
Abstract
Oral anti-hormonal drugs are essential in the treatment of breast and prostate cancer. It is well known that the interpatient variability in pharmacokinetic exposure is high for these agents and exposure–response relationships exist for many oral anti-hormonal drugs. Yet, they are still administered at fixed doses. This could lead to underdosing and thus suboptimal efficacy in some patients, while other patients could be overdosed resulting in unnecessary side effects. Therapeutic drug monitoring (TDM), individualized dosing based on measured blood concentrations of the drug, could therefore be a valid option to further optimize treatment. In this review, we provide an overview of relevant clinical pharmacokinetic and pharmacodynamic characteristics of oral anti-hormonal drugs in oncology and translate these into practical guidelines for TDM. For some agents, TDM targets are not well established yet and as a reference the median pharmacokinetic exposure could be targeted (exemestane: minimum plasma concentration (Cmin) 4.1 ng/mL and enzalutamide: Cmin 11.4 mg/L). However, for most drugs, exposure–efficacy analyses could be translated into specific targets (abiraterone: Cmin 8.4 ng/mL, anastrozole: Cmin 34.2 ng/mL, and letrozole: Cmin 85.6 ng/mL). Moreover, prospective clinical trials have shown TDM to be feasible for tamoxifen, for which the exposure–efficacy threshold of its active metabolite endoxifen is 5.97 ng/mL. Based on the available data, we therefore conclude that individualized dosing based on drug concentrations is feasible and promising for oral anti-hormonal drugs and should be developed further and implemented into clinical practice.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Although many oral anti-hormonal drugs show exposure–response relationships and the interpatient variability in pharmacokinetic exposure is high, they are still administered at fixed doses. |
This could lead to underdosing in some patients, which may result in suboptimal efficacy, while other patients might be overdosed, causing unnecessary toxicity. |
Treatment could be optimized by therapeutic drug monitoring (TDM), which is individualized dosing based on measured blood concentrations of the drug. |
In this review, we summarize the available pharmacokinetic and pharmacodynamic data, discuss exposure–toxicity and exposure–efficacy relationships, and propose pharmacokinetic targets that can be used for TDM. |
1 Introduction
Breast and prostate cancer are both highly prevalent malignancies, with breast cancer being the most commonly diagnosed malignancy in women and prostate cancer in men in the Western world. Breast and prostate cancer represent the second leading cause of cancer deaths in women and men, respectively [1]. As these tumors are often dependent on estrogens and androgens for their growth, anti-hormonal drugs are imperative in their treatment.
Even though many oral anti-hormonal drugs show exposure–response relationships and the interpatient variability in pharmacokinetic (PK) exposure is high (up to 141% for abiraterone) [2], they are still administered at fixed doses. As a result, some patients may be underdosed, which could lead to suboptimal efficacy, while other patients might be overdosed, causing unnecessary toxicity. Treatment could be optimized by therapeutic drug monitoring (TDM), which is individualized dosing based on measured blood concentrations of the drug [3,4,5,6,7,8].
Use of TDM in oncology has been previously advocated and for other targeted therapies, such as kinase inhibitors, TDM targets have been described previously [3, 4, 6, 7, 9]. The aim of this review is to summarize the available PK and pharmacodynamic (PD) data on oral anti-hormonal drugs, to discuss exposure–toxicity and exposure–efficacy relationships and to propose PK targets, which can be used for TDM.
Table 1 provides a summary of selected (steady-state) PK parameters of these drugs. The proposed targets and TDM recommendations have been summarized in Table 2.
2 Methods
Although this is not a systematic review, the literature was searched as comprehensively as possible. For all oral anti-hormonal drugs, the US FDA Clinical Pharmacology and Biopharmaceutics Review and the Committee for Medicinal Products for Human Use European Public Assessment Report were consulted. Furthermore, PubMed searches were performed using the term ‘pharmacokinetics’ in combination with the different oral anti-hormonal drugs. In addition, citation snowballing was used to find other relevant studies.
3 Practical Recommendations for Therapeutic Drug Monitoring of Oral Anti-Hormonal Drugs in Oncology
3.1 Anti-Androgens
3.1.1 Abiraterone
Abiraterone acetate (Zytiga®) is the prodrug of abiraterone, which is a steroidal irreversible inhibitor of 17α-hydroxylase [cytochrome P450 (CYP)17], thereby blocking the androgen synthesis. Abiraterone acetate is currently indicated for metastatic castration-resistant prostate cancer [10]. In the near future, this indication might be expanded to patients with locally advanced or metastatic prostate cancer who are naive to anti-hormonal treatment [11].
According to the Summary of Product Characteristics, abiraterone acetate should be ingested in a modified fasting state, which means no food 2 h before or 1 h after drug intake. Chi et al. studied the food effect of the pharmacokinetics of abiraterone acetate and found a seven- and five-fold increase in maximum plasma concentration (Cmax) and area under the plasma concentration-time curve (AUC), respectively, with a low-fat meal and a 17- and 10-fold increase in Cmax and AUC, respectively, with a high-fat meal, compared to overnight fasting in healthy subjects. However, in patients with metastatic castration-resistant prostate cancer, the food effect was compared to a modified fasting state, showing a less pronounced effect (similar exposure with low-fat meals and a twofold increase with high-fat meals). Adverse events (all grade ≤ 3) were similar in the different groups (mainly hot flashes, fatigue, and hypokalemia) [12].
Abiraterone acetate has a high interpatient variability of 41–141% for AUC from time zero to infinity and 46% for Cmin, with an intrapatient variability of 33% [2, 13]. In the population-PK model described by Stuyckens et al., food and hepatic impairment appeared to be relevant covariates that influence abiraterone exposure, while 70% of the interpatient variability remained unexplained [14].
