
Abstract
Background and Objective
Physiologically based pharmacokinetic (PBPK) models are valuable for translating in vitro absorption, distribution, metabolism, and excretion (ADME) data to predict clinical pharmacokinetics, and can enable discovery and early clinical stages of pharmaceutical research. However, in predicting pharmacokinetics of organic anion transporting polypeptide (OATP) 1B substrates based on in vitro transport and metabolism data, PBPK models typically require additional empirical in vitro-to-in vivo scaling factors (ESFs) in order to accurately recapitulate observed clinical profiles. As model simulation is very sensitive to ESFs, a critical evaluation of ESF estimation is prudent. Previously studies have applied classic ‘two-stage’ and ‘naïve pooled data’ approaches in identifying a set of compound independent ESFs. However, the ‘two-stage’ approach has the parameter identification issue in separately fitting data for individual compounds, while the ‘naïve pooled data’ approach ignores interstudy variability, leading to potentially biased ESF estimates.
Methods
In this study, we have applied a nonlinear mixed-effect approach in estimating ESF of the PBPK model and incorporated additional data from 86 runs of in vitro uptake assay and 49 clinical studies of 12 training compounds in model development to further enhance the translation of in vitro data to predict the pharmacokinetics of OATP1B substrate drugs. To test predication accuracy of the model, a ‘leave-one-out’ analysis has been performed.
Results
The established model can reasonably describe the clinical observations, with both mean values and interstudy variabilities quantified for ESF and volume of distribution parameters. The mean estimates are largely consistent with values in the previous reports. The interstudy variabilities of these parameters are estimated to be at least 50% (as coefficient of variation). Most compounds can be reasonably predicted in the ‘leave-one-out’ analysis.
Conclusion
This study improves the confidence in predicting the pharmacokinetics of OATP1B substrates in individual studies of small sample sizes, and quantifies the variability associated with the prediction.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
In predicting the pharmacokinetics of organic anion transporting polypeptide (OATP) 1B substrates based on in vitro data, physiologically based pharmacokinetic (PBPK) models typically require additional empirical scaling factors (ESFs) to accurately recapitulate observed clinical profiles. |
To identify ESFs by fitting clinical data of training compounds, ‘two-stage’ and ‘naïve pooled data’ approaches have been applied in previously published studies but may lead to biased estimates. |
This study applies a nonlinear mixed-effect approach in estimating ESFs of the PBPK model after incorporating additional training data from 86 runs of in vitro uptake assay and 49 clinical studies of 12 compounds in model development to further enhance the reliability of predictions and to quantify the variabilities associated with such predictions. |
1 Introduction
Acidic compounds of molecule weight above 400 g/mol represent a major class of small molecule therapies. According to the Extended Clearance Classification System (ECCS), organic anion transporting polypeptide (OATP) 1B-mediated hepatic uptake is commonly associated with their systemic clearance [1]. Albeit years of research for its critical role in drug discovery and development, predicting hepatic active uptake and pharmacokinetics of OATP substrates remains a challenging task. A gap that cannot be physiologically interpreted is usually found between in vitro predicted and clinically observed hepatic uptake clearances [2]. A few previous studies have suggested that such a gap is likely a result of different protein expression levels between in vitro and in vivo systems, hence after accounting for a relative expression factor (REF), in vivo hepatic uptake can be predicted with in vitro data [3, 4]. However, these studies usually only include a very limited number of testing compounds, hence it is unclear if the conclusions made in these studies can be generalized and applied to most other OATP substrates. Furthermore, quantitative proteomic studies suggest that expression levels of OATPs in the in vitro systems (e.g., primary human hepatocytes) are close to that in liver samples; thus, the protein expression differences do not solely explain the large gap between in vitro-predicted and in vivo-observed hepatic uptake clearances [2, 5].
