
Abstract
Acute heart failure (AHF) is characterized by high morbidity and mortality and high costs. Although the treatment of AHF has not changed substantially in recent decades, it is becoming clear that treatment strategies for AHF need to address both the immediate hemodynamic abnormalities giving rise to congestion as well as prevent organ damage that can influence long-term prognosis. Serelaxin, the recombinant form of human relaxin-2, a naturally occurring peptide hormone, has been found to significantly improve symptoms and signs of AHF, prevent in-hospital worsening heart failure, as well as significantly improve 180-day cardiovascular and all-cause mortality after a 48-h infusion commenced within 16 h of presentation (RELAX-AHF study). Available data suggest that the clinical benefits may be attributable to a potential combination of multiple actions of serelaxin, including improving systemic, cardiac, and renal hemodynamics, and protecting cells and organs from damage via anti-inflammatory, anti-cell death, anti-fibrotic, anti-hypertrophic, and pro-angiogenic effects. This manuscript describes the short- and long-term effects of serelaxin in AHF patients, analyzing how these effects can be explained by taking into account the range of hemodynamic and non-hemodynamic actions of serelaxin. In addition, this paper also addresses several aspects related to the role of serelaxin in the therapy of AHF that remain to be clarified and warrant further investigation.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Heart failure (HF) is thought to affect approximately 15 million patients in Europe and 5 million in the USA [1, 2]. HF places a large burden on patients and healthcare systems [1, 3, 4], and hospitalization for acute HF (AHF) contributes to much of the economic burden [1]. There are more than 1 million hospitalizations for AHF each year in the USA [2], and each hospitalization for AHF costs in the region of $US19,000 [5]. AHF is associated with high rates of subsequent mortality and hospital readmission, and is an increasing problem due to an aging population and the increasing prevalence of risk factors [1, 6–9]. Treatment of AHF has not changed substantially in recent decades, and major trials with new therapies have failed, likely due to a poor understanding of the pathophysiology of AHF, as well as an insufficient knowledge of the mechanisms of action of the new agents used to treat patients with AHF [10].
AHF is a syndrome the pathophysiology of which is complex and not fully understood, probably due to the lack of adequate experimental models of AHF. It is now known that it involves not only impaired hemodynamic function, but also neurohormonal activation, inflammation, and oxidative stress, which can promote myocardial, renal, and hepatic injury and remodeling [11–13]. This multi-organ damage may accelerate disease progression and impact long-term prognosis. Indeed, many existing and emerging markers of cardiac and extra-cardiac injury confirm that such damage is associated with worse long-term outcomes in AHF [14–30]. Therefore, it appears that treatment strategies for AHF need to address both the immediate hemodynamic abnormalities, giving rise to dyspnea and congestion, as well as prevent organ damage, by addressing the myocardial and extra-cardiac cell injury and remodeling that influence long-term prognosis.
Serelaxin is the recombinant form of human relaxin-2, a naturally occurring peptide hormone that mediates systemic hemodynamic and renal adaptive changes during pregnancy [31]. Serelaxin mediates its actions by binding to a member of the G-protein-coupled receptors known as relaxin family peptide (RXFP) receptors, that are located in heart tissue, blood vessels, and the kidneys [31, 32]. Binding of serelaxin to its RXFP1 receptor activates multiple signaling pathways promoting systemic and renal hemodynamic effects [32, 33]. However, serelaxin also appears to have multiple additional effects in the cardiovascular (CV) system and other organs, including anti-inflammatory, anti-cell injury/death, anti-fibrotic, anti-hypertrophic, and pro-angiogenic effects [33–35]. The combination of the hemodynamic and organ-protective effects of serelaxin may contribute to the short- and long-term benefits of this agent in AHF, as shown in a recent phase III study of serelaxin in AHF, the RELAX-AHF (RELAXin in Acute Heart Failure) study.
This review aims to outline the clinical data with serelaxin in patients with AHF, integrating and extrapolating the available mechanistic data that could help to explain both the short- and long-term clinical effects observed with this novel agent. Some of these mechanistic studies have investigated the effects of non-human forms of relaxin-2 instead of serelaxin (recombinant human relaxin-2), and where possible this review tries to highlight the form used (e.g. porcine relaxin-2 vs serelaxin). This review also provides a critical analysis of the issues still pending clarification regarding the role of serelaxin in the therapy of AHF.
