
Abstract
Background and Objectives
Recent analysis revealed strong associations between prostate-specific antigen (PSA) dynamics and overall survival (OS) in metastatic castration-resistant prostate cancer (mCRPC) and supported PSA dynamics as bridging surrogacy endpoints for clinical benefit from treatment with abiraterone acetate plus prednisone. This analysis aimed to investigate the abiraterone exposure–PSA dynamics relationship in mCRPC.
Methods
Abiraterone pharmacokinetics–PSA models were constructed using data from the COU-AA-301 (chemotherapy-pretreated) and COU-AA-302 (chemotherapy-naïve) trials comparing abiraterone acetate 1000 mg/day plus prednisone 5 mg twice daily with prednisone alone in mCRPC. The drug effect–PSA dynamics relationship was modeled as a function of selected pharmacokinetic measures. The influences of baseline demographic variables, laboratory values, and disease status on PSA dynamics were assessed.
Results
A tumor growth inhibition model best described PSA dynamics post-treatment with abiraterone acetate. Abiraterone acetate treatment in chemotherapy-pretreated and chemotherapy-naïve patients increased the PSA decay rate (k dec) to the same extent (1.28-fold, 95 % confidence interval [CI] 0.58–1.98; and 0.93-fold, 95 % CI 0.6–1.27, respectively). Lower baseline lactate dehydrogenase and higher baseline testosterone significantly increased k dec. Findings from our analysis suggest a maximum-effect relationship between abiraterone trough concentration and PSA dynamics in both patient populations. The majority of patients had a steady-state trough concentration greater than the estimated half maximal effective concentration.
Conclusion
The model appropriately described the exposure–response relationship between abiraterone and PSA dynamics in chemotherapy-pretreated and chemotherapy-naïve patients following oral administration of abiraterone acetate.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
The pharmacokinetics–prostate-specific antigen (PSA) model is an appropriate pharmacokinetics/pharmacodynamics model, with clinical utility in describing the time course of PSA responses following treatment with abiraterone acetate plus prednisone. |
Abiraterone exposure significantly increased PSA decay rate, with similar drug effects in chemotherapy-naïve and chemotherapy-pretreated patients with metastatic castration-resistant prostate cancer. |
Serum abiraterone concentrations above the model-predicted half maximal effective concentration were observed in the majority of the study patients. |
1 Introduction
Pharmacokinetic/pharmacodynamic modeling has been used widely to quantify exposure–response relationship for therapeutic agents [1]. Examining such relationships is instrumental for evaluating efficacy of the dosing regimens, supporting the choice of adopted dose levels, and providing information on how, and whether, exposure can be adjusted for various subsets of a given patient population [1].
Treatment of patients with abiraterone acetate, the prodrug of the androgen biosynthesis inhibitor abiraterone, results in prostate-specific antigen (PSA) decline by ≥50 % in approximately 40–50 % of chemotherapy-pretreated patients with metastatic castration-resistant prostate cancer (mCRPC) and in approximately 50–70 % of patients with chemotherapy-naive mCRPC [2–5]. These PSA declines have been directly linked to the reduction of residual testosterone concentrations and inhibition of adrenal androgens through the selective inhibition of cytochrome P450 (CYP) 17 by abiraterone [6].
Our recent PSA survival analysis of COU-AA-301 (chemotherapy-pretreated patients) [7] and COU-AA-302 (chemotherapy-naïve patients) [8] showed strong associations between abiraterone acetate–induced PSA declines and overall survival (OS), and good predictive performance of PSA for survival outcome following administration of abiraterone acetate plus prednisone [9]. A model that characterizes the pharmacokinetics–PSA relationship for abiraterone acetate–treated patients from the same studies would therefore provide an invaluable link between abiraterone exposure and survival in the patient populations. Longitudinal abiraterone pharmacokinetics and PSA data obtained in the COU-AA-301 and COU-AA-302 studies were used to quantify this exposure–response relationship.
The objective of this analysis was to investigate the relationship between abiraterone exposure and PSA dynamics through modeling the time course of PSA concentrations. We hypothesized that the magnitude of PSA reduction was related to the extent of abiraterone exposure in both chemotherapy-pretreated and chemotherapy-naïve patients with mCRPC.
