major obstacle is that chronic rapamycin treatment induces insulin resistance in animal models (Sarbassov et al. 2006; Orr et al. 2014) and human subjects (Teutonico et al. 2005; Blagosklonny 2013), likely through the inhibition of mTORC2 (Sarbassov et al. 2006). Nonetheless, investigators are continuing to devise strategies to overcome these pitfalls. For instance, since mTORC1 is more sensitive to low-dose and short-term rapamycin treatment, intermittent rapamycin dosing strategies are being employed to circumvent the negative effects of mTORC2 inhibition, and this dosing strategy has had success in both animal models (Arriola Apelo et al. 2016a, b) and humans (Mannick et al. 2014).
While the need for research and therapeutics targeting brain-specific pathologies in Alzheimer’s disease remains relevant, expanding the research focus to include common signaling pathways that are affected across organ systems offers a complementary approach to therapeutic discovery. Moreover, identifying and targeting the common molecular mechanisms linking Alzheimer’s disease with common comorbidities, such as type 2 diabetes, may provide further insight into the role of mTOR and potentially uncover additional molecules for therapeutic intervention. This promising research approach offers hope to identify treatments that mitigate brain pathologies while simultaneously alleviating comorbid medical conditions that plague patients with Alzheimer’s disease.
References
(1995) U.K. prospective diabetes study 16. Overview of 6 years’ therapy of type II diabetes: a progressive disease. U.K. Prospective Diabetes Study Group. Diabetes 44(11):1249–1258
Ailhaud G (2006) Adipose tissue as a secretory organ: from adipogenesis to the metabolic syndrome. C R Biol 329(8):570–577. discussion 653–575
Alejandro EU, Gregg B, Blandino-Rosano M, Cras-Meneur C, Bernal-Mizrachi E (2015) Natural history of beta-cell adaptation and failure in type 2 diabetes. Mol Asp Med 42:19–41
Alejandro EU, Bozadjieva N, Blandino-Rosano M, Wasan MA, Elghazi L, Vadrevu S, Satin L, Bernal-Mizrachi E (2017) Overexpression of kinase-dead mTOR impairs glucose homeostasis by regulating insulin secretion and not beta-cell mass. Diabetes 66(8):2150–2162
An WL, Cowburn RF, Li L, Braak H, Alafuzoff I, Iqbal K, Iqbal IG, Winblad B, Pei JJ (2003) Up-regulation of phosphorylated/activated p70 S6 kinase and its relationship to neurofibrillary pathology in Alzheimer's disease. Am J Pathol 163(2):591–607
Antion MD, Merhav M, Hoeffer CA, Reis G, Kozma SC, Thomas G, Schuman EM, Rosenblum K, Klann E (2008) Removal of S6K1 and S6K2 leads to divergent alterations in learning, memory, and synaptic plasticity. Learn Mem 15(1):29–38
Arai H, Lee VM, Messinger ML, Greenberg BD, Lowery DE, Trojanowski JQ (1991) Expression patterns of beta-amyloid precursor protein (beta-APP) in neural and nonneural human tissues from Alzheimer’s disease and control subjects. Ann Neurol 30(5):686–693
Ardestani A, Maedler K (2016) MST1: a promising therapeutic target to restore functional beta cell mass in diabetes. Diabetologia 59(9):1843–1849
Ardestani A, Paroni F, Azizi Z, Kaur S, Khobragade V, Yuan T, Frogne T, Tao W, Oberholzer J, Pattou F, Conte JK, Maedler K (2014) MST1 is a key regulator of beta cell apoptosis and dysfunction in diabetes. Nat Med 20(4):385–397
Arriola Apelo SI, Neuman JC, Baar EL, Syed FA, Cummings NE, Brar HK, Pumper CP, Kimple ME, Lamming DW (2016a) Alternative rapamycin treatment regimens mitigate the impact of rapamycin on glucose homeostasis and the immune system. Aging Cell 15(1):28–38
Arriola Apelo SI, Pumper CP, Baar EL, Cummings NE, Lamming DW (2016b) Intermittent administration of rapamycin extends the life span of female C57BL/6J mice. J Gerontol A Biol Sci Med Sci 71(7):876–881
Bachar E, Ariav Y, Ketzinel-Gilad M, Cerasi E, Kaiser N, Leibowitz G (2009) Glucose amplifies fatty acid-induced endoplasmic reticulum stress in pancreatic beta-cells via activation of mTORC1. PLoS One 4(3):e4954
Banko JL, Poulin F, Hou L, DeMaria CT, Sonenberg N, Klann E (2005) The translation repressor 4E-BP2 is critical for eIF4F complex formation, synaptic plasticity, and memory in the hippocampus. J Neurosci 25(42):9581–9590
Barlow AD, Nicholson ML, Herbert TP (2013) Evidence for rapamycin toxicity in pancreatic beta-cells and a review of the underlying molecular mechanisms. Diabetes 62(8):2674–2682
Bartolome A, Kimura-Koyanagi M, Asahara S, Guillen C, Inoue H, Teruyama K, Shimizu S, Kanno A, Garcia-Aguilar A, Koike M, Uchiyama Y, Benito M, Noda T, Kido Y (2014) Pancreatic beta-cell failure mediated by mTORC1 hyperactivity and autophagic impairment. Diabetes 63(9):2996–3008
Bedse G, Di Domenico F, Serviddio G, Cassano T (2015) Aberrant insulin signaling in Alzheimer’s disease: current knowledge. Front Neurosci 9:204
Bentzinger CF, Romanino K, Cloetta D, Lin S, Mascarenhas JB, Oliveri F, Xia J, Casanova E, Costa CF, Brink M, Zorzato F, Hall MN, Ruegg MA (2008) Skeletal muscle-specific ablation of raptor, but not of rictor, causes metabolic changes and results in muscle dystrophy. Cell Metab 8(5):411–424
Bitel CL, Kasinathan C, Kaswala RH, Klein WL, Frederikse PH (2012) Amyloid-beta and tau pathology of Alzheimer’s disease induced by diabetes in a rabbit animal model. J Alzheimers Dis 32(2):291–305
Blagosklonny MV (2013) TOR-centric view on insulin resistance and diabetic complications: perspective for endocrinologists and gerontologists. Cell Death Dis 4:e964
Blandino-Rosano M, Chen AY, Scheys JO, Alejandro EU, Gould AP, Taranukha T, Elghazi L, Cras-Meneur C, Bernal-Mizrachi E (2012) mTORC1 signaling and regulation of pancreatic beta-cell mass. Cell Cycle 11(10):1892–1902
Blandino-Rosano M, Barbaresso R, Jimenez-Palomares M, Bozadjieva N, Werneck-de-Castro JP, Hatanaka M, Mirmira RG, Sonenberg N, Liu M, Ruegg MA, Hall MN, Bernal-Mizrachi E (2017) Loss of mTORC1 signalling impairs beta-cell homeostasis and insulin processing. Nat Commun 8:16014
Bodine SC, Stitt TN, Gonzalez M, Kline WO, Stover GL, Bauerlein R, Zlotchenko E, Scrimgeour A, Lawrence JC, Glass DJ, Yancopoulos GD (2001) Akt/mTOR pathway is a crucial regulator of skeletal muscle hypertrophy and can prevent muscle atrophy in vivo. Nat Cell Biol 3(11):1014–1019
Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel JC, Decker H, Silverman MA, Kazi H, Melo HM, McClean PL, Holscher C, Arnold SE, Talbot K, Klein WL, Munoz DP, Ferreira ST, De Felice FG (2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease- associated Abeta oligomers. J Clin Invest 122(4):1339–1353
Boncompagni S, Moussa CE, Levy E, Pezone MJ, Lopez JR, Protasi F, Shtifman A (2012) Mitochondrial dysfunction in skeletal muscle of amyloid precursor protein-overexpressing mice. J Biol Chem 287(24):20534–20544
Boyle PA, Buchman AS, Wilson RS, Leurgans SE, Bennett DA (2009) Association of muscle strength with the risk of Alzheimer disease and the rate of cognitive decline in community-dwelling older persons. Arch Neurol 66(11):1339–1344
Brandt C, Pedersen BK (2010) The role of exercise-induced myokines in muscle homeostasis and the defense against chronic diseases. J Biomed Biotechnol 2010:520258
Briaud I, Lingohr MK, Dickson LM, Wrede CE, Rhodes CJ (2003) Differential activation mechanisms of Erk-1/2 and p70(S6K) by glucose in pancreatic beta-cells. Diabetes 52(4):974–983
Burns JM, Johnson DK, Watts A, Swerdlow RH, Brooks WM (2010) Reduced lean mass in early Alzheimer disease and its association with brain atrophy. Arch Neurol 67(4):428–433
Caccamo A, Majumder S, Richardson A, Strong R, Oddo S (2010) Molecular interplay between mammalian target of rapamycin (mTOR), amyloid-beta, and Tau: effects on cognitive impairments. J Biol Chem 285(17):13107–13120
Caccamo A, Maldonado MA, Majumder S, Medina DX, Holbein W, Magri A, Oddo S (2011) Naturally secreted amyloid-beta increases mammalian target of rapamycin (mTOR) activity via a PRAS40-mediated mechanism. J Biol Chem 286(11):8924–8932
Caccamo A, Magri A, Medina DX, Wisely EV, Lopez-Aranda MF, Silva AJ, Oddo S (2013) mTOR regulates tau phosphorylation and degradation: implications for Alzheimer’s disease and other tauopathies. Aging Cell 12(3):370–380
Caccamo A, De Pinto V, Messina A, Branca C, Oddo S (2014) Genetic reduction of mammalian target of rapamycin ameliorates Alzheimer’s disease-like cognitive and pathological deficits by restoring hippocampal gene expression signature. J Neurosci 34(23):7988–7998
Cammalleri M, Lutjens R, Berton F, King AR, Simpson C, Francesconi W, Sanna PP (2003) Time-restricted role for dendritic activation of the mTOR-p70S6K pathway in the induction of late-phase long-term potentiation in the CA1. Proc Natl Acad Sci U S A 100(24):14368–14373
Canon ME, Crimmins EM (2011) Sex differences in the association between muscle quality, inflammatory markers, and cognitive decline. J Nutr Health Aging 15(8):695–698
Cao D, Lu H, Lewis TL, Li L (2007) Intake of sucrose-sweetened water induces insulin resistance and exacerbates memory deficits and amyloidosis in a transgenic mouse model of Alzheimer disease. J Biol Chem 282(50):36275–36282