2 Clinical Effects of Serelaxin in Patients with Acute Heart Failure (AHF)
2.1 Short-Term Effects
RELAX-AHF was a phase III, multicenter, randomized, double-blind, placebo-controlled study designed to investigate the efficacy and safety of serelaxin in the treatment of AHF [36, 37]. Patients hospitalized for AHF with systolic blood pressure (SBP) >125 mmHg, mild-to-moderate renal impairment, and increased levels of N-terminal pro-B-type natriuretic peptide (NT-proBNP) were randomized within 16 h to serelaxin (30 μg/kg/day as a 48-h intravenous infusion) or placebo (both on top of standard of care). Serelaxin significantly reduced the short-term effects of AHF, including symptoms and signs of congestion during hospitalization and in-hospital outcomes, compared with placebo. Serelaxin significantly improved the primary efficacy endpoint of dyspnea relief through day 5 assessed by the visual analog scale compared with placebo (p = 0.007). It also provided a numerical, but not statistically significant, improvement in dyspnea relief assessed by the Likert scale at 6, 12, and 24 h [36]. Serelaxin-treated patients experienced significant improvements in other symptoms and signs of AHF (dyspnea on exertion, orthopnea, edema, rales) at day 2 compared with placebo-treated patients. Consistent with prompt decongestion, serelaxin treatment was associated with significantly lower NT-proBNP levels at day 2 compared with placebo (p < 0.001) [36]. In terms of in-hospital measures, significantly fewer serelaxin-treated patients experienced in-hospital worsening HF through day 5 compared with placebo (p < 0.001) and serelaxin treatment was associated with a significantly shorter length of hospital stay (p = 0.0039) [36].
2.2 Long-Term Effects
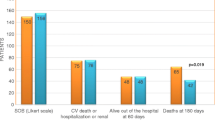
An interesting observation from the RELAX-AHF study was that although serelaxin did not reduce the composite secondary endpoint of CV death or hospitalization at day 60, it reduced, by 37 %, both CV death (p = 0.028) and all-cause mortality at day 180 (p = 0.02) [36]. All-cause mortality and CV mortality were not primary endpoints of the RELAX-AHF study and therefore these data need to be confirmed in an appropriately designed, prospective trial. However, the effect on all-cause mortality at day 180 with serelaxin was consistent with findings from the phase II pre-RELAX-AHF study (Fig. 1; [14]), which also reported a numerical reduction in 180-day CV and all-cause mortality with serelaxin versus placebo [38]. Of note, the effects of serelaxin on outcomes were similar across subgroups of patients based on pre-specified covariates (age, sex, race, geographic region, estimated glomerular filtration rate [eGFR], time from presentation to randomization, baseline SBP, history of diabetes, atrial fibrillation, ischemic heart disease, cardiac devices, intravenous nitrates at randomization) [39] or ejection fraction (either preserved or reduced) [40]. The reduction in mortality at 180 days in RELAX-AHF occurred despite serelaxin being administered for only 48 h during the episode of acute decompensation [36]. This final observation raises interesting questions about how a short-term treatment may impact long-term consequences in AHF, as discussed in the following section.

3 Potential Mechanisms of Action of Serelaxin in Patients with AHF
3.1 Rationale for Short-Term Effects
Insights into the possible mechanisms involved in these short-term effects of serelaxin are given by the interpretation of results of earlier clinical studies. In a study of stable patients with compensated HF, serelaxin provided hemodynamic improvements, including reduced pulmonary capillary wedge pressure (PCWP) and systemic vascular resistance (SVR), and increased cardiac index, suggesting it may reduce cardiac overload and improve cardiac performance [41, 42]. Similarly, reductions in SVR and PCWP and increases in cardiac index have been observed with serelaxin in patients with AHF [43]. Serelaxin has also been shown to increase renal plasma flow, and reduce renal vascular resistance in rats [44] and in healthy volunteers [45], suggesting it may increase renal perfusion. Such improvements in myocardial and renal function will decrease preload and afterload, leading to relief of the symptoms and signs of congestion and renal impairment.
The hemodynamic effects of serelaxin appear to be predominantly a consequence of receptor-mediated release of nitric oxide and a reduction in endothelin-1 levels [34, 46], although emerging evidence suggests that angiogenic growth factors (e.g. vascular endothelial growth factor [VEGF]) may also contribute to the short-term effects of serelaxin on vascular tone [47]. Interestingly, the vasorelaxant effects of serelaxin are greater in vasoconstricted blood vessels than in less constricted blood vessels [38, 48]. This has led to the suggestion that the natural hormone is a regulator of vascular tone and protects against the imbalances of vascular reactivity [49]. Indeed, unlike diuretics, which appear to activate neurohormonal systems, such as the renin-angiotensin-aldosterone system (RAAS) [50–52], serelaxin appears to counteract the activation of neurohormonal systems. For example, porcine relaxin-2 [53] and serelaxin [54] have been reported to blunt the vasoconstrictive response and increase in systemic blood pressure (BP) due to angiotensin II. In addition, serelaxin has been shown to decrease SVR in rats with angiotensin-mediated hypertension [55]. Importantly, the decline in SVR elicited by serelaxin was offset by a corresponding rise in cardiac output, thereby maintaining mean arterial pressure (MAP) [55]. Thus, serelaxin mimics the vasodilatory influences of normal pregnancy, during which MAP declines only slightly relative to the marked reduction in SVR. This property of serelaxin may be particularly useful to help preserve organ (e.g. renal) perfusion in conditions of hemodynamic compromise and RAAS activation, such as AHF.