A ‘middle-out’ approach has been previously proposed by estimating the empirical in vitro-to-in vivo scaling factors using clinical data of training compounds and applying the estimated scaling factors in predicting the pharmacokinetics of novel compounds [6, 7]. Briefly, Jones and colleagues estimated empirical scaling factors (ESFs) of seven OATP1B substrates by fitting each individual compound’s clinical data separately with a physiologically based pharmacokinetic (PBPK) model, and then taking the geometric mean of individual estimates as the scaling factor value for novel compounds [7]. However, with this classic ‘two-stage’ approach, the PBPK model can be overparameterized with limited data of individual compounds but many fitted parameters (i.e., scaling factors for hepatic active uptake, passive diffusion, metabolism, and biliary excretion). As such, the individual compound estimates and corresponding geometric mean of these estimates may not be reliable. Li and colleagues subsequently solved this issue with a ‘naïve pooled data’ approach, which simultaneously fits clinical data of all seven compounds [6]. However, this approach pools data from different clinical studies of different compounds as though they came from a single subject in one study of a single compound. It ignores potential interindividual variability by lumping it into residual errors (i.e., intraindividual variability). Essentially, now it is well known that the ‘naïve pooled data’ approach may produce imprecise estimates by improperly treating intra- and interindividual variability [8]. On the other hand, as the nonlinear mixed-effect approach that better handles variabilities is more likely to produce both accurate and precise estimates, it has been serving as the standard approach in modeling clinical pharmacokinetic data today. Arguably, most published PBPK models thus far are trained by mean clinical data digitized from publications. Hence, such models may not explicitly involve intersubject variability. However, given that the pharmacokinetic data included in PBPK model training are most likely generated from first-in-human studies of very small sample sizes, large interstudy variability is expected and cannot be ignored due to small cohorts of different subjects included in these studies. In this study, to further improve the translation of in vitro data of OATP substrates to the clinical pharmacokinetic profiles, we aimed to extend the previous PBPK evaluation to incorporate interstudy variability and apply the nonlinear mixed-effect approach in estimating scalars. To enable the nonlinear mixed-effect modeling, in vitro and clinical data from multiple in vitro uptake and in vivo pharmacokinetic studies of the training compounds are included in model building.
2 Methods
2.1 Determine In Vitro Transport Rates by Mechanistic Modeling of Sandwich-Cultured Human Hepatocyte Data with a Nonlinear Mixed-Effect Approach
Historical in-house sandwich culture human hepatocyte (SCHH) data of 12 OATP substrates are included. Some SCHH data have been previously published [7]. Although pitavastatin and telmisartan have been tested in an SCHH assay, they are excluded from the PBPK modeling due to the challenge in modeling conversion between lactone form and parent compound for pitavastatin [9], and de-glucuronidation of metabolite and associated enterohepatic recycling for pitavastatin and telmisartan [10]. Including pitavastatin and telmisartan but not including their specific disposition mechanisms involving enterohepatic recycling of metabolites will likely lead to model misspecification, while incorporating additional disposition mechanisms into the current model will likely result in overparameterization due to limited data. An experimental procedure has previously been introduced [7]. The structural model for analyzing data is a slightly modified version of the previously published model [7]. Briefly, the model (Eqs. 1–4) includes three compartments representing extracellular media (EC), intracellular space (IC), and bile canaliculi (BC). V, C, and t represents volume, concentration, and time. CL and fu represent clearance and free fraction. In the assay condition with calcium (CA), compound is secreted into the bile canaliculi through CLbile,SCHH. In the assay condition without calcium, the compound is secreted into the extracellular media through CLbile,SCHH. CA is set to 1 in the presence of calcium and zero in the absence of calcium. In the assay condition with rifamycin SV, both active processes (i.e., CLuptake,SCHH and CLbile,SCHH) are assumed to be zero [11,12,13]. Metabolism and basolateral efflux are assumed to be zero to reduce the number of floating parameters, and can be justified due to the short incubation time in SCHH studies.
When fitting data from the SCHH assay, we note that the intracellular accumulation at time zero is typically greater than zero. We assume that this phenomenon is due to a non-specific binding to the cell surface, which is proportional to the concentration of hepatocytes in the media. In fitting SCHH data, the intracellular accumulation includes both intracellular amount due to transport and intracellular binding, as well as the amount on the cell surface, which is calculated as the product of the amount in media and extracellular bound fraction. Extracellular unbound fraction was modeled with Eq. (4) (see the derivation in the electronic supplementary material [ESM]), where ASCHH presents the amount of protein per well determined in the experiment, while two empirical binding association constants, Ka,EC,1 and Ka,EC,2, are included for binding in the absence and presence of rifamycin SV.
RSV is set to 1 in the presence of rifamycin SV and zero in the absence of rifamycin SV. VEC,SCHH is determined experimentally. VIC,SCHH is calculated as the product of ASCHH, the number of cells per milligram of protein in the well (2.5 million per milligram incubation protein), and the intracellular volume of a million cells (0.0026 mL). CLuptake,SCHH, CLpassive,SCHH, CLbile,SCHH, fu,IC,SCHH, Ka,EC,1, and Ka,EC,2 are estimated by fitting experimental data. For a single compound, each run of the SCHH experiment is considered as an individual. Interrun variabilities associated with all fitted parameters (except for fu,IC,SCHH) are estimated together with their corresponding population means, assuming distributions of individual values follow log normal distributions. fu,IC,SCHH is assumed to be the same across different runs. The first-order conditional estimation (FOCE) approach is applied in parameter estimation, while the log-likelihood profiling (PsN-Toolkit 5.2.6) is performed to approximate 95% confidence intervals [14]. As CLbile,SCHH cannot be estimated with confidence for a few compounds, three models are tested, including the full model, the model with CLbile,SCHH but without variability associated with CLbile,SCHH, and the model without CLbile,SCHH. The model with minimal Akaike information criterion (AIC) is chosen as the final model. All the mathematical models presented in this article are implemented in NONMEM® 7.5 (ICON plc, Dublin, Ireland) with proportional error model.
