
Abstract
Oral anticoagulants and antiplatelet drugs are commonly prescribed to lower the risk of cardiovascular diseases, such as venous and arterial thrombosis, which represent the leading causes of mortality worldwide. A significant percentage of patients taking antithrombotics will nevertheless experience bleeding or recurrent ischemic events, and this represents a major public health issue. Cardiovascular medicine is now questioning the one-size-fits-all policy, and more personalized approaches are increasingly being considered. However, the available tools are currently limited and they are only moderately able to predict clinical events or have a significant impact on clinical outcomes. Predicting concentrations of antithrombotics in blood could be an effective means of personalization as they have been associated with bleeding and recurrent ischemia. Target concentration interventions could take advantage of physiologically based pharmacokinetic (PBPK) and population-based pharmacokinetic (POPPK) models, which are increasingly used in clinical settings and have attracted the interest of governmental regulatory agencies, to propose dosages adapted to specific population characteristics. These models have the benefit of combining parameters from different sources, such as experimental in vitro data and patients’ demographic, genetic, and physiological in vivo data, to characterize the dose–concentration relationships of compounds of interest. As such, they can be used to predict individual drug exposure. In the near future, these models could therefore be a valuable means of predicting personalized antithrombotic blood concentrations and, hopefully, of preventing clinical non-response or bleeding in a given patient. Existing approaches for personalization of antithrombotic prescriptions will be reviewed using practical examples for P2Y12 inhibitors and direct oral anticoagulants. The review will additionally focus on the existing PBPK and POPPK models for these two categories of drugs. Lastly, we address potential scenarios for their implementation in clinics, along with the main limitations and challenges.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Current approaches for the personalization of antithrombotics (biological, genetic, and clinical approaches) have shown mixed results to date. |
Pharmacokinetic predictive models can be a valuable means of predicting antithrombotic blood concentrations and of preventing clinical non-response or bleeding in a given patient. |
There are important hurdles that need to be considered for the implementation of such models in clinical practice in the near future. |
1 Introduction
Oral anticoagulants and antiplatelet drugs are commonly used alone or in combination for various indications to reduce the risk of thrombotic events [1]. Antithrombotics are required for the vast majority of cardiovascular diseases. Together, these diseases are the leading cause of mortality worldwide, with a continuous increase in their relative importance, not only in high- or middle-income countries but also in lower-middle-income countries [2]. Despite recent major advances in antithrombotic management for cardiovascular patients, one challenging issue facing many current therapies is that their clinical efficacy can vary significantly between patients for the same standardized dosage [3,4,5]. Major trials and national registries have shown that approximately 9–10% of patients under dual antiplatelet therapy (DAPT) and 1–3% of patients under direct oral anticoagulants (DOACs) still experienced thrombotic events after 1 year [5,6,7,8,9,10,11,12,13,14]. Some patients will also experience significant bleeding events, at rates ranging from 2% for DAPT to 4% per year for DOACs, according to randomized clinical trials (RCTs) and national registries [14, 15]. These percentages may be considered relatively low, but, from a population viewpoint, they represent a major public health issue. The antiplatelet drug class of P2Y12 inhibitors and anticoagulants are indeed among the most common causes of emergency department visits for drug-related adverse events (AEs; mostly bleeding) in the US [16, 17]. Unfortunately, existing tools are only moderately able to predict clinical events or have a significant impact on clinical outcomes [18,19,20,21]. Clinicians thus refer to medical history, patient’s clinical characteristics and clinical judgment to adapt therapeutic strategy, which can be challenging and hazardous [22]. In this context, the concept of precision (or personalized or individualized) medicine has been gaining ground in recent years, including via support from new European and American governmental initiatives on drug personalization [23,24,25]. Precision medicine refers to a medical model using a characterization of individual patients’ phenotypes and genotypes to tailor the right adapted therapeutic strategy and find the best benefit–risk balance [24]. Existing approaches for personalization of antithrombotic prescriptions are already available and will be reviewed using practical examples for P2Y12 inhibitors and DOAC drugs. The review will additionally focus on the physiologically based pharmacokinetic (PBPK) and population-based pharmacokinetic (POPPK) models that are promising and emergent approaches for the personalization of antithrombotics. Indeed, these models could help in identifying the sources of variability influencing drug exposure and clinical response, and could also be put to use in tools tailoring treatments to patients according to experimental in vitro data and patients’ demographic, genetic, and physiological in vivo data. We suggest here the validation of these models in clinical settings could provide clinicians with an important and original means of prescribing antithrombotics more safely and efficiently.
2 Current Personalized Approaches for P2Y12 Inhibitors
2.1 Platelet Reactivity
The guidelines from the American College of Cardiology Foundation/American Heart Association (ACCF/AHA, 2016) and the European Society of Cardiology/European Association for Cardio-Thoracic Surgery (ESC/EACTS, 2018) recommend starting DAPT with a P2Y12 inhibitor and aspirin for acute coronary syndromes (ACS) and then ideally continuing for up to 1 year, whether or not patients undergo a percutaneous coronary intervention (PCI) [8, 26]. Despite major improvement in ACS management, DAPT is associated with an increased risk of bleeding [27,28,29]. However, approved dosages by the European Medicines Agency (EMA) are strongly limited and cannot be reduced for vulnerable patients (e.g. over- or underweight patients, elderly, and chronic liver or kidney diseases), with the exception of prasugrel (5 mg once daily for patients with a body weight < 60 kg) but clinical data supporting its use are lacking (Table 1) [30,31,32].
As bleeding and ischemic events are associated with worse short- and long-term outcomes, attempts have been made to tailor antiplatelet strategy according to an individual’s biological response to P2Y12 inhibitors [33, 34]. Interindividual variability of the biological response to P2Y12 inhibitors is well established, particularly for clopidogrel, as 16–50% of patients are deemed poor responders to treatment (high on-treatment platelet reactivity [HTPR]), depending on the test used for assessing platelet reactivity (PR) [35]. There is a similar interindividual variability in PR for ticagrelor and prasugrel that is a serious concern for patients with a low or very low on-treatment platelet reactivity (LTPR) phenotype with respect to their bleeding risk [36, 37].
Several factors influence PR of patients treated with clopidogrel. The genetic variants, such as cytochrome P450 (CYP)2C19*2 that influence clopidogrel’s bioactivation and lead to a loss-of-function phenotype, were found to be involved in HTPR [38] and associated with the risk of recurrent ischemic events, but not in a consistent manner across studies [39]. On the other hand, carriers of the CYP2C19*17 allele more frequently have a low on-clopidogrel reactivity phenotype and more often develop bleeding complications [40, 41]. In addition, type 2 diabetes mellitus, chronic kidney disease, age (> 65 years), C-reactive protein, body weight, body mass index, and left ventricular function are some of the non-genetic factors that increase PR and reduce platelet response to clopidogrel [42,43,44,45,46,47,48,49]. Importantly, meta-analyses mainly based on observational studies have found a positive relationship between HTPR and subsequent thrombotic/ischemic events, particularly in cases of ACS and/or after PCI [50,51,52]. On the contrary, LTPR increases the risk of bleeding events [51].
Intervention trials of antiplatelet regimens tailored to the biological PR phenotype have so far provided contrasting results and may depend on the patient’s level of cardiovascular risk and the clinical setting [53,54,55,56,57,58,59]. To date, the ESC guidelines do not recommend PR-guided therapy [26].
2.2 De-escalation
Prasugrel and ticagrelor provide more extensive platelet inhibition and are less susceptible to genetic variation and drug–drug interactions than clopidogrel [36, 37, 60,61,62]. No genetic variants have yet been associated with a clinical outcome for ticagrelor or prasugrel [61,62,63]. Similarly to clopidogrel, prasugrel is a prodrug whose formation to its active metabolite via a two-step process initiated by plasma esterases and followed by a single cytochrome-dependent step (CYP3A4/5 and CYP2B6) is more efficient and less variable than clopidogrel [60, 64,65,66]. Ticagrelor is mainly metabolized by CYP3A4/5, and both parent and metabolite exhibit antiplatelet activity [66]. Superiority of prasugrel and ticagrelor on ischemic outcome over clopidogrel was originally established in two large, multicentric RCTs [28, 29]. However, prasugrel and ticagrelor were associated with an increased risk of major bleeding and non-coronary artery bypass graft surgery major bleeding, respectively, compared with clopidogrel [7, 8].
Since PR is higher in the early phases of ACS, and generally decreases quickly within days, strategies based on strong antiplatelet treatment in the acute phase of ACS followed by de-escalation to a less potent antiplatelet drug in the maintenance phase have been evaluated in recent promising RCTs [67]. In the TROPICAL-ACS platelet function therapy (PFT)-guided de-escalation RCT, ACS patients (n = 2610) managed with PCI and initially treated with prasugrel were switched to clopidogrel after 7 days. Their PR was then tested and poor responders to clopidogrel were switched back to prasugrel, while clopidogrel was continued in good responders. The non-inferior primary endpoint (cardiovascular death, myocardial infarction [MI], stroke, or bleeding [Bleeding Academic Research Consortium ≥ 2]) was achieved with a similar rate of combined ischemic and major bleeding events after 1 year in both groups (p for non-inferiority = 0.0004; hazard ratio 0.81, 95% confidence interval 0.62–1.06; p for superiority = 0.12) [68]. The trial met its primary endpoint as it demonstrated non-inferiority for a net clinical benefit but did not show a benefit for patients on bleeding rates. A smaller, open-label, monocentric, and unguided de-escalation randomized trial showed a benefit in terms of bleeding event rates [69]. In addition, questions regarding safety remain as the study was not powered for ischemic events alone [70].