2 Methods
2.1 Patients and Treatment
Key details on the eligibility criteria, baseline patient characteristics, and study designs for COU-AA-301 (N = 1195) [7] and COU-AA-302 (N = 1088) [8] have been previously published. In both phase III studies, oral abiraterone acetate 1000 mg once daily was administered with prednisone 5 mg twice daily (abiraterone arm) and compared with placebo plus prednisone (prednisone arm). Abiraterone acetate was taken at least 1 h before a meal or at least 2 h after a meal, as recommended.
The review boards at all participating institutions approved these studies, which were conducted in accordance with the principles set forth in the Declaration of Helsinki, the International Conference on Harmonisation, and the Guidelines for Good Clinical Practice. Written informed consent was obtained from each patient prior to participation in both trials.
2.2 Prostate-Specific Antigen (PSA) Assessment
The PSA concentrations were analyzed in a central laboratory and used for model development. Patients from the pharmacokinetic subpopulation in the abiraterone arm and all patients in the prednisone arm from each trial were included in the model development. All patients received at least one dose of study drug and had a baseline, and a minimum of one post-baseline, PSA measurement available.
The PSA measurements were obtained at prespecified times between screening visit and end-of-treatment visit, with the majority of post-dose PSA measurements obtained after at least 4 weeks of exposure to abiraterone. In COU-AA-301, PSA measurements were conducted at screening, every three cycles (day 1 of cycles 1, 4, 7, 10), and at the end-of-treatment visit. In COU-AA-302, PSA measurements were taken at screening; day 1 of cycles 1, 3, 5, 7, and 10; every three cycles after cycle 10; and at the end-of-treatment visit. The median numbers of cycles of abiraterone acetate administered were 8 and 15 for COU-AA-301 and COU-AA-302, respectively.
A median of three (range 1–13) and six (range 1–13) PSA concentrations per patient were collected from COU-AA-301 and COU-AA-302, respectively. In total, 1717 PSA measurements from 555 patients in COU-AA-301 and 3995 PSA measurements from 642 patients in COU-AA-302 were included in the analysis.
2.3 Assessment of Exposure to Abiraterone
Pharmacokinetic data, which included a maximum of seven time points per patient during the course of treatment, were obtained from 162 patients in COU-AA-301 and 103 patients in COU-AA-302. The elimination half-life of abiraterone is approximately 12 h [10]. Therefore, the pharmacokinetics of abiraterone are expected to reach steady state 3 days after abiraterone acetate administration. Post-treatment PSA measurements were obtained after at least 4 weeks of exposure to abiraterone; individual steady-state trough concentrations (C min) were used to drive the pharmacokinetics–PSA dynamics. The individual steady-state C min were predicted based on the individual parameters derived from a two-compartment pharmacokinetic model [11].
2.4 Development of the PSA Dynamic Model
The time course of PSA generally showed non-linear behavior over time, with an initial drop from the baseline value followed by a maximal change and, in some instances, late recovery toward the baseline or below baseline. Four structural models that were frequently used in modeling PSA profiles or tumor growth were compared to select the PSA model that best described the time course of PSA responses to abiraterone acetate. The investigated structural PSA model included a bi-exponential model [12], a mixed exponential and linear model [13], a mixed Weibull and linear model [14], and a tumor growth inhibition (TGI) model [15–17].
Akaike’s information criterion (AIC) was used to compare the investigated base structural models (i.e. without covariates) to take into account the trade-off between fit of the models to the data and the corresponding complexity of the models (Online Resource 1). Based on the AIC, the TGI model was found to best describe the sparse PSA concentrations over time compared with the other investigated models, including the commonly used biexponential model [12].