Importantly, region-specific vascular effects of human relaxin-2 (and, in turn, the recombinant hormone) may be influenced by the cellular localization of RXFP1 receptors within different blood vessels. In a recent study performed in rats, RXFP1 immunostaining in endothelial and vascular smooth muscle cells was found to be higher in mesenteric vessels than in other vascular beds [56]. Accordingly, subcutaneous administration of serelaxin for 5 days in rats was shown to reduce wall stiffness and increase volume compliance in mesenteric arteries but not in the other vessels examined. These changes were associated with enhanced bradykinin-mediated relaxation and modification in geometrical properties, but not compositional changes in the extracellular matrix in mesenteric arteries. Therefore, the mesenteric resistance artery appears to be a key target for the vasorelaxant actions of serelaxin. The potential relevance of this action in AHF is highlighted by the fact that increased capacitance of mesenteric vessels may facilitate a shift in volume from the systemic circulation to the splanchnic reservoir, decreasing effective circulatory volume and thereby relieving congestion [57]. This may explain why symptomatic relief occurred without significant weight loss in serelaxin-treated patients in RELAX-AHF [36].
3.2 Rationale for Long-Term Effects
The mortality benefit observed with serelaxin may be explained by various clinical and pre-clinical observations indicating that it has a unique mechanism of action that has the potential to influence the various pathological processes occurring during decompensation, such as end-organ damage, which drive long-term prognosis in AHF.
3.2.1 Effects Against Cell Injury/Death
In RELAX-AHF, serelaxin treatment reduced levels of high-sensitivity cardiac troponin T compared with standard of care, and fewer patients receiving serelaxin had increases in cardiac troponin T compared with those receiving placebo (Fig. 2 [14]). In addition, elevations in cardiac troponin T > 20 % from baseline were shown to be associated with increased 180-day mortality [14]. Thus, serelaxin appears to have cardioprotective properties.

In patients with acute heart failure in the RELAX-AHF study, serelaxin was associated with lower levels of high-sensitivity cardiac troponin levels (hs-cTnT) (a) and a reduced incidence of patients with an elevation (≥20 % from baseline) in hs-cTnT (b) compared with placebo [14]). Reprinted from Metra [14] (Fig. 3a), copyright (2013), with permission from Elsevier
Serelaxin may promote myocardial cell preservation via a number of different mechanisms. For example, serelaxin has been found to have a number of anti-inflammatory effects, including reducing inflammatory cell infiltration into myocardial tissue following ischemia [58]. In addition, serelaxin promotes cell survival by reducing oxidative stress-mediated apoptosis, calcium overload, and infarct size in animal models of ischemia/reperfusion injury (Fig. 3) [33, 58]. By stimulating angiogenesis, and improving stem cell survival and cell–cell coupling, serelaxin may also facilitate myocardial tissue healing [33]. Of interest, recent evidence shows that serelaxin promotes the maturation and proliferation of immature cardiomyocytes [59], thus suggesting that it can participate in myocardial regeneration. Consistent with these findings, in a swine model of ischemia/reperfusion, serelaxin caused a dose-related reduction of serum and tissue markers of cardiomyocyte injury/death and leukocyte recruitment (Fig. 3), with an accompanying overall improvement in cardiac performance [58, 60].

Serelaxin reduces inflammatory leukocyte recruitment and oxygen free radical-mediated cardiomyocyte injury (a), apoptosis of cardiomyocytes (assessed by caspase-3 activity and TUNEL [Terminal deoxynucleotidyl transferase dUTP nick end labeling] assay) (b) and markers of cardiomyocyte injury (creatine kinase MB isoenzyme [CK-MB] and troponin T) (c) in an in vivo porcine model of ischemia/reperfusion [58]. Sham operated animals (n = 5); all other groups (n = 8). Sham-operated = negative control; I-R = ischemia-reperfusion = positive control (vehicle only). Reprinted from Perna [58], Federation of American Societies for Experimental Biology (FASEB) copyright (2005)
Serelaxin may also provide protection against renal cell damage. In RELAX-AHF, serelaxin provided significant reductions from baseline in several markers of renal dysfunction (creatinine, blood urea nitrogen [BUN], and cystatin C) through day 5. In addition, serelaxin significantly reduced the incidence of worsening renal function (defined as an increase from baseline in cystatin C ≥0.3 mg/L or serum creatinine ≥0.3 mg/dL) compared with placebo [14]. Interestingly, these measures of worsening renal function were associated with increased 180-day all-cause mortality. This study also showed that levels of uric acid, a marker of oxidative stress, inflammation, and/or poor renal function associated with poor prognosis in AHF [14, 61, 62], were reduced in patients with AHF receiving serelaxin compared with placebo [14]. Consistent with these findings, acute serelaxin administration has been shown to induce nitric oxide signaling pathways and attenuate structural and functional injury, oxidative stress, and neutrophil activation in a rat model of kidney ischemia/reperfusion injury [63].