Unfortunately, insufficient PK data have been collected in the pivotal trial and phase II trials to evaluate the exposure–toxicity relationships [10]. Abiraterone acetate was generally well tolerated, and no dose-limiting toxicities were reported for doses up to 2000 mg once daily (QD) [15].
A model has been developed in which a relationship between pharmacokinetics and prostate-specific antigen (PSA) reduction has been established [2, 16]. Additionally, in a prospective observational study in patients with castration-resistant prostate cancer (n = 61), higher abiraterone trough concentrations (Cmin) were found in PSA responders compared to non-responders (12.0 vs. 8.0 ng/mL, p = 0.0015), in which PSA response was defined as a PSA decline of at least 50% after 3 months of treatment [13]. The most predictive Cmin cut-off for PSA response was 8.4 ng/mL according to a receiver operating characteristic curve. Using this threshold, exposure–survival analysis found a progression-free survival, defined as the time from treatment initiation to the first progression event (either PSA or radiologic progression), of 7.4 months in patients with a Cmin below 8.4 ng/mL and 12.2 months in patients with a Cmin above 8.4 ng/mL (p = 0.044). Nineteen of the 55 patients (35%) in this study had a Cmin below 8.4 ng/mL [13].
Abiraterone is converted into the active metabolite ∆4-abiraterone (D4A) by the enzyme 3β-hydroxysteroid-dehydrogenase. Although the conversion ratio of abiraterone to D4A is low (~ 5%), it targets multiple steps of the androgen receptor signaling pathway, some of them more potently than abiraterone. An exposure–efficacy relationship has not been established for D4A hitherto, but given the dual mechanism of action of D4A (both inhibition of CYP17 and blockage of the androgen receptor), it is to be expected that such a relationship exists and may be identified. Therefore, measuring this metabolite could be interesting to refine TDM-guided dosing in the future [17, 18].
Given the clear exposure–efficacy relationship, a target Cmin of ≥ 8.4 ng/mL can be recommended for abiraterone. At the currently used fixed dose of 1000 mg QD, 35% of patients do not reach this threshold, with the potential to increase progression-free survival by 4.8 months for this subpopulation. Although it is advised to administer abiraterone in a modified fasting state, in clinical practice, patients often take it after an overnight fast. Since a clinically significant food effect has been shown when compared to overnight fasting, a first step in the case of low exposure could be to administer abiraterone concomitantly with a light meal or a snack, before escalating the dose.
3.1.2 Enzalutamide
Enzalutamide (Xtandi®) is an androgen-receptor antagonist indicated for the treatment of metastatic castration-resistant prostate cancer [19]. Enzalutamide is metabolized by CYP2C8 and CYP3A4/5 to an inactive carboxylic acid (M1) and an active N-desmethyl metabolite (M2). The mean ± standard deviation Cmin of the approved 160-mg QD dose was 11.4 ± 3.0 mg/L for enzalutamide, 13.0 ± 3.8 mg/L for M2, and 8.4 ± 6.8 mg/L for M1 [20]. Since M2 has a high abundancy and similar potency to enzalutamide, and concentrations of these two compounds can differ between patients (± 25%), it could be scientifically interesting to measure both enzalutamide and its M2 metabolite [19]. Future studies should clarify the role of this M2 metabolite in TDM.
No clinically significant exposure–toxicity relationships have been found so far [19]. An exposure–efficacy analysis has been executed for enzalutamide in the pivotal phase III study, using the sum of the Cmin for enzalutamide and M2 with overall survival as the endpoint. All quartiles performed significantly better than placebo (p < 0.0001), yet no differences between the exposure quartiles could be found (p ≥ 0.5499) [20].
In the phase I/II trial, PSA decreases at 12 weeks were comparable in the different dose concentrations (range 60–600 mg) [21]. Although enzalutamide targets the androgen receptor and could therefore cause a PSA decline without reflecting tumor response, it has been shown that PSA decline after 12 weeks is still associated with progression-free survival and overall survival [22].
In the phase I trial, 16β-18F-5α-dihydrotestosterone positron emission tomography imaging in 22 patients suggested androgen receptor binding was higher in the 150-mg group (corresponding to a median Cmin of 11.4 mg/L) than in the 60-mg dose group (corresponding to a median Cmin of approximately 5 mg/L). No additional effect was seen at higher doses, suggesting a plateau at a dose of 150 mg and Cmin of 12 mg/L [21].
Given the lack of an exposure–toxicity relationship, the limited evidence for an exposure–efficacy relationship, and the small interpatient variability in exposure (26% for Cmin), enzalutamide may not be the ideal drug for TDM. In the absence of an exposure–efficacy target, the mean Cmin of 11.4 mg/L at the standard dose of 160 mg QD could be used as a reference. As this is the mean exposure, approximately 50% of patients will have concentrations below this reference. Based on the 16β-18F-5α-dihydrotestosterone data, dose increments could be considered in patients with a very low (e.g., < 5 mg/L) Cmin. Taking into account the mean exposure and standard deviation, less than 2.5% of patients will have trough concentrations < 5 mg/L.
3.2 Anti-Estrogens
3.2.1 Tamoxifen
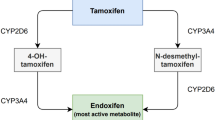
Tamoxifen (Nolvadex®) is an estrogen receptor antagonist indicated for the treatment of estrogen receptor-positive breast cancer. Tamoxifen is extensively metabolized mainly by CYP2D6 and CYP3A4 into a range of active and inactive metabolites [23]. Endoxifen is one of the most potent and abundant metabolites and, therefore, TDM of tamoxifen has focused on measuring endoxifen concentrations. Endoxifen shows a large interpatient variability in steady-state concentrations of 40–49%, while the intrapatient variability is only 11% [24,25,26].