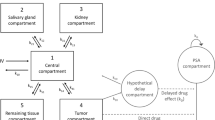
The TGI model is a differential equation approach to describe tumor growth:
where B i is the baseline PSA at time 0 for the ith patient; k prod,i is the first-order production rate of PSA for the ith patient; k dec,i is the PSA decay rate due to treatment (i.e. exposure) via tumor inhibition for the ith patient; \(e^{{ - \lambda_{i} t}}\) is an exponentially decreasing function over time to account for declining drug effect over time possibly due to the development of the potential tumor resistance [18–21], and f(Exposure i ) is a function of the drug exposure (e.g. treatment information or various pharmacokinetic metrics such as steady-state C min and area under the plasma concentration–time curve [AUC]) for the ith patient. Interindividual random effects were included in the model parameters to account for the correlation among the individual time courses of PSA responses and for the heterogeneous patterns of individual PSA time profiles. Patient-specific parameter estimates were given by, for example, k dec,i = k dec · exp(η i ), where k dec is the population mean of the PSA decay rate and η i is the difference between the individual and population mean PSA decay rate on a log scale assumed to follow a normal distribution, with a mean of zero and variance of ω 2. Log-transformed PSA data were modeled due to wide ranges of PSA concentrations commonly observed in clinical studies [12, 22]. The additive residual error for the log-transformed PSA concentrations, accounting for measurement error, model misspecifications, and other unexplained sources of variability, followed a normal distribution with a mean of zero and variance of σ 2. The time course of PSA responses from patients was analyzed by non-linear mixed-effect analysis with NONMEM® version 7.2.0 (Icon Development Solutions, Ellicott City, MD, USA).
2.5 Modeling Drug Effect on the Pharmacodynamics of PSA
The effect of abiraterone on PSA dynamics was investigated at two different levels: (i) by treatment arm to evaluate the treatment effect (abiraterone acetate + prednisone) versus prednisone alone; and (ii) by individual to evaluate the exposure–response relationship using individual predicted concentration data (e.g. C min) based on a population pharmacokinetic model for abiraterone [11]. The treatment effect θ TRT (abiraterone acetate + prednisone vs. prednisone) was incorporated as the exposure measure into the model as follows:
The concentration–effect relationship was described by a simple maximum drug effect (E max)-type function:
The individual steady-state C min (empirical Bayes estimates) for the pharmacokinetic population was predicted using the published population pharmacokinetic model [11], while the C min was set to 0 for patients from the prednisone arm, since prednisone coadministered with abiraterone acetate is an active agent and this arm provided important baseline data. It should be noted that steady-state AUC was also tested to fit the model. The steady-state C min provided A better model fit (i.e. lower AIC) than AUC, and was therefore selected as the exposure metric for the pharmacokinetic/pharmacodynamic model. The shrinkage was small (<10 %) for the primary pharmacokinetic parameter (i.e. apparent oral clearance) and has been previously reported [11].
2.6 Influence of Baseline Covariates on PSA Dynamics
The influence of baseline covariates was assessed for the pharmacokinetics–PSA dynamics model. Baseline demographics (e.g. age, body weight, race), laboratory values (e.g. albumin, hemoglobin, lactate dehydrogenase [LDH], alkaline phosphatase, testosterone, dehydroepiandrosterone, androstenedione, alanine aminotransferase, aspartate aminotransferase, total bilirubin, etc.), type of progression (e.g. radiographic progression vs. PSA-only progression), and number of prior chemotherapy regimens were evaluated for their influence on PSA dynamics. Graphical exploratory analysis of the individual empirical Bayes estimates of random effects of model parameters (i.e. k prod, k dec, and λ) was used for screening the covariates. The covariates associated with the model parameters were included in the PSA dynamic model. Continuous covariates were included in the model by use of a power function after centering on the median, while categorical variables were modeled as a single factor and compared with a selected level for each variable. Since the covariate analysis was exploratory in nature, no formal multiplicity adjustments were made.
2.7 Model Evaluation
As longitudinal data collected in survival studies are usually accompanied by a large percentage of missing data due to patient death or censoring before the end of the study (e.g. >90 % for the current studies), the use of standard model evaluation methods (e.g. residuals, visual predictive check plots, or other methods involving population model parameters) becomes problematic because their reference distribution is not directly available [23]. The present mixed-effects PSA modeling was based on ‘missing at random’, a commonly used and recommended assumption for missing data by the US FDA for analysis of longitudinal data in the clinical studies [24]. The model was evaluated using goodness-of-fit criteria. The primary diagnostic criterion was the match between individual predicted values and observed data, as used in similar studies [13], and was the main diagnostic criterion used in many recent publications for PSA dynamic models and other tumor dynamic models [25–27].