2.2 Determine In Vitro Intrinsic Metabolic Rate Based on Human Hepatocyte Stability Assay
In vitro apparent metabolic rates (CLapp,HHEP) and the corresponding hepatocyte to buffer partitioning coefficient (KP,HHEP) have been determined by the human hepatocyte (HHEP) stability assay in a previous study [15]. The CLapp,HHEP generated from this assay is likely confounded by the residual transporter activity in the suspended hepatocytes, which requires KP,HHEP determined in the same assay to correct for. The intrinsic metabolic clearance (CLint,HHEP) [Eq. 5, ESM Table S1) is calculated as previously published [15], where fu,IC,HHEP is approximated with fu,IC,SCHH.
VEC,HHEP and VIC,HHEP are the total incubation (0.9987 mL) and intracellular (0.0013 mL) volumes of the suspension hepatocyte assay, respectively.
2.3 Physiologically Based Pharmacokinetic (PBPK) Modeling of Organic Anion Transporting Polypeptide (OATP) Substrates
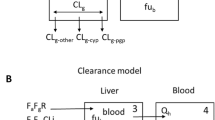
A reduced PBPK model has been developed (schematic diagram presented in Fig. 1). In the model, systemic plasma (P) and hepatic extracellular space (EC,H) are connected by hepatic blood flow (QH) [Eq. 6]. RB/P represents blood-to-plasma ratio. A full PBPK model explicitly incorporating non-liver tissues was not considered in this study because such a model requires accurate values for tissue-to-plasma partitioning coefficients (KP). KP values for PBPK models are typically predicted with mechanistic models, in silico models, or approximated with KP experimentally determined in preclinical species or in vitro assays [16]. When fixed at inaccurate values, KP of non-liver tissues are likely to confound estimates of hepatic clearance processes and corresponding in vitro to in vivo scaling factors. As such, we lump non-liver tissue distribution into a single peripheral compartment (Eq. 7), with its volume (VPERI) and distribution clearance (CLD) estimated by fitting clinical data. Renal clearance (CLR) is from the literature (ESM Table S1).

Physiologically based pharmacokinetic model structure. Black arrows are blood flows and distribution clearance. Between EC and IC, blue double-headed arrows are passive diffusion clearance and dark red single-headed arrows are active uptake clearance. Red arrows represent metabolic clearance, green arrows represent biliary excretion and bile flow, purple arrow represents renal clearance, and the light orange single-headed arrows represent mass transfer within empirical absorption compartments. EC extracellular, IC intracellular
Oral absorption is modeled as follows (Eqs. 8, 9), where kt and ka represent the empirical mass transfer rate and absorption rate in the gastrointestinal tract, and kbile is an empirical biliary flow rate for modeling enterohepatic recycling.
The liver distribution in this model is described as permeability limited with five sequential segments as previously described [7]. Every segment includes one pair of extracellular (EC) and intracellular (IC) subcompartments. There is active hepatic uptake clearance (CLuptake,H) and passive diffusion clearance (CLpassive,H) between the extracellular and intracellular subcompartments, and metabolism (CLmetabolism,H) and biliary excretion (CLbile,H) within the intracellular subcompartments (Eqs. 10–13).
In the above equations, i presents the second to the fifth segment, and fu,p and fu,IC,H represent plasma free fraction in plasma and hepatic intracellular space. The values of RB/P and fu,p have been previously determined (ESM Table S1), and fu,IC,H is approximated with fu,IC,SCHH. QH, VP, VEC,H, and VIC,H are set to the products of body weight and previously published values of 1.225 L/h per kilogram of body weight—0.04145, 0.004676, and 0.01931 L per kilogram of body weight [17]. The body weights are from original clinical studies where pharmacokinetic data are digitized, or are assumed to be 70 kg if not reported.
An empirical biliary compartment is included to enable enterohepatic recycling (Eq. 14).