No clear relationship between endoxifen concentration and toxicity has been reported in the literature. A retrospective study (n = 109) could not find evidence for an association between exposure and hot flashes, a major side effect of tamoxifen treatment [27]. In another prospective trial (n = 122), no significant correlation was found between tamoxifen metabolites and the hot flash score (p = 0.07) [8].
A retrospective analysis of 1370 patients with estrogen receptor-positive breast cancer receiving tamoxifen in the adjuvant setting found that patients in the lowest endoxifen exposure quintile (0–5.9 ng/mL) had a higher risk of recurrence than patients above this threshold (hazard ratio 0.74; 95% confidence interval 0.55–1.00). The recurrence rate was 16% for patients in the lowest quintile vs. 10.1–14.7% in the higher exposure quintiles. The investigators also explored dichotomized optimal cut-off points for the association between endoxifen concentrations and additional breast cancer events, in which an endoxifen concentration ≥ 5.97 ng/mL was the best threshold. This threshold corresponds closely to the lowest quintile [28].
With the same dataset, an anti-estrogenic activity score was developed taking into account the IC50-corrected concentrations of tamoxifen, endoxifen, 4-hydroxytamoxifen, and N-desmethyltamoxifen [29]. An anti-estrogenic activity score threshold of 1798 was associated with a hazard ratio of 0.69 (95% confidence interval 0.48–0.99). It should be noted that this anti-estrogenic activity score was dominated by endoxifen, suggesting that endoxifen can serve as a proxy for the overall anti-estrogenic effect of tamoxifen and its metabolites.
While a clear exposure–efficacy relationship has been demonstrated in the adjuvant setting, Neven et al. did not find this relationship in the neo-adjuvant and metastatic setting [30].
In a recent prospective clinical trial (n = 122), tamoxifen doses were tailored based on endoxifen concentrations [8]. Breast cancer patients with an endoxifen concentration < 5.6 ng/mL (corresponding to 15 nmol/L) received a 20-mg dose increase, while patients with endoxifen concentrations between 5.6 and 11.2 ng/mL (or 15–30 nmol/L) were recommended a dose increase of 10 mg. All patients with endoxifen concentrations ≥ 11.2 ng/mL continued treatment at the fixed dose of 20 mg of tamoxifen. In total, 68 of 122 patients had at least one dose increment, after which 96% of patients achieved an endoxifen concentration ≥ 5.6 ng/mL, compared to only 76% at baseline [8].
Although it is known that CYP2D6 intermediate and poor metabolizer phenotypes are associated with lower endoxifen concentrations, the CYP2D6 phenotype only accounts for 18–43% of the interpatient variability in endoxifen concentrations [28, 30,31,32]. Twenty-four percent of the poor metabolizers and 58% of the intermediate metabolizers still have an endoxifen concentration above the efficacy threshold, while 12% of the normal metabolizers do not reach this threshold [28]. As endoxifen concentrations cannot be adequately predicted by the CYP2D6 phenotype, we advocate endoxifen-guided dosing instead of genotype-guided dosing.
At the currently used fixed dose of 20 mg QD, 20% of patients do not reach the proposed efficacy threshold of 5.97 ng/mL, with the potential to lower the recurrence rate by 26% in this subpopulation. The presence of a large retrospective exposure–efficacy study and prospective dose individualization study supports the conclusion that it is feasible to dose tamoxifen based on measured endoxifen concentrations, using ≥ 5.97 ng/mL as a threshold, although no unequivocal evidence from a prospective trial is available yet, which demonstrates that TDM increases tamoxifen treatment efficacy.
3.2.2 Aromatase Inhibitors
Estrogens are synthesized from androgens by the aromatase enzyme complex. This enzyme system is inhibited by aromatase inhibitors (AIs). After previous use of the first- and second-generation AIs (e.g., aminoglutethimide and formestane), the third-generation AIs currently used in clinical practice are anastrozole, letrozole, and exemestane. These drugs are indicated for the treatment of postmenopausal patients with estrogen receptor-positive breast cancer, either in the (neo)adjuvant or metastatic setting [33,34,35]. Anastrozole and letrozole are non-steroidal AIs that bind reversibly to aromatase while exemestane is a steroidal AI that binds irreversibly to aromatase [36].
Since AIs inhibit the synthesis of estrogens, measuring circulating estrogen levels would be a good biomarker for efficacy. However, the sensitivity of the currently most commonly used estrogen assays is insufficient to measure the low concentrations of circulating estradiol in postmenopausal women, especially in those receiving AI treatment [37, 38]. Patients receiving anastrozole, letrozole, and exemestane treatment have median estradiol concentrations of 1.26, 0.63, and 0.63 pg/mL, respectively [39, 40]. In daily clinical practice, circulating estradiol is measured using immunoassays (optimized to measure concentrations between 40 and 2000 pg/mL [38]), while mass spectrometry would be a more sensitive method, although this method is more costly and labor intensive. Even mass spectrometry assays are not always sensitive enough to measure the low circulating concentrations of estradiol in patients receiving AI treatment, for which assays with a lower limit of quantification (LLOQ) of 0.1–0.2 pg/mL are needed [38]. A recently published article suggested measuring gonadotrophins as a possible surrogate marker for estrogen activity [41]. Future studies are needed to confirm the feasibility of gonadotrophins as a biomarker for the efficacy of AIs.