3 Results
3.1 Model for PSA Dynamics
The parameter estimates of the final dynamic model for chemotherapy-pretreated and chemotherapy-naïve patients of the pharmacokinetic subpopulation are listed in Table 1. Chemotherapy-pretreated patients had a similar population mean k prod and k dec (0.24 [0.20–0.28] month−1 and 0.20 [0.16–0.26] month−1, respectively) compared with chemotherapy-naïve patients (0.20 [0.18–0.22] month−1 and 0.20 [0.17–0.24] month−1, respectively). The estimate of the resistance rate constant was greater in chemotherapy-pretreated (0.20 [0.12–0.32] month−1) compared with chemotherapy-naïve patients (0.09 [0.07–0.10] month−1). The estimated baseline PSA was threefold higher for chemotherapy-pretreated (124 [107–141] ng/mL) than for chemotherapy-naïve patients (40.2 [35.6–44.8] ng/mL). Large (>50 %) interindividual variability in k prod, k dec, resistance rate constant, and baseline PSA was identified, indicating heterogeneous PSA dynamics.
3.2 Drug Effect on PSA Dynamics
Figure 1 depicts a higher k dec in patients in the abiraterone arm than in those in the prednisone arm. This increase in k dec was similar in the chemotherapy-pretreated and chemotherapy-naïve patients.

Box plot for interindividual variability (random effects) for individual PSA reduction rate constant from the base tumor growth inhibition models by treatment (abiraterone acetate + prednisone vs. prednisone) in the COU-AA-301 (chemotherapy-pretreated) and COU-AA-302 (chemotherapy-naïve) studies. PSA prostate-specific antigen
The model characterized the influence of exposure to abiraterone (Table 2). The estimated treatment effect (abiraterone acetate + prednisone vs. prednisone) was similar in both patient groups, i.e. a 1.28-fold (95 % confidence interval [CI] 0.58–1.98) and 0.93-fold (95 % CI 0.6–1.27) increase in k dec for chemotherapy-pretreated and chemotherapy-naïve patients, respectively. There was no treatment effect on k prod (Online Resource 2).
Further exploration of the exposure–response relationship (Fig. 2) revealed an E max association between k dec and abiraterone C min in both chemotherapy-pretreated and chemotherapy-naïve patients. The E max exposure–response relationship was similar in both patient groups. The estimated maximum effect of abiraterone on k dec was 1.72 (95 % CI 0.88–2.56) times higher than that of the prednisone arm in the chemotherapy-pretreated patients (Table 2). Similarly, the maximum effect of abiraterone was estimated at 1.12 (95 % CI 0.60–1.64) in the chemotherapy-naïve patients (Table 2). The model-estimated half maximal effective concentration (EC50) for PSA reduction was 4.75 ng/mL (95 % CI 3.09–6.41 ng/mL) and 1.56 ng/mL (95 % CI –2.61 to 5.73 ng/mL) for chemotherapy-pretreated and chemotherapy-naïve patients, respectively (Table 2). The E max relationships are deemed similar between patient populations due to overlapping CIs for both the estimated maximum effect and EC50. This was further supported by the similarity in the estimated treatment effect (abiraterone acetate + prednisone vs. prednisone) and the graphical analysis of patients in the COU-AA-301 and COU-AA-302 studies (Fig. 2), where the smoothing curves for the effect on k dec appear to be similar in both patient populations. The pharmacokinetics–PSA model demonstrated that approximately 93 and 97 % of chemotherapy-pretreated and chemotherapy-naïve patients, respectively, had a steady-state C min greater than the estimated EC50 values (Fig. 3).

Scatter plot of interindividual variability (random effects) for individual PSA decay rate constant from the base tumor growth inhibition models versus observed individual steady-state trough concentrations (C min) in the COU-AA-301 (chemotherapy-pretreated) and COU-AA-302 (chemotherapy-naïve) studies. Curves represent smoothing lines. PSA prostate-specific antigen

Box plot of predicted steady-state trough concentrations (C min) for patients with pharmacokinetic samples in the COU-AA-301 (chemotherapy-pretreated) and COU-AA-302 (chemotherapy-naïve) studies. Dotted line indicates the model-predicted estimated half maximal effective concentration
3.3 Covariate Effect on PSA Dynamics
Both LDH and testosterone were identified as baseline covariates that influenced PSA dynamics (p < 0.0001). In this analysis, baseline LDH had a negative impact on k dec (Table 1). The lower the baseline LDH concentration, the higher the k dec. The estimated power coefficient for LDH was –0.76 (95 % CI –0.95 to –0.57) and –0.29 (95 % CI –0.59 to 0.02) for the chemotherapy-pretreated and chemotherapy-naïve patients, respectively. Baseline testosterone was also identified as a significant covariate for k dec following abiraterone acetate administration. The higher the testosterone concentration, the higher the k dec (estimated power coefficient = 0.22 [95 % CI 0.11–0.33] for chemotherapy-pretreated patients) (Table 1).