Caselli RJ, Chen K, Lee W, Alexander GE, Reiman EM (2008) Correlating cerebral hypometabolism with future memory decline in subsequent converters to amnestic pre-mild cognitive impairment. Arch Neurol 65(9):1231–1236
Castets P, Lin S, Rion N, Di Fulvio S, Romanino K, Guridi M, Frank S, Tintignac LA, Sinnreich M, Ruegg MA (2013) Sustained activation of mTORC1 in skeletal muscle inhibits constitutive and starvation-induced autophagy and causes a severe, late-onset myopathy. Cell Metab 17(5):731–744
Catalan V, Gomez-Ambrosi J, Rodriguez A, Ramirez B, Andrada P, Rotellar F, Valenti V, Moncada R, Marti P, Silva C, Salvador J, Fruhbeck G (2015) Expression of S6K1 in human visceral adipose tissue is upregulated in obesity and related to insulin resistance and inflammation. Acta Diabetol 52(2):257–266
Chau GC, Im DU, Kang TM, Bae JM, Kim W, Pyo S, Moon EY, Um SH (2017) mTOR controls ChREBP transcriptional activity and pancreatic beta cell survival under diabetic stress. J Cell Biol 216(7):2091–2105
Cloetta D, Thomanetz V, Baranek C, Lustenberger RM, Lin S, Oliveri F, Atanasoski S, Ruegg MA (2013) Inactivation of mTORC1 in the developing brain causes microcephaly and affects gliogenesis. J Neurosci 33(18):7799–7810
Connor B, Young D, Yan Q, Faull RL, Synek B, Dragunow M (1997) Brain-derived neurotrophic factor is reduced in Alzheimer’s disease. Brain Res Mol Brain Res 49(1–2):71–81
Cooper GJ, Willis AC, Clark A, Turner RC, Sim RB, Reid KB (1987a) Purification and characterization of a peptide from amyloid-rich pancreases of type 2 diabetic patients. Proc Natl Acad Sci U S A 84(23):8628–8632
Cooper GJ, Willis AC, Reid KB, Clark A, Baker CA, Turner RC, Lewis CE, Morris JF, Howland K, Rothbard JB (1987b) Diabetes-associated peptide. Lancet 2(8565):966
Costa-Mattioli M, Monteggia LM (2013) mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nat Neurosci 16(11):1537–1543
Cota D (2009) Mammalian target of rapamycin complex 1 (mTORC1) signaling in energy balance and obesity. Physiol Behav 97(5):520–524
Cota D, Matter EK, Woods SC, Seeley RJ (2008) The role of hypothalamic mammalian target of rapamycin complex 1 signaling in diet-induced obesity. J Neurosci 28(28):7202–7208
Craft S, Watson GS (2004) Insulin and neurodegenerative disease: shared and specific mechanisms. Lancet Neurol 3(3):169–178
Crane PK, Walker R, Hubbard RA, Li G, Nathan DM, Zheng H, Haneuse S, Craft S, Montine TJ, Kahn SE, McCormick W, McCurry SM, Bowen JD, Larson EB (2013) Glucose levels and risk of dementia. N Engl J Med 369(6):540–548
Cruz-Jentoft AJ, Baeyens JP, Bauer JM, Boirie Y, Cederholm T, Landi F, Martin FC, Michel JP, Rolland Y, Schneider SM, Topinkova E, Vandewoude M, Zamboni M, P. European Working Group on Sarcopenia in Older (2010) Sarcopenia: European consensus on definition and diagnosis: report of the European Working Group on Sarcopenia in Older People. Age Ageing 39(4):412–423
Cukierman-Yaffe T, Gerstein HC, Williamson JD, Lazar RM, Lovato L, Miller ME, Coker LH, Murray A, Sullivan MD, Marcovina SM, Launer LJ, I. Action to Control Cardiovascular Risk in Diabetes-Memory in Diabetes (2009) Relationship between baseline glycemic control and cognitive function in individuals with type 2 diabetes and other cardiovascular risk factors: the action to control cardiovascular risk in diabetes-memory in diabetes (ACCORD-MIND) trial. Diabetes Care 32(2):221–226
Cybulski N, Hall MN (2009) TOR complex 2: a signaling pathway of its own. Trends Biochem Sci 34(12):620–627
D’Ercole AJ, Ye P, Calikoglu AS, Gutierrez-Ospina G (1996) The role of the insulin-like growth factors in the central nervous system. Mol Neurobiol 13(3):227–255
Dash PK, Orsi SA, Moore AN (2006) Spatial memory formation and memory-enhancing effect of glucose involves activation of the tuberous sclerosis complex-mammalian target of rapamycin pathway. J Neurosci 26(31):8048–8056
Dasuri K, Zhang L, Kim SOKF, Bruce-Keller AJ, Keller JN (2016) Dietary and donepezil modulation of mTOR signaling and neuroinflammation in the brain. Biochim Biophys Acta (BBA) – Mol Basis Dis 1862(2):274–283
Dazert E, Hall MN (2011) mTOR signaling in disease. Curr Opin Cell Biol 23(6):744–755
de la Monte SM (2014) Type 3 diabetes is sporadic Alzheimers disease: mini-review. Eur Neuropsychopharmacol 24(12):1954–1960
Dean PM (1973) Ultrastructural morphometry of the pancreatic -cell. Diabetologia 9(2):115–119
Debette S, Beiser A, Hoffmann U, Decarli C, O’Donnell CJ, Massaro JM, Au R, Himali JJ, Wolf PA, Fox CS, Seshadri S (2010) Visceral fat is associated with lower brain volume in healthy middle-aged adults. Ann Neurol 68(2):136–144
DeFronzo RA (2004) Dysfunctional fat cells, lipotoxicity and type 2 diabetes. Int J Clin Pract 143(Suppl):9–21
DeFronzo RA, Tripathy D (2009) Skeletal muscle insulin resistance is the primary defect in type 2 diabetes. Diabetes Care 32(Suppl 2):S157–S163
Dello Russo C, Lisi L, Tringali G, Navarra P (2009) Involvement of mTOR kinase in cytokine-dependent microglial activation and cell proliferation. Biochem Pharmacol 78(9):1242–1251
Di Domenico F, Barone E, Perluigi M, Butterfield DA (2017) The triangle of death in Alzheimer’s disease brain: the aberrant cross-talk among energy metabolism, mammalian target of rapamycin signaling, and protein homeostasis revealed by redox proteomics. Antioxid Redox Signal 26(8):364–387
Di Paolo S, Teutonico A, Leogrande D, Capobianco C, Schena PF (2006) Chronic inhibition of mammalian target of rapamycin signaling downregulates insulin receptor substrates 1 and 2 and AKT activation: a crossroad between cancer and diabetes? J Am Soc Nephrol 17(8):2236–2244
Dibbens LM, de Vries B, Donatello S, Heron SE, Hodgson BL, Chintawar S, Crompton DE, Hughes JN, Bellows ST, Klein KM, Callenbach PM, Corbett MA, Gardner AE, Kivity S, Iona X, Regan BM, Weller CM, Crimmins D, O’Brien TJ, Guerrero-Lopez R, Mulley JC, Dubeau F, Licchetta L, Bisulli F, Cossette P, Thomas PQ, Gecz J, Serratosa J, Brouwer OF, Andermann F, Andermann E, van den Maagdenberg AM, Pandolfo M, Berkovic SF, Scheffer IE (2013) Mutations in DEPDC5 cause familial focal epilepsy with variable foci. Nat Genet 45(5):546–551
Ding F, Yao J, Rettberg JR, Chen S, Brinton RD (2013) Early decline in glucose transport and metabolism precedes shift to ketogenic system in female aging and Alzheimer’s mouse brain: implication for bioenergetic intervention. PLoS One 8(11):e79977
Du H, Guo L, Yan S, Sosunov AA, McKhann GM, Yan SS (2010) Early deficits in synaptic mitochondria in an Alzheimer’s disease mouse model. Proc Natl Acad Sci U S A 107(43):18670–18675
Efeyan A, Comb WC, Sabatini DM (2015) Nutrient-sensing mechanisms and pathways. Nature 517(7534):302–310
Ehninger D (2013) From genes to cognition in tuberous sclerosis: implications for mTOR inhibitor-based treatment approaches. Neuropharmacology 68:97–105
Ehninger D, Han S, Shilyansky C, Zhou Y, Li W, Kwiatkowski DJ, Ramesh V, Silva AJ (2008) Reversal of learning deficits in a Tsc2+/− mouse model of tuberous sclerosis. Nat Med 14(8):843–848
Elghazi L, Balcazar N, Blandino-Rosano M, Cras-Meneur C, Fatrai S, Gould AP, Chi MM, Moley KH, Bernal-Mizrachi E (2010) Decreased IRS signaling impairs beta-cell cycle progression and survival in transgenic mice overexpressing S6K in beta-cells. Diabetes 59(10):2390–2399
Elghazi L, Blandino-Rosano M, Alejandro E, Cras-Meneur C, Bernal-Mizrachi E (2017) Role of nutrients and mTOR signaling in the regulation of pancreatic progenitors development. Mol Metab 6(6):560–573
Elmquist JK, Maratos-Flier E, Saper CB, Flier JS (1998) Unraveling the central nervous system pathways underlying responses to leptin. Nat Neurosci 1(6):445–450
Elovaara I, Icen A, Palo J, Erkinjuntti T (1985) CSF in Alzheimer’s disease. Studies on blood-brain barrier function and intrathecal protein synthesis. J Neurol Sci 70(1):73–80
Embury CM, Dyavarshetty B, Lu Y, Wiederin JL, Ciborowski P, Gendelman HE, Kiyota T (2017) Cathepsin B improves ss-amyloidosis and learning and memory in models of Alzheimer’s disease. J Neuroimmune Pharmacol 12(2):340–352
Emmerzaal TL, Kiliaan AJ, Gustafson DR (2015) 2003–2013: a decade of body mass index, Alzheimer’s disease, and dementia. J Alzheimers Dis 43(3):739–755
Erbsloh F, Bernsmeier A, Hillesheim H (1958) The glucose consumption of the brain & its dependence on the liver. Arch Psychiatr Nervenkr Z Gesamten Neurol Psychiatr 196(6):611–626
Fishwick KJ, Li RA, Halley P, Deng P, Storey KG (2010) Initiation of neuronal differentiation requires PI3-kinase/TOR signalling in the vertebrate neural tube. Dev Biol 338(2):215–225
Fraenkel M, Ketzinel-Gilad M, Ariav Y, Pappo O, Karaca M, Castel J, Berthault MF, Magnan C, Cerasi E, Kaiser N, Leibowitz G (2008) mTOR inhibition by rapamycin prevents beta-cell adaptation to hyperglycemia and exacerbates the metabolic state in type 2 diabetes. Diabetes 57(4):945–957