Another RCT has evaluated the safety and efficacy of PFT-guided therapy in ACS patients aged > 75 years (n = 877) [71]. Ischemic and bleeding rates were similar in the group receiving a standard dose of prasugrel (5 mg/day) versus PFT-guided escalation (prasugrel 10 mg/day) or de-escalation (clopidogrel 75 mg/day) [71]. This outcome confirms an age effect also observed in a subgroup analysis from the TROPICAL-ACS trial as the youngest patients (< 70 years of age) seemed to benefit the most from PFT-guided therapy [72]. Of importance, two ongoing trials are aiming to assess a genotype-based guided therapy (Table 2), even if subgroup analysis from the TROPICAL-ACS trial has recently failed to show the benefit of such a strategy [73]. Finally, rapid de-escalation to ticagrelor monotherapy was recently assessed in a large, multicenter, open-label RCT, but the results do not support any changes in current practice [74].
It is too early yet to conclude about the clinical utility of de-escalation, but it could be considered in specific scenarios, such as patients with high bleeding risk, as suggested by the last ESC guidelines and the results from the TROPICAL-ACS trial [26].
2.3 Value of Predictive Scores in Dual Antiplatelet Therapy
There is debate on the duration of DAPT, which may range from less than 6 months to more than 12 months [75,76,77,78,79,80]. Several predictive scores for long-term outcome were developed in the context of a need for clinicians to find the optimal duration of DAPT [19,20,21, 80], such as the PRECISE-DAPT and DAPT scores [20, 21, 26]. In patients with a score equal or superior to 2 in the DAPT trial, a reduction in the risk of thrombotic events after a prolonged 30-month DAPT was observed [20]. The increased risk of bleeding did not mitigate this reduction. The PRECISE-DAPT score suggests a shorter duration of DAPT (3–6 months) in patients at risk of bleeding (scores ≥ 25) [21]. Among these scores, it is of note that bleeding and thrombosis share several risk factors, making assessment of the balance between ischemic and bleeding risks very challenging for clinicians. The resulting C-statistics are only moderately able to predict clinical events depending on the externally validated cohort (from 0.64 to 0.70) [19,20,21]. C-statistic corresponds to the area under the receiver operating curves for diagnostic or prognostic tests and is a measure of discrimination, ranging from perfect (1 or 100%) to no better than chance (≤ 0.5 or 50%) [81]. Consequently, the C-statistic can be interpreted as the probability that a randomly chosen subject from the disease group has a higher predicted probability of having the disease than a randomly chosen subject from the disease-free group [81]. In addition, the clinical impacts of these risk-prediction models have never been assessed as part of a clinical decision-making strategy in a prospective RCT [7]. Their value in improving patient outcomes remains unproven.
2.4 Drug Monitoring
Data on the relationship between drug exposure-efficacy and safety events are scarce for P2Y12 inhibitors. Indeed, direct serum concentrations are not routinely used due to technical reasons [1]. Instead, vasodilator-stimulated phosphoprotein (VASP) assay or platelet-mediated aggregation of fibrinogen-coated polystyrene beads (VerifyNow®) are used to test PR, and correlate well with drug concentrations [82]. Among the few studies on the association between drug exposure and clinical events, a population pharmacokinetic/pharmacodynamic (PK/PD) study of 4426 patients treated with ticagrelor (within the PEGASUS-TIMI 54 trial) showed that the predicted risk of cardiovascular death/MI/stroke decreased with increasing ticagrelor exposure [83]. However, the relationship was relatively flat, indicating that a near-maximal response had already been achieved within the lower exposure range studied. Similarly, the predicted risk of major bleeding increased with increasing ticagrelor exposure, but again the relationship was relatively flat [83]. Limitations of this study included that blood samples were only collected from approximately one-third of patients and that average steady-state concentrations of ticagrelor were unavailable at low concentrations. Extrapolations outside the predicted exposure range should be interpreted with caution [83].
In conclusion, the different biological, clinical, and genetic approaches to personalizing P2Y12 inhibitors have shown mixed results to date (summarized in Table 3). No single factor can explain the observed biological variability in antiplatelet response, and predictive scores based on clinical data alone are only moderately able to predict clinical events since the risk factors for bleeding and ischemic events overlap. There is thus a need for further research to find other relevant co-factors and then build models, ideally, combining different genetic, clinical, and biological approaches.
3 Current Personalized Approaches Involving Direct Oral Anticoagulants
3.1 Dosage Adaptation According to Patients’ Clinical Characteristics
Unlike vitamin K antagonists (VKAs), DOACs have specific targets: rivaroxaban, apixaban, and edoxaban directly and specifically inhibit factor Xa, whereas dabigatran directly inhibits thrombin [84]. Based on large RCTs of patients with non-valvular atrial fibrillation (AF), DOACs were associated with similar or lower rates of ischemic stroke and major bleeding as warfarin [6]. Thus, European and American guidelines now recommend DOACs over VKAs in the vast majority of patients with non-valvular AF [85, 86]. The efficacy and safety of DOACs for the treatment of deep vein thrombosis and pulmonary embolism were compared with VKAs in six, large, phase III trials that consistently showed the non-inferiority of DOACs with regard to recurrent venous thromboembolism (VTE) and a lower risk of clinically relevant bleeding [87]. DOACs are therefore also recommended as the first-line anticoagulant treatment for VTE [88].
Different dosages of DOACs have been approved by the EMA and can be adapted according to clinical characteristics, such as renal clearance, age, body weight, and drug–drug interaction (DDI) (Table 4) [89,90,91,92]. In real-life situations, a patient can cumulate several comorbidities and medication, and it is thus currently difficult for clinicians to adjust the dosage accordingly or decide whether to start anticoagulant therapy. The recent results from the ORBIT-AF II registry highlights this difficulty since half of the patients receiving a reduced off-label dosage of DOACs have a tendency (not statistically significant) for an unfavorable clinical outcome [22].
In addition, there is a lack of robust clinical data to support DOAC prescribing in some categories of patients excluded from clinical trials, such patients at extremes of body weights [93], and patients with advanced chronic hepatic or kidney disease [94, 95]. Moreover, DOACs are associated with a higher risk profile of abnormal uterine bleeding in VTE patients (in particular Xa inhibitors, but not dabigatran) [96] and gastrointestinal bleeding (for rivaroxaban, high-dose dabigatran and edoxaban) [97]. Altogether, these evidences suggest that the current standardized dosages for DOACs do not fit all categories of patients.
3.2 Drug Monitoring
Contrary to VKAs such as warfarin, DOACs do not routinely require dose adjustment and monitoring because of their more favorable PK profile and a larger therapeutic window [98]. However, significant interindividual variability in DOAC concentrations has been observed in both RCTs and real-life settings [99]. In a multicenter study including 330 patients, drug concentrations varied by more than 20-fold among patients treated with dabigatran, by nearly 15-fold with rivaroxaban, and by 7-fold with apixaban [100]. Several clinical factors can explain this variability, such as renal and hepatic impairment, body weight, age, ethnicity, DDI involving P-glycoprotein, and CYP3A4/5 induction or inhibition [99]. Genetic variations may also be a factor but this has scarcely been investigated [101,102,103].
DOAC blood concentration is thus increasingly considered interesting since it could help to discriminate between ischemic and bleeding risk events due to this larger-than-expected variability in drug concentrations in real-life settings [104]. In addition, there is accumulating evidence that blood concentrations of DOACs are associated with major or life-threatening bleeding, as well as ischemic stroke or systemic embolism [104]. This has been shown in secondary analyses of major RCTs. Some evidence has been published, such as for dabigatran, but, for other DOACs, most of the data are only found in drug registration documents provided to the US FDA by pharmaceutical companies [105,106,107,108]. For instance, across the 10th to 90th percentile range of steady-state trough plasma concentrations, achieved with a twice-daily dose of dabigatran 150 mg, the overall risk of major bleeding during the trial ranged from approximately 2–7% for a typical 72-year-old AF patient known to have prior stroke and diabetes; this was a clinically relevant variability [108]. An inverse relationship also exists for thromboembolic events, but it is less pronounced [108]. Edoxaban also exhibits robust concentration-dependent relationships with both ischemic stroke and life-threatening/fatal bleeds [105]. An exposure–efficacy relationship was studied in a subset of patients treated with apixaban (n = 2932) whose exposure data were available in the ARISTOTLE trial. As opposed to dabigatran and edoxaban, the probability of ischemic stroke was independent of exposure to apixaban at the dose level studied. This lack of association may yet be limited by the narrow exposure range and the small number of ischemic stroke events in the PK subset (n = 27). The probability of major bleeding was found to increase with increased exposure to apixaban. In patients treated with apixaban 5 mg twice daily, a twofold increase in drug exposure increased the probability of major bleeding within 1 year, from 1.79 to 3.11% [107]. The relationship between drug exposure and clinical events has not been analyzed for rivaroxaban, but prothrombin time (PT) has been used instead [106] since it is correlated to rivaroxaban blood concentrations. As for apixaban, PT data from 7008 patients in the ROCKET per-protocol analysis dataset demonstrated that the occurrence of ischemic stroke was independent of PT in the 10–30 s range [106]; however, the risk of major bleeding increased with PT. Interestingly, the relationship between prolonged PT and major bleeding was clearly exacerbated (by at least 50%) in patients taking concomitant aspirin [106]. A recent paper including 565 patients with AF (the START laboratory registry) also showed a relationship between DOAC concentrations and clinical events in real-life settings [109]. During the 1-year follow-up, all the thromboembolic complications occurred in patients whose minimal drug concentrations were in the lowest quartile interval calculated for each drug. This study’s size was limited and will have to be confirmed in future larger-scale studies.
