
Abstract
Alzheimer’s disease (AD) and Type 2 diabetes mellitus (T2DM) are two of the most common age-related diseases. There is accumulating evidence of an overlap in the pathophysiological mechanisms of these two diseases. Studies have demonstrated insulin pathway alternation may interact with amyloid-β protein deposition and tau protein phosphorylation, two essential factors in AD. So attention to the use of anti-diabetic drugs in AD treatment has increased in recent years. In vitro, in vivo, and clinical studies have evaluated possible neuroprotective effects of anti-diabetic different medicines in AD, with some promising results. Here we review the evidence on the therapeutic potential of insulin, metformin, Glucagon-like peptide-1 receptor agonist (GLP1R), thiazolidinediones (TZDs), Dipeptidyl Peptidase IV (DPP IV) Inhibitors, Sulfonylureas, Sodium-glucose Cotransporter-2 (SGLT2) Inhibitors, Alpha-glucosidase inhibitors, and Amylin analog against AD. Given that many questions remain unanswered, further studies are required to confirm the positive effects of anti-diabetic drugs in AD treatment. So to date, no particular anti-diabetic drugs can be recommended to treat AD.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Alzheimer's disease (AD) is the most common age-related neurological disorder. More than 30 million people worldwide suffer from dementia, of whom about two-third have AD (Patterson 2018). AD is characterized by a gradual decline in memory and cognitive function, leading to premature death several years after diagnosis. The main pathological features of AD are an abnormal accumulation of amyloid-β (Aβ), misfolded and aggregated forms of tau protein, and severe loss of neurons in brain tissue called brain atrophy (Meng et al. 2020; Shieh et al. 2020). Although more than a century has passed since the first case of AD was diagnosed, there is no definitive cure for the disease. For this reason, to identify the treatment of AD, its relationship with other diseases has been investigated. It has been reported that Type 2 diabetes mellitus (T2DM) and its consequences, including hyperinsulinemia, hyperglycemia, vascular lesions, and inflammation, are independent risk factors for AD (Haan 2006; Kandimalla et al. 2017; Boccardi et al. 2019).
T2DM is a chronic metabolic disease that can damage blood vessels, nerves, eyes, and kidneys (Goodarzi et al. 2016), is characterized by insulin resistance and relative insulin deficiency (Ramos-Rodriguez et al. 2017). Further studies have shown that diabetic patients often show cognitive impairment similar to the early stages of AD (Gorska-Ciebiada et al. 2014). A survey of AD patients showed that 80% of people with AD had impaired glucose tolerance or were diabetic (Janson et al. 2004). Epidemiological, clinical, and molecular evidence also suggests that there is a significant overlap in the pathophysiological mechanisms of T2DM and AD and that people with diabetes are at higher risk for AD, so the term "type 3 diabetes" has been introduced to describe these patients (Stoeckel et al. 2016; Arnold et al. 2018). Brain atrophy, impaired glucose metabolism, insulin signaling pathway, inflammation, mitochondrial dysfunction, and oxidative stress are common pathophysiological features in AD and T2DM (De la Monte and Wands 2008; de Matoset al. 2018; Meng et al. 2020; Shieh et al. 2020).
On the other hand, many studies support the relationship between AD and T2DM at the genetic level (Hao et al. 2015; Gao et al. 2016). Although there is no definitive conclusion on how AD and T2DM are related, their similar pathogenesis has provided convincing evidence that AD could be considered a metabolic disorder, which glucose metabolism and insulin signaling are impaired in the brain. Thus, several studies and clinical trials have been conducted to assess the neuroprotective effects of anti-diabetic drugs. This review discusses the main anti-diabetic drugs that are suitable treatment candidates for AD.
Insulin as a bridge between T2DM and AD
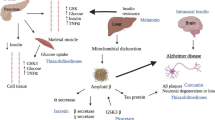
Insulin is the primary regulator of energy homeostasis and appetite, and it modulates brain activity. With its neurotrophic, neuromodulatory, and neuroprotective effects, insulin in the brain helps to control nutrient homeostasis and cognitive function (Blázquez et al. 2014). Therefore, insulin and its signaling pathway can affect many neuronal activities. Insulin is involved in maintaining nerve function by inducing the expression of the genes that are involved in acetylcholine synthesis. Consequently, suboptimal insulin values and hyposensitivity to the insulin receptor can be a biochemical link between diabetes and AD (Li and Hölscher 2007; Stanciu et al. 2020). Under normal conditions, insulin signaling can inhibit the production of Aβ and Tau phosphorylation by reducing BACE1 (Beta-Secretase 1) mRNA expression and activity, reducing Amyloid Precursor Protein (APP), and inhibiting Glycogen synthase kinase-3(GSK3β) phosphorylation (Ly et al. 2012). The insulin signaling pathway, including PI3K/AKT, is disrupted in the insulin resistance state. The PI3K / Akt pathway regulates downstream factors such as apoptotic pathway proteins, GSK3β, mTORC1, and forkhead box transcription factor (FOX) (Gabbouj et al. 2019; Hölscher 2019). Increased activation of GSK-3β in insulin resistance conditions may lead to Tau hyperphosphorylation, an important event in the formation of Neurofibrillary tangle (NFTs). Changes in Aβ production and clearance play an important role in AD. Increasing the activity of GSK3β lead to activation of presenilin 1 and finally Aβ accumulation (5). The insulin-degrading enzyme (IDE) plays an important role in clearing insulin and Aβ, and reducing its function can be important in developing both AD and diabetes. In addition to reducing IDE production in an insulin resistance state, insulin is elevated and competes with Aβ for binding to the IDE resulting in slower clearance of Aβ in the brain (Fig. 1) (Farris et al. 2003). Receptors for Advanced Glycation End Products (RAGE) have an important role in Aβ clearance. Bothe the AGEs and Aβ could bind to RAGE. Under insulin resistance conditions, AGEs compete with Aβ for binding to the RAGE. Therefore, the clearance rate of Aβ is reduced. In addition, the binding of AGEs and Aβ to RAGE increase inflammatory cytokines expression such as tumor necrosis factor (TNFα), Interleukin 6 (IL6), which accelerate the development of AD (Kong et al. 2020). Insulin Receptor Substrate 1 (IRS1), an adapter protein in the insulin signaling pathway, has been shown to be involved in the pathogenesis of AD. A recent study showed that abnormal IRS1-pS (616) phosphorylation is a pathological feature of AD (Yarchoan et al. 2014).

Insulin as a bridge between T2DM and AD. Schematic outline of neuronal insulin signaling in the AD brain. Insulin is involved in maintaining nerve function by inducing acetylcholine synthesis gene expression. Under normal conditions, insulin signaling can inhibit the production of Aβ and Tau phosphorylation by reducing BACE1 mRNA expression and activity, reducing APP, and inhibiting GSK3β phosphorylation. Under insulin resistance conditions, APP cleavage by γ secretase in trance membrane domain and release insoluble Aβ into extracellular space that can further intensify insulin resistance (a, b, c). insulin resistance leads to inhibition of IDE and decreased Aβ clearance (d). Increased activation of GSK-3β in insulin resistance conditions may lead to Tau hyperphosphorylation (e)
Therapeutic potential of type 2 diabetes drugs against AD
Although it is not clear how AD and T2DM are related, similar pathogenesis and common pathophysiological pathways in both diseases have led to an exponential increase in the number of studies examining the therapeutic effects of anti-diabetic drugs against AD (Cao et al. 2018). These studies may lead to the discovery of an effective drug against AD. Therefore, reviewing previous studies can provide valuable clues for future studies. In this review, we will discuss the therapeutic potential of several anti-diabetic drugs such as insulin, metformin, Glucagon-like peptide-1 receptor (GLP1R) agonist, thiazolidinediones (TZDs), Dipeptidyl Peptidase IV (DPP IV) inhibitors, Sulfonylureas, Sodium-glucose Cotransporter-2 (SGLT2) inhibitors, Alpha-glucosidase inhibitors and Amylin analog against AD. We will also discuss the mechanism of the action of these anti-diabetic drugs in AD pathology and analyze the results of different studies from animal and human studies.
Insulin
Insulin is a peptide hormone that affects the metabolism of carbohydrates, fats, and proteins. The expression of the insulin receptor and insulin-sensitive transmitters such as GLUT4 on the surface of the hypothalamus and hippocampus indicates insulin's role in the brain's physiological function (Reger et al. 2006). For example, by inducing the expression of N-methyl-D-aspartate (NMDA) receptors, insulin induces long-term potentiation (LTP) (Skeberdis et al. 2001), and by modulating neurotransmitters such as acetylcholine and norepinephrine, it improves cognitive-behavioral performance (Figlewicz et al. 1993; Reger et al. 2006). Therefore, given the increasing AD prevalence in insulin resistance and diabetes patients, there has been a rise in studies on the effects of insulin on AD. In these studies, the effects of insulin on improving AD, intravenously and intranasal, were investigated. Studies on a 3xTg-AD mouse model fed a high-fat diet showed that obesity and insulin resistance reduced memory and cognitive function and improved memory following intravenous insulin treatment. Insulin reduced the production of Aβ in mice through mechanisms such as the reduction of BCAE1. This finding has also been confirmed by in vitro studies (Pandini et al. 2013). Insulin also decreased Aβ production and improved AD disorders by increasing x11α and decreasing the LC3I / LC3II ratio (Vandal et al. 2014). In addition to Aβ, insulin affects the amount of tau-phosphorylated protein in the brain. In a model of diabetic mice with high and low doses of Streptozotocin (STZ), intravenous injection of insulin led to a decrease in phosphatase 2A protein and a decrease in the amount of tau-phosphorylated protein (Gratuze et al. 2017). Despite the evident effects of intravenous insulin on cognitive, functional, and memory behaviors of patients with AD disease, due to the long-term hypoglycemic effects and limitation in crossing the blood–brain barrier (Craft et al. 1996; Kern et al. 2001; Morris and Burns 2012), intranasal use of the insulin has been recently introduced.
In Alzheimer's rodent models, intranasal insulin use improves the pathological features of AD, as well as the short-term and long-term memory (Barone et al. 2019). In a study, 6 weeks of intranasal insulin treatment improved cognitive function and decreased tau-phosphorylated protein (Thr205, Ser262, and Ser396) by reducing the kinases of GSK-3, extracellular signal‑regulated protein kinase (ERK1 / 2), and Ca2 + /Calmodulin protein kinase II (CaMKII) in the hippocampus of Alzheimer's rats (Guo et al. 2017). Neuritis characterized by activation of Astroglia and Microglia is a pathological feature of AD that occurs before the formation of amyloid plaques and NFTs (Prickaerts et al. 1999; Mrak and Griffin 2001). Intranasal insulin treatment also decreases the activation markers of astrocytes (Glial fibrillary acidic protein) and microglia (Ionized calcium-binding adaptor molecule 1 (Iba1) in the hippocampus of rats with the intraventricular injection of STZ. Insulin can also be an effective treatment for AD by increasing the doublecortin (DCX) neurogenesis marker and inducing insulin signaling by increasing AKT expression (Guo et al. 2017). In addition to improving learning and memory, insulin has been shown to increase IRS-1-PI3K-Akt-GSK3β pathway activity through the olfactory bulb – subventricular zone – subgranular zone (OB-SVZ-SGZ) in the hippocampus and subclavian region (Lv et al. 2020). Long-term treatment with insulin and GLP1 agonist (Exenatide) has been reported to reduce IRSP gene expression, improve insulin signaling pathway, and reduce Aβ by 30–30% in the Tg2576 model of AD mice (Robinson et al. 2019). It has also been observed that daily intranasal insulin administration improves cognitive and memory impairments after decreasing the level of consciousness caused by propofol (Zhang et al. 2016). The results of these studies are inconsistent with those of Elizabeth M, et al. as intranasal administration of insulin to SAMP8 mice had no effects on the expression of insulin signaling pathway but it could be a candidate in treatment of AD disease by modulating the other pathways including inflammatory pathways (Rhea et al. 2019).