Hypothetically, one could imagine the dosing of AIs could be personalized using a pharmacodynamic biomarker, such as measured estradiol concentrations or gonadotrophin levels. In the absence of these data, individualized dosing based on pharmacokinetics is more within reach.
3.2.2.1 Anastrozole
Ingle et al. reported a high variability in anastrozole concentrations at the standard dose of 1 mg QD, with a median of 33.2 ng/mL, interquartile range of 23.5–44.8 ng/mL, and a range from LLOQ (0.1 ng/mL) to 132.1 ng/mL (n = 649) [42], while the intrapatient variability is small (7–12%) [43].
To our knowledge, no exposure–toxicity relationship has been described for anastrozole. In phase I studies, patients received repeated doses up to 10 mg QD and single doses up to 60 mg QD, which were well tolerated and did not cause any serious adverse events [44]. A linear dose–exposure relationship was found for doses of 0.5 up to 10 mg [44].
Dose–efficacy and exposure–efficacy relationships have only been studied with estrogen suppression as a surrogate marker of effect. Although previous studies showed estradiol suppression to below the limits of detection (LLOQ 2 pg/mL) for doses of 1 mg or higher [44, 45], it could still be possible that higher doses suppress estradiol to a greater extent, which could not have been quantified with these assays.
In a prospective study (n = 649), Ingle et al. reported significantly lower anastrozole concentrations in patients with stable or increased estradiol concentrations compared with patients with decreased estradiol concentrations (LLOQ 0.625 pg/mL) after the start of anastrozole treatment (26.7 vs. 34.2 ng/mL, p < 0.001) [42]. This indicates that TDM could be of value for anastrozole. However, because not all patients with decreased estradiol concentrations compared to baseline necessarily have sufficient estrogen suppression, higher anastrozole concentrations might be needed to attain adequate estrogen suppression.
No definitive exposure–efficacy target has been proposed yet for anastrozole. However, based on the available data, dose escalation could be considered for patients with a Cmin < 34.2 ng/mL [42]. Since the median exposure is 33.2 ng/mL, approximately 50% of patients will have a Cmin below this threshold at the currently used fixed dose of 1 mg QD. Future studies should further investigate the relationship of anastrozole plasma concentrations with both circulating estrogen levels and progression-free and recurrence-free survival.
3.2.2.2 Exemestane
Although exemestane is extensively metabolized, 17-hydroxy-exemestane is the only active metabolite. However, because the 17-hydroxy-exemestane concentration is ten times lower than the exemestane concentration and 17-hydroxy-exemestane is 2.6 times less potent than exemestane, its additional anti-estrogenic effect is limited [35].
Estradiol and exemestane share the same steroidal backbone. This structural resemblance can lead to falsely elevated estradiol concentrations in immunoassays. Therefore, measuring estradiol concentrations with liquid chromatography–tandem mass spectrometry instead of immunoassays is the preferred option [46].
Hertz et al. reported a median Cmax of exemestane of 7.7 ng/mL (range 2.5–72.0, n = 246) at the standard daily dose of 25 mg. Higher exemestane concentrations have been associated with the CYP3A4*22 variant, white race, elevated liver enzymes, renal insufficiency, lower body mass index (BMI), and not having received prior chemotherapy (all p < 0.05). However, these factors explained less than 10% of the overall interpatient variability in exemestane concentrations [47].
No exposure–toxicity relationship has been shown for exemestane. In general, exemestane is well tolerated, with single doses up to 800 mg and multiple doses up to 200 mg administered in phase I studies [35].
Exposure increases proportionally with increasing dose. Estrogen suppression was maximal at a dose of 25 mg (used assay is not mentioned) [35]. However, exemestane concentrations were not significantly different in patients who did and did not achieve estradiol suppression to undetectable concentrations (LLOQ 1.25 pg/mL) [48].
No exposure–efficacy analyses have been reported yet for exemestane. Future studies need to explore any relationship between exemestane exposure and clinical response. In the absence of an exposure–efficacy target, the median Cmax of 7.7 ng/mL could be used as a reference for TDM, corresponding to a calculated trough concentration of 4.1 ng/mL [49]. As this is the median exposure, approximately 50% of patients will have trough concentrations below this proposed reference.
3.2.2.3 Letrozole
Desta et al. reported a high interpatient variability, with a median steady-state exposure of 88.4 ng/mL [range: LLOQ (7 ng/mL) – 349.2 ng/mL)] at the standard dose of 2.5 mg QD [50]. Higher exposure was associated with increasing age, lower BMI, and CYP2A6 genetic variations. The lower exposure with increasing BMI can be explained by the fact that letrozole is a highly lipophilic drug with a large volume of distribution (183 L), which increases with increasing BMI. These three variables explain only 32.3% of the interpatient variability, thus a large proportion remains to be elucidated [50].
In phase I studies, single doses up to 30 mg and repeated doses up to 5 mg were well tolerated. Higher exposure did not cause increased toxicity [34]. Exposure increases approximately linearly with doses up to the standard dose of 2.5 mg QD, while at higher doses the exposure increases non-linearly [34].
No significant relationship was found between dose (range 0.5–5.0 mg) and estrogen suppression, albeit the assay used may not have been sensitive enough (LLOQ 2.5 pg/mL) [34]. Furthermore, Hertz et al. found that median steady-state concentrations of letrozole were comparable in patients who did and did not achieve E2 suppression to undetectable concentrations (LLOQ 1.25 pg/mL, 88.8 vs. 105.7 ng/mL, respectively, p = 0.63) [48].