To date, no trial has compared the results of DOAC therapy with or without drug monitoring, and there are no guidelines on the steps to follow to improve the quality of anticoagulation therapy [98]. Current recommendations from the International Society on Thrombosis and Haemostasis on measuring the anticoagulant effects of DOACs include specific scenarios such as bleeding or before an unplanned surgery or invasive procedure [98].
3.3 Value of Predictive Scores for Anticoagulants
Balancing the individual risk of thrombotic events and bleeding has always been, and remains, challenging. Scores for evaluating the risk of cardiac embolism in AF patients have been developed to help clinicians decide whether to initiate oral anticoagulation therapy. Since anticoagulation is associated with a bleeding risk, the benefit of anticoagulation should exceed this risk. The CHA2DS2-VASc and CHADS2 scores are the most frequently used risk models, and they consider various recognized predictors such as hypertension, diabetes, age, sex, history of stroke, vasculopathy, and cardiac insufficiency [110, 111]. The CHA2DS2-VASc score is preferred for its ability to recategorize low-risk patients into a risk group for which anticoagulation is recommended [112]. It is important to note that these models have a limited ability to predict risk since the C-statistics are, at best, 0.67 and 0.69 for the CHA2DS2-VASc and CHADS2 scores, respectively [18]. Part of the risk spectrum is thus not covered by the score. The recent P2-CHA2DS2-VASc score, which includes abnormal p-wave axis, has shown a significant improvement in ischemic stroke prediction in AF but has not yet been implemented in clinical routine [113]. The risk of bleeding for AF patients is assessed using three main scores, among which the HAS-BLED is the most popular [114,115,116]. Unfortunately, and as is the case for platelet inhibitors, the individual components of these scores are similar to the individual components of the CHA2DS2-VASc and CHADS2 scores (hypertension, age, previous stroke, diabetes). It is thus not surprising that higher bleeding risk is found among patients with higher thrombotic risks [117]. This overlap makes clinical decisions harder since patients with a high risk of AF are often also at a high risk of bleeding. Most of these scores have been derived and/or validated in patients taking VKAs, but not DOACs. The real-world performance of these scores for DOACs is low since C-statistics at 1-year follow-up for ATRIA and HAS-BLED are approximately 0.59 [118]. Highlighting these limitations, the ESC does not recommend a particular bleeding score [85].
As observed for antiplatelet drugs, there are only limited approaches for personalizing DOACs (summarized in Table 5). In addition, the approved dosages do not cover some categories of patients. An individualized prediction of DOAC exposure could represent an interesting option to better identify these higher-risk patients as it appears to be a reliable means of predicting the probabilities of clinical events.
4 Are Pharmacokinetic (PK) Modeling Approaches Promising for Antithrombotic Personalization?
As discussed above, there is currently no reliable tool to help clinicians predict whether a given patient, in a given clinical setting, is receiving the drug and/or dose that will optimally manage the risk of under- or overdosing dependent on that individual’s risk profile. Because antithrombotic blood concentrations have been associated with clinical events, predicting their concentrations would represent a convenient means of identifying probable non-responder patients or patients at high risk of bleeding, but without the need for blood sampling and monitoring.
Blood concentration prediction could be achieved by taking advantage of PK modeling such as POPPK approaches and recent advances in PBPK modeling [119].
4.1 Population-Based PK Models
POPPK allows for identifying and quantifying factors affecting drug disposition, such as demographic, pathophysiological, environmental, or genetic factors. POPPK is widely used for dosage optimization and constitutes the basis of Bayesian-based therapeutic drug monitoring [120,121,122,123]. POPPK is now increasingly use for drug optimization in various areas, including infectious disease [124], and has been prospectively validated to optimize peak and trough concentrations of amikacin in neonates [125]. Several studies have demonstrated the population approach’s potential for optimizing VKA dosing based on concentrations or biomarker monitoring tools [126, 127]. A mechanism-based decision support tool was proposed for warfarin dose adjustment before starting therapy by predicting the most probable warfarin dose to reach an optimal international normalized ratio (INR), or during treatment to guide dose adjustment [126]. This population PK/PD (POPPK/PD)-based tool performed well in both adults and children by predicting dose per day and per week, as well as the corresponding INR prediction. In another POPPK/PD-based study, a nomogram was developed for warfarin dose adjustment at the initiation phase based on CYP2C9 and VKORC1 polymorphisms, at a maintenance phase based on genetic and clinical factors, and previous INR data [127]. Using in silico clinical trial simulations with this model, a therapeutic INR was reached in a population of diverse ethnic and genetic groups within 1 week. These two studies showed that by shortening the period to reach a stable INR with warfarin, and by reducing the interindividual variability, these models could participate in improving patients’ clinical outcomes, but this remains to be challenged in prospective studies. POPPK models have been used to characterize the PK profile of all DOACs marketed to date in different populations [128,129,130,131,132,133,134]. These POPPK models have been used for various purposes: to describe renal influence on PKs; to quantify bleeding risk using exposure–response analysis; and, more recently, for dosage optimization. Nonetheless, validated POPPK models for P2Y12 inhibitors are needed [135]. Indeed, the lack of information on blood concentrations for this class of drug is a limitation for model-based dosage optimization.
4.2 Physiologically-Based PK Models
PBPK models rely on a physiologically realistic compartmental structure into which input parameters from different sources (e.g. in vitro studies, intrinsic properties of the compound of interest, and demographic data such as ethnicity, age, clearance organ function, body weight or body mass index, polymedication, and genetics) are combined to predict plasma concentration–time profiles [136, 137]. This so-called bottom–up approach—the model is built from first principles, literature, or in vitro data—is classically opposite to the top–down approach, where all parameters are estimated from in vivo data [138]. PBPK does not exclude the use of in vivo data via a top–down approach, such as POPPK, and both approaches are increasingly seen as complementary [139]. PBPK models successfully predicted relevant DDI or PK profiles in populations at risk, such as patients with renal insufficiencies or children [136, 140]. At least 20 approved drugs have used PBPK simulations in regulatory agency submissions, including submissions to the FDA [136, 140]. A recent review in Europe showed that pharmaceutical companies had submitted 67 procedures including one or more PBPK models to the EMA [141]. The growing trend of using PBPK models in drug submissions to regulators, for a variety of purposes, led the EMA and FDA to publish specific guidelines in 2017 for their use in support of regulatory approval [142, 143].
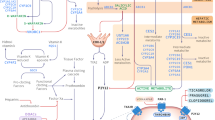
PBPK models have already been built for antithrombotics but few have been clinically validated. For instance, a PBPK model for ticagrelor has been clinically validated in a DDI context involving HIV patients taking antiretroviral drugs boosted with ritonavir or cobicistat. Using a simulated interaction between ticagrelor 180 mg and ritonavir 100 mg, a lower dose (25% of the regular dose) of ticagrelor was predicted to obtain the same PK and platelet inhibition as ticagrelor administered alone in human volunteers. This PBPK model could be used prospectively to broaden the use of ticagrelor in patients with HIV treated using ritonavir, regardless of the CYP3A4/5 inhibition (Fig. 1) [144]. Another recent PBPK absorption model was able to predict the PK profile of prasugrel’s active metabolite in the presence of a proton pump inhibitor [145]. For DOACs, existing models are related to rivaroxaban. For instance, a DDI model of patients with various degrees of renal impairment and who were receiving a regimen of rivaroxaban with CYP/efflux transporter inhibitors was constructed using a PBPK model. This model predicted a clinically significant increase in rivaroxaban exposure, such that FDA reviewers recognized the potential effects of concurrent renal impairment and the use of a moderate/strong CYP3A4/5 inhibitor on rivaroxaban exposure that had not been evaluated by the applicant [136]. Another PBPK model was successfully developed to predict rivaroxaban’s PK profile at different doses in healthy subjects and patients with hepatic or renal dysfunction [146]. Promising results outside the field of antithrombotics have shown, as a proof of concept, that a PBPK model works in a real-life setting [147]. Polasek et al. successfully predicted olanzapine drug exposure in 14 patients using a PBPK approach. This result, obtained from standardized conditions of clinical trials, is really encouraging to test PBPK models in real conditions.