Human studies have provided evidence regarding the effectiveness of insulin in the treatment of AD. Investigations using functional magnetic resonance imaging (MRI), electroencephalogram (EEG), magnetoencephalography (MEG) have shown the positive effects of intranasal insulin on the improvement of central nervous system function (Kullmann et al. 2016). A study on healthy individuals found that 8 weeks of intranasal insulin administration reduced anger thresholds, increased mood and self-esteem, and improved memory. In addition, receiving insulin in this way did not affect systemic insulin and blood glucose levels. (Benedict et al. 2004, 2007). Two studies on the elderly with AD showed that short-term (21 days) intranasal insulin administration improved memory function, decreased cortisol levels, and increased Aβ 40 solution form (Reger et al. 2008; Claxton et al. 2015). In another randomized, double-blind, placebo-controlled trial, the effects of intranasal insulin at doses of 20 and 40 IU for 4 months on 104 patients with cognitive impairment and AD were investigated. This study showed that insulin in both amounts of 20 and 40 IU improves AD by improving the Scale-Cognitive subscale (ADAS-Cog12) (Craft et al. 2012). Another study showed that insulin's effects on memory function occur at high doses, and the APOE-ε4 genotype enhances the effects of insulin (Claxton et al. 2015). In this regard, additional studies showed that the effects of intranasal insulin might be improved by gender and ApoE ε4 genotype (Claxton et al. 2013). A placebo-controlled double-blind, randomized trial showed improved memory and decreased tau-P181 / Aβ42 ratio following regular insulin therapy for 2 and 4 months (Craft et al. 2017). Although it is difficult to demonstrate the therapeutic activity of Large molecular weight compounds like proteins and peptides due to some physiological barriers such as BBB, enzymatic degradation, and clearance, numerous studies have shown successful impacts of insulin in the preclinical and clinical stages of AD treatment. However, none of them has been approved for AD treatment to date. Therefore, it appears that more data are required to establish the actual value of insulin appropriately in the treatment of AD.
Metformin
Metformin has recently attracted considerable scholarly attention in the treatment of AD. In the mice model of AD, metformin causes memory improvement and reduces the pathological factors of Aβ and tau phosphorylation (Chen et al. 2021). Low-dose metformin has been reported to reduce scopolamine-induced cognitive impairment and significantly reduce inflammation and oxidative stress in rats (Mostafa et al. 2016). Also, the administration of metformin in the Senescence Accelerated Mouse (SAMP8) mouse model improves learning ability and memory by reducing APPc99 and pTau levels (Farr et al. 2019). These effects of metformin may be mediated through activating the AMPK, inhibiting the P65 NF-κB pathways, and reducing the expression of BACE1 protein (Ou et al. 2018). In addition, metformin protects neurons against apoptosis by reducing JNK hyperphosphorylation (Chen et al. 2016; Chenet al. 2019). A study on the AD mouse model reported conflicting results despite the above data. Activating the AMPK by metformin increases the severity of memory impairment in males, whereas it has protective effects in females (DiTacchio et al. 2015). In a placebo-controlled randomized trial, 80 non-diabetic participants with mild cognitive impairment received metformin or placebo daily for 12 months. Although promising results were obtained for the memory, no differences were observed for ADAS-Cog12, CSF, Aβ42, and cerebral glucose metabolism (Luchsinger et al. 2016). A placebo-controlled clinical trial study showed that metformin is safe and tolerable in patients with AD. This drug didn't have significant effects on, Aβ42, total tau, and p-tau in CSF and language and motor speed but improved learning/memory and attention (Koenig et al. 2017).
In contrast to the above reports, a cohort study of 4651 diabetic patients showed that long-term metformin administration increased the risk of all-cause dementia, vascular dementia, and AD (HR: 2.13, 95% CI = 1.20–3.79) (Kuan et al. 2017). These findings are in line with previous results indicating that long-term use of metformin increases the risk of AD (Moore et al. 2013). One study has examined the effects of anti-diabetic drugs on the brain and the risk of AD. The results of the research showed that compared to metformin, glyburide had the highest score, whereas GLP-1 receptor agonists and rosiglitazone had the lowest score for risk of AD (Akimoto et al. 2020). Furthermore, long-term use of sulfonylureas, thiazolidinediones, or insulin has a lower AD risk than metformin (Imfeld et al. 2012). The paradoxical effect of metformin on the risk of AD was studied in a transgenic mouse model with tauopathy. Although metformin decreases tau phosphorylation in the cerebral cortex and hippocampus through AMPK / mTOR and PP2A, it also increases the insoluble form of tau and Aβ plates in the brain. Metformin also stimulates caspase-3 to increase the cleavage of Tau proteins and disrupts synaptic communication (Barini et al. 2016). Metformin-induced accumulation of autophagosomes leads to increased γ-secretase activity and Aβ generation (Son et al. 2016). Although most of the data on the use of metformin in dementia / AD with or without T2DM are promising, it should be noted that the effect of metformin might depend on complex underlying pathological processes. In some cases, metformin has even been shown to have harmful effects. Some studies have failed to provide convincing evidence of the effect of metformin on mild cognitive impairment or AD, which may be due to a short period of use or duration of studies. Therefore, to determine the effects of metformin in the control and treatment of dementia/AD, it is necessary to define broader cohort studies with long-term use of metformin and larger study groups.
Thiazolidinediones
Thiazolidinediones (glitazones) are an important family of anti-diabetic drugs that exert their anti-inflammatory and insulin-sensitizing effects by stimulating peroxisome proliferator-activated receptor gamma (PPARγ). At the molecular level, PPAR-γ plays a critical role in regulating glucose and lipid metabolism and inflammatory responses. The Thiazolidinediones family improves insulin resistance in adipose and muscle tissues via activating PPAR-γ. Thiazolidinediones also inhibit hepatic gluconeogenesis, a process involved in regulating blood glucose (Hurren and Dunham 2021; Long et al. 2021). In recent years, various studies have demonstrated that thiazolidinediones can effectively treat neurodegenerative diseases, especially AD, by reducing and delaying the risk of neurodegeneration (Miller et al. 2011; Pérez and Quintanilla 2015; Galimberti and Scarpini 2017; Meng et al. 2020). Thiazolidinediones have been reported to treat AD by decreasing inflammatory cytokines, oxidative stress, Aβ deposits, glial activation, and Tau protein phosphorylation (Pérez and Quintanilla 2015). In the following section, we will discuss the effects of pioglitazone and rosiglitazone on the treatment of AD in-vivo and in-vitro and clinical trial studies.
Pioglitazone
Pioglitazone has been approved since 1999 as a supplement to control blood glucose in T2DM patients (Al-Majed et al. 2016). In animal models, the preventive effects of pioglitazone on AD have been identified. Chen et al. observed that pioglitazone suppressed hyper-activation of cyclin-dependent kinase 5 (Cdk5) in the hippocampus of mice with the mutated APP / PS1 gene by reducing P35 protein expression (Chen et al. 2015). Pioglitazone reduced astroglial activation and cortical cholinergic innervation. It also reversed cerebral blood flow (CBF) and cerebral glucose uptake (CGU) responses to increased neural activity but failed to improve spatial memory. Hence, chronic administration of pioglitazone overcomes cerebrovascular dysfunction and alters neurometabolic coupling, and counteracts oxidative stress in the brain, glial activation, and to some extent, cholinergic denervation (Nicolakakis et al. 2008). It has also been indicated that 7-day oral treatment of APPV717I mice with pioglitazone reduced the number of active microglia and reactive astrocytes in the hippocampus and cortex. Moreover, pioglitazone decreased the expression of the proinflammatory genes such as cyclooxygenase 2 (COX2) and nitric oxide synthase (iNOS) and reduced mRNA and protein BACE1(Heneka et al. 2005). In the mouse model of AD treated with pioglitazone, a remarkable decrease in the level of soluble and insoluble Aβ was observed in the brain, which was associated with the loss of both diffuse and dense-core plaques within the brain cortex (Mandrekar-Colucci et al. 2012). Furthermore, due to the anti-inflammatory effects of pioglitazone, treatment with this drug resulted in a phenotypic change of microglial cells from proinflammatory M1 to an M2 anti-inflammatory state, a phenomenon correlated with increased phagocytosis of deposited Aβ (Mandrekar-Colucci et al. 2012; Galimberti and Scarpini 2017). Recently, it has been reported that the levels of collapsin response mediator protein-2 (CRMP-2) and p35 protein were increased in the cerebellum of APP / PS1 mice with AD. Pioglitazone normalized the levels of these proteins in the cerebellum and attenuated impaired motor coordination ability and long-term depression in the pre-Aβ accumulation stage (Toba et al. 2016). Yang et al. illustrated that pioglitazone improves insulin sensitivity and attenuates Aβ42 accumulation in rats with diet-induced insulin resistance by modulation of AKT/GSK3β activation(Yang et al. 2017). In another study, four months of treatment with pioglitazone at 18 mg/Kg/day in the triple transgenic mouse model of AD (3xTg-AD) led to improving learning on the active avoidance task, lower Aβ and tau deposits in the hippocampus, and increase of short- and long-term plasticity. (Searcy et al. 2012). Meanwhile, Xiang et al. found that pioglitazone had a neuroprotective effect against scopolamine-induced cholinergic system deficit and cognitive impairment by decreased acetylcholine levels and reduced choline acetyltransferase activity, and elevated acetylcholinesterase activity in the hippocampus or cortex (Xiang et al. 2012). Recently, a study investigated the effects of chronic administration of pioglitazone on learning and memory in an STZ-induced AD rat model. The findings elucidated that pioglitazone impaired spatial learning and memory in normal rats but improved learning and memory in STZ-induced AD rats(Aali et al. 2020).
There is clinical evidence for the effects of pioglitazone on AD. For instance, in clinical trials, a pilot prospective randomized, open-controlled study revealed that pioglitazone improved metabolism and cognition in AD and mild cognitive impairment (MCI) patients (Hanyu et al. 2009). A further randomized, open-controlled trial indicated that cognitive improvement was correlated with the decline of TNF-α status after pioglitazone therapy (Hanyu et al. 2010). Additionally, it was reported that pioglitazone treatment improved cognition and regional cerebral blood flow (rCBF) in the parietal lobe and displayed stabilization of the disease in mild AD accompanied with T2DM patients compared with control (Hanyu et al. 2010). Another double-blind, placebo-controlled, randomized controlled trial demonstrated that pioglitazone was well tolerated, and no unanticipated safety problems in AD patients without DM were observed via an 18-month treatment duration (Geldmacher et al. 2011).
Taken together, although this drug helps improve the cognition of diabetic patients with AD and mild Cognitive Impairment (MCI), contradictory results have been reported from in-vivo and clinical trials studies. The discrepancies between the studies could be due to the different length of treatment, sample size, inclusion and exclusion criteria, and patient selection methods. Therefore, more basic and clinical investigations are needed in this regard.