Frolich L, Blum-Degen D, Bernstein HG, Engelsberger S, Humrich J, Laufer S, Muschner D, Thalheimer A, Turk A, Hoyer S, Zochling R, Boissl KW, Jellinger K, Riederer P (1998) Brain insulin and insulin receptors in aging and sporadic Alzheimer’s disease. J Neural Transm (Vienna) 105(4–5):423–438
Gafford GM, Parsons RG, Helmstetter FJ (2013) Memory accuracy predicts hippocampal mTOR pathway activation following retrieval of contextual fear memory. Hippocampus 23(9):842–847
Gangloff YG, Mueller M, Dann SG, Svoboda P, Sticker M, Spetz JF, Um SH, Brown EJ, Cereghini S, Thomas G, Kozma SC (2004) Disruption of the mouse mTOR gene leads to early postimplantation lethality and prohibits embryonic stem cell development. Mol Cell Biol 24(21):9508–9516
Geuna E, Roda D, Rafii S, Jimenez B, Capelan M, Rihawi K, Montemurro F, Yap TA, Kaye SB, De Bono JS, Molife LR, Banerji U (2015) Complications of hyperglycaemia with PI3K–AKT–mTOR inhibitors in patients with advanced solid tumours on phase I clinical trials. Br J Cancer 113(11):1541–1547
Grinberg LT, Rueb U, Heinsen H (2011) Brainstem: neglected locus in neurodegenerative diseases. Front Neurol 2:42
Gu Y, Lindner J, Kumar A, Yuan W, Magnuson MA (2011) Rictor/mTORC2 is essential for maintaining a balance between beta-cell proliferation and cell size. Diabetes 60(3):827–837
Guertin DA, Stevens DM, Thoreen CC, Burds AA, Kalaany NY, Moffat J, Brown M, Fitzgerald KJ, Sabatini DM (2006) Ablation in mice of the mTORC components raptor, rictor, or mLST8 reveals that mTORC2 is required for signaling to Akt-FOXO and PKCalpha, but not S6K1. Dev Cell 11(6):859–871
Haaksma ML, Vilela LR, Marengoni A, Calderon-Larranaga A, Leoutsakos JS, Olde Rikkert MGM, Melis RJF (2017) Comorbidity and progression of late onset Alzheimer’s disease: a systematic review. PLoS One 12(5):e0177044
Hamada S, Hara K, Hamada T, Yasuda H, Moriyama H, Nakayama R, Nagata M, Yokono K (2009) Upregulation of the mammalian target of rapamycin complex 1 pathway by Ras homolog enriched in brain in pancreatic beta-cells leads to increased beta-cell mass and prevention of hyperglycemia. Diabetes 58(6):1321–1332
Han J, Wang B, Xiao Z, Gao Y, Zhao Y, Zhang J, Chen B, Wang X, Dai J (2008) Mammalian target of rapamycin (mTOR) is involved in the neuronal differentiation of neural progenitors induced by insulin. Mol Cell Neurosci 39(1):118–124
Hara K, Maruki Y, Long X, Yoshino K, Oshiro N, Hidayat S, Tokunaga C, Avruch J, Yonezawa K (2002) Raptor, a binding partner of target of rapamycin (TOR), mediates TOR action. Cell 110(2):177–189
Hara T, Nakamura K, Matsui M, Yamamoto A, Nakahara Y, Suzuki-Migishima R, Yokoyama M, Mishima K, Saito I, Okano H, Mizushima N (2006) Suppression of basal autophagy in neural cells causes neurodegenerative disease in mice. Nature 441(7095):885–889
Hardie DG, Ashford ML (2014) AMPK: regulating energy balance at the cellular and whole body levels. Physiology (Bethesda) 29(2):99–107
Harrington LS, Findlay GM, Gray A, Tolkacheva T, Wigfield S, Rebholz H, Barnett J, Leslie NR, Cheng S, Shepherd PR, Gout I, Downes CP, Lamb RF (2004) The TSC1-2 tumor suppressor controls insulin-PI3K signaling via regulation of IRS proteins. J Cell Biol 166(2):213–223
Harrison DE, Strong R, Sharp ZD, Nelson JF, Astle CM, Flurkey K, Nadon NL, Wilkinson JE, Frenkel K, Carter CS, Pahor M, Javors MA, Fernandez E, Miller RA (2009) Rapamycin fed late in life extends lifespan in genetically heterogeneous mice. Nature 460(7253):392–395
Harvey J, Solovyova N, Irving A (2006) Leptin and its role in hippocampal synaptic plasticity. Prog Lipid Res 45(5):369–378
Hasselbalch SG, Knudsen GM, Videbaek C, Pinborg LH, Schmidt JF, Holm S, Paulson OB (1999) No effect of insulin on glucose blood-brain barrier transport and cerebral metabolism in humans. Diabetes 48(10):1915–1921
Hatanaka M, Maier B, Sims EK, Templin AT, Kulkarni RN, Evans-Molina C, Mirmira RG (2014) Palmitate induces mRNA translation and increases ER protein load in islet beta-cells via activation of the mammalian target of rapamycin pathway. Diabetes 63(10):3404–3415
Hauptmann S, Scherping I, Drose S, Brandt U, Schulz KL, Jendrach M, Leuner K, Eckert A, Muller WE (2009) Mitochondrial dysfunction: an early event in Alzheimer pathology accumulates with age in AD transgenic mice. Neurobiol Aging 30(10):1574–1586
Henningsen J, Rigbolt KT, Blagoev B, Pedersen BK, Kratchmarova I (2010) Dynamics of the skeletal muscle secretome during myoblast differentiation. Mol Cell Proteomics 9(11):2482–2496
Henry FE, Hockeimer W, Chen A, Mysore SP, Sutton MA (2017) Mechanistic target of rapamycin is necessary for changes in dendritic spine morphology associated with long-term potentiation. Mol Brain 10(1):50
Ho AJ, Raji CA, Becker JT, Lopez OL, Kuller LH, Hua X, Lee S, Hibar D, Dinov ID, Stein JL, Jack CR Jr, Weiner MW, Toga AW, Thompson PM, S. Cardiovascular Health and Adni (2010) Obesity is linked with lower brain volume in 700 AD and MCI patients. Neurobiol Aging 31(8):1326–1339
Ho AJ, Raji CA, Saharan P, DeGiorgio A, Madsen SK, Hibar DP, Stein JL, Becker JT, Lopez OL, Toga AW, Thompson PM, I. Alzheimer’s Disease Neuroimaging (2011) Hippocampal volume is related to body mass index in Alzheimer’s disease. Neuroreport 22(1):10–14
Hofer MM, Barde YA (1988) Brain-derived neurotrophic factor prevents neuronal death in vivo. Nature 331(6153):261–262
Howarth C, Gleeson P, Attwell D (2012) Updated energy budgets for neural computation in the neocortex and cerebellum. J Cereb Blood Flow Metab 32(7):1222–1232
Hoyer S, Nitsch R, Oesterreich K (1991) Predominant abnormality in cerebral glucose utilization in late-onset dementia of the Alzheimer type: a cross-sectional comparison against advanced late-onset and incipient early-onset cases. J Neural Transm Park Dis Dement Sect 3(1):1–14
Huang W, Zhu PJ, Zhang S, Zhou H, Stoica L, Galiano M, Krnjevic K, Roman G, Costa-Mattioli M (2013) mTORC2 controls actin polymerization required for consolidation of long-term memory. Nat Neurosci 16(4):441–448
Hull RL, Westermark GT, Westermark P, Kahn SE (2004) Islet amyloid: a critical entity in the pathogenesis of type 2 diabetes. J Clin Endocrinol Metab 89(8):3629–3643
Inoki K, Ouyang H, Zhu T, Lindvall C, Wang Y, Zhang X, Yang Q, Bennett C, Harada Y, Stankunas K, Wang CY, He X, MacDougald OA, You M, Williams BO, Guan KL (2006) TSC2 integrates Wnt and energy signals via a coordinated phosphorylation by AMPK and GSK3 to regulate cell growth. Cell 126(5):955–968
Jacinto E, Loewith R, Schmidt A, Lin S, Ruegg MA, Hall A, Hall MN (2004) Mammalian TOR complex 2 controls the actin cytoskeleton and is rapamycin insensitive. Nat Cell Biol 6(11):1122–1128
Jacinto E, Facchinetti V, Liu D, Soto N, Wei S, Jung SY, Huang Q, Qin J, Su B (2006) SIN1/MIP1 maintains rictor-mTOR complex integrity and regulates Akt phosphorylation and substrate specificity. Cell 127(1):125–137
Janson J, Laedtke T, Parisi JE, O’Brien P, Petersen RC, Butler PC (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53(2):474–481
Janssen I, Heymsfield SB, Wang ZM, Ross R (2000) Skeletal muscle mass and distribution in 468 men and women aged 18-88 yr. J Appl Physiol (1985) 89(1):81–88
Jia K, Chen D, Riddle DL (2004) The TOR pathway interacts with the insulin signaling pathway to regulate C. elegans larval development, metabolism and life span. Development 131(16):3897–3906
Johnston O, Rose CL, Webster AC, Gill JS (2008) Sirolimus is associated with new-onset diabetes in kidney transplant recipients. J Am Soc Nephrol 19(7):1411–1418
Jolivalt CG, Hurford R, Lee CA, Dumaop W, Rockenstein E, Masliah E (2010) Type 1 diabetes exaggerates features of Alzheimer’s disease in APP transgenic mice. Exp Neurol 223(2):422–431
Jossin Y, Goffinet AM (2007) Reelin signals through phosphatidylinositol 3-kinase and Akt to control cortical development and through mTor to regulate dendritic growth. Mol Cell Biol 27(20):7113–7124
Jung HS, Chung KW, Won Kim J, Kim J, Komatsu M, Tanaka K, Nguyen YH, Kang TM, Yoon KH, Kim JW, Jeong YT, Han MS, Lee MK, Kim KW, Shin J, Lee MS (2008) Loss of autophagy diminishes pancreatic beta cell mass and function with resultant hyperglycemia. Cell Metab 8(4):318–324
Kaduszkiewicz H, Zimmermann T, Beck-Bornholdt HP, van den Bussche H (2005) Cholinesterase inhibitors for patients with Alzheimer’s disease: systematic review of randomised clinical trials. BMJ 331(7512):321–327
Kaeberlein M, Powers RW 3rd, Steffen KK, Westman EA, Hu D, Dang N, Kerr EO, Kirkland KT, Fields S, Kennedy BK (2005) Regulation of yeast replicative life span by TOR and Sch9 in response to nutrients. Science 310(5751):1193–1196
Kahn SE, Haffner SM, Heise MA, Herman WH, Holman RR, Jones NP, Kravitz BG, Lachin JM, O’Neill MC, Zinman B, Viberti G, A. S. Group (2006) Glycemic durability of rosiglitazone, metformin, or glyburide monotherapy. N Engl J Med 355(23):2427–2443
Kakumoto K, Ikeda J, Okada M, Morii E, Oneyama C (2015) mLST8 promotes mTOR-mediated tumor progression. PLoS One 10(4):e0119015