In vivo CLuptake,H, CLpassive,H, and CLmetabolism,H are connected with in vitro CLuptake,SCHH, CLpassive,SCHH, CLint,HHEP with both physiological scaling factors (i.e., 120 million cells per gram of liver tissue [18]) and empirical scaling factors (ESF) [Eqs. 15–18].
Three ESFs, VPREI, CLD, ka,2, kt,1, and kt,2 for seven OATP substrates are estimated simultaneously by fitting clinical data of all compounds. The ka,1 value is not identifiable and is hence fixed at zero. Interstudy variabilities are estimated for all fitted parameters, assuming distributions of individual values follow log normal distributions. In total, forty-nine clinical studies (66 groups of different participants) are included in the current analysis. Given that all clinical data included in this study are digitized from previous publications (ESM Table S2) where results were presented as mean exposures, only interstudy, but not intersubject, variability can be accessed. All data from the same group of participants are treated as data from a single individual. The Importance Sampling approach (IMP method in NONMEM®) is applied in estimating population mean and variability. The 95% confidence intervals are approximated by the Markov Chain Monte Carlo approach for accessing uncertainty of estimates (BAYES method in NONMEM®), but not the estimated covariance matrix considering that confidence regions are irregular for nonlinear multiparameter PBPK models [6]. As the current study focuses on predicting hepatic clearance processes, oral absorption in population predictions is simulated with individually fitted values, so that the prediction is not confounded by improperly assumed values for absorption parameters.
To test predication accuracy of the model, a ‘leave-one-out’ analysis has been performed as previously described by predicting the pharmacokinetic profile of one individual compound each time it is taken out of the training set [6], although with interstudy variability incorporated into the current prediction. Median prediction and 95% intervals for testing compounds are generated based on 2000 simulated studies, with mean and interstudy variability estimated using training compounds.
3 Results
3.1 Modeling In Vitro Sandwich Culture Human Hepatocyte Data
The proposed model structure can reasonably describe the data generated from SCHH studies (ESM Fig. S1). Although the mean CLuptake,SCHH can be precisely determined, relatively large interrun variability has been observed for a few compounds (Table 1), highlighting the importance of generating data from repeated runs to be used as reliable inputs for PBPK modeling. The CLbile,SCHH values can be confidently estimated for most compounds, except for cerivastatin, glyburide, PF-05089771, and repaglinide, for which biliary excretions in PBPK modeling are fixed at zero.
3.2 PBPK Modeling of 12 OATP Substrates
The proposed PBPK model structure is robust enough to describe all pharmacokinetic data included in the current analysis (individual fits in Fig. 2 and ESM Fig. S2). Albeit variabilities in the in vitro and clinical studies, population simulations still reasonably match the data for most compounds (Fig. 2 and ESM Fig. S2). The estimated mean values of ESFs (Table 2) are largely consistent with previous estimates [6]. The estimated interstudy variabilities (expressed as coefficients of variation) are > 50% for these parameters. Large uncertainties are seen for two biliary parameters, potentially because of insufficient training data for biliary excretion. As to estimates for individual compounds and individual studies, except for repaglinide and PF-05241328, it is difficult to see an apparent correlation between estimated values and compounds (Fig. 3). PF-05241328 data for analysis are only available from two groups in a single study. Hence, we cannot rule out the possibility that the estimates for PF-05241328 are just outliers that do not represent population means. In the ‘leave-one-out’ analysis, it is hard to identify a singular influencer (Table 3). Most compounds can be reasonably predicted, with the 95% prediction intervals covering the observations (Fig. 4).