Rosiglitazone
Rosiglitazone is another important insulin-sensitizing drug in the treatment of T2DM. Evidence from in-vitro and in-vivo studies suggests that rosiglitazone may improve cognition and the pathology of Aβ in AD patients. For example, rosiglitazone enhanced learning and memory impairments in Tg2576 mice by affecting brain levels of IDE and Aβ42 (Pedersen et al. 2006). In addition, Aβ accumulation was decreased in 7-month-old APP / PS1 mice following 4 weeks of treatment with rosiglitazone (O’Reilly and Lynch 2012). Treatment of APPswe / PSEN1DE9 mice with rosiglitazone reduced spatial memory impairment induced by amyloid burden; Aβ aggregates and Aβ oligomers; and astrocytic and microglia activation. Besides, rosiglitazone treatment prevents changes in presynaptic and postsynaptic markers (Toledo and Inestrosa 2010). One of the important pathways in AD is the Wnt signaling (Inestrosa et al. 2021). Rosiglitazone was shown to increase β-catenin and inhibit GSK3β resulting in reducing various neuropathological markers of AD (Toledo and Inestrosa 2010).
Interestingly, in mice with T2DM and AD, treatment with rosiglitazone increased IDE expression levels, reduced Aβ40 and Aβ42 accumulation, and learning and spatial cognition disorders by activating the PPARγ / AMPK signaling pathway (Zhou et al. 2018). Rosiglitazone has been elucidated to improve cognitive ability, learning, and memory impairment through modulating the insulin-dependent signaling pathway Akt-GSK3β followed by reducing hyper-phosphorylation of Tau and neurofilament proteins (Tokutake et al. 2012).
Clinical trials have reported conflicting results regarding the effect of rosiglitazone on AD. In a preliminary study, 30 subjects with mild AD or amnestic mild cognitive impairment were randomly selected, with 20 treated with rosiglitazone (4 mg daily) and 10 treated with placebo for 6 months. Rosiglitazone exhibited better delayed recall (months 4 and 6) and selective attention (month 6) than those in the placebo group. At the 6th month, plasma Aβ levels remained unchanged in subjects receiving rosiglitazone, but it decreased in those receiving placebo (Watson et al. 2005). Another study using three different doses of rosiglitazone (2, 4, or 8 mg for 6 months) indicated a significant improvement in the primary endpoint (change of ADASCog from the beginning) only in APOE4 patients receiving 8 mg of rosiglitazone (Risner et al. 2006). However, the clinical trial results were inconsistent with the evidence from the previous two studies. This discrepancy seems to be due to the duration of rosiglitazone treatment and the type of subjects included in the study regarding APOE positive or negative. Although rosiglitazone has beneficial effects on insulin resistance, glucose and lipid metabolism, its weak penetration into the brain and its association with severe cardiotoxicity limit its clinical applications. Today, only pioglitazone is approved for the treatment of T2DM, while rosiglitazone is not prescribed due to the high incidence of cardiovascular complications.
Sulfonylureas
Sulfonylureas (e.g., glimepiride, tolbutamide, glyburide) are commonly used to treat diabetes. Their molecular mechanism is to stimulate endogenous insulin secretion by binding to specific receptors on pancreatic β cells, leading to closing ATP-sensitive potassium channels and increasing intracellular calcium levels (Costello and Shivkumar 2020). These channels are expressed in the pituitary, microglia, and nerve cells in various brain areas, including the hippocampus, frontal cortex, amygdala, and hypothalamus. Molecular studies have shown that ATP-sensitive potassium channels consist of four kir pore-forming subunits and four sulfonylurea regulatory subunits. Three sulfonylurea receptors (SUR) isoforms (SUR1, SUR2A, SUR2B) have been identified in the pancreas, heart, and arteries, respectively. The Kir subunit can also exist in two forms, Kir6.1 and Kir6.2 (Lefer et al. 2009; Principalli et al. 2015). Recent studies have shown that potassium ATP channels of the central nervous system play an essential role in neuroprotection, synaptic transmission, neuroplasticity, and neurobehavioral disorders such as anxiety, depression, learning, and memory loss. In addition, these channels have been documented to play an important role in the pathogenesis of AD (Zubov et al. 2020). Therefore, targeting these channels could be potentially a strategy to treat AD.
The SUR1 subunit can interact with the transient receptor potential cation channel subfamily M member 4 (TRPM4) channels. The expression of SUR1-TRPM4 increases following the damage or inflammation in nerve cells. It also activates microglia cells in the CNS following inflammation and causes cognitive decline. Opening of SUR1-TRPM4 channels due to edema and cell death causes a cytotoxic effect. It has been suggested that glibenclamide inhibited the SUR1-TRPM4 channel by binding directly to the SUR1 subunit and thus preventing CNS cell death (Simard et al. 2006; Kurland et al. 2016). Glibenclamide is a known K-ATP channel blocker that can cross blood–brain barrier systems and affects brain nerve cells (Wen et al. 2021).
Moreover, it has been demonstrated that Aβ25-35 leads to over activation of the hypothalamic–pituitary–adrenal-(HPA) axis and an increase of the symptoms of depression and anxiety in rats. The blockage of K-ATP channels with glibenclamide reduces behaviors related to depression and anxiety in Aβ25-treated rats by normalizing HPA axis activity (Esmaeili et al. 2018). In addition, improvement in anxiety-like symptoms following treatment with glyburide was observed in high-fat-diet-induced obesity mice (Gainey et al. 2016). Recent studies in animal models of AD have shown evidence for a two-way pathway between Aβ and the K-ATP channel. For example, Aβ was found to increase the expression of KATP channel subunits in primary neurons (Ma et al. 2009). In contrast, the K-ATP channel modulates Aβ production (Kong et al. 2015). Tolbutamide inhibits Aβ1-42-induced memory impairment and synaptic plasticity alterations in the hippocampus (105), a K-ATP channel blocker.
Sulfonylureas affect ABC transporters, especially ABCA1. ABCA1 is expressed in various nerve cells and is involved in regulating cholesterol levels. Impairment of ABC1 leads to increased inflammatory responses and impaired synaptic transmission. In a mice model of AD, inactivation of ABCA1 increased soluble and insoluble forms of amyloid and produced amyloid plaques and Lewy bodies. On the other hand, due to the interaction of glibenclamide with ABCA1, it can also be involved in the processes described (Koldamova et al. 2014; van Deijk et al. 2017).
Although in vitro and in vivo studies have reported the preventive effects of sulfonylurea drugs against AD, clinical studies have reported conflicting results. One study reported that long-term use of sulfonylureas was not associated with a changed risk of developing AD (Imfeld et al. 2012). In another study by Hsu et al., combined treatment of metformin with sulfonylurea reduced the risk of dementia by 35% over 8 years (Hsu et al. 2011). Interestingly, in a meta-analysis of 5 cohort studies, it was found that there was no significant relationship between the use of metformin or sulfonylurea and brain function and structure outcomes (Weinstein et al. 2019). From the results of these studies, it can be concluded that more clinical studies using larger sample sizes are needed to accurately investigate the mechanism of the action of sulfonylurea in AD.
DPP4 inhibitors
DPP4, as a serine exopeptidase, is ubiquitously expressed on the surface of a variety of cells. This exopeptidase selectively cleaves N-terminal dipeptides from various substrates, including cytokines, growth factors, neuropeptides, and the incretin hormones. Accordingly, DPP4 exerts multifunctional effects on different signaling pathways such as inflammation, glucose metabolism, neurophysiological and neuroendocrine processes, food intake, pain, and vascular modulation. It has been suggested that the suppression of DPP-4 activity may increase the anti-inflammatory and neuroprotective effects in the brain (Cheng et al. 2020). Studies have reported that DPP4 inhibitors improve mitochondrial dysfunction and neuroinflammation, prevent Aβ deposition and inhibit total tau protein formation and phosphorylation (Chalichem et al. 2018; Wu et al. 2020). Commonly available DPP-4 inhibitors include linagliptin, vildagliptin, and saxagliptin. In this section, we summarize in-vivo, in-vitro, and clinical studies on the effects of DPP4 inhibitor family drugs on the neuropathophysiology of AD.
Linagliptin
Linagliptin is one of the most potent inhibitors of DPP-4, which shows greater selectivity for DPP-4 than the other DPPs and related proteases (e.g., DPP8 and DPP9). Approved by the FDA in 2011, this drug does not pass through the BBB, but by peripherally inhibiting DPP-4 triples the incretin level so that it can pass through the BBB (Röhrborn et al. 2015; Cheng et al. 2020). Recent in vitro and in vivo studies have shown that linagliptin can significantly inhibit the neurodegeneration process observed in AD through several molecular mechanisms. For example, Aβ disrupts insulin signaling and kills SK-N-MC cells. Linagliptin protects SK-N-MC cells against Aβ-induced toxicity, and restoring insulin signaling inhibits GSK3b activation and Tau protein hyperphosphorylation. In addition, it reduces Aβ-induced mitochondrial dysfunction and intracellular ROS production, which may be due to the activation of AMPK-Sirt1 signaling (Kornelius et al. 2015). In another study, linagliptin was reported to improve behavioral abnormalities, oxidative stress, inflammation, and cuprizone-induced demyelination by modulating the AMPK / SIRT1 and JAK2 / STAT3 / NF-κB signaling pathways (Elbaz et al. 2018). In addition, 8-week administration of linagliptin has been shown to reduce cognitive deficits, brain incretin levels, Aβ deposition, tau protein phosphorylation, and neuroinflammation in 3xTg-AD mice (Kosaraju et al. 2017). Nakaoka et al. observed that PS19 mice treated with linagliptin have higher levels of GLP1 and lower fasting blood glucose than the controls. They reported that linagliptin significantly restored spatial reference memory and increased CBF without affecting the rate of tau or eNOS protein phosphorylation in the brain. In fact, it improves cognitive deterioration caused by a high-fat diet in tauopathy by increasing brain perfusion through an eNOS-independent pathway (Nakaoku et al. 2019). It has been found that linagliptin improved cognitive impairment in STZ-induced diabetic mice by inhibiting oxidative stress, reducing NADPH oxidase and TNF-α expression, and microglial activation (Ide et al. 2020). It is of note that to date no experimental study has been performed to evaluate the effects of linagliptin on improving the neurophysiopathological conditions of AD, and such studies are necessary.
Saxagliptin
Saxagliptin is one of the oral drugs from the DPP4 family of inhibitors that is well tolerated with minimal side effects and is used to treat diabetic patients (Men et al. 2018). To date, only one study has examined the efficacy of saxagliptin in a rat model of AD. Three months after the onset of AD, these rats were treated orally with saxagliptin (0.25, 0.5, and 1 mg/kg) for 60 days. This study showed a reduction in Aβ deposition, tau phosphorylation, and inflammatory markers following treatment with saxagliptin. Saxagliptin also improved hippocampal GLP-1 and memory retention (Kosaraju et al. 2013). Due to the small number of studies on the effect of saxagliptin on neurological processes in AD, more cellular, animal, and clinical trials are needed to determine the beneficial effects and the exact mechanism of this drug.