Kalyani RR, Saudek CD, Brancati FL, Selvin E (2010) Association of diabetes, comorbidities, and A1C with functional disability in older adults: results from the National Health and Nutrition Examination Survey (NHANES), 1999–2006. Diabetes Care 33(5):1055–1060
Kapahi P, Zid BM, Harper T, Koslover D, Sapin V, Benzer S (2004) Regulation of lifespan in Drosophila by modulation of genes in the TOR signaling pathway. Curr Biol 14(10):885–890
Kassai H, Sugaya Y, Noda S, Nakao K, Maeda T, Kano M, Aiba A (2014) Selective activation of mTORC1 signaling recapitulates microcephaly, tuberous sclerosis, and neurodegenerative diseases. Cell Rep 7(5):1626–1639
Ke YD, Delerue F, Gladbach A, Gotz J, Ittner LM (2009) Experimental diabetes mellitus exacerbates tau pathology in a transgenic mouse model of Alzheimer’s disease. PLoS One 4(11):e7917
Kennedy BK, Lamming DW (2016) The mechanistic target of rapamycin: the grand ConducTOR of metabolism and aging. Cell Metab 23(6):990–1003
Khamzina L, Veilleux A, Bergeron S, Marette A (2005) Increased activation of the mammalian target of rapamycin pathway in liver and skeletal muscle of obese rats: possible involvement in obesity-linked insulin resistance. Endocrinology 146(3):1473–1481
Kim DH, Sarbassov DD, Ali SM, King JE, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2002) mTOR interacts with raptor to form a nutrient-sensitive complex that signals to the cell growth machinery. Cell 110(2):163–175
Kim BW, Choi M, Kim YS, Park H, Lee HR, Yun CO, Kim EJ, Choi JS, Kim S, Rhim H, Kaang BK, Son H (2008) Vascular endothelial growth factor (VEGF) signaling regulates hippocampal neurons by elevation of intracellular calcium and activation of calcium/calmodulin protein kinase II and mammalian target of rapamycin. Cell Signal 20(4):714–725
Klein S, Coppack SW, Mohamed-Ali V, Landt M (1996) Adipose tissue leptin production and plasma leptin kinetics in humans. Diabetes 45(7):984–987
Kleinert M, Sylow L, Fazakerley DJ, Krycer JR, Thomas KC, Oxboll AJ, Jordy AB, Jensen TE, Yang G, Schjerling P, Kiens B, James DE, Ruegg MA, Richter EA (2014) Acute mTOR inhibition induces insulin resistance and alters substrate utilization in vivo. Mol Metab 3(6):630–641
Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, Ueno T, Koike M, Uchiyama Y, Kominami E, Tanaka K (2006) Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature 441(7095):880–884
Kovac A, Zilka N, Kazmerova Z, Cente M, Zilkova M, Novak M (2011) Misfolded truncated protein tau induces innate immune response via MAPK pathway. J Immunol 187(5):2732–2739
Krabbe KS, Nielsen AR, Krogh-Madsen R, Plomgaard P, Rasmussen P, Erikstrup C, Fischer CP, Lindegaard B, Petersen AM, Taudorf S, Secher NH, Pilegaard H, Bruunsgaard H, Pedersen BK (2007) Brain-derived neurotrophic factor (BDNF) and type 2 diabetes. Diabetologia 50(2):431–438
Krebs M, Brunmair B, Brehm A, Artwohl M, Szendroedi J, Nowotny P, Roth E, Furnsinn C, Promintzer M, Anderwald C, Bischof M, Roden M (2007) The mammalian target of rapamycin pathway regulates nutrient-sensitive glucose uptake in man. Diabetes 56(6):1600–1607
Kucejova B, Duarte J, Satapati S, Fu X, Ilkayeva O, Newgard CB, Brugarolas J, Burgess SC (2016) Hepatic mTORC1 opposes impaired insulin action to control mitochondrial metabolism in obesity. Cell Rep 16(2):508–519
Kumar A, Harris TE, Keller SR, Choi KM, Magnuson MA, Lawrence JC Jr (2008) Muscle-specific deletion of rictor impairs insulin-stimulated glucose transport and enhances Basal glycogen synthase activity. Mol Cell Biol 28(1):61–70
Kuo YM, Kokjohn TA, Watson MD, Woods AS, Cotter RJ, Sue LI, Kalback WM, Emmerling MR, Beach TG, Roher AE (2000) Elevated abeta42 in skeletal muscle of Alzheimer disease patients suggests peripheral alterations of AbetaPP metabolism. Am J Pathol 156(3):797–805
Kwon G, Marshall CA, Pappan KL, Remedi MS, McDaniel ML (2004) Signaling elements involved in the metabolic regulation of mTOR by nutrients, incretins, and growth factors in islets. Diabetes 53(Suppl 3):S225–S232
Lamming DW, Sabatini DM (2013) A central role for mTOR in lipid homeostasis. Cell Metab 18(4):465–469
Lamming DW, Ye L, Katajisto P, Goncalves MD, Saitoh M, Stevens DM, Davis JG, Salmon AB, Richardson A, Ahima RS, Guertin DA, Sabatini DM, Baur JA (2012) Rapamycin-induced insulin resistance is mediated by mTORC2 loss and uncoupled from longevity. Science 335(6076):1638
Laplante M, Sabatini DM (2012) mTOR signaling in growth control and disease. Cell 149(2):274–293
Laske C, Stransky E, Leyhe T, Eschweiler GW, Wittorf A, Richartz E, Bartels M, Buchkremer G, Schott K (2006) Stage-dependent BDNF serum concentrations in Alzheimer’s disease. J Neural Transm (Vienna) 113(9):1217–1224
Lavan BE, Fantin VR, Chang ET, Lane WS, Keller SR, Lienhard GE (1997a) A novel 160-kDa phosphotyrosine protein in insulin-treated embryonic kidney cells is a new member of the insulin receptor substrate family. J Biol Chem 272(34):21403–21407
Lavan BE, Lane WS, Lienhard GE (1997b) The 60-kDa phosphotyrosine protein in insulin-treated adipocytes is a new member of the insulin receptor substrate family. J Biol Chem 272(17):11439–11443
Leibowitz G, Bachar E, Shaked M, Sinai A, Ketzinel-Gilad M, Cerasi E, Kaiser N (2010) Glucose regulation of beta-cell stress in type 2 diabetes. Diabetes Obes Metab 12(Suppl 2):66–75
Leibson CL, Rocca WA, Hanson VA, Cha R, Kokmen E, O’Brien PC, Palumbo PJ (1997) Risk of dementia among persons with diabetes mellitus: a population-based cohort study. Am J Epidemiol 145(4):301–308
Li YH, Werner H, Puschel AW (2008) Rheb and mTOR regulate neuronal polarity through Rap1B. J Biol Chem 283(48):33784–33792
Li D, Liu F, Yang T, Jin T, Zhang H, Luo X, Wang M (2016a) Rapamycin protects against neuronal death and improves neurological function with modulation of microglia after experimental intracerebral hemorrhage in rats. Cell Mol Biol (Noisy-le-Grand) 62(11):67–75
Li D, Wang C, Yao Y, Chen L, Liu G, Zhang R, Liu Q, Shi FD, Hao J (2016b) mTORC1 pathway disruption ameliorates brain inflammation following stroke via a shift in microglia phenotype from M1 type to M2 type. FASEB J 30(10):3388–3399
Liang H, Tantiwong P, Sriwijitkamol A, Shanmugasundaram K, Mohan S, Espinoza S, Defronzo RA, Dube JJ, Musi N (2013) Effect of a sustained reduction in plasma free fatty acid concentration on insulin signalling and inflammation in skeletal muscle from human subjects. J Physiol 591(11):2897–2909
Lieb W, Beiser AS, Vasan RS, Tan ZS, Au R, Harris TB, Roubenoff R, Auerbach S, DeCarli C, Wolf PA, Seshadri S (2009) Association of plasma leptin levels with incident Alzheimer disease and MRI measures of brain aging. JAMA 302(23):2565–2572
Lim YA, Rhein V, Baysang G, Meier F, Poljak A, Raftery MJ, Guilhaus M, Ittner LM, Eckert A, Gotz J (2010) Abeta and human amylin share a common toxicity pathway via mitochondrial dysfunction. Proteomics 10(8):1621–1633
Liu K, Lu Y, Lee JK, Samara R, Willenberg R, Sears-Kraxberger I, Tedeschi A, Park KK, Jin D, Cai B, Xu B, Connolly L, Steward O, Zheng B, He Z (2010) PTEN deletion enhances the regenerative ability of adult corticospinal neurons. Nat Neurosci 13(9):1075–1081
Liu D, Bordicchia M, Zhang C, Fang H, Wei W, Li JL, Guilherme A, Guntur K, Czech MP, Collins S (2016a) Activation of mTORC1 is essential for beta-adrenergic stimulation of adipose browning. J Clin Invest 126(5):1704–1716
Liu Z, Gan L, Liu G, Chen Y, Wu T, Feng F, Sun C (2016b) Sirt1 decreased adipose inflammation by interacting with Akt2 and inhibiting mTOR/S6K1 pathway in mice. J Lipid Res 57(8):1373–1381
Lopes DH, Colin C, Degaki TL, de Sousa AC, Vieira MN, Sebollela A, Martinez AM, Bloch C Jr, Ferreira ST, Sogayar MC (2004) Amyloidogenicity and cytotoxicity of recombinant mature human islet amyloid polypeptide (rhIAPP). J Biol Chem 279(41):42803–42810
Lourenco MV, Clarke JR, Frozza RL, Bomfim TR, Forny-Germano L, Batista AF, Sathler LB, Brito-Moreira J, Amaral OB, Silva CA, Freitas-Correa L, Espirito-Santo S, Campello-Costa P, Houzel JC, Klein WL, Holscher C, Carvalheira JB, Silva AM, Velloso LA, Munoz DP, Ferreira ST, De Felice FG (2013) TNF-alpha mediates PKR-dependent memory impairment and brain IRS-1 inhibition induced by Alzheimer’s beta-amyloid oligomers in mice and monkeys. Cell Metab 18(6):831–843
Ma YQ, Wu DK, Liu JK (2013) mTOR and tau phosphorylated proteins in the hippocampal tissue of rats with type 2 diabetes and Alzheimer’s disease. Mol Med Rep 7(2):623–627
Ma Y, Wu D, Zhang W, Liu J, Chen S, Hua B (2015) Investigation of PI3K/PKB/mTOR/S6K1 signaling pathway in relationship of type 2 diabetes and Alzheimer’s disease. Int J Clin Exp Med 8(10):18581–18590
Magdalon J, Chimin P, Belchior T, Neves RX, Vieira-Lara MA, Andrade ML, Farias TS, Bolsoni-Lopes A, Paschoal VA, Yamashita AS, Kowaltowski AJ, Festuccia WT (2016) Constitutive adipocyte mTORC1 activation enhances mitochondrial activity and reduces visceral adiposity in mice. Biochim Biophys Acta 1861(5):430–438