3.4 Model Evaluation
Individual observed PSA concentrations along with individual and population predictions of PSA dynamics for 25 randomly sampled patients from the final model are shown in Online Resource 3. Flexibility of the TGI model of PSA dynamics in describing various types of individual profiles, such as linear increase/decrease, sigmoid decrease, bell-shaped time course, and exponential decrease, is also illustrated. Diagnostic plots of the observed versus predicted PSA concentrations for individual patients (Online Resource 4) provide evidence that the data in this model were uniformly and closely distributed around the line of identity, and demonstrated an overall excellent model fit of individual PSA concentrations.
4 Discussion
We report for the first time the relationship between exposure and PSA dynamics for abiraterone in chemotherapy-pretreated and chemotherapy-naïve patients with mCRPC using an exposure–PSA modeling framework. In the vast majority of mCRPC patients, the abiraterone C min was greater than the model-estimated efficacious concentration (i.e. EC50), following repeated oral administration of abiraterone acetate at the recommended dose of 1000 mg/day. Taken together with our previous PSA survival analysis of the same patients treated with abiraterone [9], we have established a link between abiraterone exposure and survival in mCRPC.
The TGI model exhibited the versatility to address the heterogeneity observed in PSA dynamics and the robustness to provide precise parameter estimates from datasets available. The model adequately linked abiraterone exposure to tumor inhibition, represented by reductions in PSA after treatment. The assumption of the model was that the change in tumor burden during therapy is indicated by the change in PSA following treatment [28]. Tumor growth can be divided into two phases: an initial exponential phase (first-order growth) followed by a linear growth phase [29]. In oncology clinical trials, patients are usually switched to other subsequent therapies after disease progression and then only followed for the evaluation of a few potential endpoints (e.g. survival). Therefore, the tumor growth data collected in clinical studies often consist of only the initial exponential growth phase, making the first-order growth assumption applicable. This may explain why the TGI model and its assumptions of first-order production has been widely used for describing data from clinical studies [15, 17, 22, 27, 30].
As anticipated, both higher tumor resistance rate constant and baseline PSA levels in chemotherapy-pretreated patients compared with chemotherapy-naïve patients were confirmed through this analysis. As PSA dynamics are dependent on the extent of disease, a large interindividual variability in PSA dynamics was expected. Also as expected and despite the significant heterogenous relationship of PSA to disease extent, exposure to abiraterone led to PSA reductions. The effect of abiraterone acetate + prednisone on k dec was approximately twofold the effect of prednisone alone. The exposure–response relationship was similar in the chemotherapy-pretreated and chemotherapy-naïve patient populations.
Baseline testosterone and LDH were identified as significant covariates for k dec. As the baseline testosterone levels increased, k dec also increased. The effect of testosterone on PSA dynamics is related to the mechanism of action of abiraterone in inhibiting the production of testosterone and other steroidal hormones [2, 6]. Smaller k dec was associated with higher baseline LDH levels, which usually reflect higher disease burden and are included in prognostic models [31]. Further exploratory analysis showed that tumor cell count is positively associated with LDH levels (data not shown), confirming the relationship between LDH and disease burden.
Our previous analysis of COU-AA-301 and COU-AA-302 demonstrated that survival outcomes in both chemotherapy-pretreated and chemotherapy-naïve patients can be adequately predicted through PSA kinetics, and that the Prentice criteria for surrogacy were met on the individual patient level [9]. The current analysis can also be adapted to provide key information for conducting future meta-analyses across different compounds to facilitate investigation of the surrogacy of PSA in mCRPC at compound and trial levels.