In an exposure–efficacy analysis, patients were divided into groups reaching different letrozole plasma concentrations. This analysis showed a tendency to an increase in the time to tumor progression for those patients with a letrozole plasma concentration ≥ 85.6 ng/mL [34]. Future studies need to confirm this exposure–efficacy relationship. Until then, the most appropriate target for TDM of letrozole is 85.6 ng/mL. Because the median exposure is 88.4 ng/mL, slightly less than 50% of patients will not reach this target at the currently used fixed dose of 2.5 mg QD.
4 Discussion
The data presented in this review highlight clear opportunities to improve and to optimize treatment with anti-hormonal agents in oncology through TDM. However, the evidence for this is not equally strong for all agents. Because of this, we evaluated the available evidence and proposed TDM recommendations, which we considered either exploratory, promising, or viable, as presented in Table 2. The provided TDM recommendation is considered promising if a PK TDM target is available or viable if a prospective TDM study has been conducted. Otherwise the recommendations are considered exploratory.
Future studies are needed to explore exposure–efficacy relationships for those oral anti-hormonal drugs that are classified as exploratory (anastrozole, enzalutamide, and exemestane). In addition, prospective clinical studies should be performed to demonstrate the safety and feasibility of TDM for those oral anti-hormonal drugs that are classified as promising (abiraterone and letrozole). Ideally, for those drugs for which TDM is viable (tamoxifen), randomized controlled trials comparing TDM and fixed dosing with regard to relevant clinical efficacy endpoints such as progression-free survival and overall survival would be needed. Then, TDM could be fully integrated in clinical practice and become the standard of care. However, given the large patient numbers needed to conduct adequately powered randomized controlled trials, especially in the adjuvant setting, this will be a major challenge. Instead, future research could focus on prospective clinical studies strengthening the evidence of the PK target and confirming the safety and feasibility of TDM [51].
Currently, exposure–efficacy and exposure–toxicity analyses are pivotal parts of the drug development process [52]. However, in the era in which most of the older oral anti-hormonal drugs were registered, this was uncommon, resulting in a paucity of PK–PD data for these agents. Nonetheless, a PK target could be identified for four of the seven included agents. Overall, these targets amounted to 85% (± 19%) of the mean population exposure (Fig. 1). This is in accordance with the data for kinase inhibitors, as reported previously of 82% (± 17%) [3, 4]. Thus, targeting the mean exposure, in the absence of exposure–efficacy analyses, generally leads to adequate concentrations.

Proposed therapeutic drug monitoring (TDM) thresholds as percentages of the average exposure, on average the threshold amounted to 85% (± 19%) of the population average (indicated by the dotted line)
While awaiting a TDM target based on exposure–response analyses, measuring drug concentrations and collecting data on the efficacy and toxicity in routine patient care can provide us with valuable data on exposure–efficacy and exposure–toxicity relationships, comparable to safety monitoring as part of post-marketing surveillance.
To measure drug concentrations of oral anti-hormonal drugs, validated bio-analytical assays are needed. Our methods for the quantification of abiraterone, enzalutamide, and endoxifen have been previously published [53,54,55]. Additionally, methods on the quantification of anastrozole, letrozole, and exemestane have been published by other investigators [56,57,58]. Currently, we are validating an assay for the simultaneous measurement of all mentioned oral anti-hormonal drugs, which makes this a suitable assay for TDM in clinical practice.
Apart from the apparent advantages of TDM, another potential advantage for anti-hormonal drugs could be the monitoring of medication adherence, as it is has been shown that compliance decreases with long-term treatment [59]. Additionally, TDM could help in detecting drug–drug interactions.
Since many of the anti-hormonal drugs have considerably long elimination half-lives, PK sampling for TDM should be timed appropriately to ensure that steady-state concentration has been achieved. In Table 1, the time until steady-state concentration has been reached is specified for the different compounds.
In disciplines other than oncology, TDM is being broadly applied, for example in patients using antibiotics, antiretroviral drugs, and immunosuppressants. An important difference, however, is the fact that in oncology we are reluctant to reduce the dose in the case of high drug concentrations because tumor progression is irreversible. For this reason, we advise to increase the dose in the case of low drug concentrations, while reducing doses only in the case of toxicity.
5 Conclusion
This review has summarized the clinical PK and PD properties of oral anti-hormonal drugs used in daily oncology practice and aimed to translate these data into practical guidelines for TDM. For abiraterone, anastrozole, and letrozole, PK targets for TDM could be identified. Furthermore, for tamoxifen, a prospective clinical trial has already demonstrated the feasibility of individualizing the dose based on the endoxifen concentration. However, prospective studies to correlate individualized dosing with tumor response or outcome parameters, such as progression-free survival and overall survival, are lacking. To conclude, the data presented in this review highlight clear opportunities to study and improve treatment with oral anti-hormonal agents in oncology via TDM.
References
Siegel RL, Miller KD, Jemal A. Cancer statistics. CA Cancer J Clin. 2016;66:7–30.
Committee for Medicinal Products for Human Use European Medicines Agency. Abiraterone European public assessment report. 2011. http://www.ema.europa.eu/docs/en_GB/document_library/EPAR_-_Product_Information/human/002321/WC500112858.pdf. Accessed 26 May 2018.
Verheijen R, Yu H, Schellens J, Beijnen J, Steeghs N, Huitema A. Practical recommendations for therapeutic drug monitoring of kinase inhibitors in oncology. Clin Pharmacol Ther. 2017;102:765–76.
Yu H, Steeghs N, Nijenhuis C, Schellens J, Beijnen J, Huitema A. Practical guidelines for therapeutic drug monitoring of anticancer tyrosine kinase inhibitors: focus on the pharmacokinetic targets. Clin Pharmacokinet. 2014;53:305–25.