Vildagliptin
Vildagliptin under the chemical name (1-[[3-hydroxy-1-adamantyl) amino] acetyl]-2-cyano-(S)-pyrrolidine) and the Galvus proprietary name was introduced in 2007 as one of the key drugs in the DPP4 inhibitor family for treatment T2DM (De Oliveira et al. 2017). In terms of the mechanism of the action, cellular and animal studies have demonstrated that vildagliptin has positive effects on neurological processes and cognitive functions in AD by increasing the expression and the activity of GLP-1 in peripheral blood and reducing Aβ deposition, phosphorylated tau protein, and inflammation in brain tissue (Ma et al. 2018; Khalaf et al. 2019; Yossef et al. 2020). For example, in a study, it was reported that vildagliptin reduced GSK3b and Tau phosphorylation levels by reducing apoptosis-related proteins (caspase-3 and caspase-9) in SH-SY5Y cells. Decreased expression of PSEN1 and PSEN2 also exerts a protective effect against Aβ (Dokumacı and Aycan 2019). Khalaf et al. observed that vildagliptin alone and combined with memantine, a subfamily of glutamate receptors involved in brain function, decreased blood glucose, HOMA-IR, lipid profile, homocysteine, malondialdehyde, acetylcholinesterase, and increased apolipoprotein E. In addition, treatment of mice with combined diabetes AD using vildagliptin, alone and in combination with memantine, reduces the expression of amyloid precursor protein and phosphorylated tau protein (Khalaf et al. 2019). Treatment with vildagliptin has been reported to improve memory deficits and reduce neuronal apoptosis in the hippocampus. In addition, treating Alzheimer's rats with Vildagliptin increased BCL2 expression levels, while the expression levels of caspase-3, Bcl-2-associated protein (Bcl ‑ 2 associated X protein), and AD-associated proteins in the hippocampus were reduced (Ma et al. 2018). Moreover, vildagliptin was able to reduce the levels of tau phosphorylated proteins, amyloid precursor protein (APPs), and p-GSKs, and increase the expression levels of the PSD 95 proteins, synaptophysin, and p-Akt, with positive effects on AD recovery (Ma et al. 2018). Pipatpiboon et al. observed that vildagliptin prevented neuronal insulin resistance by restoring long-term insulin-induced depression, neuronal IR phosphorylation, IRS-1 phosphorylation, and Akt / PKB-ser phosphorylation. It also improved mitochondrial dysfunction and cognitive function (Pipatpiboon et al. 2013). In a recent study, vildagliptin was shown to reduce inflammatory markers (TNF-α), apoptosis (caspase 3), and oxidative stress (FOXO1) in the hippocampus, BCL2 associated X (BAX), and inhibit JAK2 / STAT3 signaling pathway along with restoration of metabolic disorders (Yossef et al. 2020). Apart from in-vivo and in vitro studies, only one clinical study investigated the effects of vildagliptin on cognitive function in elderly patients with T2DM. This study showed that the addition of vildagliptin to the treatment of diabetic patients improves cognitive function and metabolic control in elderly patients with T2DM(Bulut et al. 2020). Overall, all cell and animal studies have confirmed that vildagliptin has beneficial effects on cognitive function and neurological processes in AD models. However, more studies are needed to confirm this.
GLP-1 agonists
Reports have shown that stimulation of Glucagon-like peptide-1 receptor (GLP-1) can play neurotrophic and neuroprotective roles in many types of cellular and animal neurodegeneration models (Salcedo et al. 2012). Therefore, synthetic analogs of GLP-1 can have beneficial effects in neurodegenerative diseases such as AD (Meng et al. 2020). In the following, we will focus on the drugs of the GLP-1 agonists family.
Liraglutide
Liraglutide is a synthetic analog of GLP-1 with 97% homology. This drug has various effects on AD by crossing the blood–brain barrier (Hunter and Hölscher 2012), including reducing the amount of Aβ plaques, attenuating insulin receptor and synaptic pathology, and reducing cognitive disorders (Long-Smith et al. 2013; Batista et al. 2018; Holubová et al. 2019). Different signaling pathways have been proposed for these effects, addressed below.
Synaptic plasticity acts as a link in the brain between type 2 diabetes and AD. Liraglutide in amyloid precursor protein (APP) / PS1 mice regulates synaptic plasticity and can increase neuron proliferation and differentiation (Parthsarathy and Hölscher 2013). This was confirmed by a recent study by Hamilton et al. They found that Liraglutide increased the number of progenitor cells or doublecortin-positive young neurons in the dentate gyrus of diabetic mice (Hamilton et al. 2011). In the studies of McClean et al., Liraglutide has been shown to improve spatial learning and memory and prevent impairment in synaptic plasticity in the CA1 area of the hippocampus (Llorens-Martín et al. 2014). It also reduces chronic inflammation and increases long-term potentiation (LTP) in Alzheimer's mice model (McClean et al. 2010, 2011, 2015). In addition, treatment with Liraglutide significantly increases memory retention and total hippocampal CA1 pyramidal neuron numbers (Hansen et al. 2015). In the hyperhomocysteinemia rat model, Liraglutide reduced tau hyperphosphorylation and Aβ overproduction, which may alleviate AD-like cognitive impairment (Zhang et al. 2019). Different groups have shown that Liraglutide can reduce the level of hyperphosphorylation of tau and neurofilaments through the glycogen synthase kinase-3β (GSK-3β) c-Jun N terminal kinase (JNK), extracellular signal-regulated kinase (ERK), and cAMP signaling pathways (Hunter and Hölscher 2012; Han et al. 2013; Xiong et al. 2013; Yang et al. 2013; Qi et al. 2016, 2017; Chen et al. 2017).
The effects of this drug have also been observed in clinical trial studies. It has been observed that 12 weeks of this drug in diabetic patients with a family history of AD led to a significant improvement in connectivity between bilateral hippocampal and anterior medial frontal structure (Watson et al. 2019). A clinical pilot study has also shown that Liraglutide improves chronic depression and brain volume on magnetic resonance imaging (MRI) scans (Mansur et al. 2017). Another study found that six months of Liraglutide administration inhibited the reduction of glucose metabolism in the brain according to [(18) F] FDG positron electron tomography (FDG-PET) scans. However, no change in amyloid plaque was observed (Gejl et al. 2016). On the other hand, in their animal studies on APP / PS1 mouse models of AD, Hansen et al. found that long-term use of Liraglutide did not affect amyloid plaque levels (Hansen et al. 2016).
Exenatide
Exenatide is another drug in the GLP-1 agonists family that has been shown to have potential therapeutic effects in AD. Exenatide can increase progenitor cells or doublecortin-positive young neurons in the dentate gyrus (Hamilton et al. 2011). Studies have also suggested the influential role of this drug in neuroprotective pathways. Accordingly, Exenatide has protective effects against tau hyperphosphorylation in mice model of AD through downregulation of GSK-3β activity (Chen et al. 2012; Xu et al. 2015). Four-weeks treatment with this drug in APP / PS1 mice can rescue memory deficits and neuropathological changes and reduce acetylglucosaminyltransferase III (GnT-III) expression through Akt / GSK-3β / β-catenin pathway in neurons (Wang et al. 2018). It has been shown that the Exenatide could reduce hippocampal IRS-1pSer and reduce JNK / TNF-α pathway activity and TNF-α levels. It also improves memory and cognitive behaviors (Bomfim et al. 2012; Solmaz et al. 2015). Studies have also reported that the improvement in cognitive function of the brain caused by treatment with Exenatide may be due to increased anaerobic glycolysis of the brain. However, these results were observed only in PS1-KI mice and not in 3xTg-AD mice (Bomba et al. 2013). In addition, Exenatide can reduce the harmful induction of Aβ42 in the rat hippocampal CA1 region and increase the cAMP signaling pathway (Wang et al. 2016). Clinical trial studies for further investigation about Exenatide in this issue are suggested.
Lixisenatide
Lixisenatide enhances neuronal progenitor proliferation in the dentate gyrus, neurogenesis in the brain, and increases cAMP levels in the brain. Lixisenatide can cross the blood–brain barrier easily and even at lower concentrations than Liraglutide (Hunter and Hölscher 2012). In the meantime, it has been reported that lixisenatide brings about cognitive and pathological improvements in AD at lower doses compared to Liraglutide. It also increases long-term potentiation, prevents synaptic number reduction in APP / PS1 mice, and decreases the load of amyloid plaque and chronic inflammation (McClean and Hölscher 2014). The results of Cai et al., study have shown that this drug can prevent reducing spatial learning and memory ability by injecting Aβ25-35 into the hippocampus of rats, and this effect is mediated through the PI3K-Akt-GSK3β pathway (Cai et al. 2014).
Dulaglutide
Dulaglutide is another member of this family of drugs that have shown effects against AD. Dulaglutide has a beneficial impact on cognitive ability and memory impairment in the streptozocin-induced AD mouse model. In addition, it reduces the phosphorylation of tau proteins and neurofilament. These results suggest that the neuroprotective role of dulaglutide may be through the PI3K / Akt / GSK3β signaling pathway (Zhou et al. 2019).
SGLT2 inhibitors (empagliflozin, canagliflozin, dapagliflozin)
Sodium-glucose co-transporter 2 inhibitors (SGLT2i) are oral anti-hyperglycemic drugs approved to treat type 2 diabetes. Some studies point to their potential effects on the central nervous system's neuroprotective properties.
The study by Hierro-Bujalance et al. has shown that empagliflozin can reduce amyloid β (Aβ) levels, reduce the density of senile plaques in the cortex, and hypothalamus and cause positive cognitive effects (Hierro-Bujalance et al. 2020). Lin et al. also obtained confirmatory results in this regard. They observed that treatment with empagliflozin in db/db mice inhibited cognitive dysfunction associated with a reduction in cerebral oxidative stress and increased cerebral brain-derived neurotrophic factor (BDNF) (Lin et al. 2014). BDNF has important and different roles in the nervous system, including differentiation, maturation, and survival of nerve cells and neuroprotective effects in glutamatergic stimulation, cerebral ischemia, neurotoxicity, and hypoglycemia situations (Wiciński et al. 2020). The protective effect of empagliflozin has also been suggested to be through inhibition of apoptosis. According to a study by Abdel et al., empagliflozin reduced caspase-3 and infarct size and increased HIF-1α / VEGF level in rats with cerebral ischemia / reperfusion (I / R) (Abdel-latif et al. 2020). Interestingly, elderly diabetic rats treated with empagliflozin experienced positive effects on their brain structure and demonstrated protection against abnormalities in the neurovascular units (Hayden et al. 2019).
Canagliflozin from the SGLTi family had favorable effects in scopolamine-induced memory impairment rats. This effect can be attributed to the potential inhibitory properties of its acetylcholine esterase activity (Arafa et al. 2017). These results appear to be consistent with another study that confirms that canagliflozin has dual inhibitor properties for both AChE and SGLT2 (Syed et al. 2014).
Other drugs in this class dapagliflozin, has been shown to have inhibitory effects on acetylcholinesterase. Based on molecular docking, it has been shown that this drug can bind to the AChE receptor with low binding energy, similar to the energy required to bind to SGLT2 (Shaikh et al. 2016). Other drugs in this family, such as ertugliflozin and sotagliflozin, show similar properties (Shakil 2017). Another study on mice receiving a high-fat diet leading to insulin resistance and cognitive decline showed that dapagliflozin could improve hippocampal synaptic plasticity and prevent cognitive decline (Sa-nguanmoo et al. 2017). In addition, SGLT2 inhibitors can enhance angiogenesis and neurogenesis and prevent ischemia-related cerebral damage (Wiciński et al. 2020). Eventually, despite the positive effects of SGLT2 inhibitors on AD, there is still insufficient evidence in this area and various in-vitro and in-vivo studies are required.