A practical example of the successful development and validation of a PBPK model in healthy volunteers. Development of a PBPK model requires a bottom-up procedure relying on a physiologically realistic compartmental structure into which input parameters from different sources are combined to predict plasma concentration–time profiles. System component includes parameters related to human physiology; drug-dependent parameters include properties related to the drug itself; extrinsic factors include environmental parameters such as DDI and toxic exposure; and intrinsic factors include genetics, sex, or disease. Marsousi et al. [144] created a model like this to simulate the interaction between ticagrelor 180 mg and ritonavir 100 mg. In doing so, it was calculated that a lower dose of ticagrelor, when coadministered with ritonavir, could result in the same PK and platelet inhibition as ticagrelor administered alone. A clinical study was then conducted in healthy volunteers. The model successfully predicted the observed PK profiles of ticagrelor and its active metabolite. Adapted from Zhao et al. [136], Marsousi et al. [144] and Darwich et al. [148]. PBPK physiologically based pharmacokinetic, DDI drug–drug interaction, PK pharmacokinetics
4.3 Limitations of PK Models
There remain important limitations to the implementation of such models. The first is the lack of available models for some compounds and the need to obtain the critical, but not necessarily easily available, data from several (academic and industrial) sources in order to build the models. In addition, models must be externally validated since there is little evidence of their use and impact on a large scale in clinical care settings [147, 148]. Evidence-based efficacy and cost-benefit analyses are also pivotal to seeing broader implementation [148]. Finally, PBPK and POPPK modeling requires strong interdisciplinary coordination between clinical pharmacologists, physicians, academic and industrial researchers, and patient groups, which does not occur widely [148].
The challenge is especially great in cardiovascular medicine since patients often have multiple comorbidities and comedications, which increase a model’s complexity. However, as discussed above, very few options offer the possibility of integrating so many parameters from different sources at the same time. An ideal model would be based on all the available information about the patient and the disease they are being treated for, comorbid diseases, medication they are receiving, and cytochrome genotyping and phenotyping as these become increasingly available [148]. In the future, an ideal tool could help clinicians prospectively manage and identify patients at risk of bleeding or thrombosis, and it could be implemented on individual electronic patient record systems. However, dose adjustment seems to be more challenging since dosages are limited to those tested in clinical trials and provided by pharmaceutical companies, and are based on clinical characteristics (body weight, age, and renal clearance), not drug concentrations [104]. However, this could represent an interesting option, as a recent model suggested that a dose reduction of rivaroxaban could reduce bleeding-associated events and mortality [134].
In the meantime, another approach would be to allow the identification of antithrombotics that are associated with unacceptably high rates of patient bleeding or stroke (e.g. 90th or 10th percentile of the drug exposure distribution), while considering the available population data for antithrombotic blood concentrations (Fig. 2); the clinician would then select the antithrombotic with the best efficacy–safety profile for a given patient. Given that the costs for CYP phenotyping and genotyping are decreasing very fast and would require no more than the available information of individual patients (e.g. standard laboratory data, drug information from admission notes), the cost of this informed antithrombotic selection approach is expected to be low. Ideally, the model would need to be dynamic and improvable over time, with permanent feedback between predicted and observed results.

Possible implementation of a PBPK model for antithrombotic blood concentration predictions. A validated PBPK prediction model would allow for a reliable prediction of blood concentration for each antithrombotic in a given patient according to her or his individual characteristics and comedication. This would enable selection of the most appropriate DOAC and its dosing regimen depending on the patient’s clinical risk profiles for thrombosis and bleeding. In this example, DOAC n°4 would be the most appropriate for the patient with specific concerns on the risk of bleeding as his or her predicted concentration would be situated in the 70th percentile (green tick) of population distribution of real concentrations for this same molecule. The other predicted concentrations would place the patient at higher risk for bleeding as they would be situated at the highest extremes of the population distribution (red crosses). DOAC direct oral anticoagulant, PBPK physiologically based pharmacokinetic
5 Complementary Approaches: Clinical Decision Support and Alerts
Other complementary approaches to the risk management of antithrombotic AEs based on clinical decision support systems have been developed [149, 150] and implemented within hospital information systems [151, 152]. Automated detection of potential AEs may prove useful and is less labor-intensive and faster [150]. However, automated detection of AEs generates many false-positive alerts, targets inappropriate prescriptions instead of clinically relevant AEs, and considers neither the type of hospital or unit (e.g. medical, surgical) nor the patients’ medical characteristics [149, 150]. Owing to their limitations and to the amount of structured and unstructured information contained in electronic medical records, new AE detection and monitoring systems are currently being developed based on multiple data sources and methods involving structured data mining and natural language processing [153,154,155,156], which, in the latter case, although being strongly language dependent, has demonstrated its power to support AE detection. The specificity of alerts will soon improve; notifications will be prioritized and will offer detailed advice. These decision support systems are heading towards patient-centered decision support, but the most important research question remains as to whether they will be able to improve patient outcomes rather than just processes [151].
6 Conclusions
Despite the recent advances of cardiovascular medicine in reducing the risk of bleeding and thrombosis, and thanks to the development of new compounds such as DOACs and P2Y12 inhibitors, some individuals still cannot benefit from these agents because these individuals do not fit the standardized patient profiles. The absence of clinical response or the AEs associated with this category of patients represents a challenging public health issue. Current approaches for the personalization of antithrombotics (biological, genetic, and clinical approaches) have shown mixed results to date. In parallel with the real need to improve our knowledge regarding the different co-factors influencing treatment response, PK predictive models represent a new approach to antithrombotic therapies that is ready to be tested in clinical settings. Successfully implementing such models would help clinicians and patients to share clinical decision making thanks to reliable information on the benefits and risks of various personalized treatment strategies.
References
Sibbing D, Spannagl M. Direct oral anticoagulants and antiplatelet agents. Clinical relevance and options for laboratory testing. Hamostaseologie. 2014;34(1):78–84.
World Health Organization. The top 10 causes of death. 2017 (updated). https://www.who.int/news-room/fact-sheets/detail/the-top-10-causes-of-death. Accessed 1 Jun 2019.
Houser SR. The American Heart Association’s New Institute for Precision Cardiovascular Medicine. Circulation. 2016;134(24):1913–4.
Lee CW, Ahn JM, Park DW, Kang SJ, Lee SW, Kim YH, et al. Optimal duration of dual antiplatelet therapy after drug-eluting stent implantation: a randomized, controlled trial. Circulation. 2014;129(3):304–12.
Larsen TB, Skjoth F, Nielsen PB, Kjaeldgaard JN, Lip GY. Comparative effectiveness and safety of non-VKA oral anticoagulants and warfarin in patients with atrial fibrillation: propensity weighted nationwide cohort study. BMJ. 2016;353:i3189.
Ntaios G, Papavasileiou V, Diener HC, Makaritsis K, Michel P. Nonvitamin-K-antagonist oral anticoagulants in patients with atrial fibrillation and previous stroke or transient ischemic attack: a systematic review and meta-analysis of randomized controlled trials. Stroke. 2012;43(12):3298–304.
Valgimigli M, Bueno H, Byrne RA, Collet JP, Costa F, Jeppsson A, et al. 2017 ESC focused update on dual antiplatelet therapy in coronary artery disease developed in collaboration with EACTS: the Task Force for dual antiplatelet therapy in coronary artery disease of the European Society of Cardiology (ESC) and of the European Association for Cardio-Thoracic Surgery (EACTS). Eur Heart J. 2018;39(3):213–60.
Levine GN, Bates ER, Bittl JA, Brindis RG, Fihn SD, Fleisher LA, et al. 2016 ACC/AHA guideline focused update on duration of dual antiplatelet therapy in patients with coronary artery disease: a report of the American College of Cardiology/American Heart Association Task Force on clinical practice guidelines: an update of the 2011 ACCF/AHA/SCAI Guideline for Percutaneous Coronary Intervention, 2011 ACCF/AHA Guideline for Coronary Artery Bypass Graft Surgery, 2012 ACC/AHA/ACP/AATS/PCNA/SCAI/STS Guideline for the diagnosis and management of patients with stable ischemic heart disease, 2013 ACCF/AHA guideline for the management of ST-elevation myocardial infarction, 2014 AHA/ACC guideline for the management of patients with non-ST-elevation acute coronary syndromes, and 2014 ACC/AHA guideline on perioperative cardiovascular evaluation and management of patients undergoing noncardiac surgery. Circulation. 2016;134(10):e123–55.
Lee CH, Cheng CL, Kao Yang YH, Chao TH, Chen JY, Li YH. Cardiovascular and bleeding risks in acute myocardial infarction newly treated with ticagrelor vs. clopidogrel in Taiwan. Circ J. 2018;82(3):747–56.
Zeymer U, Widimsky P, Danchin N, Lettino M, Bardaji A, Barrabes JA, et al. P2Y12 receptor inhibitors in patients with non-ST-elevation acute coronary syndrome in the real world: use, patient selection, and outcomes from contemporary European registries. Eur Heart J Cardiovasc Pharmacother. 2016;2(4):229–43.
Alexopoulos D, Xanthopoulou I, Deftereos S, Hamilos M, Sitafidis G, Kanakakis I, et al. Contemporary antiplatelet treatment in acute coronary syndrome patients undergoing percutaneous coronary intervention: 1-year outcomes from the GReek AntiPlatElet (GRAPE) Registry. J Thromb Haemost. 2016;14(6):1146–54.
Ako J, Morino Y, Okuizumi K, Usami M, Nakamura M. Japanese postmarketing surveillance of clopidogrel in patients with non-ST-segment elevation acute coronary syndrome, stable angina, old myocardial infarction, and ST-segment elevation myocardial infarction after percutaneous coronary intervention in a real-life setting: the final report (J-PLACE Final). Cardiovasc Interv Ther. 2016;31(2):101–13.
Beyer-Westendorf J, Forster K, Pannach S, Ebertz F, Gelbricht V, Thieme C, et al. Rates, management, and outcome of rivaroxaban bleeding in daily care: results from the Dresden NOAC registry. Blood. 2014;124(6):955–62.