Beumer JH. Without therapeutic drug monitoring, there is no personalized cancer care. Clin Pharmacol Ther. 2013;93:228–30.
Paci A, Veal G, Bardin C, Levêque D, Widmer N, Beijnen J, et al. Review of therapeutic drug monitoring of anticancer drugs part 1: cytotoxics. Eur J Cancer. 2014;50:2010–9.
Widmer N, Bardin C, Chatelut E, Paci A, Beijnen J, Levêque D, et al. Review of therapeutic drug monitoring of anticancer drugs part two: targeted therapies. Eur J Cancer. 2014;50:2020–36.
Fox P, Balleine RL, Lee C, Gao B, Balakrishnar B, Menzies AM, et al. Dose escalation of tamoxifen in patients with low endoxifen level: evidence for therapeutic drug monitoring: the TADE study. Clin Cancer Res. 2016;22:3164–71.
De Wit D, Guchelaar HJ, Den Hartigh J, Gelderblom H, Van Erp NP. Individualized dosing of tyrosine kinase inhibitors: are we there yet? Drug Discov Today. 2015;20:18–36.
US Food and Drug Administration. Center for Drug Evaluation and Research. Abiraterone clinical pharmacology and biopharmaceutics review. 2011. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2011/202379Orig1s000ClinPharmR.pdf. Accessed 26 May 2018.
James ND, de Bono JS, Spears MR, Clarke NW, Mason MD, Dearnaley DP, et al. Abiraterone for prostate cancer not previously treated with hormone therapy. N Engl J Med. 2017;377:338–51.
Chi KN, Spratlin J, Kollmannsberger C, North S, Pankras C, Gonzalez M, et al. Food effects on abiraterone pharmacokinetics in healthy subjects and patients with metastatic castration-resistant prostate cancer. J Clin Pharmacol. 2015;55:1406–14.
Carton E, Noe G, Huillard O, Golmard L, Giroux J, Cessot A, et al. Relation between plasma trough concentration of abiraterone and prostate-specific antigen response in metastatic castration-resistant prostate cancer patients. Eur J Cancer. 2017;72:54–61.
Stuyckens K, Saad F, Xu XS, Ryan CJ, Smith MR, Griffin TW, et al. Population pharmacokinetic analysis of abiraterone in chemotherapy-naïve and docetaxel-treated patients with metastatic castration-resistant prostate cancer. Clin Pharmacokinet. 2014;53:1149–60.
Attard G, Reid AHM, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71.
Steven X, Charles X, Kim JR, Matthew S, Saad F, Griffin TW, et al. Modeling the relationship between exposure to abiraterone and prostate-specific antigen dynamics in patients with metastatic castration-resistant prostate cancer. Clin Pharmacokinet. 2017;56:55–63.
Li Z, Bishop AC, Alyamani M, Garcia JA, Dreicer R, Bunch D, et al. Conversion of abiraterone to D4A drives anti-tumour activity in prostate cancer. Nature. 2015;523:347–51.
Emamekhoo H, Li Z, Sharifi N. Clinical significance of D4A in prostate cancer therapy with abiraterone. Cell Cycle. 2015;14:3213–4.
US Food and Drug Administration. Center for Drug Evaluation and Research. Enzalutamide clinical pharmacology and biopharmaceutics review. 2012. http://www.accessdata.fda.gov/drugsatfda_docs/nda/2012/203415Orig1s000ClinPharmR.pdf. Accessed 26 May 2018.
Gibbons JA, Ouatas T, Krauwinkel W, Ohtsu Y, van der Walt J-S, Beddo V, et al. Clinical pharmacokinetic studies of enzalutamide. Clin Pharmacokinet. 2015;54:1043–55.
Scher HI, Anand A, Rathkopf D, Shelkey J, Morris MJ, Danila DC, et al. Antitumour activity of MDV3100 in castration-resistant prostate cancer: a phase 1-2 study. Lancet. 2010;375:1437–46.
Armstrong AJ, Saad F, Phung D, Dmuchowski C, Shore ND, Fizazi K, et al. Clinical outcomes and survival surrogacy studies of prostate-specific antigen declines following enzalutamide in men with metastatic castration-resistant prostate cancer previously treated with docetaxel. Cancer. 2017;123:2303–11.
de Vries Schultink AHM, Zwart W, Linn SC, Beijnen JH, Huitema ADR. Effects of pharmacogenetics on the pharmacokinetics and pharmacodynamics of tamoxifen. Clin Pharmacokinet. 2015;54:797–810.
Jager NGL, Rosing H, Schellens JHM, Linn SC, Beijnen JH. Tamoxifen dose and serum concentrations of tamoxifen and six of its metabolites in routine clinical outpatient care. Breast Cancer Res Treat. 2014;143:477–83.
Borges S, Desta Z, Li L, Skaar TC, Ward BA, Nguyen A, et al. Quantitative effect of CYP2D6 genotype and inhibitors on tamoxifen metabolism: implication for optimization of breast cancer treatment. Clin Pharmacol Ther. 2006;80:61–74.
Mürdter TE, Schroth W, Bacchus-Gerybadze L, Winter S, Heinkele G, Simon W, et al. Activity levels of tamoxifen metabolites at the estrogen receptor and the impact of genetic polymorphisms of phase I and II enzymes on their concentration levels in plasma. Clin Pharmacol Ther. 2011;89:1–10.
Jager N, Koornstra R, Vincent A, van Schaik R, Huitema A, Korse C, et al. Hot flashes are not predictive for serum concentrations of tamoxifen and its metabolites. BMC Cancer. 2013;13:612.