Meglitinides (repaglinide, nateglinide, mitiglinide)
Meglitinides are oral non-sulfonylurea drugs for type 2 diabetes that facilitate insulin secretion, and their primary function is inhibiting ATP-sensitive potassium channels in pancreatic beta cells. One of the most common manifestations of neurodegenerative diseases, including AD, is a disorder in the regulation of intracellular calcium concentrations in neurons. Downstream Regulatory Element Antagonist Modulator (DREAM) is a calcium-binding protein in neurons that has a specific function in protein–protein and DNA–protein reactions. Studies have shown that rapaglinide can reduce the half-life of DREAM and disrupt the PS-2 and DREAM reactions, suggesting that rapaglinide may be involved in pathways associated with AD (Naranjo et al. 2018; Santiago et al. 2019). Another manifestation of these drugs is their neuroprotective effects, which may not be directly related to AD. Rapaglinide has been shown to reduce the destructive effects of Kanic acid, in the CA3 region of the hypothalamus, whereas nateglinide does not show such effects (Kim et al. 2014a, b). In addition, Xiao et al., showed that rapaglinide can inhibit the proliferation and migration of Glioblastoma multiforme cells and inhibit the expression of Bcl-2, Beclin-1 and PD-L1 in orthotopic glioma cells (Xiao et al. 2017). On the other hand, it has been shown that nateglinide can also exert its neuroprotective effects in different ways. According to Saad et al. study, nateglinide have different effects such as inhibition of motor activity deterioration through Cav-1, anti-inflammatory property through JAK2 / STAT3 pathway and anti-apoptotic and antioxidant actions by inhibition of Caspase3 and NO and MPO, respectively in the hypothalamus of rats treated with this drug (Saad et al. 2019). According to our search of literature, no study was found on mitiglinide in this area. In general, more studies in the meglitinides family are required to establish the actual mechanism of these medicines on AD.
Conclusion
Based on accumulating evidence that indicates strong association and similar pathological mechanisms between AD and T2DM we discussed the potential of anti-diabetic drugs against AD in currently available animal and clinical studies. Based on data collection in Table 1, the use of intranasal insulin, rosiglitazone, glibenclamide, and GLP-1 agonists was found to have positive effects in AD treatment. In addition, although pioglitazone and sulfonylureas improved diabetic conditions and cognitive impairment, but contradictory results have been seen in the in-vivo and clinical studies. Rosiglitazone has beneficial effects on insulin resistance, glucose, and lipid metabolism, but due to weak penetration in the brain and cardiotoxicity association, it is not recommended for AD treatment. GLP1 agonist (Exenatide) and DPP4 inhibitors (Vidagliptin) have neuroprotective effects. However, more studies are needed to confirm it. Since that metformin is the first line of treatment in diabetes and the most widely used drug in the treatment of diabetes, it is necessary to study its effects on AD. Although most of the data on the use of metformin in AD with or without T2DM are promising, it should be noted that the effect of metformin might depend on complex underlying pathological processes. In some cases, metformin has even been shown to have harmful effects. Therefore, to determine the effects of metformin in the control and treatment of dementia/AD, it is necessary to define broader cohort studies with long-term use of metformin and larger study groups.
Data availability
The data used to support the findings of this study are included in the article.
References
Aali E, Esmaeili MH, Mahmodi SS, Solimani P (2020) Effects of chronic administration of pioglitazone on learning and memory in rat model of streptozotocin-induced Alzheimer’s disease. J Inflamm Dis 24(4):294–307
Abdel-latif RG et al (2020) Empagliflozin alleviates neuronal apoptosis induced by cerebral ischemia/reperfusion injury through HIF-1α/VEGF signaling pathway. Arch Pharmacal Res 43(5):514–525
Akimoto H, Negishi A, Oshima S, Wakiyama H, Okita M, Horii N et al (2020) Antidiabetic drugs for the risk of alzheimer disease in patients with type 2 DM using FAERS. Am J Alzheimer’s Dis Other Dementias 35:1533317519899546
Al-Majed A et al (2016) Pioglitazone. Profiles Drug Subst Excipients Relat Methodol 41:379–438
Arafa NMS, Ali EHA, Hassan MK (2017) Canagliflozin prevents scopolamine-induced memory impairment in rats: Comparison with galantamine hydrobromide action. Chem Biol Interact 277:195–203
Arnold SE et al (2018) Brain insulin resistance in type 2 diabetes and Alzheimer disease: concepts and conundrums. Nat Rev Neurol 14(3):168
Barini E, Antico O, Zhao Y, Asta F, Tucci V, Catelani T et al (2016) Metformin promotes tau aggregation and exacerbates abnormal behavior in a mouse model of tauopathy. Mol Neurodegener 11(1):1–20
Barone E, Tramutola A, Triani F, Calcagnini S, Di Domenico F, Ripoli C et al (2019) Biliverdin reductase-A mediates the beneficial effects of intranasal insulin in Alzheimer disease. Mol Neurobiol 56(4):2922–2943
Batista AF, Forny-Germano L, Clarke JR, Lyra e Silva NM, Brito-Moreira J, Boehnke SE et al (2018) The diabetes drug liraglutide reverses cognitive impairment in mice and attenuates insulin receptor and synaptic pathology in a non-human primate model of Alzheimer’s disease. J Pathol 245(1):85–100
Benedict C, Hallschmid M, Hatke A, Schultes B, Fehm HL, Born J et al (2004) Intranasal insulin improves memory in humans. Psychoneuroendocrinology 29(10):1326–1334
Benedict C, Hallschmid M, Schmitz K, Schultes B, Ratter F, Fehm HL et al (2007) Intranasal insulin improves memory in humans: superiority of insulin aspart. Neuropsychopharmacology 32(1):239–243
Blázquez E et al (2014) Insulin in the brain: its pathophysiological implications for States related with central insulin resistance, type 2 diabetes and Alzheimer’s disease. Front Endocrinol 5:161
Boccardi V et al (2019) Diabetes drugs in the fight against Alzheimer’s disease. Ageing Res Rev 54:100936
Bomba M, Ciavardelli D, Silvestri E, Canzoniero LM, Lattanzio R, Chiappini P et al (2013) Exenatide promotes cognitive enhancement and positive brain metabolic changes in PS1-KI mice but has no effects in 3xTg-AD animals. Cell Death Dis 4(5):e612-e
Bomfim TR, Forny-Germano L, Sathler LB, Brito-Moreira J, Houzel J-C, Decker H et al (2012) An anti-diabetes agent protects the mouse brain from defective insulin signaling caused by Alzheimer’s disease–associated Aβ oligomers. J Clin Investig 122(4):1339–1353
Bulut EA, Alak ZYS, Dokuzlar O, Kocyigit SE, Soysal P, Smith L et al (2020) Cognitive and metabolic outcomes of vildagliptin addition to the therapy in patients with type 2 diabetes mellitus: 26 week follow-up study. Arch Gerontol Geriatr 88:104013
Cai HY, Hölscher C, Yue XH, Zhang SX, Wang XH, Qiao F et al (2014) Lixisenatide rescues spatial memory and synaptic plasticity from amyloid β protein-induced impairments in rats. Neuroscience 277:6–13
Cao B et al (2018) Comparative efficacy and acceptability of antidiabetic agents for Alzheimer’s disease and mild cognitive impairment: A systematic review and network meta-analysis. Diabetes Obes Metab 20(10):2467–2471
Chalichem NSS et al (2018) Possible role of DPP4 inhibitors to promote hippocampal neurogenesis in Alzheimer’s disease. J Drug Target 26(8):670–675
Chen S, Liu A-r, An F-m, Yao W-b, Gao X-d (2012) Amelioration of neurodegenerative changes in cellular and rat models of diabetes-related Alzheimer’s disease by exendin-4. AGE 34(5):1211–24
Chen J, Li S, Sun W, Li J (2015) Anti-diabetes drug pioglitazone ameliorates synaptic defects in AD transgenic mice by inhibiting cyclin-dependent kinase5 activity. PLoS ONE 10(4):e0123864
Chen S, Sun J, Zhao G, Guo A, Chen Y, Fu R et al (2017) Liraglutide improves water maze learning and memory performance while reduces hyperphosphorylation of tau and neurofilaments in APP/PS1/Tau triple transgenic mice. Neurochem Res 42(8):2326–2335
Chen J-L, Luo C, Pu D, Zhang G-Q, Zhao Y-X, Sun Y et al (2019) Metformin attenuates diabetes-induced tau hyperphosphorylation in vitro and in vivo by enhancing autophagic clearance. Exp Neurol 311:44–56
Chen Y, Zhao S, Fan Z, Li Z, Zhu Y, Shen T et al (2021) Metformin attenuates plaque-associated tau pathology and reduces amyloid-β burden in APP/PS1 mice. Alzheimer’s Res Ther 13(1):1–13
Chen B, Teng Y, Zhang X, Lv X, Yin Y (2016) Metformin alleviated aβ-induced apoptosis via the suppression of jnk mapk signaling pathway in cultured hippocampal neurons. Biomed Res Int. 2016:1421430. https://doi.org/10.1155/2016/1421430
Cheng Q et al (2020) Can dipeptidyl peptidase-4 inhibitors treat cognitive disorders? Pharmacol Ther 212:107559
Claxton A, Baker LD, Wilkinson CW, Trittschuh EH, Chapman D, Watson G et al (2013) Sex and ApoE genotype differences in treatment response to two doses of intranasal insulin in adults with mild cognitive impairment or Alzheimer’s disease. J Alzheimers Dis 35(4):789–797
Claxton A, Baker LD, Hanson A, Trittschuh EH, Cholerton B, Morgan A et al (2015) Long-acting intranasal insulin detemir improves cognition for adults with mild cognitive impairment or early-stage Alzheimer’s disease dementia. J Alzheimers Dis 44(3):897–906
Costello RA, Shivkumar A (2020) Sulfonylureas. StatPearls [Internet]
Craft S et al (1996) Memory improvement following induced hyperinsulinemia in Alzheimer’s disease. Neurobiol Aging 17(1):123–130
Craft S, Baker LD, Montine TJ, Minoshima S, Watson GS, Claxton A et al (2012) Intranasal insulin therapy for Alzheimer disease and amnestic mild cognitive impairment: a pilot clinical trial. Arch Neurol 69(1):29–38
Craft S, Claxton A, Baker LD, Hanson AJ, Cholerton B, Trittschuh EH et al (2017) Effects of regular and long-acting insulin on cognition and Alzheimer’s disease biomarkers: a pilot clinical trial. J Alzheimers Dis 57(4):1325–1334
De la Monte SM, Wands JR (2008) Alzheimer’s disease is type 3 diabetes—evidence reviewed. J Diabetes Sci Technol 2(6):1101–1113