Many recent modeling studies offer thoughtful and detailed analyses of the relationship between PSA dynamics and survival in prostate cancer [25, 26, 32]; however, these analyses do not consider the exposure–response relationship, only use either simulated data or a previously developed pharmacokinetic model based on pooled data from unrelated, smaller or earlier-phase clinical trials, or are focused on earlier prostate cancer disease states. Our model builds on the previous literature by evaluating the PSA dynamics for an agent targeting androgen biosynthesis for CRPC treatment, a therapeutic strategy that has become increasingly important for this patient population and for which an exposure–survival relationship through PSA dynamics has not been fully described.
A number of limitations may have affected the conclusions drawn from this modeling analysis. The CI associated with the estimated EC50 (–2.61 to 5.73 ng/mL) in chemotherapy-naïve patients suggests large uncertainty in the exposure response in this patient population, which was probably caused by the limited available data for this study. To further confirm the non-linear E max models, linear concentration–response models were also tested in this analysis. The E max models fit the data much better than the linear models as the AIC values based on the E max models were 31 points lower than those based on the linear models for both chemotherapy-pretreated and chemotherapy-naïve patients. It is worth mentioning that despite the wide CI around the estimated EC50 value for the chemotherapy-naïve patients, the majority of these patients (approximately 80 %) still had a steady-state C min greater than the upper bound of the CI (5.73 ng/mL). In addition, numerical difficulties were encountered in the concentration–response modeling for chemotherapy-naïve patients as the model did not converge with the first-order conditional estimation algorithm used for this analysis. Furthermore, in most chemotherapy-naïve patients, baseline testosterone concentrations were below the lower limit of detection, potentially attributed to the use of a different testosterone assay with varying sensitivities in the castration range for COU-AA-301 [33, 34], so the effect of baseline testosterone on k dec could not be estimated.
5 Conclusions
A TGI model best described the time course of PSA pharmacodynamic response following treatment with abiraterone acetate. Exposure to abiraterone significantly increased the rate of PSA reduction, with similar treatment effect observed in both chemotherapy-naïve and chemotherapy-pretreated patients with mCRPC. The pharmacokinetic/pharmacodynamic model showed that the majority of patients in both studies had a steady-state C min greater than the model-estimated efficacious concentration. This developed model can potentially be used with our PSA survival outcome framework to connect exposure and survival through PSA dynamics for mCPRC patients treated with abiraterone acetate, and can also potentially be adapted for other therapeutic strategies that target the androgen receptor axis in mCRPC.
References
US Food and Drug Administration. Guidance for industry: exposure-response relationships. Available at: http://www.fda.gov/downloads/drugs/guidancecomplianceregulatoryinformation/guidances/ucm072109.pdf. Accessed 7 Apr 2015.
Attard G, Reid AH, A’Hern R, Parker C, Oommen NB, Folkerd E, et al. Selective inhibition of CYP17 with abiraterone acetate is highly active in the treatment of castration-resistant prostate cancer. J Clin Oncol. 2009;27:3742–8.
Danila DC, Morris MJ, de Bono JS, Ryan CJ, Denmeade SR, Smith MR, et al. Phase II multicenter study of abiraterone acetate plus prednisone therapy in patients with docetaxel-treated castration-resistant prostate cancer. J Clin Oncol. 2010;28:1496–501.
Reid AH, Attard G, Danila DC, Oommen NB, Olmos D, Fong PC, et al. Significant and sustained antitumor activity in post-docetaxel, castration-resistant prostate cancer with the CYP17 inhibitor abiraterone acetate. J Clin Oncol. 2010;28:1489–95.
Ryan CJ, Smith MR, Fong L, Rosenberg JE, Kantoff P, Raynaud F, et al. Phase I clinical trial of the CYP17 inhibitor abiraterone acetate demonstrating clinical activity in patients with castration-resistant prostate cancer who received prior ketoconazole therapy. J Clin Oncol. 2010;28:1481–8.
Attard G, Reid AH, Yap TA, Raynaud F, Dowsett M, Settatree S, et al. Phase I clinical trial of a selective inhibitor of CYP17, abiraterone acetate, confirms that castration-resistant prostate cancer commonly remains hormone driven. J Clin Oncol. 2008;26:4563–71.