Maj M, Gartner W, Ilhan A, Neziri D, Attems J, Wagner L (2010) Expression of TAU in insulin-secreting cells and its interaction with the calcium-binding protein secretagogin. J Endocrinol 205(1):25–36
Maj M, Hoermann G, Rasul S, Base W, Wagner L, Attems J (2016) The microtubule-associated protein tau and its relevance for pancreatic beta cells. J Diabetes Res 2016:1964634
Majumder S, Richardson A, Strong R, Oddo S (2011) Inducing autophagy by rapamycin before, but not after, the formation of plaques and tangles ameliorates cognitive deficits. PLoS One 6(9):e25416
Majumder S, Caccamo A, Medina DX, Benavides AD, Javors MA, Kraig E, Strong R, Richardson A, Oddo S (2012) Lifelong rapamycin administration ameliorates age-dependent cognitive deficits by reducing IL-1beta and enhancing NMDA signaling. Aging Cell 11(2):326–335
Malagelada C, Lopez-Toledano MA, Willett RT, Jin ZH, Shelanski ML, Greene LA (2011) RTP801/REDD1 regulates the timing of cortical neurogenesis and neuron migration. J Neurosci 31(9):3186–3196
Mannaa M, Kramer S, Boschmann M, Gollasch M (2013) mTOR and regulation of energy homeostasis in humans. J Mol Med (Berl) 91(10):1167–1175
Mannick JB, Del Giudice G, Lattanzi M, Valiante NM, Praestgaard J, Huang B, Lonetto MA, Maecker HT, Kovarik J, Carson S, Glass DJ, Klickstein LB (2014) mTOR inhibition improves immune function in the elderly. Sci Transl Med 6(268):268ra179
Manning BD, Toker A (2017) AKT/PKB signaling: navigating the network. Cell 169(3):381–405
Marfaing P, Penicaud L, Broer Y, Mraovitch S, Calando Y, Picon L (1990) Effects of hyperinsulinemia on local cerebral insulin binding and glucose utilization in normoglycemic awake rats. Neurosci Lett 115(2–3):279–285
Masini M, Bugliani M, Lupi R, del Guerra S, Boggi U, Filipponi F, Marselli L, Masiello P, Marchetti P (2009) Autophagy in human type 2 diabetes pancreatic beta cells. Diabetologia 52(6):1083–1086
Matthews VB, Astrom MB, Chan MH, Bruce CR, Krabbe KS, Prelovsek O, Akerstrom T, Yfanti C, Broholm C, Mortensen OH, Penkowa M, Hojman P, Zankari A, Watt MJ, Bruunsgaard H, Pedersen BK, Febbraio MA (2009) Brain-derived neurotrophic factor is produced by skeletal muscle cells in response to contraction and enhances fat oxidation via activation of AMP-activated protein kinase. Diabetologia 52(7):1409–1418
Mazure CM, Swendsen J (2016) Sex differences in Alzheimer’s disease and other dementias. Lancet Neurol 15(5):451–452
McDaniel ML, Marshall CA, Pappan KL, Kwon G (2002) Metabolic and autocrine regulation of the mammalian target of rapamycin by pancreatic beta-cells. Diabetes 51(10):2877–2885
McKhann G, Drachman D, Folstein M, Katzman R, Price D, Stadlan EM (1984) Clinical diagnosis of Alzheimer’s disease: report of the NINCDS-ADRDA Work Group under the auspices of Department of Health and Human Services Task Force on Alzheimer’s Disease. Neurology 34(7):939–944
Miklossy J, Qing H, Radenovic A, Kis A, Vileno B, Laszlo F, Miller L, Martins RN, Waeber G, Mooser V, Bosman F, Khalili K, Darbinian N, McGeer PL (2010) Beta amyloid and hyperphosphorylated tau deposits in the pancreas in type 2 diabetes. Neurobiol Aging 31(9):1503–1515
Mir SU, George NM, Zahoor L, Harms R, Guinn Z, Sarvetnick NE (2015) Inhibition of autophagic turnover in beta-cells by fatty acids and glucose leads to apoptotic cell death. J Biol Chem 290(10):6071–6085
Miura P, Amirouche A, Clow C, Belanger G, Jasmin BJ (2012) Brain-derived neurotrophic factor expression is repressed during myogenic differentiation by miR-206. J Neurochem 120(2):230–238
Moloney AM, Griffin RJ, Timmons S, O'Connor R, Ravid R, O’Neill C (2010) Defects in IGF-1 receptor, insulin receptor and IRS-1/2 in Alzheimer’s disease indicate possible resistance to IGF-1 and insulin signalling. Neurobiol Aging 31(2):224–243
Monteiro-Cardoso VF, Castro M, Oliveira MM, Moreira PI, Peixoto F, Videira RA (2015) Age-dependent biochemical dysfunction in skeletal muscle of triple-transgenic mouse model of Alzheimer’s disease. Curr Alzheimer Res 12(2):100–115
Moon HY, Becke A, Berron D, Becker B, Sah N, Benoni G, Janke E, Lubejko ST, Greig NH, Mattison JA, Duzel E, van Praag H (2016) Running-induced systemic cathepsin B secretion is associated with memory function. Cell Metab 24(2):332–340
Mori H, Inoki K, Opland D, Munzberg H, Villanueva EC, Faouzi M, Ikenoue T, Kwiatkowski DJ, Macdougald OA, Myers MG Jr, Guan KL (2009) Critical roles for the TSC-mTOR pathway in beta-cell function. Am J Physiol Endocrinol Metab 297(5):E1013–E1022
Morris JK, Burns JM (2012) Insulin: an emerging treatment for Alzheimer’s disease dementia? Curr Neurol Neurosci Rep 12(5):520–527
Mosconi L, Pupi A, De Leon MJ (2008) Brain glucose hypometabolism and oxidative stress in preclinical Alzheimer’s disease. Ann N Y Acad Sci 1147:180–195
Mosconi L, Mistur R, Switalski R, Tsui WH, Glodzik L, Li Y, Pirraglia E, De Santi S, Reisberg B, Wisniewski T, de Leon MJ (2009) FDG-PET changes in brain glucose metabolism from normal cognition to pathologically verified Alzheimer’s disease. Eur J Nucl Med Mol Imaging 36(5):811–822
Mullins GR, Wang L, Raje V, Sherwood SG, Grande RC, Boroda S, Eaton JM, Blancquaert S, Roger PP, Leitinger N, Harris TE (2014) Catecholamine-induced lipolysis causes mTOR complex dissociation and inhibits glucose uptake in adipocytes. Proc Natl Acad Sci U S A 111(49):17450–17455
Muoio DM, Newgard CB (2008) Mechanisms of disease: molecular and metabolic mechanisms of insulin resistance and beta-cell failure in type 2 diabetes. Nat Rev Mol Cell Biol 9(3):193–205
Murakami M, Ichisaka T, Maeda M, Oshiro N, Hara K, Edenhofer F, Kiyama H, Yonezawa K, Yamanaka S (2004) mTOR is essential for growth and proliferation in early mouse embryos and embryonic stem cells. Mol Cell Biol 24(15):6710–6718
Neff F, Flores-Dominguez D, Ryan DP, Horsch M, Schroder S, Adler T, Afonso LC, Aguilar-Pimentel JA, Becker L, Garrett L, Hans W, Hettich MM, Holtmeier R, Holter SM, Moreth K, Prehn C, Puk O, Racz I, Rathkolb B, Rozman J, Naton B, Ordemann R, Adamski J, Beckers J, Bekeredjian R, Busch DH, Ehninger G, Graw J, Hofler H, Klingenspor M, Klopstock T, Ollert M, Stypmann J, Wolf E, Wurst W, Zimmer A, Fuchs H, Gailus-Durner V, Hrabe de Angelis M, Ehninger D (2013) Rapamycin extends murine lifespan but has limited effects on aging. J Clin Invest 123(8):3272–3291
Ni Q, Gu Y, Xie Y, Yin Q, Zhang H, Nie A, Li W, Wang Y, Ning G, Wang W, Wang Q (2017) Raptor regulates functional maturation of murine beta cells. Nat Commun 8:15755
Nie D, Di Nardo A, Han JM, Baharanyi H, Kramvis I, Huynh T, Dabora S, Codeluppi S, Pandolfi PP, Pasquale EB, Sahin M (2010) Tsc2-Rheb signaling regulates EphA-mediated axon guidance. Nat Neurosci 13(2):163–172
Nixon RA (2013) The role of autophagy in neurodegenerative disease. Nat Med 19(8):983–997
O’Neill C (2013) PI3-kinase/Akt/mTOR signaling: impaired on/off switches in aging, cognitive decline and Alzheimer’s disease. Exp Gerontol 48(7):647–653
Oddo S (2012) The role of mTOR signaling in Alzheimer disease. Front Biosci (Schol Ed) 4:941–952
Ohara T, Doi Y, Ninomiya T, Hirakawa Y, Hata J, Iwaki T, Kanba S, Kiyohara Y (2011) Glucose tolerance status and risk of dementia in the community: the Hisayama study. Neurology 77(12):1126–1134
Olofsson CS, Gopel SO, Barg S, Galvanovskis J, Ma X, Salehi A, Rorsman P, Eliasson L (2002) Fast insulin secretion reflects exocytosis of docked granules in mouse pancreatic B-cells. Pflugers Arch 444(1–2):43–51
Omura T, Sano M, Omura K, Hasegawa T, Doi M, Sawada T, Nagano A (2005) Different expressions of BDNF, NT3, and NT4 in muscle and nerve after various types of peripheral nerve injuries. J Peripher Nerv Syst 10(3):293–300
Opie EL (1901) On the relation of chronic interstitial pancreatitis to the islands of langerhans and to diabetes melutus. J Exp Med 5(4):397–428
Orr ME, Oddo S (2013) Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther 5(5):53
Orr ME, Salinas A, Buffenstein R, Oddo S (2014) Mammalian target of rapamycin hyperactivity mediates the detrimental effects of a high sucrose diet on Alzheimer’s disease pathology. Neurobiol Aging 35(6):1233–1242
Orr ME, Sullivan AC, Frost B (2017) A brief overview of tauopathy: causes, consequences, and therapeutic strategies. Trends Pharmacol Sci 38(7):637–648
Oshiro N, Takahashi R, Yoshino K, Tanimura K, Nakashima A, Eguchi S, Miyamoto T, Hara K, Takehana K, Avruch J, Kikkawa U, Yonezawa K (2007) The proline-rich Akt substrate of 40 kDa (PRAS40) is a physiological substrate of mammalian target of rapamycin complex 1. J Biol Chem 282(28):20329–20339
Ostrowski K, Rohde T, Asp S, Schjerling P, Pedersen BK (1999) Pro- and anti-inflammatory cytokine balance in strenuous exercise in humans. J Physiol 515(Pt 1):287–291
Ott A, Stolk RP, van Harskamp F, Pols HA, Hofman A, Breteler MM (1999) Diabetes mellitus and the risk of dementia: the Rotterdam Study. Neurology 53(9):1937–1942
Ozcelik S, Fraser G, Castets P, Schaeffer V, Skachokova Z, Breu K, Clavaguera F, Sinnreich M, Kappos L, Goedert M, Tolnay M, Winkler DT (2013) Rapamycin attenuates the progression of tau pathology in P301S tau transgenic mice. PLoS One 8(5):e62459