Examples of observed and simulated clinical pharmacokinetic time-course profiles of OATP substrates. Black circles and lines represent observations and individual fittings, and red lines represent population simulations. Additional data and simulations are given in electronic supplementary material Fig. S2. OATP organic anion transporting polypeptide, IV intravenous, PO oral

Estimated parameter values of individual studies by the nonlinear mixed-effect modeling

Leave-one-out analysis. The compounds shown in the titles of subplots are the testing compounds not included in modeling training. Black circles represent observations, and solid lines and grey bands represent the 50, 2.5, and 97.5% percentiles of 2000 simulations. IV intravenous, PO oral
4 Discussion
As gaps are frequently seen between in vitro scaled versus in vivo observed hepatic clearance for OATP1B substrates, a set of ESFs are usually required for PBPK models to describe clinical pharmacokinetics. The source of ESF is unknown and is not explored in this study, but could be a result of low in vitro protein expression, low in vitro transporter activity, in vitro experimental conditions inconsistent with in vivo conditions, a misspecified structural PBPK model that cannot accurately describe in vivo transporter-mediated processes, and/or other unknown key physiological processes not captured by the PBPK model [2, 19]. For instance, there are several recent reports debating the potential ‘albumin-mediated uptake’ to recover the in vitro to in vivo disconnect in predicting transporter-mediated hepatic clearance [20,21,22,23]. Given some knowledge gaps, translation of transporter data based on current in vitro assays has been a challenge, and thus identifying ESF values will be the center of building a PBPK model for predicting transporter-mediated clearance. ESFs are initially included as compound-dependent parameters in an effort to build ‘top-down’ PBPK models for OATP substrates [2]. Using compound-dependent ESF may substantially reduce the PBPK model’s prediction ability for novel compounds in drug discovery as discussed previously [2, 6]. To address this issue, compound-independent ESFs are developed in a ‘middle-out’ modeling approach by fitting clinical pharmacokinetic data of multiple OATP substrates with ‘two-stage’ and ‘naïve pooled data’ approaches [6, 7]. The ‘two-stage’ approach can be limiting due to parameter identification issue in fitting individual compounds, while the ‘naïve pooled data’ approach cannot capture variability and will likely lead to bias in estimating the population mean of ESF across multiple studies and multiple compounds [8]. In addition, previous studies [6, 7] only used data from a limited number of in vitro and clinical studies for global ESF estimation. Considering relatively large interstudy variability observed, data from additional studies may be required to increase the reliability of ESF estimates. To further improve the ‘middle-out’ PBPK model for OATP substrates, in this study we applied the nonlinear mixed-effect approach in estimating values of both in vitro transport parameters and in vivo PBPK parameters. Coupled with the nonlinear mixed-effect approach, a large amount of in vitro SCHH data (3–16 replicates per compound) and clinical pharmacokinetic data from multiple independent studies were also included in the current study.
In this study, relatively large interstudy variability is noted for most estimated parameters of the in vitro SCHH and in vivo PBPK models. This highlights the importance of including data from multiple studies for unbiased parameter estimation in building ‘middle-out’ models. By checking distinct PBPK parameter values for individual studies estimated in the nonlinear mixed-effect modeling (Fig. 3), we note that most compounds share similar ESF values, and that the interstudy variabilities for parameters are more likely due to participants of different physiological profiles included in clinical studies of small sample sizes, quality of in vitro and digitized clinical data, and potentially a parameter identifiability issue in fitting a PBPK model to data from individual studies, but less likely due to an ‘intrinsic’ difference in ESF values for different compounds. In other words, the variability seen among different studies of different compounds is likely an interstudy variability but not an intercompound variability. The observation supports the use of multiple training compounds to identify a set of uniform ESFs in predicting the pharmacokinetics of novel OATP substrates. By incorporating quantified parameter variability into PBPK prediction (Fig. 4), it is now possible to know how well the pharmacokinetics from individual studies of small sample sizes can be predicted. This can be critical in designing an optimal dosing regimen in first-in-human studies. The prediction intervals tend to become wider towards the end of the study, indicating less confidence in predicting low concentrations in the terminal phase of the plasma concentration-time profiles. However, for compounds with efficacy and adverse effects likely driven by high concentrations, low confident predictions at a low concentration range may be less concerning (see the leave-one-out analysis result plotted in normal scale in ESM Fig. S3). Due to the empirical nature of ESF, it is challenging to identify the source of variability, although it could be a result of polymorphism of OATPs and other hepatic uptake transporters [24]. Although hepatic uptake of all training compounds in the current study are mediated mainly through OATP1B, other transporters may also contribute to this process to various degrees, which may require scaling values different from OATP1B, and potentially result in differences in ESF values observed among different compounds. Variabilities in fixed parameters (e.g., hepatic blood flow) are also lumped into the variabilities of ESF and other fitted parameters.