Fizazi K, Scher HI, Molina A, Logothetis CJ, Chi KN, Jones RJ, et al. Abiraterone acetate for treatment of metastatic castration-resistant prostate cancer: final overall survival analysis of the COU-AA-301 randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2012;13:983–92.
Ryan CJ, Smith MR, Fizazi K, Saad F, Mulders PF, Sternberg CN, et al. Abiraterone acetate plus prednisone versus placebo plus prednisone in chemotherapy-naive men with metastatic castration-resistant prostate cancer (COU-AA-302): final overall survival analysis of a randomised, double-blind, placebo-controlled phase 3 study. Lancet Oncol. 2015;16:152–60.
Xu XS, Ryan CJ, Stuyckens K, Smith MR, Saad F, Griffin TW, et al. Correlation between prostate-specific antigen kinetics and overall survival in abiraterone acetate-treated castration-resistant prostate cancer patients. Clin Cancer Res. 2015;21:3170–7.
Zytiga® (abiraterone acetate) [US prescribing information]. Horsham: Janssen Biotech Inc.; 2011.
Stuyckens K, Saad F, Xu XS, Ryan CJ, Smith MR, Griffin TW, et al. Population pharmacokinetic analysis of abiraterone in chemotherapy-naive and docetaxel-treated patients with metastatic castration-resistant prostate cancer. Clin Pharmacokinet. 2014;53:1149–60.
Subtil F, Rabilloud M. Robust non-linear mixed modelling of longitudinal PSA levels after prostate cancer treatment. Stat Med. 2010;29:573–87.
Wang Y, Sung C, Dartois C, Ramchandani R, Booth BP, Rock E, et al. Elucidation of relationship between tumor size and survival in non-small-cell lung cancer patients can aid early decision making in clinical drug development. Clin Pharmacol Ther. 2009;86:167–74.
Gomeni R, Merlo-Pich E. Bayesian modelling and ROC analysis to predict placebo responders using clinical score measured in the initial weeks of treatment in depression trials. Br J Clin Pharmacol. 2007;63:595–613.
Bruno R, Claret L. On the use of change in tumor size to predict survival in clinical oncology studies: toward a new paradigm to design and evaluate phase II studies. Clin Pharmacol Ther. 2009;86:136–8.
Bruno R, Lu JF, Sun YN, Claret L. A modeling and simulation framework to support early clinical drug development decisions in oncology. J Clin Pharmacol. 2011;51:6–8.
Claret L, Girard P, Hoff PM, Van CE, Zuideveld KP, Jorga K, et al. Model-based prediction of phase III overall survival in colorectal cancer on the basis of phase II tumor dynamics. J Clin Oncol. 2009;27:4103–8.
Chen Y, Clegg NJ, Scher HI. Anti-androgens and androgen-depleting therapies in prostate cancer: new agents for an established target. Lancet Oncol. 2009;10:981–91.
Hu R, Dunn TA, Wei S, Isharwal S, Veltri RW, Humphreys E, et al. Ligand-independent androgen receptor variants derived from splicing of cryptic exons signify hormone-refractory prostate cancer. Cancer Res. 2009;69:16–22.
Ribba B, Holford NH, Magni P, Troconiz I, Gueorguieva I, Girard P, et al. A review of mixed-effects models of tumor growth and effects of anticancer drug treatment used in population analysis. CPT Pharmacomet Syst Pharmacol. 2014;3:e113.
Giacinti S, Bassanelli M, Aschelter AM, Milano A, Roberto M, Marchetti P. Resistance to abiraterone in castration-resistant prostate cancer: a review of the literature. Anticancer Res. 2014;34:6265–9.
Frances N, Claret L, Bruno R, Iliadis A. Tumor growth modeling from clinical trials reveals synergistic anticancer effect of the capecitabine and docetaxel combination in metastatic breast cancer. Cancer Chemother Pharmacol. 2011;68:1413–9.
Rizopoulos D, Verbeke G, Molenberghs G. Multiple-imputation-based residuals and diagnostic plots for joint models of longitudinal and survival outcomes. Biometrics. 2010;66:20–9.
Panel on Handling Missing Data in Clinical Trials, National Research Council. The prevention and treatment of missing data in clinical trials. Washington, DC: National Academies Press; 2010.