Graham DJ, Reichman ME, Wernecke M, Hsueh YH, Izem R, Southworth MR, et al. Stroke, bleeding, and mortality risks in elderly medicare beneficiaries treated with dabigatran or rivaroxaban for nonvalvular atrial fibrillation. JAMA Intern Med. 2016;176(11):1662–71.
Sahlen A, Varenhorst C, Lagerqvist B, Renlund H, Omerovic E, Erlinge D, et al. Outcomes in patients treated with ticagrelor or clopidogrel after acute myocardial infarction: experiences from SWEDEHEART registry. Eur Heart J. 2016;37(44):3335–42.
Shehab N, Lovegrove MC, Geller AI, Rose KO, Weidle NJ, Budnitz DS. US emergency department visits for outpatient adverse drug events, 2013–2014. JAMA. 2016;316(20):2115–25.
Shehab N, Sperling LS, Kegler SR, Budnitz DS. National estimates of emergency department visits for hemorrhage-related adverse events from clopidogrel plus aspirin and from warfarin. Arch Intern Med. 2010;170(21):1926–33.
Van Staa TP, Setakis E, Di Tanna GL, Lane DA, Lip GY. A comparison of risk stratification schemes for stroke in 79,884 atrial fibrillation patients in general practice. J Thromb Haemost. 2011;9(1):39–48.
Matteau A, Yeh RW, Camenzind E, Steg PG, Wijns W, Mills J, et al. Balancing long-term risks of ischemic and bleeding complications after percutaneous coronary intervention with drug-eluting stents. Am J Cardiol. 2015;116(5):686–93.
Yeh RW, Secemsky EA, Kereiakes DJ, Normand SL, Gershlick AH, Cohen DJ, et al. Development and validation of a prediction rule for benefit and harm of dual antiplatelet therapy beyond 1 year after percutaneous coronary intervention. JAMA. 2016;315(16):1735–49.
Costa F, van Klaveren D, James S, Heg D, Raber L, Feres F, et al. Derivation and validation of the predicting bleeding complications in patients undergoing stent implantation and subsequent dual antiplatelet therapy (PRECISE-DAPT) score: a pooled analysis of individual-patient datasets from clinical trials. Lancet. 2017;389(10073):1025–34.
Steinberg BA, Shrader P, Pieper K, Thomas L, Allen LA, Ansell J, et al. Frequency and outcomes of reduced dose non-vitamin K antagonist anticoagulants: results from ORBIT-AF II (The Outcomes Registry for Better Informed Treatment of Atrial Fibrillation II). J Am Heart Assoc. 2018;7(4):e007633.
Parikh RB, Schwartz JS, Navathe AS. Beyond genes and molecules—a precision delivery initiative for precision medicine. N Engl J Med. 2017;376(17):1609–12.
Nimmesgern E, Benediktsson I, Norstedt I. Personalized Medicine in Europe. Clin Transl Sci. 2017;10(2):61–3.
Jameson JL, Longo DL. Precision medicine–personalized, problematic, and promising. N Engl J Med. 2015;372(23):2229–34.
Neumann FJ, Sousa-Uva M, Ahlsson A, Alfonso F, Banning AP, Benedetto U, et al. 2018 ESC/EACTS Guidelines on myocardial revascularization. Eur Heart J. 2019;40(2):87–165.
Yusuf S, Zhao F, Mehta SR, Chrolavicius S, Tognoni G, Fox KK, et al. Effects of clopidogrel in addition to aspirin in patients with acute coronary syndromes without ST-segment elevation. N Engl J Med. 2001;345(7):494–502.
Wiviott SD, Braunwald E, McCabe CH, Montalescot G, Ruzyllo W, Gottlieb S, et al. Prasugrel versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2007;357(20):2001–15.
Wallentin L, Becker RC, Budaj A, Cannon CP, Emanuelsson H, Held C, et al. Ticagrelor versus clopidogrel in patients with acute coronary syndromes. N Engl J Med. 2009;361(11):1045–57.
European Medecines Agency. Plavix product information. Last updated 5 Dec 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/plavix#product-information-section. Accessed 1 Jun 2019.
European Medecines Agency. Brilique product information. Last updated 30 May 2017. https://www.ema.europa.eu/en/medicines/human/EPAR/brilique#product-information-section. Accessed 1 Jun 2019.
European Medecines Agency. Efient product information. Last updated 18 Jan 2019. https://www.ema.europa.eu/en/medicines/human/EPAR/efient#product-information-section. Accessed 1 Jun 2019.
Eikelboom JW, Mehta SR, Anand SS, Xie C, Fox KA, Yusuf S. Adverse impact of bleeding on prognosis in patients with acute coronary syndromes. Circulation. 2006;114(8):774–82.
Secemsky EA, Matteau A, Yeh RW, Steg PG, Camenzind E, Wijns W, et al. Comparison of short- and long-term cardiac mortality in early versus late stent thrombosis (from Pooled PROTECT Trials). Am J Cardiol. 2015;115(12):1678–84.
Mallouk N, Labruyere C, Reny JL, Chapelle C, Piot M, Fontana P, et al. Prevalence of poor biological response to clopidogrel: a systematic review. Thromb Haemost. 2012;107(3):494–506.
Wallentin L, Varenhorst C, James S, Erlinge D, Braun OO, Jakubowski JA, et al. Prasugrel achieves greater and faster P2Y12receptor-mediated platelet inhibition than clopidogrel due to more efficient generation of its active metabolite in aspirin-treated patients with coronary artery disease. Eur Heart J. 2008;29(1):21–30.
Gurbel PA, Bliden KP, Butler K, Tantry US, Gesheff T, Wei C, et al. Randomized double-blind assessment of the ONSET and OFFSET of the antiplatelet effects of ticagrelor versus clopidogrel in patients with stable coronary artery disease: the ONSET/OFFSET study. Circulation. 2009;120(25):2577–85.
Hulot JS, Bura A, Villard E, Azizi M, Remones V, Goyenvalle C, et al. Cytochrome P450 2C19 loss-of-function polymorphism is a major determinant of clopidogrel responsiveness in healthy subjects. Blood. 2006;108(7):2244–7.
Bauer T, Bouman HJ, van Werkum JW, Ford NF, ten Berg JM, Taubert D. Impact of CYP2C19 variant genotypes on clinical efficacy of antiplatelet treatment with clopidogrel: systematic review and meta-analysis. BMJ. 2011;343:d4588.
Grosdidier C, Quilici J, Loosveld M, Camoin L, Moro PJ, Saut N, et al. Effect of CYP2C19*2 and *17 genetic variants on platelet response to clopidogrel and prasugrel maintenance dose and relation to bleeding complications. Am J Cardiol. 2013;111(7):985–90.
Cuisset T, Loosveld M, Morange PE, Quilici J, Moro PJ, Saut N, et al. CYP2C19*2 and *17 alleles have a significant impact on platelet response and bleeding risk in patients treated with prasugrel after acute coronary syndrome. JACC Cardiovasc Interv. 2012;5(12):1280–7.
Angiolillo DJ, Badimon JJ, Saucedo JF, Frelinger AL, Michelson AD, Jakubowski JA, et al. A pharmacodynamic comparison of prasugrel vs. high-dose clopidogrel in patients with type 2 diabetes mellitus and coronary artery disease: results of the Optimizing anti-Platelet Therapy In diabetes MellitUS (OPTIMUS)-3 Trial. Eur Heart J. 2011;32(7):838–46.
Muller C, Caillard S, Jesel L, El Ghannudi S, Ohlmann P, Sauleau E, et al. Association of estimated GFR with platelet inhibition in patients treated with clopidogrel. Am J Kidney Dis. 2012;59(6):777–85.
Mavrakanas TA, Alam A, Reny JL, Fontana P. Platelet reactivity in stable cardiovascular patients with chronic kidney disease. Platelets. 2018;29(5):455–62.
Gurbel PA, Bliden KP, Tantry US. Effect of clopidogrel with and without eptifibatide on tumor necrosis factor-alpha and C-reactive protein release after elective stenting: results from the CLEAR PLATELETS 1b study. J Am Coll Cardiol. 2006;48(11):2186–91.
Fontana P, Senouf D, Mach F. Biological effect of increased maintenance dose of clopidogrel in cardiovascular outpatients and influence of the cytochrome P450 2C19*2 allele on clopidogrel responsiveness. Thromb Res. 2008;121(4):463–8.
Geisler T, Grass D, Bigalke B, Stellos K, Drosch T, Dietz K, et al. The residual platelet aggregation after deployment of intracoronary stent (PREDICT) score. J Thromb Haemost. 2008;6(1):54–61.
Shuldiner AR, O’Connell JR, Bliden KP, Gandhi A, Ryan K, Horenstein RB, et al. Association of cytochrome P450 2C19 genotype with the antiplatelet effect and clinical efficacy of clopidogrel therapy. JAMA. 2009;302(8):849–57.
Fontana P, Berdague P, Castelli C, Nolli S, Barazer I, Fabbro-Peray P, et al. Clinical predictors of dual aspirin and clopidogrel poor responsiveness in stable cardiovascular patients from the ADRIE study. J Thromb Haemost. 2010;8(12):2614–23.
Combescure C, Fontana P, Mallouk N, Berdague P, Labruyere C, Barazer I, et al. Clinical implications of clopidogrel non-response in cardiovascular patients: a systematic review and meta-analysis. J Thromb Haemost. 2010;8(5):923–33.