Madlensky L, Natarajan L, Tchu S, Pu M, Mortimer J, Flatt SW, et al. Tamoxifen metabolite concentrations, CYP2D6 genotype, and breast cancer outcomes. Clin Pharmacol Ther. 2011;89:718–25.
de Vries Schultink AHM, Alexi X, van Werkhoven E, Madlensky L, Natarajan L, Flatt SW, et al. An antiestrogenic activity score for tamoxifen and its metabolites is associated with breast cancer outcome. Breast Cancer Res Treat. 2017;161(3):567–74.
Neven P, Jongen L, Lintermans A, Van Asten K, Blomme C, Lambrechts D, et al. Tamoxifen metabolism and efficacy in breast cancer: a prospective multicentre trial. Clin Cancer Res. 2018;24(10):2312–8.
Jin Y, Desta Z, Stearns V, Ward B, Ho H, Lee KH, et al. CYP2D6 genotype, antidepressant use, and tamoxifen metabolism during adjuvant breast cancer treatment. J Natl Cancer Inst. 2005;97:30–9.
Dezentje V, den Hartigh J, Guchelaar H, Hessing T, van der Straaten T, Vletter-Bogaartz J. Association between endoxifen serum concentration and predicted CYP2D6 phenotype in a prospective cohort of patients with early-stage breast cancer. J Clin Oncol. 2011;15(Suppl.):562.
US Food and Drug Administration. Center for Drug Evaluation and Research. Anastrozole clinical pharmacology and biopharmaceutics review. 2000. https://www.accessdata.fda.gov/drugsatfda_docs/nda/2000/20-541S006_Arimidex_biopharmr.pdf. Accessed 26 May 2018.
US Food and Drug Administration. Center for Drug Evaluation and Research. Letrozole clinical pharmacology and biopharmaceutics review. https://www.accessdata.fda.gov/drugsatfda_docs/nda/97/20726_FEMARA 2.5MG_BIOPHARMR.PDF. Accessed 26 May 2018.
US Food and Drug Administration. Center for Drug Evaluation and Research. Exemestane clinical pharmacology and biopharmaceutics review. 1999. https://www.accessdata.fda.gov/drugsatfda_docs/nda/99/20-753_Aromasin_biopharmr_P1.pdf. Accessed 26 May 2018.
Kelly CM, Buzdar AU. Anastrozole. Expert Opin Drug Saf. 2010;9:995–1003.
Pauwels S, Lintermans A, Neven P, Verhaeghe J, Jans I, Billen J, et al. Need for estradiol assays with a lower functional sensitivity in clinical studies examining postmenopausal women treated with aromatase inhibitors. J Clin Oncol. 2013;31:509.
Ketha H, Girtman A, Singh RJ. Estradiol assays: the path ahead. Steroids. 2015;99:39–44.
Ingle JN, Buzdar AU, Schaid DJ, Goetz MP, Batzler A, Robson ME, et al. Variation in anastrozole metabolism and pharmacodynamics in women with early breast cancer. Cancer Res. 2010;70:3278–86.
Folkerd EJ, Dixon JM, Renshaw L, A’Hern RP, Dowsett M. Suppression of plasma estrogen levels by letrozole and anastrozole is related to body mass index in patients with breast cancer. J Clin Oncol. 2012;30:2977–80.
Oberguggenberger A, Meraner V, Sztankay M, Beer B, Weigel G, Oberacher H, et al. Can we use gonadotropin plasma concentration as surrogate marker for BMI-related incomplete estrogen suppression in breast cancer patients receiving anastrozole? BMC Cancer. 2017;17:1–7.
Ingle JN, Kalari KR, Buzdar AU, Robson ME, Goetz MP, Desta Z, et al. Estrogens and their precursors in postmenopausal women with early breast cancer receiving anastrozole. Steroids. 2015;99:32–8.
Micheal F, Saranya S, Aparna N, Sridevi N, Chithra R, Judith MP. Concepts of bioequivalence and its impact on truncated area under curve (AUC) of drugs with long half life in point estimate and intra-subject variability. J Pharm Sci Res. 2012;4:1890–6.
Plourde P, Dyroff M, Dukes M. Arimidex: a potent and selective fourth-generation aromatase inhibitor. Breast Cancer Res Treat. 1994;30:103–11.
Geisler J, King N, Dowsett M, Ottestad L, Lundgren S, Walton P, et al. Influence of anastrozole (Arimidex), a selective, non-steroidal aromatase inhibitor, on in vivo aromatisation and plasma oestrogen levels in postmenopausal women with breast cancer. Br J Cancer. 1996;74:1286–91.
Mandic S, Kratzsch J, Mandic D, Debeljak Z, Lukic I, Horvat V, et al. Falsely elevated serum oestradiol due to exemestane therapy. Ann Clin Biochem. 2017;54(3):402–5.
Hertz DL, Kidwell KM, Seewald NJ, Gersch CL, Desta Z, Flockhart DA, et al. Polymorphisms in drug-metabolizing enzymes and steady-state exemestane concentration in postmenopausal patients with breast cancer. Pharmacogenom J. 2017;17(6):521–7.
Hertz DL, Speth KA, Kidwell KM, Gersch CL, Desta Z, Storniolo AM, et al. Variable aromatase inhibitor plasma concentrations do not correlate with circulating estrogen concentrations in post-menopausal breast cancer patients. Breast Cancer Res Treat. 2017;165(3):659–68.