Although deviations between data and population simulations are typically expected for nonlinear mixed-effect modeling simply due to interindividual variability (interstudy variability in this analysis), such deviations for bosentan, glyburide, PF-05241328, and repaglinide are larger than deviations for other training compounds (ESM Fig. S2). Similarly, bosentan, repaglinide, and PF-05241328 are not well-predicted in the leave-one-out analysis (Fig. 4), potentially due to disposition mechanisms specific for these compounds that are not well captured either in the current PBPK model structure or by SCHH assay. Although in this study we have tried to incorporate as many SCHH runs as possible and have estimated values of SCHH parameters with a nonlinear mixed-effect modeling approach, the estimated in vitro transport clearances may still deviate from ‘true’ population means given the high variability and relatively small sample size of SCHH runs. The potentially biased in vitro inputs may contribute to deviations between PBPK fitting and clinical observations. Bosentan is known for its nonlinear pharmacokinetics and potentially saturated clearance at high concentration [25], however this may not be the reason for underpredictions as the individual fitting assuming linear processes can describe the data (i.e., the bosentan pharmacokinetic data included in the current analysis are still in the linear range). The PBPK model overpredicts plasma concentrations of repaglinide and PF-05241328 in humans. The difficulty in capturing active uptake of repaglinide by in vitro assay has been previously documented [26], hence it is possible that the SCHH assay underpredicts its active uptake in the current study. As for PF-05241328, in-house data from an intravenous (1 mg/kg) bile duct cannulated rat study suggests that approximately 72% dosing amount is excreted as unchanged parent in rat bile over 6 h. However, in the current PBPK model, biliary secretion for PF-05241328 is not sufficiently high based on SCHH input (Table 2, ESM Fig. S1). Hence, it is likely that the current PBPK model does not capture the biliary secretion of PF-05241328 and thus projected a lower overall systemic clearance. For the majority of other compounds, the model predictions align with the clinical pharmacokinetics observed across multiple studies.
A reduced PBPK model is employed in the current study instead of a full-body PBPK model. A full-body PBPK model requires tissue-to-plasma partition coefficients (KP) for all non-liver tissues, which are typically predicted by mechanistic models based on physiochemical properties of the compounds [27]. However, it has been previously noted that predicted KP may not reliably represent compound distribution into non-liver tissues [10, 16]. The estimated ESFs for hepatic clearance will be biased if KP for non-liver tissues are fixed at misspecified values. Additional scalers can be applied to correct the predicted KP values, however the use of compound-dependent empirical scalers makes the physiological distribution model a complicated empirical distribution model. As such, we decided to use the simple empirical distribution model to reduce the computational burden in data fitting. The estimated VPERI and CLD are largely consistent across different compounds (Fig. 3) and can be used in predicting novel OATP1B substrates, typically seen with ECCS class 1B/3B drugs (Fig. 4). The estimated mean VPERI value of 0.114 L/kg is also consistent with current understanding of volume of distribution for acids [28]. For any future compound with a non-liver tissue distribution significantly different from the training compounds, the current empirical distribution model can be easily replaced with a physiological model, an animal-based model, or any other distribution model without changing liver-related equations and parameters.
In the current model, a five-segment liver is applied following previous publications [6, 7]. The five-segment model that approximates the dispersion model is typically required for in vitro to in vivo scaling of compounds with high hepatic extraction ratios [29]. Considering that most training compounds in the current study may not have extremely high hepatic extraction ratios, the liver model with segments less than five may also be able to describe the training data. However, estimated ESF values are specific to the liver model. Using the five-segment model enables the prediction of the pharmacokinetics of compounds with high hepatic extraction in future without re-estimating ESF, or incorporating such compounds into the training set without changing the existing model structure. All floating parameters in the current PBPK model are estimated with interstudy variability, to avoid making an arbitrary assumption that the observed variability in pharmacokinetic data is only due to variabilities in some parameters but not others. However, given that most literature pharmacokinetic data included in this study are following oral dosing, the variabilities in absorption parameters may not be precisely separated from the variabilities associated with hepatic clearance parameters and distribution parameters. This may confound the estimated values of ESF. As more training compounds with intravenous data are likely incorporated into this modeling platform in future, part of the oral data may be removed from the training set by then. Although values of absorption parameters are also estimated in the fitting process (ESM Table S4), we do not recommend using these values to predict oral absorption of any future OATP substrates, as the values of empirical absorption parameters estimated in this study may not be universally applicable to other OATP substrates.
In Table 1, the fu,IC,SCHH value for pravastatin is estimated to be > 1. In the current SCHH model and similar previously published models [7], the fu,IC,SCHH value is essentially determined as the ratio of unbound media concentration to total intracellular concentration at equilibrium in the absence of active transport. Hence, its value is affected by all factors that contribute to intracellular concentration. These factors include intracellular binding, pH partitioning, and permeation of ionized and neutral molecules [30]. Although intrinsic unbound fraction due to intracellular binding is bounded by 1, fu,IC,SCHH is not necessarily < 1. We prefer using fu,IC,SCHH over intrinsic unbound fraction in SCHH and PBPK models because it implicitly incorporates factors contributing to intracellular concentration without explicitly modeling complicated processes of pH partitioning and permeation of various species of different ionization status.