Tantry US, Bonello L, Aradi D, Price MJ, Jeong YH, Angiolillo DJ, et al. Consensus and update on the definition of on-treatment platelet reactivity to adenosine diphosphate associated with ischemia and bleeding. J Am Coll Cardiol. 2013;62(24):2261–73.
Aradi D, Kirtane A, Bonello L, Gurbel PA, Tantry US, Huber K, et al. Bleeding and stent thrombosis on P2Y12-inhibitors: collaborative analysis on the role of platelet reactivity for risk stratification after percutaneous coronary intervention. Eur Heart J. 2015;36(27):1762–71.
Bonello L, Camoin-Jau L, Armero S, Com O, Arques S, Burignat-Bonello C, et al. Tailored clopidogrel loading dose according to platelet reactivity monitoring to prevent acute and subacute stent thrombosis. Am J Cardiol. 2009;103(1):5–10.
Price MJ, Berger PB, Teirstein PS, Tanguay JF, Angiolillo DJ, Spriggs D, et al. Standard- vs high-dose clopidogrel based on platelet function testing after percutaneous coronary intervention: the GRAVITAS randomized trial. JAMA. 2011;305(11):1097–105.
Collet JP, Cuisset T, Range G, Cayla G, Elhadad S, Pouillot C, et al. Bedside monitoring to adjust antiplatelet therapy for coronary stenting. N Engl J Med. 2012;367(22):2100–9.
Trenk D, Stone GW, Gawaz M, Kastrati A, Angiolillo DJ, Muller U, et al. A randomized trial of prasugrel versus clopidogrel in patients with high platelet reactivity on clopidogrel after elective percutaneous coronary intervention with implantation of drug-eluting stents: results of the TRIGGER-PCI (Testing Platelet Reactivity In Patients Undergoing Elective Stent Placement on Clopidogrel to Guide Alternative Therapy With Prasugrel) study. J Am Coll Cardiol. 2012;59(24):2159–64.
Gurbel PA, Jeong YH, Navarese EP, Tantry US. Platelet-mediated thrombosis: from bench to bedside. Circ Res. 2016;118(9):1380–91.
Reny JL, Berdague P, Poncet A, Barazer I, Nolli S, Fabbro-Peray P, et al. Antiplatelet drug response status does not predict recurrent ischemic events in stable cardiovascular patients: results of the Antiplatelet Drug Resistances and Ischemic Events study. Circulation. 2012;125(25):3201–10.
Zhou Y, Wang Y, Wu Y, Huang C, Yan H, Zhu W, et al. Individualized dual antiplatelet therapy based on platelet function testing in patients undergoing percutaneous coronary intervention: a meta-analysis of randomized controlled trials. BMC Cardiovasc Disord. 2017;17(1):157.
Norgard NB, Dinicolantonio JJ. Clopidogrel, prasugrel, or ticagrelor? a practical guide to use of antiplatelet agents in patients with acute coronary syndromes. Postgrad Med. 2013;125(4):91–102.
Varenhorst C, Eriksson N, Johansson A, Barratt BJ, Hagstrom E, Akerblom A, et al. Effect of genetic variations on ticagrelor plasma levels and clinical outcomes. Eur Heart J. 2015;36(29):1901–12.
Franken CC, Kaiser AF, Kruger JC, Overbeck K, Mugge A, Neubauer H. Cytochrome P450 2B6 and 2C9 genotype polymorphism–a possible cause of prasugrel low responsiveness. Thromb Haemost. 2013;110(1):131–40.
Wallentin L, James S, Storey RF, Armstrong M, Barratt BJ, Horrow J, et al. Effect of CYP2C19 and ABCB1 single nucleotide polymorphisms on outcomes of treatment with ticagrelor versus clopidogrel for acute coronary syndromes: a genetic substudy of the PLATO trial. Lancet. 2010;376(9749):1320–8.
Payne CD, Li YG, Small DS, Ernest CS 2nd, Farid NA, Jakubowski JA, et al. Increased active metabolite formation explains the greater platelet inhibition with prasugrel compared to high-dose clopidogrel. J Cardiovasc Pharmacol. 2007;50(5):555–62.
Hagihara K, Kazui M, Kurihara A, Yoshiike M, Honda K, Okazaki O, et al. A possible mechanism for the differences in efficiency and variability of active metabolite formation from thienopyridine antiplatelet agents, prasugrel and clopidogrel. Drug Metab Dispos. 2009;37(11):2145–52.
Ferri N, Corsini A, Bellosta S. Pharmacology of the new P2Y12 receptor inhibitors: insights on pharmacokinetic and pharmacodynamic properties. Drugs. 2013;73(15):1681–709.
Deharo P, Cuisset T. Monitoring platelet function: what have we learned from randomized clinical trials? Cardiovasc Diagn Ther. 2018;8(5):621–9.
Sibbing D, Aradi D, Jacobshagen C, Gross L, Trenk D, Geisler T, et al. Guided de-escalation of antiplatelet treatment in patients with acute coronary syndrome undergoing percutaneous coronary intervention (TROPICAL-ACS): a randomised, open-label, multicentre trial. Lancet. 2017;390(10104):1747–57.
Cuisset T, Deharo P, Quilici J, Johnson TW, Deffarges S, Bassez C, et al. Benefit of switching dual antiplatelet therapy after acute coronary syndrome: the TOPIC (timing of platelet inhibition after acute coronary syndrome) randomized study. Eur Heart J. 2017;38(41):3070–8.
Kupka D, Sibbing D. De-escalation of P2Y12 receptor inhibitor therapy after acute coronary syndromes in patients undergoing percutaneous coronary intervention. Korean Circ J. 2018;48(10):863–72.
Cayla G, Cuisset T, Silvain J, Leclercq F, Manzo-Silberman S, Saint-Etienne C, et al. Platelet function monitoring to adjust antiplatelet therapy in elderly patients stented for an acute coronary syndrome (ANTARCTIC): an open-label, blinded-endpoint, randomised controlled superiority trial. Lancet. 2016;388(10055):2015–22.
Sibbing D, Gross L, Trenk D, Jacobshagen C, Geisler T, Hadamitzky M, et al. Age and outcomes following guided de-escalation of antiplatelet treatment in acute coronary syndrome patients undergoing percutaneous coronary intervention: results from the randomized TROPICAL-ACS trial. Eur Heart J. 2018;39(29):2749–58.
Gross L, Trenk D, Jacobshagen C, Krieg A, Gawaz M, Massberg S, et al. Genotype-phenotype association and impact on outcomes following guided de-escalation of anti-platelet treatment in acute coronary syndrome patients: the TROPICAL-ACS genotyping substudy. Thromb Haemost. 2018;118(9):1656–67.
Vranckx P, Valgimigli M, Juni P, Hamm C, Steg PG, Heg D, et al. Ticagrelor plus aspirin for 1 month, followed by ticagrelor monotherapy for 23 months vs aspirin plus clopidogrel or ticagrelor for 12 months, followed by aspirin monotherapy for 12 months after implantation of a drug-eluting stent: a multicentre, open-label, randomised superiority trial. Lancet. 2018;392(10151):940–9.
Rozemeijer R, Voskuil M, Greving JP, Bots ML, Doevendans PA, Stella PR. Short versus long duration of dual antiplatelet therapy following drug-eluting stents: a meta-analysis of randomised trials. Neth Heart J. 2018;26(5):242–51.
Collet JP, Silvain J, Barthelemy O, Range G, Cayla G, Van Belle E, et al. Dual-antiplatelet treatment beyond 1 year after drug-eluting stent implantation (ARCTIC-Interruption): a randomised trial. Lancet. 2014;384(9954):1577–85.
Park SJ, Park DW, Kim YH, Kang SJ, Lee SW, Lee CW, et al. Duration of dual antiplatelet therapy after implantation of drug-eluting stents. N Engl J Med. 2010;362(15):1374–82.
Valgimigli M, Campo G, Monti M, Vranckx P, Percoco G, Tumscitz C, et al. Short- versus long-term duration of dual-antiplatelet therapy after coronary stenting: a randomized multicenter trial. Circulation. 2012;125(16):2015–26.
Mauri L, Kereiakes DJ, Yeh RW, Driscoll-Shempp P, Cutlip DE, Steg PG, et al. Twelve or 30 months of dual antiplatelet therapy after drug-eluting stents. N Engl J Med. 2014;371(23):2155–66.
Costa F, Valgimigli M. The optimal duration of dual antiplatelet therapy after coronary stent implantation: to go too far is as bad as to fall short. Cardiovasc Diagn Ther. 2018;8(5):630–46.
Petrie A, Sabin C. Medical statistics at a glance. 3rd ed. New York: Wiley Blackwell; 2009.
Patrono C. The P2Y12 receptor: no active metabolite, no party. Nat Rev Cardiol. 2009;6(4):271–2.
Roshammar D, Nyberg J, Andersson T, Stanski D, Storey RF, Hamren B. Exposure-response analyses supporting ticagrelor dosing recommendation in patients with prior myocardial infarction. J Clin Pharmacol. 2017;57(5):573–83.
Nutescu EA, Burnett A, Fanikos J, Spinler S, Wittkowsky A. Pharmacology of anticoagulants used in the treatment of venous thromboembolism. J Thromb Thrombolysis. 2016;41(1):15–31.