Panja D, Dagyte G, Bidinosti M, Wibrand K, Kristiansen AM, Sonenberg N, Bramham CR (2009) Novel translational control in Arc-dependent long term potentiation consolidation in vivo. J Biol Chem 284(46):31498–31511
Park KK, Liu K, Hu Y, Smith PD, Wang C, Cai B, Xu B, Connolly L, Kramvis I, Sahin M, He Z (2008) Promoting axon regeneration in the adult CNS by modulation of the PTEN/mTOR pathway. Science 322(5903):963–966
Parsons RG, Gafford GM, Helmstetter FJ (2006) Translational control via the mammalian target of rapamycin pathway is critical for the formation and stability of long-term fear memory in amygdala neurons. J Neurosci 26(50):12977–12983
Paschoal VA, Amano MT, Belchior T, Magdalon J, Chimin P, Andrade ML, Ortiz-Silva M, Castro E, Yamashita AS, Rosa Neto JC, Camara NO, Festuccia WT (2017) mTORC1 inhibition with rapamycin exacerbates adipose tissue inflammation in obese mice and dissociates macrophage phenotype from function. Immunobiology 222(2):261–271
Paz-Filho G, Wong ML, Licinio J (2010) Leptin levels and Alzheimer disease. JAMA 303(15):1478. author reply 1478–1479
Pedersen L, Hojman P (2012) Muscle-to-organ cross talk mediated by myokines. Adipocyte 1(3):164–167
Pedros I, Petrov D, Artiach G, Abad S, Ramon-Duaso C, Sureda F, Pallas M, Beas-Zarate C, Folch J, Camins A (2015) Adipokine pathways are altered in hippocampus of an experimental mouse model of Alzheimer’s disease. J Nutr Health Aging 19(4):403–412
Pei JJ, Hugon J (2008) mTOR-dependent signalling in Alzheimer’s disease. J Cell Mol Med 12(6B):2525–2532
Pei JJ, Bjorkdahl C, Zhang H, Zhou X, Winblad B (2008) p70 S6 kinase and tau in Alzheimer’s disease. J Alzheimers Dis 14(4):385–392
Pende M, Kozma SC, Jaquet M, Oorschot V, Burcelin R, Le Marchand-Brustel Y, Klumperman J, Thorens B, Thomas G (2000) Hypoinsulinaemia, glucose intolerance and diminished beta-cell size in S6K1-deficient mice. Nature 408(6815):994–997
Perez-Gonzalez R, Antequera D, Vargas T, Spuch C, Bolos M, Carro E (2011) Leptin induces proliferation of neuronal progenitors and neuroprotection in a mouse model of Alzheimer’s disease. J Alzheimers Dis 24(Suppl 2):17–25
Peterson TR, Laplante M, Thoreen CC, Sancak Y, Kang SA, Kuehl WM, Gray NS, Sabatini DM (2009) DEPTOR is an mTOR inhibitor frequently overexpressed in multiple myeloma cells and required for their survival. Cell 137(5):873–886
Plaschke K, Kopitz J, Siegelin M, Schliebs R, Salkovic-Petrisic M, Riederer P, Hoyer S (2010) Insulin-resistant brain state after intracerebroventricular streptozotocin injection exacerbates Alzheimer-like changes in Tg2576 AbetaPP-overexpressing mice. J Alzheimers Dis 19(2):691–704
Polak P, Cybulski N, Feige JN, Auwerx J, Ruegg MA, Hall MN (2008) Adipose-specific knockout of raptor results in lean mice with enhanced mitochondrial respiration. Cell Metab 8(5):399–410
Polito VA, Li H, Martini-Stoica H, Wang B, Yang L, Xu Y, Swartzlander DB, Palmieri M, di Ronza A, Lee VM, Sardiello M, Ballabio A, Zheng H (2014) Selective clearance of aberrant tau proteins and rescue of neurotoxicity by transcription factor EB. EMBO Mol Med 6(9):1142–1160
Popken GJ, Hodge RD, Ye P, Zhang J, Ng W, O’Kusky JR, D’Ercole AJ (2004) In vivo effects of insulin-like growth factor-I (IGF-I) on prenatal and early postnatal development of the central nervous system. Eur J Neurosci 19(8):2056–2068
Powers RW 3rd, Kaeberlein M, Caldwell SD, Kennedy BK, Fields S (2006) Extension of chronological life span in yeast by decreased TOR pathway signaling. Genes Dev 20(2):174–184
Prentki M, Nolan CJ (2006) Islet beta cell failure in type 2 diabetes. J Clin Invest 116(7):1802–1812
Rachdi L, Balcazar N, Osorio-Duque F, Elghazi L, Weiss A, Gould A, Chang-Chen KJ, Gambello MJ, Bernal-Mizrachi E (2008) Disruption of Tsc2 in pancreatic beta cells induces beta cell mass expansion and improved glucose tolerance in a TORC1-dependent manner. Proc Natl Acad Sci U S A 105(27):9250–9255
Raclot T, Groscolas R (1993) Differential mobilization of white adipose tissue fatty acids according to chain length, unsaturation, and positional isomerism. J Lipid Res 34(9):1515–1526
Raskind MA, Peskind ER, Halter JB, Jimerson DC (1984) Norepinephrine and MHPG levels in CSF and plasma in Alzheimer’s disease. Arch Gen Psychiatry 41(4):343–346
Rhodes CJ, White MF, Leahy JL, Kahn SE (2013) Direct autocrine action of insulin on beta-cells: does it make physiological sense? Diabetes 62(7):2157–2163
Riahi Y, Wikstrom JD, Bachar-Wikstrom E, Polin N, Zucker H, Lee MS, Quan W, Haataja L, Liu M, Arvan P, Cerasi E, Leibowitz G (2016) Autophagy is a major regulator of beta cell insulin homeostasis. Diabetologia 59(7):1480–1491
Ristow M (2004) Neurodegenerative disorders associated with diabetes mellitus. J Mol Med (Berl) 82(8):510–529
Rivera EJ, Goldin A, Fulmer N, Tavares R, Wands JR, de la Monte SM (2005) Insulin and insulin-like growth factor expression and function deteriorate with progression of Alzheimer’s disease: link to brain reductions in acetylcholine. J Alzheimers Dis 8(3):247–268
Robertson RP, Harmon J, Tran PO, Poitout V (2004) Beta-cell glucose toxicity, lipotoxicity, and chronic oxidative stress in type 2 diabetes. Diabetes 53(Suppl 1):S119–S124
Roczniak-Ferguson A, Petit CS, Froehlich F, Qian S, Ky J, Angarola B, Walther TC, Ferguson SM (2012) The transcription factor TFEB links mTORC1 signaling to transcriptional control of lysosome homeostasis. Sci Signal 5(228):ra42
Rodriguez-Casado A, Toledano-Diaz A, Toledano A (2017) Defective insulin signalling, mediated by inflammation, connects obesity to Alzheimer disease; relevant pharmacological therapies and preventive dietary interventions. Curr Alzheimer Res 14(8):894–911
Roh C, Han J, Tzatsos A, Kandror KV (2003) Nutrient-sensing mTOR-mediated pathway regulates leptin production in isolated rat adipocytes. Am J Physiol Endocrinol Metab 284(2):E322–E330
Rotschafer SE, Razak KA (2014) Auditory processing in fragile x syndrome. Front Cell Neurosci 8:19
Russo E, Follesa P, Citraro R, Camastra C, Donato A, Isola D, Constanti A, De Sarro G, Donato G (2014) The mTOR signaling pathway and neuronal stem/progenitor cell proliferation in the hippocampus are altered during the development of absence epilepsy in a genetic animal model. Neurol Sci 35(11):1793–1799
Ruvinsky I, Sharon N, Lerer T, Cohen H, Stolovich-Rain M, Nir T, Dor Y, Zisman P, Meyuhas O (2005) Ribosomal protein S6 phosphorylation is a determinant of cell size and glucose homeostasis. Genes Dev 19(18):2199–2211
Saha AK, Xu XJ, Balon TW, Brandon A, Kraegen EW, Ruderman NB (2011) Insulin resistance due to nutrient excess: is it a consequence of AMPK downregulation? Cell Cycle 10(20):3447–3451
Sancak Y, Thoreen CC, Peterson TR, Lindquist RA, Kang SA, Spooner E, Carr SA, Sabatini DM (2007) PRAS40 is an insulin-regulated inhibitor of the mTORC1 protein kinase. Mol Cell 25(6):903–915
Sancak Y, Peterson TR, Shaul YD, Lindquist RA, Thoreen CC, Bar-Peled L, Sabatini DM (2008) The Rag GTPases bind raptor and mediate amino acid signaling to mTORC1. Science 320(5882):1496–1501
Santacruz K, Lewis J, Spires T, Paulson J, Kotilinek L, Ingelsson M, Guimaraes A, DeTure M, Ramsden M, McGowan E, Forster C, Yue M, Orne J, Janus C, Mariash A, Kuskowski M, Hyman B, Hutton M, Ashe KH (2005) Tau suppression in a neurodegenerative mouse model improves memory function. Science 309(5733):476–481
Sarbassov DD, Ali SM, Kim DH, Guertin DA, Latek RR, Erdjument-Bromage H, Tempst P, Sabatini DM (2004) Rictor, a novel binding partner of mTOR, defines a rapamycin-insensitive and raptor-independent pathway that regulates the cytoskeleton. Curr Biol 14(14):1296–1302
Sarbassov DD, Guertin DA, Ali SM, Sabatini DM (2005) Phosphorylation and regulation of Akt/PKB by the rictor-mTOR complex. Science 307(5712):1098–1101
Sarbassov DD, Ali SM, Sengupta S, Sheen JH, Hsu PP, Bagley AF, Markhard AL, Sabatini DM (2006) Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol Cell 22(2):159–168
Saxton RA, Sabatini DM (2017a) mTOR signaling in growth, metabolism, and disease. Cell 168(6):960–976
Saxton RA, Sabatini DM (2017b) mTOR signaling in growth, metabolism, and disease. Cell 169(2):361–371
Schicknick H, Schott BH, Budinger E, Smalla KH, Riedel A, Seidenbecher CI, Scheich H, Gundelfinger ED, Tischmeyer W (2008) Dopaminergic modulation of auditory cortex-dependent memory consolidation through mTOR. Cereb Cortex 18(11):2646–2658
Schnyder S, Handschin C (2015) Skeletal muscle as an endocrine organ: PGC-1alpha, myokines and exercise. Bone 80:115–125
Schuh RA, Jackson KC, Schlappal AE, Spangenburg EE, Ward CW, Park JH, Dugger N, Shi GL, Fishman PS (2014) Mitochondrial oxygen consumption deficits in skeletal muscle isolated from an Alzheimer’s disease-relevant murine model. BMC Neurosci 15:24
Schwartz MW, Peskind E, Raskind M, Boyko EJ, Porte D Jr (1996) Cerebrospinal fluid leptin levels: relationship to plasma levels and to adiposity in humans. Nat Med 2(5):589–593