5 Conclusion
By incorporating in vitro SCHH data of repeated runs and clinical pharmacokinetic data from multiple studies, we have applied the nonlinear mixed-effect approach in estimating ESFs and other parameters in a PBPK framework for OATP1B substrates. The new method presented here provides a less biased and more reliable approach in translating in vitro transporter data into clinical pharmacokinetic predictions.
References
Varma MV, El-Kattan AF, Feng B, Steyn SJ, Maurer TS, Scott DO, et al. Extended Clearance Classification System (ECCS) informed approach for evaluating investigational drugs as substrates of drug transporters. Clin Pharmacol Ther. 2017;102(1):33–6.
Li R, Barton HA, Varma MV. Prediction of pharmacokinetics and drug-drug interactions when hepatic transporters are involved. Clin Pharmacokinet. 2014;53(8):659–78.
Yin M, Balhara A, Marie S, Tournier N, Gaborik Z, Unadkat JD. Successful prediction of human hepatic concentrations of transported drugs using the proteomics-informed relative expression factor approach. Clin Pharmacol Ther. 2024;115(3):595–605.
Bosgra S, van de Steeg E, Vlaming ML, Verhoeckx KC, Huisman MT, Verwei M, et al. Predicting carrier-mediated hepatic disposition of rosuvastatin in man by scaling from individual transfected cell-lines in vitro using absolute transporter protein quantification and PBPK modeling. Eur J Pharm Sci. 2014;65:156–66.
Alluri RV, Li R, Varma MVS. Transporter-enzyme interplay and the hepatic drug clearance: what have we learned so far? Expert Opin Drug Metab Toxicol. 2020;16(5):387–401.
Li R, Barton HA, Yates PD, Ghosh A, Wolford AC, Riccardi KA, et al. A “middle-out” approach to human pharmacokinetic predictions for OATP substrates using physiologically-based pharmacokinetic modeling. J Pharmacokinet Pharmacodyn. 2014;41(3):197–209.
Jones HM, Barton HA, Lai Y, Bi YA, Kimoto E, Kempshall S, et al. Mechanistic pharmacokinetic modeling for the prediction of transporter-mediated disposition in humans from sandwich culture human hepatocyte data. Drug Metab Dispos. 2012;40(5):1007–17.
Sheiner LB, Beal SL. Evaluation of methods for estimating population pharmacokinetics parameters. I. Michaelis–Menten model: routine clinical pharmacokinetic data. J Pharmacokinet Biopharm. 1980;8(6):553–71.
Fujino H, Yamada I, Shimada S, Yoneda M, Kojima J. Metabolic fate of pitavastatin, a new inhibitor of HMG-CoA reductase: human UDP-glucuronosyltransferase enzymes involved in lactonization. Xenobiotica. 2003;33(1):27–41.
Li R, Ghosh A, Maurer TS, Kimoto E, Barton HA. Physiologically based pharmacokinetic prediction of telmisartan in human. Drug Metab Dispos. 2014;42(10):1646–55.
Bi YA, Costales C, Mathialagan S, West M, Eatemadpour S, Lazzaro S, et al. Quantitative contribution of six major transporters to the hepatic uptake of drugs: “SLC-Phenotyping” using primary human hepatocytes. J Pharmacol Exp Ther. 2019;370(1):72–83.
US Food and Drug Administration Center for Drug Evaluation and Research. NDA 210910 multi-discipline review. US FDA; 2018.
Morgan RE, van Staden CJ, Chen Y, Kalyanaraman N, Kalanzi J, Dunn RT 2nd, et al. A multifactorial approach to hepatobiliary transporter assessment enables improved therapeutic compound development. Toxicol Sci. 2013;136(1):216–41.
Lindbom L, Pihlgren P, Jonsson EN. PsN-Toolkit—a collection of computer intensive statistical methods for non-linear mixed effect modeling using NONMEM. Comput Methods Progr Biomed. 2005;79(3):241–57.
Riccardi KA, Tess DA, Lin J, Patel R, Ryu S, Atkinson K, et al. A novel unified approach to predict human hepatic clearance for both enzyme- and transporter-mediated mechanisms using suspended human hepatocytes. Drug Metab Dispos. 2019;47(5):484–92.
Mathew S, Tess D, Burchett W, Chang G, Woody N, Keefer C, et al. Evaluation of prediction accuracy for volume of distribution in rat and human using in vitro, in vivo, PBPK and QSAR methods. J Pharm Sci. 2021;110(4):1799–823.
Shah DK, Betts AM. Towards a platform PBPK model to characterize the plasma and tissue disposition of monoclonal antibodies in preclinical species and human. J Pharmacokinet Pharmacodyn. 2012;39(1):67–86.