In RELAX-AHF, serelaxin administration was associated with reduced levels of transaminases [14], suggestive of a hepatoprotective effect. Of interest, increases in serum transaminases of ≥20 % from baseline to day 2 were associated with increased 180-day all-cause mortality [14]. In addition, patients treated with serelaxin experienced fewer adverse events classified as hepatobiliary disorders than those treated with placebo (0.2 vs. 1.8 %, respectively; odds ratio 0.10 [95 % confidence interval (CI) 0.01–0.77]) [14]. Such benefits may be explained not only by an offloading effect (i.e. alleviating hepatic congestion), but also by a direct cytoprotective effect in the liver. Experiments to simulate liver transplantation in rats, for example, have shown that addition of serelaxin to the preservation solution could reduce markers of cell damage compared with controls [64, 65]. Using a model of isolated perfused rat liver, the same group has demonstrated that serelaxin administration improved oxygen supply (as assessed by spectrophotometric imaging) and decreased myeloperoxidase and malondialdehyde activities (as assessed by immunohistochemistry) compared with control [64]. These findings indicated that serelaxin can protect the liver by improving oxygen distribution and reducing inflammation and oxidative stress, an effect that could also be beneficial in AHF.
3.2.2 Effects Against Tissue Fibrosis
By protecting cells and organs from damage, serelaxin may indirectly reduce the consequent reparative response and fibrosis. In addition, serelaxin may have direct anti-fibrotic effects, and preclinical studies have shown that serelaxin can inhibit collagen synthesis and promote collagen breakdown of cardiac fibroblasts (Fig. 4) [66–68]. In a study using relaxin gene-knockout (RLX-KO) mice, the RLX-KO mice underwent progressive increases in cardiac and renal collagen content compared with wild-type animals. However, serelaxin treatment for 14 days in RLX-KO mice in the early or developed stages of fibrosis caused the reversal of collagen deposition in the heart and kidneys [69]. In a mouse model of reparative fibrosis after a myocardial infarction, treatment with a serelaxin infusion for 7 or 30 days significantly reduced collagen density in the infarcted and border zones, without increasing the risk of ventricular rupture due to collagen breakdown, compared with mice treated with vehicle control (Fig. 4) [70, 71]. In addition, serelaxin treatment was associated with reduced cardiac fibroblast differentiation and cardiomyocyte apoptosis, as well as the induction of de novo blood vessel growth [70]. This suggests that serelaxin may regulate reparative fibrosis without adverse consequences, enabling subsequent myocardial healing. The effect of serelaxin on fibrosis appears to be achieved via Notch-1-mediated inhibition of the pro-fibrogenic effects of the transforming growth factor (TGF)-β/p-Smad axis [72]. Serelaxin appears to upregulate matrix metalloproteinases (MMPs) and downregulate tissue inhibitors of metalloproteinases (TIMP) [67, 68], thus facilitating the removal of fibrotic tissue. This effect of serelaxin on MMPs appears to occur via inducible nitric oxide synthase signaling, as evidenced by studies in TGF-β1-stimulated human dermal fibroblasts and primary renal myofibroblasts isolated from injured rats that were incubated with serelaxin or control for 72 h [73]. In contrast to these findings, a study in spontaneously hypertensive rats suggests that, although 2-weeks of treatment with serelaxin reverses myocardial fibrosis, this effect is not associated with increased expression of the gelatinases, MMP-2 or MMP-9 [74].