Elishmereni M, Kheifetz Y, Shukrun I, Bevan GH, Nandy D, McKenzie KM, et al. Predicting time to castration resistance in hormone sensitive prostate cancer by a personalization algorithm based on a mechanistic model integrating patient data. Prostate. 2016;76:48–57.
van Hasselt JG, Gupta A, Hussein Z, Beijnen JH, Schellens JH, Huitema AD. Disease progression/clinical outcome model for castration-resistant prostate cancer in patients treated with eribulin. CPT Pharmacomet Syst Pharmacol. 2015;4:386–95.
Jonsson F, Ou Y, Claret L, Siegel D, Jagannath S, Vij R, et al. A tumor growth inhibition model based on M-protein levels in subjects with relapsed/refractory multiple myeloma following single-agent carfilzomib use. CPT Pharmacomet Syst Pharmacol. 2015;4:711–9.
Stein WD, Gulley JL, Schlom J, Madan RA, Dahut W, Figg WD, et al. Tumor regression and growth rates determined in five intramural NCI prostate cancer trials: the growth rate constant as an indicator of therapeutic efficacy. Clin Cancer Res. 2011;17:907–17.
Benzekry S, Lamont C, Beheshti A, Tracz A, Ebos JM, Hlatky L, et al. Classical mathematical models for description and prediction of experimental tumor growth. PLoS Comput Biol. 2014;10:e1003800.
Claret L, Gupta M, Han K, Joshi A, Sarapa N, He J, et al. Evaluation of tumor-size response metrics to predict overall survival in Western and Chinese patients with first-line metastatic colorectal cancer. J Clin Oncol. 2013;31:2110–4.
Halabi S, Small EJ, Kantoff PW, Kattan MW, Kaplan EB, Dawson NA, et al. Prognostic model for predicting survival in men with hormone-refractory metastatic prostate cancer. J Clin Oncol. 2003;21:1232–7.
Desmee S, Mentre F, Veyrat-Follet C, Guedj J. Nonlinear mixed-effect models for prostate-specific antigen kinetics and link with survival in the context of metastatic prostate cancer: a comparison by simulation of two-stage and joint approaches. AAPS J. 2015;17:691–9.
Ryan CJ, Molina A, Li J, Kheoh T, Small EJ, Haqq CM, et al. Serum androgens as prognostic biomarkers in castration-resistant prostate cancer: results from an analysis of a randomized phase III trial. J Clin Oncol. 2013;31:2791–8.
Ryan CJ, Peng W, Kheoh T, Welkowsky E, Haqq CM, Chandler DW, et al. Androgen dynamics and serum PSA in patients treated with abiraterone acetate. Prostate Cancer Prostatic Dis. 2014;17:192–8.
Acknowledgments
This study was sponsored by Janssen Research & Development, Raritan, NJ, USA. Writing assistance was provided by S. Thomas, PhD, of PAREXEL, and was funded by Janssen Global Services, LLC.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Xu Steven Xu, Kim Stuyckens, Thomas W. Griffin, Youn C. Park, Margaret K. Yu, An Vermeulen, Peter De Porre, Italo Poggesi, and Partha Nandy are employees of Janssen Research & Development and own stock in Johnson & Johnson. Fred Saad has served as a consultant and speaker for, and has received research funding from, Janssen Research & Development. Charles J. Ryan has served as a speaker for, and has received research funding from, Janssen Research & Development. Matthew R. Smith has served as a consultant for, and has received research funding from, Janssen Research & Development.
Ethical approval
The review boards at all participating institutions approved these studies. All procedures performed in studies involving human participants were in accordance with the ethical standards of the institutional and/or national research committee and with the 1964 Helsinki declaration and its later amendments or comparable ethical standards.
Informed consent
Informed consent was obtained from all individual participants included in both studies.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Xu, X.S., Ryan, C.J., Stuyckens, K. et al. Modeling the Relationship Between Exposure to Abiraterone and Prostate-Specific Antigen Dynamics in Patients with Metastatic Castration-Resistant Prostate Cancer. Clin Pharmacokinet 56, 55–63 (2017). https://doi.org/10.1007/s40262-016-0425-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40262-016-0425-0