Wang Y, Chia Y, Nedelman J, Schran H, Mahon F, Molimard M. A therapeutic drug monitoring algorithm for refining the imatinib trough level obtained at different sampling times. Ther Drug Monit. 2009;31:579–84.
Desta Z, Kreutz Y, Nguyen AT, Li L, Skaar T, Kamdem LK, et al. Plasma letrozole concentrations in postmenopausal women with breast cancer are associated with CYP2A6 genetic variants, body mass index, and age. Clin Pharmacol Ther. 2011;90:693–700.
De Jonge ME, Huitema ADR, Schellens JHM, Rodenhuis S, Beijnen JH. Individualised cancer chemotherapy: strategies and performance of prospective studies on therapeutic drug monitoring with dose adaptation: a review. Clin Pharmacokinet. 2005;44:147–73.
US Food and Drug Administration. Center for Drug Evaluation and Research. Guidance for industry: exposure–response relationships: study design, data analysis and regulatory applications. FDA Guide. 2003;1–25. https://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072109.pdf.
van Nuland M, Hillebrand MJX, Rosing H, Schellens JHM, Beijnen JH. Development and validation of an LC–MS/MS method for the simultaneous quantification of abiraterone, enzalutamide, and their major metabolites in human plasma. Ther Drug Monit. 2017;39:243–51.
de Krou S, Rosing H, Nuijen B, Schellens JHM, Beijnen JH. Fast and adequate liquid chromatography-tandem mass spectrometric determination of Z-endoxifen serum levels for terapeutic drug monitoring. Ther Drug Monit. 2017;39:132–7.
van Nuland M, Rosing H, de Vries J, Ovaa H, Schellens JHM, Beijnen JH. An LC–MS/MS method for quantification of the active abiraterone metabolite Δ(4)-abiraterone (D4A) in human plasma. J Chromatogr B Anal Technol Biomed Life Sci. 2017;1068–10699:119–24.
Shao R, Yu L, Lou H, Ruan Z, Jiang B, Chen J. Development and validation of a rapid LC–MS/MS method to quantify letrozole in human plasma and its application to therapeutic drug monitoring. Biomed Chromatogr. 2016;30:632–7.
Yu J, He J, Zhang Y, Qin F, Xiong Z, Li F. Development of a liquid chromatography–tandem mass spectrometry method for determination of butoconazole nitrate in human plasma and its application to a pharmacokinetic study. Biomed Chromatogr. 2011;25:511–6.
Wang L-Z, Goh S-H, Wong AL-A, Thuya W-L, Lau J-YA, Wan S-C, et al. Validation of a rapid and sensitive LC–MS/MS method for determination of exemestane and its metabolites, 17beta-hydroxyexemestane and 17beta-hydroxyexemestane-17-O-beta-d-glucuronide: application to human pharmacokinetics study. PLoS One. 2015;10(3):e0118553.
Cardoso E, Csajka C, Schneider MP, Widmer N. Effect of adherence on pharmacokinetic/pharmacodynamic relationships of oral targeted anticancer drugs. Clin Pharmacokinet. 2018;57(1):1–6.
Gervasini G, Jara C, Olier C, Romero N, Martinez R, Carrillo JA. Polymorphisms in ABCB1 and CYP19A1 genes affect anastrozole plasma concentrations and clinical outcomes in postmenopausal breast cancer patients. Br J Clin Pharmacol. 2017;83:562–71.
Dowsett M, Cuzick J, Howell A. Jackson I; ATAC Trialists’ Group. Pharmacokinetics of anastrozole and tamoxifen alone, and in combination, during adjuvant endocrine therapy for early breast cancer in postmenopausal women: a sub-protocol of the “Arimidex™ and tamoxifen alone or in combination” (ATAC) trial. Br J Cancer. 2001;85:317–24.
Hubalek M, Oberguggenberger A, Beer B, Meraner V, Sztankay M, Oberacher H, et al. Does obesity interfere with anastrozole treatment? Positive association between body mass index and anastrozole plasma levels. Clin Breast Cancer. 2014;14:291–6.
Committee for Medicinal Products for Human Use European Medicines Agency. Public assessment report: scientific discussion exemestane. 2010. https://db.cbg-meb.nl/Pars/h104327.pdf%0A. Accessed 26 May 2018.
Bisagni G, Cocconi G, Scaglione F, Fraschini F, Pfister C, Trunet PF. Letrozole, a new oral non-steroidal aromatase inhibitor in treating postmenopausal patients with advanced breast cancer: a pilot study. Ann Oncol. 1996;7:99–102.
Binkhorst L, Kloth JSL, de Wit AS, de Bruijn P, Lam MH, Chaves I, et al. Circadian variation in tamoxifen pharmacokinetics in mice and breast cancer patients. Breast Cancer Res Treat. 2015;152:119–28.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Funding
No funding was received for the preparation of this article.
Conflict of interest
Remy B. Verheijen is currently a full-time employee of AstraZeneca, Cambridge, UK. Although Jan H. M. Schellens is involved in Modra Pharmaceuticals, this article does not contain information that poses a conflict of interest as it does not examine any product of Modra Pharmaceuticals or products related to this spinout company. Stefanie L. Groenland, Merel van Nuland, Jos H. Beijnen, Alwin D. R. Huitema, and Neeltje Steeghs have no conflicts of interest directly relevant to the content of this article.
Rights and permissions
About this article

Cite this article
Groenland, S.L., van Nuland, M., Verheijen, R.B. et al. Therapeutic Drug Monitoring of Oral Anti-Hormonal Drugs in Oncology. Clin Pharmacokinet 58, 299–308 (2019). https://doi.org/10.1007/s40262-018-0683-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-018-0683-0