Serelaxin modulates cardiac collagen deposition in vitro (a) and in vivo (b). a Collagen content of in vitro fibroblasts, untreated/treated with serelaxin (100 ng/mL) alone or with transforming growth factor-β (2 ng/mL) or angiotensin II (10–7 M), for 72 h of culture. Results are mean data from 3–4 separate experiments [66]. Reprinted with permission from Samuel [66] with permission from the Endocrine Society, copyright (2004). b Collagen density (mean ± SE) estimated by computer-aided morphometry and MMP-13 density (expressed as a percentage per field) from eight fields representing the non-infarct region, border region, and infarct region from the lower mid-zone of hearts from a mouse model of myocardial infarction (n = 9–12/group) [70]. Reprinted by permission from Macmillan Publishers Ltd: Laboratory Investigation [70], copyright (2011). Ang angiotensin, BR border region, IR infarct region, MI myocardial infarction, MMP matrix metalloproteinase, NIR non-infarct region, SE standard error, TGF transforming growth factor
Serelaxin may have anti-fibrotic effects in the liver. Increased expression of the RXFP1 receptors has been observed in fibrotic liver disease [75]. In human cultured hepatic stellate cells, a 72-h treatment with serelaxin has been reported to induce an anti-fibrogenic phenotype [75]. Furthermore, treatment of activated cultured hepatic stellate cells from rat liver with porcine relaxin-2 for 24–48 h reduces collagen, increases secretion of TIMP-1, TIMP-2, and MMP-13, and inhibits the synthesis of new collagen compared with cells not treated with relaxin-2 [76]. In an in vivo mouse model of hepatic fibrosis, 1 week of treatment with porcine relaxin-2 was reported to reduce collagen and smooth muscle actin content and significantly reduce serum levels of hepatic transaminases [77]. A subsequent study utilizing the same model has shown that reductions in fibrosis with 4 weeks of treatment with serelaxin were associated with increases in MMP-13, decreases in TIMP-2, and impaired TGF-β signaling compared with vehicle [78].
Whether this anti-fibrotic effect of serelaxin is clinically important in AHF remains to be clarified. It is notable that many of the preclinical studies showing anti-fibrotic effects of serelaxin in vivo occurred when serelaxin was administered for durations longer than the 48-h infusion used in AHF clinical studies [66, 70, 74, 78]. However, in vitro studies have shown that anti-fibrotic actions of serelaxin can manifest even within 24–72 h [66, 68, 75].
4 Unresolved Issues
Notwithstanding the promising findings of the RELAX-AHF study, there are several issues still pending to be clarified for a better understanding of the role of serelaxin in the therapy of AHF.
4.1 Pharmacological Issues
In the RELAX-AHF study, congestion and biomarkers of end-organ damage, which were strongly associated with increased 180-day mortality, were relieved by an early 48-h intravenous infusion of serelaxin, and 180-day mortality was reduced. Hence, it is possible that early treatment of AHF, including decongestion and prevention of damage to the heart, kidneys, and liver, is critical to preventing long-term mortality in AHF. The emerging question is how an early short-term administration of serelaxin is translated into long-term clinical benefit in AHF patients. The ability of a short-term infusion to have a beneficial long-term effect on outcomes has already been clearly shown in the area of thrombolytics in acute coronary syndromes [79]. In addition, it is also possible that the effects of serelaxin persist beyond the initial infusion, providing a greater window of decongestion and organ protection during the acute event. The terminal half-life of an intravenous bolus of serelaxin is reported to be 4.6 h in healthy individuals [80], and studies in patients with AHF have shown that reductions in SVR with serelaxin were present up to 4 h, and changes in blood pressure and NT-proBNP were present up to 24 h, after cessation of the infusion [43]. Similarly, a study in patients with chronic HF has also reported that hemodynamic effects can persist, lasting up to 8 h after termination of the infusion [42].
Therefore, the possibility exists that the ability of serelaxin to intercept hemodynamic and end-organ damaging mechanisms activated at earlier stages of AHF is translated to further reduction in the burden of long-term mortality. Indeed, the beneficial effects observed with serelaxin in preclinical studies (Table 1) suggest that, when it is administered even for a short (48-h) period early during AHF, serelaxin may interrupt the cascade of pathophysiological processes that are triggered during an acute decompensation and drive the long-term consequences of AHF (Table 1).
Unfortunately, there are no good experimental models of AHF to better understand the pathophysiology of this syndrome or assess the effects of new therapies. In the absence of good models, information has been extrapolated from observations in other experimental models that may be of relevance to AHF. Given these limitations, it is unclear what the relative contributions are for the various vasorelaxant, anti-inflammatory, anti-oxidative stress, cytoprotective, anti-fibrotic, and angiogenic mechanisms in contributing to the effect of serelaxin on long-term outcomes in AHF. While it is possible that some of these effects are secondary to vasorelaxation and improved organ perfusion, it is worthwhile to note that nitrate vasodilators have not been shown to improve long-term outcomes in AHF [81, 82]. Moreover, cytoprotective, anti-inflammatory, and anti-fibrotic effects of serelaxin have been observed in vitro [68, 83–87], suggesting that there could be a component of these actions that is not dependent upon modulation of vascular tone. The need for further exploration in this area is clearly warranted.