de Matos AM et al (2018) Bridging type 2 diabetes and Alzheimer’s disease: assembling the puzzle pieces in the quest for the molecules with therapeutic and preventive potential. Med Res Rev 38(1):261–324
De Oliveira GLA et al (2017) Cost-effectiveness of vildagliptin for people with type 2 diabetes mellitus in Brazil; findings and implications. Expert Rev Pharmacoecon Outcomes Res 17(2):109–119
DiTacchio KA et al (2015) Metformin treatment alters memory function in a mouse model of Alzheimer’s disease. J Alzheimers Dis 44(1):43–48
Dokumacı AH, Aycan MBY (2019) Vildagliptine protects SH-SY5Y human neuron-like cells from A β 1–42 induced toxicity, in vitro. Cytotechnology 71(2):635–646
Elbaz EM, Senousy MA, El-Tanbouly DM, Sayed RH (2018) Neuroprotective effect of linagliptin against cuprizone-induced demyelination and behavioural dysfunction in mice: a pivotal role of AMPK/SIRT1 and JAK2/STAT3/NF-κB signalling pathway modulation. Toxicol Appl Pharmacol 352:153–161
Esmaeili MH, Bahari B, Salari A-A (2018) ATP-sensitive potassium-channel inhibitor glibenclamide attenuates HPA axis hyperactivity, depression-and anxiety-related symptoms in a rat model of Alzheimer’s disease. Brain Res Bull 137:265–276
Farr SA, Roesler E, Niehoff ML, Roby DA, McKee A, Morley JE (2019) Metformin improves learning and memory in the SAMP8 mouse model of Alzheimer’s disease. J Alzheimers Dis 68(4):1699–1710
Farris W et al (2003) Insulin-degrading enzyme regulates the levels of insulin, amyloid β-protein, and the β-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci 100(7):4162–4167
Figlewicz DP, Szot P, Israel PA, Payne C, Dorsa DM (1993) Insulin reduces norepinephrine transporter mRNA in vivo in rat locus coeruleus. Brain Res 602(1):161–164
Gabbouj S et al (2019) Altered insulin signaling in Alzheimer’s disease brain–special emphasis on PI3K-Akt pathway. Front Neurosci 13:629
Gainey SJ, Kwakwa KA, Bray JK, Pillote MM, Tir VL, Towers AE et al (2016) Short-term high-fat diet (HFD) induced anxiety-like behaviors and cognitive impairment are improved with treatment by glyburide. Front Behav Neurosci 10:156
Galimberti D, Scarpini E (2017) Pioglitazone for the treatment of Alzheimer’s disease. Expert Opin Investig Drugs 26(1):97–101
Gao L et al (2016) Shared genetic etiology between type 2 diabetes and Alzheimer’s disease identified by bioinformatics analysis. J Alzheimers Dis 50(1):13–17
Gejl M, Gjedde A, Egefjord L, Møller A, Hansen SB, Vang K et al (2016) In Alzheimer’s disease, 6-month treatment with GLP-1 analog prevents decline of brain glucose metabolism: randomized, placebo-controlled, double-blind clinical trial. Front Aging Neurosci 8:108
Geldmacher DS, Fritsch T, McClendon MJ, Landreth G (2011) A randomized pilot clinical trial of the safety of pioglitazone in treatment of patients with Alzheimer disease. Arch Neurol 68(1):45–50
Gold M, Alderton C, Zvartau-Hind M, Egginton S, Saunders AM, Irizarry M et al (2010) Rosiglitazone monotherapy in mild-to-moderate Alzheimer’s disease: results from a randomized, double-blind, placebo-controlled phase III study. Dement Geriatr Cogn Disord 30(2):131–146
Goodarzi G et al (2016) The effect of the glycolipoprotein extract (G-90) from earthworm Eisenia foetida on the wound healing process in alloxan-induced diabetic rats. Cell Biochem Funct 34(4):242–249
Gorska-Ciebiada M, et al (2014) Mild cognitive impairment and depressive symptoms in elderly patients with diabetes: prevalence, risk factors, and comorbidity. J Diabetes Res 2014:179648
Gratuze M, Julien J, Petry FR, Morin F, Planel E (2017) Insulin deprivation induces PP2A inhibition and tau hyperphosphorylation in hTau mice, a model of Alzheimer’s disease-like tau pathology. Sci Rep 7(1):1–13
Guo Z, Chen Y, Mao Y-F, Zheng T, Jiang Y, Yan Y et al (2017) Long-term treatment with intranasal insulin ameliorates cognitive impairment, tau hyperphosphorylation, and microglial activation in a streptozotocin-induced Alzheimer’s rat model. Sci Rep 7(1):1–12
Haan MN (2006) Therapy Insight: type 2 diabetes mellitus and the risk of late-onset Alzheimer’s disease. Nat Clin Pract Neurol 2(3):159–166
Hamilton A, Patterson S, Porter D, Gault VA, Holscher C (2011) Novel GLP-1 mimetics developed to treat type 2 diabetes promote progenitor cell proliferation in the brain. J Neurosci Res 89(4):481–489
Han W-N, Hölscher C, Yuan L, Yang W, Wang X-H, Wu M-N et al (2013) Liraglutide protects against amyloid-β protein-induced impairment of spatial learning and memory in rats. Neurobiol Aging 34(2):576–588
Hansen HH, Fabricius K, Barkholt P, Niehoff ML, Morley JE, Jelsing J et al (2015) The GLP-1 receptor agonist liraglutide improves memory function and increases hippocampal CA1 neuronal numbers in a senescence-accelerated mouse model of Alzheimer’s disease. J Alzheimers Dis 46(4):877–888
Hansen HH, Fabricius K, Barkholt P, Kongsbak-Wismann P, Schlumberger C, Jelsing J et al (2016) Long-Term treatment with liraglutide, a Glucagon-Like Peptide-1 (GLP-1) receptor agonist, has no effect on β-amyloid plaque load in two transgenic APP/PS1 mouse models of Alzheimer’s disease. PLoS ONE 11(7):e0158205
Hanyu H et al (2009) Pioglitazone improved cognition in a pilot study on patients with Alzheimer’s disease and mild cognitive impairment with diabetes mellitus. J Am Geriatr Soc 57(1):177–179
Hanyu H et al (2010) The role of tumor necrosis factor-alpha in cognitive improvement after peroxisome proliferator-activator receptor gamma agonist pioglitazone treatment in alzheimer’s disease. J Am Geriatr Soc 58(5):1000–1001
Hao K et al (2015) Shared genetic etiology underlying Alzheimer’s disease and type 2 diabetes. Mol Aspects Med 43:66–76
Hayden MR, Grant DG, Aroor AR, DeMarco VG (2019) Empagliflozin Ameliorates Type 2 Diabetes-Induced Ultrastructural Remodeling of the Neurovascular Unit and Neuroglia in the Female db/db Mouse. Brain Sci 9(3):57
Heneka MT, Sastre M, Dumitrescu-Ozimek L, Hanke A, Dewachter I, Kuiperi C et al (2005) Acute treatment with the PPARγ agonist pioglitazone and ibuprofen reduces glial inflammation and Aβ1–42 levels in APPV717I transgenic mice. Brain 128(6):1442–1453
Hierro-Bujalance C, Infante-Garcia C, del Marco A, Herrera M, Carranza-Naval MJ, Suarez J et al (2020) Empagliflozin reduces vascular damage and cognitive impairment in a mixed murine model of Alzheimer’s disease and type 2 diabetes. Alzheimer’s Res Ther 12(1):40
Hölscher C (2019) Insulin signaling impairment in the brain as a risk factor in Alzheimer’s disease. Front Aging Neurosci 11:88
Holubová M, Hrubá L, Popelová A, Bencze M, Pražienková V, Gengler S et al (2019) Liraglutide and a lipidized analog of prolactin-releasing peptide show neuroprotective effects in a mouse model of β-amyloid pathology. Neuropharmacology 144:377–387
Hsu C-C, Wahlqvist ML, Lee M-S, Tsai H-N (2011) Incidence of dementia is increased in type 2 diabetes and reduced by the use of sulfonylureas and metformin. J Alzheimers Dis 24(3):485–493
Hunter K, Hölscher C (2012) Drugs developed to treat diabetes, liraglutide and lixisenatide, cross the blood brain barrier and enhance neurogenesis. BMC Neurosci 13(1):33
Hurren KM, Dunham MW (2021) Are thiazolidinediones a preferred drug treatment for type 2 diabetes? Taylor & Francis
Ide M, Sonoda N, Inoue T, Kimura S, Minami Y, Makimura H et al (2020) The dipeptidyl peptidase-4 inhibitor, linagliptin, improves cognitive impairment in streptozotocin-induced diabetic mice by inhibiting oxidative stress and microglial activation. PLoS ONE 15(2):e0228750
Imfeld P, Bodmer M, Jick SS, Meier CR (2012) Metformin, other antidiabetic drugs, and risk of Alzheimer’s disease: a population-based case–control study. J Am Geriatr Soc 60(5):916–921
Inestrosa NC et al (2021) WNT signaling is a key player in alzheimer’s disease. pharmacology of the wnt signaling system. G. Schulte and P. Kozielewicz. Cham, Springer International Publishing: 357–382
Janson J et al (2004) Increased risk of type 2 diabetes in Alzheimer disease. Diabetes 53(2):474–481
Ju Y-J, Kim N, Gee MS, Jeon SH, Lee D, Do J et al (2020) Glibenclamide modulates microglial function and attenuates Aβ deposition in 5XFAD mice. Eur J Pharmacol 884:173416
Kandimalla R et al (2017) Is Alzheimer’s disease a type 3 diabetes? A critical appraisal. Biochim Biophys Acta (BBA)-Mol Basis Dis 1863(5):1078–1089
Kern W, Peters A, Fruehwald-Schultes B, Deininger E, Born J, Fehm HL (2001) Improving influence of insulin on cognitive functions in humans. Neuroendocrinology 74(4):270–280
Khalaf SS, Hafez MM, Mehanna ET, Mesbah NM, Abo-Elmatty DM (2019) Combined vildagliptin and memantine treatment downregulates expression of amyloid precursor protein, and total and phosphorylated tau in a rat model of combined Alzheimer’s disease and type 2 diabetes. Naunyn Schmiedebergs Arch Pharmacol 392(6):685–695
Kim C-H et al (2014a) Effects of nateglinide and repaglinide administered intracerebroventricularly on the CA3 hippocampal neuronal cell death and hyperglycemia induced by kainic acid in mice. Brain Res Bull 104:36–41
Kim C-H, Park S-H, Sim Y-B, Kim S-S, Kim S-J, Lim S-M et al (2014b) Effect of tolbutamide, glyburide and glipizide administered supraspinally on CA3 hippocampal neuronal cell death and hyperglycemia induced by kainic acid in mice. Brain Res 1564:33–40
Koenig AM, Mechanic-Hamilton D, Xie SX, Combs MF, Cappola AR, Xie L et al (2017) Effects of the insulin sensitizer metformin in Alzheimer’s disease: Pilot data from a randomized placebo-controlled crossover study. Alzheimer Dis Assoc Disord 31(2):107
Koldamova R et al (2014) ATP-binding cassette transporter A1: from metabolism to neurodegeneration. Neurobiol Dis 72:13–21
Kong J-J et al (2015) Nicorandil inhibits oxidative stress and amyloid-β precursor protein processing in SH-SY5Y cells overexpressing APPsw. Int J Clin Exp Med 8(2):1966
Kong Y et al (2020) Pathological mechanisms linking diabetes mellitus and Alzheimer’s disease: the receptor for advanced glycation end products (RAGE). Front Aging Neurosci 12:217