Bayliss MK, Bell JA, Jenner WN, Park GR, Wilson K. Utility of hepatocytes to model species differences in the metabolism of loxtidine and to predict pharmacokinetic parameters in rat, dog and man. Xenobiotica. 1999;29(3):253–68.
Zamek-Gliszczynski MJ, Lee CA, Poirier A, Bentz J, Chu X, Ellens H, et al. ITC recommendations for transporter kinetic parameter estimation and translational modeling of transport-mediated PK and DDIs in humans. Clin Pharmacol Ther. 2013;94(1):64–79.
Kim SJ, Lee KR, Miyauchi S, Sugiyama Y. Extrapolation of in vivo hepatic clearance from in vitro uptake clearance by suspended human hepatocytes for anionic drugs with high binding to human albumin: improvement of in vitro-to-in vivo extrapolation by considering the “Albumin-Mediated” hepatic uptake mechanism on the basis of the “Facilitated-Dissociation Model.” Drug Metab Dispos. 2019;47(2):94–103.
Li N, Badrinarayanan A, Ishida K, Li X, Roberts J, Wang S, et al. Albumin-mediated uptake improves human clearance prediction for hepatic uptake transporter substrates aiding a mechanistic in vitro-in vivo extrapolation (IVIVE) strategy in discovery research. AAPS J. 2020;23(1):1.
Bi YA, Ryu S, Tess DA, Rodrigues AD, Varma MVS. Effect of human plasma on hepatic uptake of organic anion-transporting polypeptide 1B substrates: studies using transfected cells and primary human hepatocytes. Drug Metab Dispos. 2021;49(1):72–83.
Yin M, Storelli F, Unadkat JD. Is the protein-mediated uptake of drugs by organic anion transporting polypeptides a real phenomenon or an artifact? Drug Metab Dispos. 2022;50(9):1132–41.
Li R, Barton HA, Maurer TS. Toward prospective prediction of pharmacokinetics in OATP1B1 genetic variant populations. CPT Pharmacometr Syst Pharmacol. 2014;3(12): e151.
Weber C, Schmitt R, Birnboeck H, Hopfgartner G, van Marle SP, Peeters PA, et al. Pharmacokinetics and pharmacodynamics of the endothelin-receptor antagonist bosentan in healthy human subjects. Clin Pharmacol Ther. 1996;60(2):124–37.
Kalliokoski A, Niemi M. Impact of OATP transporters on pharmacokinetics. Br J Pharmacol. 2009;158(3):693–705.
Rodgers T, Rowland M. Physiologically based pharmacokinetic modelling 2: predicting the tissue distribution of acids, very weak bases, neutrals and zwitterions. J Pharm Sci. 2006;95(6):1238–57.
Di L, Kerns EH. Chapter 19—Pharmacokinetics. In: Di L, Kerns EH, editors. Drug-like properties. 2nd ed. Boston: Academic Press; 2016. p. 267–81.
Watanabe T, Kusuhara H, Maeda K, Shitara Y, Sugiyama Y. Physiologically based pharmacokinetic modeling to predict transporter-mediated clearance and distribution of pravastatin in humans. J Pharmacol Exp Ther. 2009;328(2):652–62.
Chien HC, Zur AA, Maurer TS, Yee SW, Tolsma J, Jasper P, et al. Rapid method to determine intracellular drug concentrations in cellular uptake assays: application to metformin in organic cation transporter 1-transfected human embryonic kidney 293 cells. Drug Metab Dispos. 2016;44(3):356–64.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
This research was funded by Pfizer Inc.
Conflict of interest
Rui Li, Emi Kimoto, Yi-An Bi, David Tess, and Manthena V.S. Varma were employees of Pfizer, Inc. at the time this research was performed.
Availability of data
All data used in the analysis are either obtained from the literature (as cited) or included in the ESM.
Code availability
NONMEM code is included in the ESM.
Ethics approval
Not applicable.
Consent to participate
Not applicable.
Consent for publication
Not applicable.
Author contributions
All authors contributed to the study conception and design. The in vitro studies were performed by EK and YB, and the modeling was performed by RL. The first draft of the manuscript was written by RL and the manuscript was reviewed and edited by EK, MV, and RL. All authors reviewed and approved the final manuscript.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Li, R., Kimoto, E., Bi, YA. et al. Physiologically Based Pharmacokinetic Model of OATP1B Substrates with a Nonlinear Mixed Effect Approach: Estimating Empirical In Vitro-to-In Vivo Scaling Factors. Clin Pharmacokinet 63, 1177–1189 (2024). https://doi.org/10.1007/s40262-024-01408-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-024-01408-w