4.2 Pathophysiological Issues
Increased plasma levels of relaxin have been reported in patients with chronic HF of different etiologies [88–90]. Interestingly, patients with chronic HF, but not controls, show a transcardiac relaxin gradient [91], as well as a gradient of relaxin from the coronary blood to the peripheral blood [90]. Collectively, these data suggest that the failing heart is a source of the excess of circulating relaxin present in patients with chronic HF. Of interest, RXFP receptors do not internalize following prolonged exposure to relaxin [92], and it may be anticipated that the physiological actions of the peptide/receptor complex will be preserved, even in the presence of high systemic levels of relaxin/serelaxin. Further studies are required to ascertain whether reduced cardiac synthesis and secretion of relaxin precedes and triggers AHF, thus explaining the favorable effects of serelaxin in patients with this condition.
4.3 Clinical Issues
Although treatment with serelaxin did reduce CV and all-cause mortality by 37 % at 180 days, it did not reduce the incidence of readmission to hospital owing to HF, renal failure, or CV death, nor did it significantly increase survival, up to 60 days after hospital discharge. Although data from classical studies suggested that rehospitalization has historically been regarded as a risk factor for mortality [93–96], recent data have shown a poor correlation between the two [97]. Indeed, mortality may represent a competing risk factor for rehospitalization [98, 99]. In addition, short-term rehospitalizations might also be related to non-modifiable factors, such as social support, geographic location, and socioeconomics, so that there is a discrepancy between early readmissions and post-discharge mortality [100]. On the other hand, it must be remarked that the RELAX-AHF trial was neither designed nor powered to assess mortality as an endpoint. Therefore, because the mortality data in the RELAX-AHF trial emerge as an hypothesis-generating issue, an appropriately powered trial has been undertaken to replicate them (the RELAX-AHF-2 study comprising >6,000 patients with AHF) [101].
5 Conclusions and Perspectives
Current therapies are moderately effective at relieving the symptoms and signs of congestion by addressing the hemodynamic changes associated with AHF, but have not demonstrated any benefit on long-term outcomes, presumably because they have limited effects on the underlying pathophysiology and fail to protect organs from damage in AHF. Recently, serelaxin, the recombinant form of human relaxin-2, was shown to relieve symptoms and signs of AHF, as well as reducing CV and all-cause mortality in the phase III RELAX-AHF study in patients with AHF. This is particularly promising, as serelaxin is the only agent demonstrated to improve mortality in a large randomized controlled trial of patients with AHF, although this was not a primary endpoint of the study.
Initial data suggest that the clinical benefits observed in RELAX-AHF may be explained by the effect of serelaxin on the multiple pathophysiological processes that drive the short- and long-term consequences of AHF. Indeed, myocardial cell injury/death and fibrosis may be triggered during an AHF episode, which may lead to further cardiac dysfunction and poor prognosis in the long term. Pre-clinical data suggest that serelaxin can interrupt these processes, protecting organs from damage and improving hemodynamics, which may explain the clinical observation that short-term (48-h) administration of serelaxin early in the AHF episode reduced 180-day mortality compared with placebo.
Further studies with serelaxin are warranted to both translate the experimental information of its actions to humans, and to confirm its beneficial added value in the treatment of patients with AHF.
References
Dickstein K, Cohen-Solal A, Filippatos G, et al. ESC Guidelines for the diagnosis and treatment of acute and chronic heart failure 2008: the Task Force for the Diagnosis and Treatment of Acute and Chronic Heart Failure 2008 of the European Society of Cardiology. Developed in collaboration with the Heart Failure Association of the ESC (HFA) and endorsed by the European Society of Intensive Care Medicine (ESICM). Eur Heart J. 2008;29:2388–442.
Go AS, Mozaffarian D, Roger VL, et al. Executive summary: heart disease and stroke statistics—2013 update: a report from the American Heart Association. Circulation. 2013;127:143–52.
Grady KL. Quality of life in patients with chronic heart failure. Crit Care Nurs Clin North Am. 1993;5:661–70.
Heidenreich PA, Albert NM, Allen LA, et al. Forecasting the impact of heart failure in the United States: a policy statement from the American Heart Association. Circ Heart Fail. 2013;6:606–19.
Ng TM, Dasta JF, Durtschi AJ, McLaughlin TP, Feldman DS. Characteristics, drug therapy, and outcomes from a database of 500,000 hospitalized patients with a discharge diagnosis of heart failure. Congest Heart Fail. 2008;14:202–10.
Gheorghiade M, Pang PS. Acute heart failure syndromes. J Am Coll Cardiol. 2009;53:557–73.