Kirchhof P, Benussi S, Kotecha D, Ahlsson A, Atar D, Casadei B, et al. 2016 ESC guidelines for the management of atrial fibrillation developed in collaboration with EACTS. Eur Heart J. 2016;37(38):2893–962.
January CT, Wann LS, Calkins H, Chen LY, Cigarroa JE, Cleveland JC Jr, et al. AHA/ACC/HRS focused update of the 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation. Circulation. 2019. https://doi.org/10.1161/cir.0000000000000665(Epub 28 Jan 2019).
Di Nisio M, van Es N, Buller HR. Deep vein thrombosis and pulmonary embolism. Lancet. 2016;388(10063):3060–73.
Kearon C, Akl EA, Ornelas J, Blaivas A, Jimenez D, Bounameaux H, et al. Antithrombotic therapy for VTE disease: CHEST guideline and expert panel report. Chest. 2016;149(2):315–52.
European Medecines Agency. Pradaxa product information. Last updated 15 June 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/pradaxa#product-information-section. Accessed 1 Jun 2019.
European Medecines Agency. Xarelo product information. Last updated 6 Sep 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/xarelto#product-information-section. Accessed 1 Jun 2019.
European Medecines Agency. Eliquis product information. Last updated 3 Sept 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/eliquis#product-information-section. Accessed 1 Jun 2019.
European Medecines Agency. Lixiana product information. Last updated 8 Oct 2018. https://www.ema.europa.eu/en/medicines/human/EPAR/lixiana#product-information-section. Accessed 1 Jun 2019.
De Caterina R, Lip GYH. The non-vitamin K antagonist oral anticoagulants (NOACs) and extremes of body weight-a systematic literature review. Clin Res Cardiol. 2017;106(8):565–72.
Dhar A, Mullish BH, Thursz MR. Anticoagulation in chronic liver disease. J Hepatol. 2017;66(6):1313–26.
Weber J, Olyaei A, Shatzel J. The efficacy and safety of direct oral anticoagulants in patients with chronic renal insufficiency: a review of the literature. Eur J Haematol. 2019;102(4):312–8.
Huisman MV, Ferreira M, Feuring M, Fraessdorf M, Klok FA. Less abnormal uterine bleeding with dabigatran than warfarin in women treated for acute venous thromboembolism. J Thromb Haemost. 2018;16(9):1775–8.
Cheung KS, Leung WK. Gastrointestinal bleeding in patients on novel oral anticoagulants: risk, prevention and management. World J Gastroenterol. 2017;23(11):1954–63.
Ramos-Esquivel A. Monitoring anticoagulant therapy with new oral agents. World J Methodol. 2015;5(4):212–5.
Gong IY, Kim RB. Importance of pharmacokinetic profile and variability as determinants of dose and response to dabigatran, rivaroxaban, and apixaban. Can J Cardiol. 2013;29(7 Suppl):S24–33.
Testa S, Tripodi A, Legnani C, Pengo V, Abbate R, Dellanoce C, et al. Plasma levels of direct oral anticoagulants in real life patients with atrial fibrillation: results observed in four anticoagulation clinics. Thromb Res. 2016;137:178–83.
Storelli F, Daali Y, Desmeules J, Reny JL, Fontana P. Pharmacogenomics of oral antithrombotic drugs. Curr Pharm Des. 2016;22(13):1933–49.
Pare G, Eriksson N, Lehr T, Connolly S, Eikelboom J, Ezekowitz MD, et al. Genetic determinants of dabigatran plasma levels and their relation to bleeding. Circulation. 2013;127(13):1404–12.
Ueshima S, Hira D, Fujii R, Kimura Y, Tomitsuka C, Yamane T, et al. Impact of ABCB1, ABCG2, and CYP3A5 polymorphisms on plasma trough concentrations of apixaban in Japanese patients with atrial fibrillation. Pharmacogenet Genomics. 2017;27(9):329–36.
Eikelboom JW, Connolly SJ, Bosch J, Dagenais GR, Hart RG, Shestakovska O, et al. Rivaroxaban with or without aspirin in stable cardiovascular disease. N Engl J Med. 2017;377(14):1319–30.
US Food and Drug Administration. Briefing information Set, for the October 30, 2014, meeting of the Cardiovascular and Renal Drugs Advisory Committee. https://wayback.archive-it.org/7993/20170405211258/https:/www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM421612.pdf. Accessed 1 Jun 2019.
US Food and Drug Administration. Briefing information Xrt, for the September 8, 2011, meeting of the Cardiovascular and Renal Drugs Advisory Committee. https://wayback.archive-it.org/7993/20170405211959/https:/www.fda.gov/downloads/AdvisoryCommittees/CommitteesMeetingMaterials/Drugs/CardiovascularandRenalDrugsAdvisoryCommittee/UCM270796.pdf. Accessed 1 Jun 2019.
Center for Drug Evaluation and Research. Clinical pharmacology review NDA 202-155, December 2012. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/202155Orig1s000ClinPharmR.pdf. Accessed 1 Jun 2019.
Reilly PA, Lehr T, Haertter S, Connolly SJ, Yusuf S, Eikelboom JW, et al. The effect of dabigatran plasma concentrations and patient characteristics on the frequency of ischemic stroke and major bleeding in atrial fibrillation patients: the RE-LY Trial (Randomized Evaluation of Long-Term Anticoagulation Therapy). J Am Coll Cardiol. 2014;63(4):321–8.
Testa S, Paoletti O, Legnani C, Dellanoce C, Antonucci E, Cosmi B, et al. Low drug levels and thrombotic complications in high-risk atrial fibrillation patients treated with direct oral anticoagulants. J Thromb Haemost. 2018;16(5):842–8.
Camm AJ, Lip GY, De Caterina R, Savelieva I, Atar D, Hohnloser SH, et al. 2012 focused update of the ESC guidelines for the management of atrial fibrillation: an update of the 2010 ESC Guidelines for the management of atrial fibrillation–developed with the special contribution of the European Heart Rhythm Association. Europace. 2012;14(10):1385–413.
January CT, Wann LS, Alpert JS, Calkins H, Cigarroa JE, Cleveland JC Jr, et al. 2014 AHA/ACC/HRS guideline for the management of patients with atrial fibrillation: executive summary: a report of the American College of Cardiology/American Heart Association Task Force on practice guidelines and the Heart Rhythm Society. Circulation. 2014;130(23):2071–104.
Olesen JB, Lip GY, Hansen ML, Hansen PR, Tolstrup JS, Lindhardsen J, et al. Validation of risk stratification schemes for predicting stroke and thromboembolism in patients with atrial fibrillation: nationwide cohort study. BMJ. 2011;342:d124.
Maheshwari A, Norby FL, Roetker NS, Soliman EZ, Koene RJ, Rooney MR, et al. Refining prediction of atrial fibrillation-related stroke using the P2-CHA2DS2-VASc score. Circulation. 2019;139(2):180–91.
Gage BF, Yan Y, Milligan PE, Waterman AD, Culverhouse R, Rich MW, et al. Clinical classification schemes for predicting hemorrhage: results from the National Registry of Atrial Fibrillation (NRAF). Am Heart J. 2006;151(3):713–9.
Pisters R, Lane DA, Nieuwlaat R, de Vos CB, Crijns HJ, Lip GY. A novel user-friendly score (HAS-BLED) to assess 1-year risk of major bleeding in patients with atrial fibrillation: the Euro Heart Survey. Chest. 2010;138(5):1093–100.
Fang MC, Go AS, Chang Y, Borowsky LH, Pomernacki NK, Udaltsova N, et al. A new risk scheme to predict warfarin-associated hemorrhage: the ATRIA (Anticoagulation and Risk Factors in Atrial Fibrillation) study. J Am Coll Cardiol. 2011;58(4):395–401.
Gomes T, Mamdani MM, Holbrook AM, Paterson JM, Hellings C, Juurlink DN. Rates of hemorrhage during warfarin therapy for atrial fibrillation. CMAJ. 2013;185(2):E121–7.
Lip GYH, Skjoth F, Nielsen PB, Kjaeldgaard JN, Larsen TB. The HAS-BLED, ATRIA, and ORBIT Bleeding scores in atrial fibrillation patients using non-vitamin K antagonist oral anticoagulants. Am J Med. 2018;131(5):574.e13–27.
Rostami-Hodjegan A. Physiologically based pharmacokinetics joined with in vitro–in vivo extrapolation of ADME: a marriage under the arch of systems pharmacology. Clin Pharmacol Ther. 2012;92(1):50–61.
Wilbaux M, Fuchs A, Samardzic J, Rodieux F, Csajka C, Allegaert K, et al. Pharmacometric approaches to personalize use of primarily renally eliminated antibiotics in preterm and term neonates. J Clin Pharmacol. 2016;56(8):909–35.
Csajka C, Crettol S, Guidi M, Eap CB. Population genetic-based pharmacokinetic modeling of methadone and its relationship with the QTc interval in opioid-dependent patients. Clin Pharmacokinet. 2016;55(12):1521–33.
Nekka F, Csajka C, Wilbaux M, Sanduja S, Li J, Pfister M. Pharmacometrics-based decision tools facilitate mHealth implementation. Expert Rev Clin Pharmacol. 2017;10(1):39–46.