Kornelius E, Lin CL, Chang HH, Li HH, Huang WN, Yang YS et al (2015) DPP-4 inhibitor linagliptin attenuates Aβ-induced cytotoxicity through activation of AMPK in neuronal cells. CNS Neurosci Ther 21(7):549–557
Kosaraju J, Gali CC, Khatwal RB, Dubala A, Chinni S, Holsinger RD et al (2013) Saxagliptin: a dipeptidyl peptidase-4 inhibitor ameliorates streptozotocin induced Alzheimer’s disease. Neuropharmacology 72:291–300
Kosaraju J, Holsinger RD, Guo L, Tam KY (2017) Linagliptin, a dipeptidyl peptidase-4 inhibitor, mitigates cognitive deficits and pathology in the 3xTg-AD mouse model of Alzheimer’s disease. Mol Neurobiol 54(8):6074–6084
Kuan Y-C, Huang K-W, Lin C-L, Hu C-J, Kao C-H (2017) Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog Neuropsychopharmacol Biol Psychiatry 79:77–83
Kullmann S et al (2016) Brain insulin resistance at the crossroads of metabolic and cognitive disorders in humans. Physiol Rev 96(4):1169–1209
Kurland DB et al (2016) The Sur1-Trpm4 channel regulates NOS2 transcription in TLR4-activated microglia. J Neuroinflammation 13(1):1–23
Lefer DJ et al (2009) Sulfonylurea receptor 1 subunits of ATP-sensitive potassium channels and myocardial ischemia/reperfusion injury. Trends Cardiovasc Med 19(2):61–67
Li L, Hölscher C (2007) Common pathological processes in Alzheimer disease and type 2 diabetes: a review. Brain Res Rev 56(2):384–402
Lin B, Koibuchi N, Hasegawa Y, Sueta D, Toyama K, Uekawa K et al (2014) Glycemic control with empagliflozin, a novel selective SGLT2 inhibitor, ameliorates cardiovascular injury and cognitive dysfunction in obese and type 2 diabetic mice. Cardiovasc Diabetol 13(1):148
Llorens-Martín M et al (2014) Selective alterations of neurons and circuits related to early memory loss in Alzheimer’s disease. Front Neuroanat 8:38
Long N et al (2021) Thiazolidinediones: an in–depth study of their synthesis and application to medicinal chemistry in the treatment of diabetes mellitus. ChemMedChem 16(11):1717
Long-Smith CM, Manning S, McClean PL, Coakley MF, O’Halloran DJ, Holscher C et al (2013) The diabetes drug liraglutide ameliorates aberrant insulin receptor localisation and signalling in parallel with decreasing both amyloid-β plaque and glial pathology in a mouse model of Alzheimer’s disease. NeuroMol Med 15(1):102–114
Luchsinger JA, Perez T, Chang H, Mehta P, Steffener J, Pradabhan G et al (2016) Metformin in amnestic mild cognitive impairment: results of a pilot randomized placebo controlled clinical trial. J Alzheimers Dis 51(2):501–514
Lv H, Tang L, Guo C, Jiang Y, Gao C, Wang Y et al (2020) Intranasal insulin administration may be highly effective in improving cognitive function in mice with cognitive dysfunction by reversing brain insulin resistance. Cogn Neurodyn 14(3):323
Ly PTT, Yili W, Haiyan Z, Ruitao W, Weihui Z, Ayae K, Mingming Zhang et al. Inhibition of GSK3β-mediated BACE1 expression reduces Alzheimer-associated phenotypes. J Clin Investigation 123(1): 224–235
Ma G et al (2009) Diazoxide reverses the enhanced expression of K ATP subunits in cholinergic neurons caused by exposure to Aβ 1–42. Neurochem Res 34(12):2133–2140
Ma QH, Jiang LF, Mao JL, Xu WX, Huang M (2018) Vildagliptin prevents cognitive deficits and neuronal apoptosis in a rat model of Alzheimer’s disease. Mol Med Rep 17(3):4113–4119
Mandrekar-Colucci S, Karlo JC, Landreth GE (2012) Mechanisms underlying the rapid peroxisome proliferator-activated receptor-γ-mediated amyloid clearance and reversal of cognitive deficits in a murine model of Alzheimer’s disease. J Neurosci 32(30):10117–10128
Mansur RB, Zugman A, Ahmed J, Cha DS, Subramaniapillai M, Lee Y et al (2017) Treatment with a GLP−1R agonist over four weeks promotes weight loss-moderated changes in frontal-striatal brain structures in individuals with mood disorders. Eur Neuropsychopharmacol 27(11):1153–1162
McClean PL, Hölscher C (2014) Lixisenatide, a drug developed to treat type 2 diabetes, shows neuroprotective effects in a mouse model of Alzheimer’s disease. Neuropharmacology 86:241–258
McClean PL, Gault VA, Harriott P, Hölscher C (2010) Glucagon-like peptide-1 analogues enhance synaptic plasticity in the brain: a link between diabetes and Alzheimer’s disease. Eur J Pharmacol 630(1):158–162
McClean PL, Parthsarathy V, Faivre E, Hölscher C (2011) The diabetes drug liraglutide prevents degenerative processes in a mouse model of Alzheimer’s disease. J Neurosci 31(17):6587–6594
McClean PL, Jalewa J, Hölscher C (2015) Prophylactic liraglutide treatment prevents amyloid plaque deposition, chronic inflammation and memory impairment in APP/PS1 mice. Behav Brain Res 293:96–106
Men P et al (2018) Efficacy and safety of saxagliptin in patients with type 2 diabetes: a systematic review and meta-analysis. PLoS ONE 13(5):e0197321
Meng L et al (2020) Type 2 diabetes mellitus drugs for Alzheimer’s disease: current evidence and therapeutic opportunities. Trends Mol Med 26(6):597–614
Miller BW et al (2011) Rosiglitazone and pioglitazone for the treatment of Alzheimer’s disease. Ann Pharmacother 45(11):1416–1424
Moore EM, Mander AG, Ames D, Kotowicz MA, Carne RP, Brodaty H et al (2013) Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care 36(10):2981–2987
Morris JK, Burns JM (2012) Insulin: an emerging treatment for Alzheimer’s disease dementia? Curr Neurol Neurosci Rep 12(5):520–527
Mostafa DK, Ismail CA, Ghareeb DA (2016) Differential metformin dose-dependent effects on cognition in rats: role of Akt. Psychopharmacology 233(13):2513–2524
Mrak RE, Griffin WST (2001) Interleukin-1, neuroinflammation, and Alzheimer’s disease. Neurobiol Aging 22(6):903–908
Nakaoku Y, Saito S, Yamamoto Y, Maki T, Takahashi R, Ihara M (2019) The dipeptidyl peptidase-4 inhibitor linagliptin ameliorates high-fat induced cognitive decline in tauopathy model mice. Int J Mol Sci 20(10):2539
Naranjo R et al (2018) Inhibition of the neuronal calcium sensor DREAM modulates presenilin-2 endoproteolysis. Front Mol Neurosci 11:449
Nicolakakis N, Aboulkassim T, Ongali B, Lecrux C, Fernandes P, Rosa-Neto P et al (2008) Complete rescue of cerebrovascular function in aged Alzheimer’s disease transgenic mice by antioxidants and pioglitazone, a peroxisome proliferator-activated receptor gamma agonist. J Neurosci 28(37):9287–9296
O’Reilly J-A, Lynch M (2012) Rosiglitazone improves spatial memory and decreases insoluble Aβ 1–42 in APP/PS1 mice. J Neuroimmune Pharmacol 7(1):140–144
Osborne C, West E, Nolan W, McHale-Owen H, Williams A, Bate C (2016) Glimepiride protects neurons against amyloid-β-induced synapse damage. Neuropharmacology 101:225–236
Ou Z, Kong X, Sun X, He X, Zhang L, Gong Z et al (2018) Metformin treatment prevents amyloid plaque deposition and memory impairment in APP/PS1 mice. Brain Behav Immun 69:351–363
Pandini G et al (2013) Insulin has multiple antiamyloidogenic effects on human neuronal cells. Endocrinology 154(1):375–387
Parthsarathy V, Hölscher C (2013) Chronic treatment with the GLP1 analogue liraglutide increases cell proliferation and differentiation into neurons in an AD mouse model. PLoS ONE 8(3):e58784
Patterson C (2018) World alzheimer report 2018
Pedersen WA, McMillan PJ, Kulstad JJ, Leverenz JB, Craft S, Haynatzki GR (2006) Rosiglitazone attenuates learning and memory deficits in Tg2576 Alzheimer mice. Exp Neurol 199(2):265–273
Pérez MJ, Quintanilla RA (2015) Therapeutic actions of the thiazolidinediones in alzheimer’s disease. PPAR Research 2015: 957248
Pipatpiboon N, Pintana H, Pratchayasakul W, Chattipakorn N, Chattipakorn SC (2013) DPP 4-inhibitor improves neuronal insulin receptor function, brain mitochondrial function and cognitive function in rats with insulin resistance induced by high-fat diet consumption. Eur J Neurosci 37(5):839–849
Prickaerts J et al (1999) Cognitive performance and biochemical markers in septum, hippocampus and striatum of rats after an icv injection of streptozotocin: a correlation analysis. Behav Brain Res 102(1–2):73–88
Principalli MA et al (2015) Kir6. 2 activation by sulfonylurea receptors: a different mechanism of action for SUR 1 and SUR 2A subunits via the same residues. Physiol Rep 3(9):e12533
Qi L, Ke L, Liu X, Liao L, Ke S, Liu X et al (2016) Subcutaneous administration of liraglutide ameliorates learning and memory impairment by modulating tau hyperphosphorylation via the glycogen synthase kinase-3β pathway in an amyloid β protein induced Alzheimer disease mouse model. Eur J Pharmacol 783:23–32
Qi LQ, Chen Z, Wang YP, Liu XY, Liu XH, Ke LF et al (2017) Subcutaneous liraglutide ameliorates methylglyoxal-induced alzheimer-like tau pathology and cognitive impairment by modulating tau hyperphosphorylation and glycogen synthase kinase-3β. Am J Transl Res 9(2):247–260
Ramos-Rodriguez JJ et al (2017) Progressive neuronal pathology and synaptic loss induced by prediabetes and type 2 diabetes in a mouse model of Alzheimer’s disease. Mol Neurobiol 54(5):3428–3438
Reger M, Watson G, Frey Ii W, Baker L, Cholerton B, Keeling M et al (2006) Effects of intranasal insulin on cognition in memory-impaired older adults: modulation by APOE genotype. Neurobiol Aging 27(3):451–458
Reger MA, Watson GS, Green PS, Wilkinson CW, Baker LD, Cholerton B et al (2008) Intranasal insulin improves cognition and modulates β-amyloid in early AD. Neurology 70(6):440–448
Rhea EM, Nirkhe S, Nguyen S, Pemberton S, Bammler TK, Beyer R et al (2019) Molecular mechanisms of intranasal insulin in SAMP8 mice. J Alzheimers Dis 71(4):1361–1373
Risner M, Saunders A, Altman J, Ormandy G, Craft S, Foley I et al (2006) Efficacy of rosiglitazone in a genetically defined population with mild-to-moderate Alzheimer’s disease. Pharmacogenomics J 6(4):246–254
Robinson A, Lubitz I, Atrakchi-Baranes D, Licht-Murava A, Katsel P, Leroith D et al (2019) Combination of insulin with a GLP1 agonist is associated with better memory and normal expression of insulin receptor pathway genes in a mouse model of Alzheimer’s disease. J Mol Neurosci 67(4):504–510
Röhrborn D et al (2015) DPP4 in diabetes. Front Immunol 6:386