Seaquist ER, Damberg GS, Tkac I, Gruetter R (2001) The effect of insulin on in vivo cerebral glucose concentrations and rates of glucose transport/metabolism in humans. Diabetes 50(10):2203
Selman C, Tullet JM, Wieser D, Irvine E, Lingard SJ, Choudhury AI, Claret M, Al-Qassab H, Carmignac D, Ramadani F, Woods A, Robinson IC, Schuster E, Batterham RL, Kozma SC, Thomas G, Carling D, Okkenhaug K, Thornton JM, Partridge L, Gems D, Withers DJ (2009) Ribosomal protein S6 kinase 1 signaling regulates mammalian life span. Science 326(5949):140–144
Sengupta S, Peterson TR, Laplante M, Oh S, Sabatini DM (2010) mTORC1 controls fasting-induced ketogenesis and its modulation by ageing. Nature 468(7327):1100–1104
Settembre C, Zoncu R, Medina DL, Vetrini F, Erdin S, Erdin S, Huynh T, Ferron M, Karsenty G, Vellard MC, Facchinetti V, Sabatini DM, Ballabio A (2012) A lysosome-to-nucleus signalling mechanism senses and regulates the lysosome via mTOR and TFEB. EMBO J 31(5):1095–1108
Shah OJ, Wang Z, Hunter T (2004) Inappropriate activation of the TSC/Rheb/mTOR/S6K cassette induces IRS1/2 depletion, insulin resistance, and cell survival deficiencies. Curr Biol 14(18):1650–1656
Shan T, Zhang P, Jiang Q, Xiong Y, Wang Y, Kuang S (2016) Adipocyte-specific deletion of mTOR inhibits adipose tissue development and causes insulin resistance in mice. Diabetologia 59(9):1995–2004
Sharma A, Hoeffer CA, Takayasu Y, Miyawaki T, McBride SM, Klann E, Zukin RS (2010) Dysregulation of mTOR signaling in fragile X syndrome. J Neurosci 30(2):694–702
Sharp ZD, Strong R (2010) The role of mTOR signaling in controlling mammalian life span: what a fungicide teaches us about longevity. J Gerontol A Biol Sci Med Sci 65(6):580–589
Shigeyama Y, Kobayashi T, Kido Y, Hashimoto N, Asahara S, Matsuda T, Takeda A, Inoue T, Shibutani Y, Koyanagi M, Uchida T, Inoue M, Hino O, Kasuga M, Noda T (2008) Biphasic response of pancreatic beta-cell mass to ablation of tuberous sclerosis complex 2 in mice. Mol Cell Biol 28(9):2971–2979
Shiota C, Woo JT, Lindner J, Shelton KD, Magnuson MA (2006) Multiallelic disruption of the rictor gene in mice reveals that mTOR complex 2 is essential for fetal growth and viability. Dev Cell 11(4):583–589
Sinagoga KL, Stone WJ, Schiesser JV, Schweitzer JI, Sampson L, Zheng Y, Wells JM (2017) Distinct roles for the mTOR pathway in postnatal morphogenesis, maturation and function of pancreatic islets. Development 144(13):2402–2414
Song J, Choi SM, Kim BC (2017) Adiponectin regulates the polarization and function of microglia via PPAR-gamma signaling under amyloid beta toxicity. Front Cell Neurosci 11:64
Soumya SJ, Binu S, Helen A, Reddanna P, Sudhakaran PR (2013) 15(S)-HETE-induced angiogenesis in adipose tissue is mediated through activation of PI3K/Akt/mTOR signaling pathway. Biochem Cell Biol 91(6):498–505
Sparks DL, Scheff SW, Hunsaker JC 3rd, Liu H, Landers T, Gross DR (1994) Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 126(1):88–94
Spilman P, Podlutskaya N, Hart MJ, Debnath J, Gorostiza O, Bredesen D, Richardson A, Strong R, Galvan V (2010) Inhibition of mTOR by rapamycin abolishes cognitive deficits and reduces amyloid-beta levels in a mouse model of Alzheimer’s disease. PLoS One 5(4):e9979
Stamateris RE, Sharma RB, Kong Y, Ebrahimpour P, Panday D, Ranganath P, Zou B, Levitt H, Parambil NA, O’Donnell CP, Garcia-Ocana A, Alonso LC (2016) Glucose induces mouse beta-cell proliferation via IRS2, MTOR, and cyclin D2 but not the insulin receptor. Diabetes 65(4):981–995
Stienstra R, Haim Y, Riahi Y, Netea M, Rudich A, Leibowitz G (2014) Autophagy in adipose tissue and the beta cell: implications for obesity and diabetes. Diabetologia 57(8):1505–1516
Stoica L, Zhu PJ, Huang W, Zhou H, Kozma SC, Costa-Mattioli M (2011) Selective pharmacogenetic inhibition of mammalian target of Rapamycin complex I (mTORC1) blocks long-term synaptic plasticity and memory storage. Proc Natl Acad Sci U S A 108(9):3791–3796
Studenski S, Perera S, Patel K, Rosano C, Faulkner K, Inzitari M, Brach J, Chandler J, Cawthon P, Connor EB, Nevitt M, Visser M, Kritchevsky S, Badinelli S, Harris T, Newman AB, Cauley J, Ferrucci L, Guralnik J (2011) Gait speed and survival in older adults. JAMA 305(1):50–58
Sun XJ, Rothenberg P, Kahn CR, Backer JM, Araki E, Wilden PA, Cahill DA, Goldstein BJ, White MF (1991) Structure of the insulin receptor substrate IRS-1 defines a unique signal transduction protein. Nature 352(6330):73–77
Sun XJ, Wang LM, Zhang Y, Yenush L, Myers MG Jr, Glasheen E, Lane WS, Pierce JH, White MF (1995) Role of IRS-2 in insulin and cytokine signalling. Nature 377(6545):173–177
Takagi D, Hirano H, Watanabe Y, Edahiro A, Ohara Y, Yoshida H, Kim H, Murakami K, Hironaka S (2017) Relationship between skeletal muscle mass and swallowing function in patients with Alzheimer’s disease. Geriatr Gerontol Int 17(3):402–409
Takeda S, Sato N, Uchio-Yamada K, Sawada K, Kunieda T, Takeuchi D, Kurinami H, Shinohara M, Rakugi H, Morishita R (2010) Diabetes-accelerated memory dysfunction via cerebrovascular inflammation and Abeta deposition in an Alzheimer mouse model with diabetes. Proc Natl Acad Sci U S A 107(15):7036–7041
Takei N, Nawa H (2014) mTOR signaling and its roles in normal and abnormal brain development. Front Mol Neurosci 7:28
Talbot K, Wang HY, Kazi H, Han LY, Bakshi KP, Stucky A, Fuino RL, Kawaguchi KR, Samoyedny AJ, Wilson RS, Arvanitakis Z, Schneider JA, Wolf BA, Bennett DA, Trojanowski JQ, Arnold SE (2012) Demonstrated brain insulin resistance in Alzheimer’s disease patients is associated with IGF-1 resistance, IRS-1 dysregulation, and cognitive decline. J Clin Invest 122(4):1316–1338
Tang SJ, Reis G, Kang H, Gingras AC, Sonenberg N, Schuman EM (2002) A rapamycin-sensitive signaling pathway contributes to long-term synaptic plasticity in the hippocampus. Proc Natl Acad Sci U S A 99(1):467–472
Teixeira AL, Diniz BS, Campos AC, Miranda AS, Rocha NP, Talib LL, Gattaz WF, Forlenza OV (2013) Decreased levels of circulating adiponectin in mild cognitive impairment and Alzheimer’s disease. Neuromol Med 15(1):115–121
Teutonico A, Schena PF, Di Paolo S (2005) Glucose metabolism in renal transplant recipients: effect of calcineurin inhibitor withdrawal and conversion to sirolimus. J Am Soc Nephrol 16(10):3128–3135
Thedieck K, Polak P, Kim ML, Molle KD, Cohen A, Jeno P, Arrieumerlou C, Hall MN (2007) PRAS40 and PRR5-like protein are new mTOR interactors that regulate apoptosis. PLoS One 2(11):e1217
Thomanetz V, Angliker N, Cloetta D, Lustenberger RM, Schweighauser M, Oliveri F, Suzuki N, Ruegg MA (2013) Ablation of the mTORC2 component rictor in brain or Purkinje cells affects size and neuron morphology. J Cell Biol 201(2):293–308
Toots A, Rosendahl E, Lundin-Olsson L, Nordstrom P, Gustafson Y, Littbrand H (2013) Usual gait speed independently predicts mortality in very old people: a population-based study. J Am Med Dir Assoc 14(7):529.e521–529.e526
Tournissac M, Vandal M, Francois A, Planel E, Calon F (2017) Old age potentiates cold-induced tau phosphorylation: linking thermoregulatory deficit with Alzheimer’s disease. Neurobiol Aging 50:25–29
Tramutola A, Triplett JC, Di Domenico F, Niedowicz DM, Murphy MP, Coccia R, Perluigi M, Butterfield DA (2015) Alteration of mTOR signaling occurs early in the progression of Alzheimer disease (AD): analysis of brain from subjects with pre-clinical AD, amnestic mild cognitive impairment and late-stage AD. J Neurochem 133(5):739–749
Tremblay F, Marette A (2001) Amino acid and insulin signaling via the mTOR/p70 S6 kinase pathway. A negative feedback mechanism leading to insulin resistance in skeletal muscle cells. J Biol Chem 276(41):38052–38060
Tremblay F, Brule S, Hee Um S, Li Y, Masuda K, Roden M, Sun XJ, Krebs M, Polakiewicz RD, Thomas G, Marette A (2007) Identification of IRS-1 Ser-1101 as a target of S6K1 in nutrient- and obesity-induced insulin resistance. Proc Natl Acad Sci U S A 104(35):14056–14061
Tsai S, Sitzmann JM, Dastidar SG, Rodriguez AA, Vu SL, McDonald CE, Academia EC, O’Leary MN, Ashe TD, La Spada AR, Kennedy BK (2015) Muscle-specific 4E-BP1 signaling activation improves metabolic parameters during aging and obesity. J Clin Invest 125(8):2952–2964
Tsai SY, Rodriguez AA, Dastidar SG, Del Greco E, Carr KL, Sitzmann JM, Academia EC, Viray CM, Martinez LL, Kaplowitz BS, Ashe TD, La Spada AR, Kennedy BK (2016) Increased 4E-BP1 expression protects against diet-induced obesity and insulin resistance in male mice. Cell Rep 16(7):1903–1914
Tyler WJ, Alonso M, Bramham CR, Pozzo-Miller LD (2002) From acquisition to consolidation: on the role of brain-derived neurotrophic factor signaling in hippocampal-dependent learning. Learn Mem 9(5):224–237
Tzatsos A, Kandror KV (2006) Nutrients suppress phosphatidylinositol 3-kinase/Akt signaling via raptor-dependent mTOR-mediated insulin receptor substrate 1 phosphorylation. Mol Cell Biol 26(1):63–76