Klopp-Schulze L, Joerger M, Wicha SG, Ter Heine R, Csajka C, Parra-Guillen ZP, et al. Exploiting pharmacokinetic models of tamoxifen and endoxifen to identify factors causing subtherapeutic concentrations in breast cancer patients. Clin Pharmacokinet. 2018;57(2):229–42.
de Velde F, Mouton JW, de Winter BCM, van Gelder T, Koch BCP. Clinical applications of population pharmacokinetic models of antibiotics: challenges and perspectives. Pharmacol Res. 2018;134:280–8.
Smits A, De Cock RF, Allegaert K, Vanhaesebrouck S, Danhof M, Knibbe CA. Prospective evaluation of a model-based dosing regimen for amikacin in preterm and term neonates in clinical practice. Antimicrob Agents Chemother. 2015;59(10):6344–51.
Hamberg AK, Hellman J, Dahlberg J, Jonsson EN, Wadelius M. A Bayesian decision support tool for efficient dose individualization of warfarin in adults and children. BMC Med Inform Decis Mak. 2015;15:7.
Deng J, Vozmediano V, Rodriguez M, Cavallari LH, Schmidt S. Genotype-guided dosing of warfarin through modeling and simulation. Eur J Pharm Sci. 2017;109S:S9–14.
Liesenfeld KH, Lehr T, Dansirikul C, Reilly PA, Connolly SJ, Ezekowitz MD, et al. Population pharmacokinetic analysis of the oral thrombin inhibitor dabigatran etexilate in patients with non-valvular atrial fibrillation from the RE-LY trial. J Thromb Haemost. 2011;9(11):2168–75.
Girgis IG, Patel MR, Peters GR, Moore KT, Mahaffey KW, Nessel CC, et al. Population pharmacokinetics and pharmacodynamics of rivaroxaban in patients with non-valvular atrial fibrillation: results from ROCKET AF. J Clin Pharmacol. 2014;54(8):917–27.
Leil TA, Frost C, Wang X, Pfister M, LaCreta F. Model-based exposure-response analysis of apixaban to quantify bleeding risk in special populations of subjects undergoing orthopedic surgery. CPT Pharmacomet Syst Pharmacol. 2014;3:e136.
Niebecker R, Jonsson S, Karlsson MO, Miller R, Nyberg J, Krekels EH, et al. Population pharmacokinetics of edoxaban in patients with symptomatic deep-vein thrombosis and/or pulmonary embolism–the Hokusai-VTE phase 3 study. Br J Clin Pharmacol. 2015;80(6):1374–87.
Chang M, Yu Z, Shenker A, Wang J, Pursley J, Byon W, et al. Effect of renal impairment on the pharmacokinetics, pharmacodynamics, and safety of apixaban. J Clin Pharmacol. 2016;56(5):637–45.
Koretsune Y, Yamashita T, Kimura T, Fukuzawa M, Abe K, Yasaka M. Short-term safety and plasma concentrations of edoxaban in Japanese patients with non-valvular atrial fibrillation and severe renal impairment. Circ J. 2015;79(7):1486–95.
Yoshioka H, Sato H, Hatakeyama H, Hisaka A. Model-based meta-analysis to evaluate optimal doses of direct oral factor Xa inhibitors in atrial fibrillation patients. Blood Adv. 2018;2(10):1066–75.
Moser BA, LaBell ES, Chigutsa E, Jakubowski JA, Small DS. Population pharmacokinetic and exposure-response analyses of prasugrel in pediatric patients with sickle cell anemia. Clin Pharmacokinet. 2018;57(2):243–54.
Zhao P, Zhang L, Grillo JA, Liu Q, Bullock JM, Moon YJ, et al. Applications of physiologically based pharmacokinetic (PBPK) modeling and simulation during regulatory review. Clin Pharmacol Ther. 2011;89(2):259–67.
Djebli N, Fabre D, Boulenc X, Fabre G, Sultan E, Hurbin F. Physiologically based pharmacokinetic modeling for sequential metabolism: effect of CYP2C19 genetic polymorphism on clopidogrel and clopidogrel active metabolite pharmacokinetics. Drug Metab Dispos. 2015;43(4):510–22.
Shebley M, Sandhu P, Emami Riedmaier A, Jamei M, Narayanan R, Patel A, et al. Physiologically based pharmacokinetic model qualification and reporting procedures for regulatory submissions: a consortium perspective. Clin Pharmacol Ther. 2018;104(1):88–110.
Rostami-Hodjegan A. Reverse translation in PBPK and QSP: going backwards in order to go forward with confidence. Clin Pharmacol Ther. 2018;103(2):224–32.
Jamei M. Recent advances in development and application of physiologically-based pharmacokinetic (PBPK) models: a transition from academic curiosity to regulatory acceptance. Curr Pharmacol Rep. 2016;2:161–9.
Luzon E, Blake K, Cole S, Nordmark A, Versantvoort C, Berglund EG. Physiologically based pharmacokinetic modeling in regulatory decision-making at the European Medicines Agency. Clin Pharmacol Ther. 2017;102(1):98–105.
European Medecines Agency. Guideline on the qualification and reporting of physiologically based pharmacokinetic (PBPK) modelling and simulation simulation. http://www.ema.europa.eu/docs/en_GB/document_library/Scientific_guideline/2016/07/WC500211315.pdf. 2016. Accessed 1 Jun 2019.
US Food and Drug Administration. Physiologically based pharmacokinetic analyses—format and content guidance for industry. 2018. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/physiologically-based-pharmacokinetic-analyses-format-and-content-guidance-industry. Accessed 1 Jun 2019.
Marsousi N, Samer CF, Fontana P, Reny JL, Rudaz S, Desmeules JA, et al. Coadministration of ticagrelor and ritonavir: toward prospective dose adjustment to maintain an optimal platelet inhibition using the PBPK approach. Clin Pharmacol Ther. 2016;100(3):295–304.
Fan J, Zhang X, Zhao L. Utility of physiologically based pharmacokinetic absorption modeling to predict the impact of salt-to-base conversion on prasugrel HCl product bioequivalence in the presence of proton pump inhibitors. AAPS J. 2017;19(5):1479–86.
Xu R, Ge W, Jiang Q. Application of physiologically based pharmacokinetic modeling to the prediction of drug-drug and drug-disease interactions for rivaroxaban. Eur J Clin Pharmacol. 2018;74(6):755–65.
Polasek TM, Tucker GT, Sorich MJ, Wiese MD, Mohan T, Rostami-Hodjegan A, et al. Prediction of olanzapine exposure in individual patients using physiologically based pharmacokinetic modelling and simulation. Br J Clin Pharmacol. 2018;84(3):462–76.
Darwich AS, Ogungbenro K, Vinks AA, Powell JR, Reny JL, Marsousi N, et al. Why has model-informed precision dosing not yet become common clinical reality? Lessons from the past and a roadmap for the future. Clin Pharmacol Ther. 2017;101(5):646–56.
Yourman L, Concato J, Agostini JV. Use of computer decision support interventions to improve medication prescribing in older adults: a systematic review. Am J Geriatr Pharmacother. 2008;6(2):119–29.
Handler SM, Altman RL, Perera S, Hanlon JT, Studenski SA, Bost JE, et al. A systematic review of the performance characteristics of clinical event monitor signals used to detect adverse drug events in the hospital setting. J Am Med Inform Assoc. 2007;14(4):451–8.
Beeler PE, Bates DW, Hug BL. Clinical decision support systems. Swiss Med Wkly. 2014;144:w14073.
Fritz D, Ceschi A, Curkovic I, Huber M, Egbring M, Kullak-Ublick GA, et al. Comparative evaluation of three clinical decision support systems: prospective screening for medication errors in 100 medical inpatients. Eur J Clin Pharmacol. 2012;68(8):1209–19.
Chazard E, Preda C, Merlin B, Ficheur G, Consortium P, Beuscart R. Data-mining-based detection of adverse drug events. Stud Health Technol Inform. 2009;150:552–6.
Ohno-Machado L, Nadkarni P, Johnson K. Natural language processing: algorithms and tools to extract computable information from EHRs and from the biomedical literature. J Am Med Inform Assoc. 2013;20(5):805.
Polepalli Ramesh B, Belknap SM, Li Z, Frid N, West DP, Yu H. Automatically recognizing medication and adverse event information from food and drug administration’s adverse event reporting system narratives. JMIR Med Inform. 2014;2(1):e10.
Iqbal E, Mallah R, Jackson RG, Ball M, Ibrahim ZM, Broadbent M, et al. Identification of adverse drug events from free text electronic patient records and information in a large mental health case register. PLoS One. 2015;10(8):e0134208.
Sibbing D, Angiolillo DJ, Huber K. Antithrombotic therapy for acute coronary syndrome: past, present and future. Thromb Haemost. 2017;117(7):1240–8.
Acknowledgements
This work was performed within the framework of the Swiss National Science Foundation’s PNR74 Project 407440_167381 on the ‘Automated detection of adverse drug events from older inpatients’ electronic medical records using structured data’.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No external funding was used in the preparation of this manuscript.
Conflict of interest
Jean Terrier, Youssef Daali, Pierre Fontana, Chantal Csajka, and Jean-Luc Reny declare that they have no potential conflicts of interest that may be relevant to the contents of this manuscript.
Rights and permissions
About this article

Cite this article
Terrier, J., Daali, Y., Fontana, P. et al. Towards Personalized Antithrombotic Treatments: Focus on P2Y12 Inhibitors and Direct Oral Anticoagulants. Clin Pharmacokinet 58, 1517–1532 (2019). https://doi.org/10.1007/s40262-019-00792-y
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-019-00792-y