Hunt SA, Abraham WT, Chin MH, et al. 2009 Focused update incorporated into the ACC/AHA 2005 Guidelines for the Diagnosis and Management of Heart Failure in Adults A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines Developed in Collaboration With the International Society for Heart and Lung Transplantation. J Am Coll Cardiol. 2009;53:e1–90.
Harjola VP, Follath F, Nieminen MS, et al. Characteristics, outcomes, and predictors of mortality at 3 months and 1 year in patients hospitalized for acute heart failure. Eur J Heart Fail. 2010;12:239–48.
Siirilä-Waris K, Lassus J, Melin J, et al. Characteristics, outcomes, and predictors of 1-year mortality in patients hospitalized for acute heart failure. Eur Heart J. 2006;27:3011–7.
Felker GM, Pang PS, Adams KF, et al. Clinical trials of pharmacological therapies in acute heart failure syndromes: lessons learned and directions forward. Circ Heart Fail. 2010;3:314–25.
Cotter G, Felker GM, Adams KF, Milo-Cotter O, O’Connor CM. The pathophysiology of acute heart failure—is it all about fluid accumulation? Am Heart J. 2008;155:9–18.
Cotter G, Metra M, Milo-Cotter O, Dittrich HC, Gheorghiade M. Fluid overload in acute heart failure—re-distribution and other mechanisms beyond fluid accumulation. Eur J Heart Fail. 2008;10:165–9.
Cotter G, Milo O, Davison BA. Increased mortality after an acute heart failure episode: new pathophysiological insights from the RELAX-AHF study and beyond. Curr Heart Fail Rep. 2014;11:19–30.
Metra M, Cotter G, Davison BA, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the Relaxin in Acute Heart Failure (RELAX-AHF) development program: correlation with outcomes. J Am Coll Cardiol. 2013;61:196–206.
Kociol RD, Pang PS, Gheorghiade M, Fonarow GC, O’Connor CM, Felker GM. Troponin elevation in heart failure: prevalence, mechanisms, and clinical implications. J Am Coll Cardiol. 2010;56:1071–8.
Metra M, Bettari L, Pagani F, et al. Troponin T levels in patients with acute heart failure: clinical and prognostic significance of their detection and release during hospitalisation. Clin Res Cardiol. 2012;101:663–72.
Peacock WF 4th, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. N Engl J Med. 2008;358:2117–26.
Del Carlo CH, Pereira-Barretto AC, Cassaro-Strunz C, Latorre Mdo R, Ramires JA. Serial measure of cardiac troponin T levels for prediction of clinical events in decompensated heart failure. J Card Fail. 2004;10:43–8.
La Vecchia L, Mezzena G, Zanolla L, et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. J Heart Lung Transpl. 2000;19:644–52.
Parenti N, Bartolacci S, Carle F, Angelo F. Cardiac troponin I as prognostic marker in heart failure patients discharged from emergency department. Intern Emerg Med. 2008;3:43–7.
You JJ, Austin PC, Alter DA, Ko DT, Tu JV. Relation between cardiac troponin I and mortality in acute decompensated heart failure. Am Heart J. 2007;153:462–70.
Felker GM, Hasselblad V, Tang WH, et al. Troponin I in acute decompensated heart failure: insights from the ASCEND-HF study. Eur J Heart Fail. 2012;14:1257–64.
Braga JR, Tu JV, Austin PC, et al. Outcomes and care of patients with acute heart failure syndromes and cardiac troponin elevation. Circ Heart Fail. 2013;6:193–202.
Perna ER, Macín SM, Cimbaro Canella JP, et al. Minor myocardial damage detected by troponin T is a powerful predictor of long-term prognosis in patients with acute decompensated heart failure. Int J Cardiol. 2005;99:253–61.
Perna ER, Aspromonte N, Cimbaro Canella JP, et al. Minor myocardial damage is a prevalent condition in patients with acute heart failure syndromes and preserved systolic function with long-term prognostic implications: a report from the CIAST-HF (Collaborative Italo-Argentinean Study on cardiac Troponin T in Heart Failure) study. J Card Fail. 2012;18:822–30.
Metra M, Cotter G, Gheorghiade M, Dei Cas L, Voors AA. The role of the kidney in heart failure. Eur Heart J. 2012;33:2135–42.
Maisel AS, Choudhary R. Biomarkers in acute heart failure—state of the art. Nat Rev Cardiol. 2012;9:478–90.
Maisel AS, Mueller C, Fitzgerald R, et al. Prognostic utility of plasma neutrophil gelatinase-associated lipocalin in patients with acute heart failure: the NGAL EvaLuation Along with B-type NaTriuretic Peptide in acutely decompensated heart failure (GALLANT) trial. Eur J Heart Fail. 2011;13:846–51.