Ueno M, Carvalheira JB, Tambascia RC, Bezerra RM, Amaral ME, Carneiro EM, Folli F, Franchini KG, Saad MJ (2005) Regulation of insulin signalling by hyperinsulinaemia: role of IRS-1/2 serine phosphorylation and the mTOR/p70 S6K pathway. Diabetologia 48(3):506–518
Um SH, Frigerio F, Watanabe M, Picard F, Joaquin M, Sticker M, Fumagalli S, Allegrini PR, Kozma SC, Auwerx J, Thomas G (2004) Absence of S6K1 protects against age- and diet-induced obesity while enhancing insulin sensitivity. Nature 431(7005):200–205
Umegaki H, Kawamura T, Mogi N, Umemura T, Kanai A, Sano T (2008) Glucose control levels, ischaemic brain lesions, and hyperinsulinaemia were associated with cognitive dysfunction in diabetic elderly. Age Ageing 37(4):458–461
Une K, Takei YA, Tomita N, Asamura T, Ohrui T, Furukawa K, Arai H (2011) Adiponectin in plasma and cerebrospinal fluid in MCI and Alzheimer’s disease. Eur J Neurol 18(7):1006–1009
Vander Haar E, Lee SI, Bandhakavi S, Griffin TJ, Kim DH (2007) Insulin signalling to mTOR mediated by the Akt/PKB substrate PRAS40. Nat Cell Biol 9(3):316–323
Varshney R, Gupta S, Roy P (2017) Cytoprotective effect of kaempferol against palmitic acid-induced pancreatic beta-cell death through modulation of autophagy via AMPK/mTOR signaling pathway. Mol Cell Endocrinol 448:1–20
Vellai T, Takacs-Vellai K, Zhang Y, Kovacs AL, Orosz L, Muller F (2003) Genetics: influence of TOR kinase on lifespan in C. elegans. Nature 426(6967):620
Vernier S, Chiu A, Schober J, Weber T, Nguyen P, Luer M, McPherson T, Wanda PE, Marshall CA, Rohatgi N, McDaniel ML, Greenberg AS, Kwon G (2012) Beta-cell metabolic alterations under chronic nutrient overload in rat and human islets. Islets 4(6):379–392
Voisin T, Lugardon S, Balardy L, Vellas B (2003) Vascular risk factors and Alzheimer’s disease. Rev Med Interne 24(Suppl 3):288s–291s
Wan Z, Mah D, Simtchouk S, Klegeris A, Little JP (2014) Globular adiponectin induces a pro-inflammatory response in human astrocytic cells. Biochem Biophys Res Commun 446(1):37–42
Wan Z, Mah D, Simtchouk S, Kluftinger A, Little JP (2015) Human adipose tissue conditioned media from lean subjects is protective against H2O2 induced neurotoxicity in human SH-SY5Y neuronal cells. Int J Mol Sci 16(1):1221–1231
Wang X, Proud CG (2009) Nutrient control of TORC1, a cell-cycle regulator. Trends Cell Biol 19(6):260–267
Wang JP, Zhang MY (2017) Role for target of rapamycin (mTOR) signal pathway in regulating neuronal injury after intracerebral hemorrhage. Cell Physiol Biochem 41(1):145–153
Wang L, Harris TE, Lawrence JC Jr (2008) Regulation of proline-rich Akt substrate of 40 kDa (PRAS40) function by mammalian target of rapamycin complex 1 (mTORC1)-mediated phosphorylation. J Biol Chem 283(23):15619–15627
Wang L, Liu Y, Yan Lu S, Nguyen KT, Schroer SA, Suzuki A, Mak TW, Gaisano H, Woo M (2010) Deletion of Pten in pancreatic ss-cells protects against deficient ss-cell mass and function in mouse models of type 2 diabetes. Diabetes 59(12):3117–3126
Wellen KE, Thompson CB (2010) Cellular metabolic stress: considering how cells respond to nutrient excess. Mol Cell 40(2):323–332
Werner ED, Lee J, Hansen L, Yuan M, Shoelson SE (2004) Insulin resistance due to phosphorylation of insulin receptor substrate-1 at serine 302. J Biol Chem 279(34):35298–35305
Westermark P, Wernstedt C, Wilander E, Sletten K (1986) A novel peptide in the calcitonin gene related peptide family as an amyloid fibril protein in the endocrine pancreas. Biochem Biophys Res Commun 140(3):827–831
Westermark P, Wernstedt C, O’Brien TD, Hayden DW, Johnson KH (1987) Islet amyloid in type 2 human diabetes mellitus and adult diabetic cats contains a novel putative polypeptide hormone. Am J Pathol 127(3):414–417
Whitmer RA, Gunderson EP, Barrett-Connor E, Quesenberry CP Jr, Yaffe K (2005) Obesity in middle age and future risk of dementia: a 27 year longitudinal population based study. BMJ 330(7504):1360
Whitmer RA, Gunderson EP, Quesenberry CP Jr, Zhou J, Yaffe K (2007) Body mass index in midlife and risk of Alzheimer disease and vascular dementia. Curr Alzheimer Res 4(2):103–109
Wilson DM, Binder LI (1997) Free fatty acids stimulate the polymerization of tau and amyloid beta peptides. In vitro evidence for a common effector of pathogenesis in Alzheimer’s disease. Am J Pathol 150(6):2181–2195
Wisse BE, Schwartz MW (2003) The skinny on neurotrophins. Nat Neurosci 6(7):655–656
Woods SC, Seeley RJ, Cota D (2008) Regulation of food intake through hypothalamic signaling networks involving mTOR. Annu Rev Nutr 28:295–311
Wullschleger S, Loewith R, Hall MN (2006) TOR signaling in growth and metabolism. Cell 124(3):471–484
Xiang X, Lan H, Tang H, Yuan F, Xu Y, Zhao J, Li Y, Zhang W (2015) Tuberous sclerosis complex 1-mechanistic target of rapamycin complex 1 signaling determines brown-to-white adipocyte phenotypic switch. Diabetes 64(2):519–528
Xie J, El Sayed NM, Qi C, Zhao X, Moore CE, Herbert TP (2014a) Exendin-4 stimulates islet cell replication via the IGF1 receptor activation of mTORC1/S6K1. J Mol Endocrinol 53(1):105–115
Xie L, Sun F, Wang J, Mao X, Xie L, Yang S-H, Su D-M, Simpkins JW, Greenberg DA, Jin K (2014b) mTOR signaling inhibition modulates macrophage/microglia-mediated neuroinflammation and secondary injury via regulatory T cells after focal ischemia. J Immunol 192(12):6009
Yamazaki H, Jin Y, Tsuchiya A, Kanno T, Nishizaki T (2015) Adipose-derived stem cell-conditioned medium ameliorates antidepression-related behaviors in the mouse model of Alzheimer’s disease. Neurosci Lett 609:53–57
Yang Q, Inoki K, Ikenoue T, Guan KL (2006) Identification of Sin1 as an essential TORC2 component required for complex formation and kinase activity. Genes Dev 20(20):2820–2832
Yang SB, Lee HY, Young DM, Tien AC, Rowson-Baldwin A, Shu YY, Jan YN, Jan LY (2011) Rapamycin induces glucose intolerance in mice by reducing islet mass, insulin content, and insulin sensitivity. J Mol Med (Berl) 2012 May, 90(5):575–585
Yang Z, Liu F, Qu H, Wang H, Xiao X, Deng H (2015) 1, 25(OH)2D3 protects beta cell against high glucose-induced apoptosis through mTOR suppressing. Mol Cell Endocrinol 414:111–119
Yarchoan M, Toledo JB, Lee EB, Arvanitakis Z, Kazi H, Han LY, Louneva N, Lee VM, Kim SF, Trojanowski JQ, Arnold SE (2014) Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol 128(5):679–689
Yoshitake T, Kiyohara Y, Kato I, Ohmura T, Iwamoto H, Nakayama K, Ohmori S, Nomiyama K, Kawano H, Ueda K et al (1995) Incidence and risk factors of vascular dementia and Alzheimer’s disease in a defined elderly Japanese population: the Hisayama study. Neurology 45(6):1161–1168
Yuan T, Rafizadeh S, Gorrepati KD, Lupse B, Oberholzer J, Maedler K, Ardestani A (2017) Reciprocal regulation of mTOR complexes in pancreatic islets from humans with type 2 diabetes. Diabetologia 60(4):668–678
Zahr E, Molano RD, Pileggi A, Ichii H, Jose SS, Bocca N, An W, Gonzalez-Quintana J, Fraker C, Ricordi C, Inverardi L (2007) Rapamycin impairs in vivo proliferation of islet beta-cells. Transplantation 84(12):1576–1583
Zhao WQ, De Felice FG, Fernandez S, Chen H, Lambert MP, Quon MJ, Krafft GA, Klein WL (2008a) Amyloid beta oligomers induce impairment of neuronal insulin receptors. FASEB J 22(1):246–260
Zhao Y, Kuo TC, Weir S, Kramer MS, Ash AS (2008b) Healthcare costs and utilization for Medicare beneficiaries with Alzheimer’s. BMC Health Serv Res 8:108
Zhou J, Blundell J, Ogawa S, Kwon CH, Zhang W, Sinton C, Powell CM, Parada LF (2009) Pharmacological inhibition of mTORC1 suppresses anatomical, cellular, and behavioral abnormalities in neural-specific Pten knock-out mice. J Neurosci 29(6):1773–1783
Zhu X, Hu R, Brissova M, Stein RW, Powers AC, Gu G, Kaverina I (2015) Microtubules negatively regulate insulin secretion in pancreatic beta cells. Dev Cell 34(6):656–668
Zoncu R, Efeyan A, Sabatini DM (2011) mTOR: from growth signal integration to cancer, diabetes and ageing. Nat Rev Mol Cell Biol 12(1):21–35
Zurlo F, Larson K, Bogardus C, Ravussin E (1990) Skeletal muscle metabolism is a major determinant of resting energy expenditure. J Clin Invest 86(5):1423–1427
Zurlo F, Nemeth PM, Choksi RM, Sesodia S, Ravussin E (1994) Whole-body energy metabolism and skeletal muscle biochemical characteristics. Metabolism 43(4):481–486
Author information
Authors and Affiliations
Corresponding author
Editor information
Editors and Affiliations
Rights and permissions
Copyright information
© 2019 Springer Nature Singapore Pte Ltd.
About this chapter

Cite this chapter
Liang, H., Nie, J., Van Skike, C.E., Valentine, J.M., Orr, M.E. (2019). Mammalian Target of Rapamycin at the Crossroad Between Alzheimer’s Disease and Diabetes. In: Nakabeppu, Y., Ninomiya, T. (eds) Diabetes Mellitus. Advances in Experimental Medicine and Biology, vol 1128. Springer, Singapore. https://doi.org/10.1007/978-981-13-3540-2_10
Download citation
DOI: https://doi.org/10.1007/978-981-13-3540-2_10
Published:
Publisher Name: Springer, Singapore
Print ISBN: 978-981-13-3539-6
Online ISBN: 978-981-13-3540-2
eBook Packages: Biomedical and Life SciencesBiomedical and Life Sciences (R0)