Saad, MAEl-L et al (2019) Nateglinide exerts neuroprotective effects via downregulation of HIF-1α/TIM-3 inflammatory pathway and promotion of caveolin-1 expression in the rat’s hippocampus subjected to focal cerebral ischemia/reperfusion injury. Inflammation 43(2020):401–416
Salcedo I et al (2012) Neuroprotective and neurotrophic actions of glucagon-like peptide-1: an emerging opportunity to treat neurodegenerative and cerebrovascular disorders. Br J Pharmacol 166(5):1586–1599
Sa-nguanmoo P, Tanajak P, Kerdphoo S, Jaiwongkam T, Pratchayasakul W, Chattipakorn N et al (2017) SGLT2-inhibitor and DPP-4 inhibitor improve brain function via attenuating mitochondrial dysfunction, insulin resistance, inflammation, and apoptosis in HFD-induced obese rats. Toxicol Appl Pharmacol 333:43–50
Santiago MD et al (2019) Neuronal calcium sensor DREAM interactions with insulinotropic agent repaglinide. Biophys J 116(3):471a
Sato T, Hanyu H, Hirao K, Kanetaka H, Sakurai H, Iwamoto T (2011) Efficacy of PPAR-γ agonist pioglitazone in mild Alzheimer disease. Neurobiol Aging 32(9):1626–1633
Searcy JL, Phelps JT, Pancani T, Kadish I, Popovic J, Anderson KL et al (2012) Long-term pioglitazone treatment improves learning and attenuates pathological markers in a mouse model of Alzheimer’s disease. J Alzheimer’s Dis 30(4):943–961
Shaikh S et al (2016) Forxiga (dapagliflozin): Plausible role in the treatment of diabetes-associated neurological disorders. Biotechnol Appl Biochem 63(1):145–150
Shakil S (2017) Molecular Interaction of Anti-Diabetic Drugs With Acetylcholinesterase and Sodium Glucose Co-Transporter 2. J Cell Biochem 118(11):3855–3865
Shieh JC-C et al (2020) Alzheimer’s disease and diabetes: insulin signaling as the bridge linking two pathologies. Mol Neurobiol 57(4):1966–1977
Simard JM et al (2006) Newly expressed SUR1-regulated NC Ca-ATP channel mediates cerebral edema after ischemic stroke. Nat Med 12(4):433–440
Skeberdis VA et al (2001) Insulin promotes rapid delivery of N-methyl-D-aspartate receptors to the cell surface by exocytosis. Proc Natl Acad Sci 98(6):3561–3566
Solmaz V, Çınar BP, Yiğittürk G, Çavuşoğlu T, Taşkıran D, Erbaş O (2015) Exenatide reduces TNF-α expression and improves hippocampal neuron numbers and memory in streptozotocin treated rats. Eur J Pharmacol 765:482–487
Son SM, Shin H-J, Byun J, Kook SY, Moon M, Chang YJ et al (2016) Metformin facilitates amyloid-β generation by β-and γ-secretases via autophagy activation. J Alzheimers Dis 51(4):1197–1208
Stanciu GD et al (2020) Link between diabetes and Alzheimer’s disease due to the shared amyloid aggregation and deposition involving both neurodegenerative changes and neurovascular damages. J Clin Med 9(6):1713
Stoeckel LE, Arvanitakis Z, Gandy S, Small D, Kahn CR, Pascual-Leone A, Pawlyk A, Sherwin R, Smith P (2016) Complex mechanisms linking neurocognitive dysfunction to insulin resistance and other metabolic dysfunction. F1000Res 5:353. https://doi.org/10.12688/f1000research.8300.2
Syed MDR et al (2014) Invokana (Canagliflozin) as a dual inhibitor of acetylcholinesterase and sodium glucose Co-transporter 2: advancement in Alzheimer’s disease- diabetes type 2 linkage via an enzoinformatics study. CNS Neurol Disord - Drug Targets 13(3):447–451
Toba J et al (2016) PPARγ agonist pioglitazone improves cerebellar dysfunction at pre-Aβ deposition stage in APPswe/PS1dE9 Alzheimer’s disease model mice. Biochem Biophys Res Commun 473(4):1039–1044
Tokutake T et al (2012) Hyperphosphorylation of Tau induced by naturally secreted amyloid-β at nanomolar concentrations is modulated by insulin-dependent Akt-GSK3β signaling pathway. J Biol Chem 287(42):35222–35233
Toledo E, Inestrosa N (2010) Activation of Wnt signaling by lithium and rosiglitazone reduced spatial memory impairment and neurodegeneration in brains of an APPswe/PSEN1ΔE9 mouse model of Alzheimer’s disease. Mol Psychiatry 15(3):272–285
van Deijk ALF et al (2017) Astrocyte lipid metabolism is critical for synapse development and function in vivo. Glia 65(4):670–682
Vandal M, White PJ, Tremblay C, St-Amour I, Chevrier G, Emond V et al (2014) Insulin reverses the high-fat diet–induced increase in brain Aβ and improves memory in an animal model of Alzheimer disease. Diabetes 63(12):4291–4301
Wang X, Wang L, Xu Y, Yu Q, Li L, Guo Y (2016) Intranasal administration of Exendin-4 antagonizes Aβ31–35-induced disruption of circadian rhythm and impairment of learning and memory. Aging Clin Exp Res 28(6):1259–1266
Wang Y, Chen S, Xu Z, Chen S, Yao W, Gao X (2018) GLP-1 receptor agonists downregulate aberrant GnT-III expression in Alzheimer’s disease models through the Akt/GSK-3β/β-catenin signaling. Neuropharmacology 131:190–199
Watson GS et al (2005) Preserved cognition in patients with early Alzheimer disease and amnestic mild cognitive impairment during treatment with rosiglitazone: a preliminary study. Am J Geriatr Psychiatry 13(11):950–958
Watson KT, Wroolie TE, Tong G, Foland-Ross LC, Frangou S, Singh M et al (2019) Neural correlates of liraglutide effects in persons at risk for Alzheimer’s disease. Behav Brain Res 356:271–278
Weinstein G, Davis-Plourde KL, Conner S, Himali JJ, Beiser AS, Lee A et al (2019) Association of metformin, sulfonylurea and insulin use with brain structure and function and risk of dementia and Alzheimer’s disease: pooled analysis from 5 cohorts. PLoS ONE 14(2):e0212293
Wen L et al (2021) Effectiveness and safety of glibenclamide for stroke: protocol for a systematic review and meta-analysis. BMJ Open 11(5):e043585
Wiciński M et al (2020) Perspective of SGLT2 inhibition in treatment of conditions connected to neuronal loss: focus on alzheimer’s disease and Ischemia-related brain injury. Pharmaceuticals 13(11):379
Wu CY et al (2020) Relationships between memory decline and the use of metformin or DPP4 inhibitors in people with type 2 diabetes with normal cognition or Alzheimer’s disease, and the role APOE carrier status. Alzheimers Dement 16(12):1663–1673
Xiang GQ, Tang SS, Jiang LY, Hong H, Li Q, Wang C et al (2012) PPAR γ agonist pioglitazone improves scopolamine-induced memory impairment in mice. J Pharm Pharmacol 64(4):589–596
Xiao ZX et al (2017) Identification of repaglinide as a therapeutic drug for glioblastoma multiforme. Biochem Biophys Res Commun 488(1):33–39
Xiong H, Zheng C, Wang J, Song J, Zhao G, Shen H et al (2013) The Neuroprotection of Liraglutide on Alzheimer-Like Learning and Memory Impairment by Modulating the Hyperphosphorylation of Tau and Neurofilament Proteins and Insulin Signaling Pathways in Mice. J Alzheimers Dis 37:623–635
Xu W, Yang Y, Yuan G, Zhu W, Ma D, Hu S (2015) Exendin-4, a glucagon-like peptide-1 receptor agonist, reduces alzheimer disease-associated tau hyperphosphorylation in the hippocampus of rats with type 2 diabetes. J Investig Med 63(2):267–272
Xu F, Shen G, Su Z, He Z, Yuan L (2019) Glibenclamide ameliorates the disrupted blood–brain barrier in experimental intracerebral hemorrhage by inhibiting the activation of NLRP3 inflammasome. Brain and Behavior 9(4):e01254
Yang Y, Zhang J, Ma D, Zhang M, Hu S, Shao S et al (2013) Subcutaneous administration of liraglutide ameliorates Alzheimer-associated tau hyperphosphorylation in rats with type 2 diabetes. J Alzheimers Dis 37(3):637–648
Yang S, Chen Z, Cao M, Li R, Wang Z, Zhang M (2017) Pioglitazone ameliorates Aβ42 deposition in rats with diet-induced insulin resistance associated with AKT/GSK3β activation. Mol Med Rep 15(5):2588–2594
Yarchoan M et al (2014) Abnormal serine phosphorylation of insulin receptor substrate 1 is associated with tau pathology in Alzheimer’s disease and tauopathies. Acta Neuropathol 128(5):679–689
Yossef RR, Al-Yamany MF, Saad MA, El-Sahar AE (2020) Neuroprotective effects of vildagliptin on drug induced Alzheimer’s disease in rats with metabolic syndrome: role of hippocampal klotho and AKT signaling pathways. Eur J Pharmacol 889:173612
Zhang Y, Dai C-l, Chen Y, Iqbal K, Liu F, Gong C-X (2016) Intranasal insulin prevents anesthesia-induced spatial learning and memory deficit in mice. Sci Rep 6(1):1–10
Zhang Y, Xie J-Z, Xu X-Y, Hu J, Xu T, Jin S et al (2019) Liraglutide ameliorates hyperhomocysteinemia-induced Alzheimer-like pathology and memory deficits in rats via multi-molecular targeting. Neurosci Bull 35(4):724–734
Zhou J et al (2018) Metformin: an old drug with new applications. Int J Mol Sci 19(10):2863
Zhou M, Chen S, Peng P, Gu Z, Yu J, Zhao G et al (2019) Dulaglutide ameliorates STZ induced AD-like impairment of learning and memory ability by modulating hyperphosphorylation of tau and NFs through GSK3β. Biochem Biophys Res Commun 511(1):154–160
Zubov A, Muruzheva Z, Tikhomirova M, Karpenko M (2022) Glibenclamide as aneuroprotective antidementia drug, Archives of Physiology and Biochemistry, 128:6, 1693–1696. https://doi.org/10.1080/13813455.2020.1789170
Acknowledgements
The authors would like to thank Tehran University of Medical Sciences for kind supports.This project was financially supported by a grant from the Deputy of Research, Asadabad University of Medical Sciences.
Author information
Authors and Affiliations
Contributions
Golnaz goodarzi, Sadra Samavarchi Tehrani, and Saeed Ebrahimi Fana, Hemen moradi-sardareh: having participated in writing the article.
Mahmood Maniatie: edit the English language
Ghodratollah Panahi: completed the review and editing
Reza meshkani: supervised and approved the final manuscript
Corresponding author
Ethics declarations
Ethics approval and consent to participate
This article does not contain any studies with human participants or animals performed by any of the authors.
Consent for publication
All of the authors consent for publication.
Conflict of interests
The authors declare that there is no conflict of interests.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Goodarzi, G., Tehrani, S.S., Fana, S.E. et al. Crosstalk between Alzheimer’s disease and diabetes: a focus on anti-diabetic drugs. Metab Brain Dis 38, 1769–1800 (2023). https://doi.org/10.1007/s11011-023-01225-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s11011-023-01225-3