Damman K, Van Veldhuisen DJ, Navis G, et al. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart. 2010;96:1297–302.
Ambrosy AP, Vaduganathan M, Huffman MD, et al. Clinical course and predictive value of liver function tests in patients hospitalized for worsening heart failure with reduced ejection fraction: an analysis of the EVEREST trial. Eur J Heart Fail. 2012;14:302–11.
Bathgate RAD, Halls ML, van der Westhuizen ET, Callander GE, Kocan M, Summers RJ. Relaxin family peptides and their receptors. Physiol Rev. 2013;93:405–80.
Teichman SL, Unemori E, Dschietzig T, et al. Relaxin, a pleiotropic vasodilator for the treatment of heart failure. Heart Fail Rev. 2009;14:321–9.
Du XJ, Bathgate RA, Samuel CS, Dart AM, Summers RJ. Cardiovascular effects of relaxin: from basic science to clinical therapy. Nat Rev Cardiol. 2010;7:48–58.
Teichman SL, Unemori E, Teerlink JR, Cotter G, Metra M. Relaxin: review of biology and potential role in treating heart failure. Curr Heart Fail Rep. 2010;7:75–82.
Bennett RG. Relaxin and its role in the development and treatment of fibrosis. Transl Res. 2009;154:1–6.
Teerlink JR, Cotter G, Davison BA, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomised, placebo-controlled trial. Lancet. 2013;381:29–39.
Ponikowski P, Metra M, Teerlink JR, et al. Design of the RELAXin in acute heart failure study. Am Heart J. 2012;163:149–55.
Teerlink JR, Metra M, Felker GM, et al. Relaxin for the treatment of patients with acute heart failure (Pre-RELAX-AHF): a multicentre, randomised, placebo-controlled, parallel-group, dose-finding phase IIb study. Lancet. 2009;373:1429–39.
Metra M, Ponikowski P, Cotter G, et al. Effects of serelaxin in subgroups of patients with acute heart failure: results from RELAX-AHF. Eur Heart J. 2013;34:3128–36.
Filippatos G, Teerlink JR, Farmakis D, et al. Serelaxin in acute heart failure patients with preserved left ventricular ejection fraction: results from the RELAX-AHF trial. Eur Heart J. 2013 (Epub ahead of print).
Dschietzig T, Teichman S, Unemori E, et al. First clinical experience with intravenous recombinant human relaxin in compensated heart failure. Ann N Y Acad Sci. 2009;1160:387–92.
Dschietzig T, Teichman S, Unemori E, et al. Intravenous recombinant human relaxin in compensated heart failure: a safety, tolerability, and pharmacodynamic trial. J Card Fail. 2009;15:182–90.
Ponikowski P, Mitrovic V, Ruda M, et al. A randomized, double-blind, placebo-controlled, multicentre study to assess haemodynamic effects of serelaxin in patients with acute heart failure. Eur Heart J. 2014;35:431–41.
Danielson LA, Kercher LJ, Conrad KP. Impact of gender and endothelin on renal vasodilation and hyperfiltration induced by relaxin in conscious rats. Am J Physiol Regul Integr Comp Physiol. 2000;279:R1298–304.
Smith MC, Danielson LA, Conrad KP, Davison JM. Influence of recombinant human relaxin on renal hemodynamics in healthy volunteers. J Am Soc Nephrol. 2006;17:3192–7.
Conrad KP. Unveiling the vasodilatory actions and mechanisms of relaxin. Hypertension. 2010;56:2–9.
McGuane JT, Danielson LA, Debrah JE, Rubin JP, Novak J, Conrad KP. Angiogenic growth factors are new and essential players in the sustained relaxin vasodilatory pathway in rodents and humans. Hypertension. 2011;57:1151–60.
Debrah DO, Conrad KP, Danielson LA, Shroff SG. Effects of relaxin on systemic arterial hemodynamics and mechanical properties in conscious rats: sex dependency and dose response. J Appl Physiol. 2005;98:1013–20.
Bani D. Relaxin as a natural agent for vascular health. Vasc Health Risk Manag. 2008;4:515–24.
Missouris CG, Grouzmann E, Buckley MG, Barron J, MacGregor GA, Singer DR. How does treatment influence endocrine mechanisms in acute severe heart failure? Effects on cardiac natriuretic peptides, the renin system, neuropeptide Y and catecholamines. Clin Sci (Lond). 1998;94:591–9.
Lazzarini V, Felker GM. Management of the cardiorenal syndrome in acute heart failure. Curr Treat Options Cardiovasc Med. 2012;14:342–55.
Broqvist M, Dahlström U, Karlberg BE, Karlsson E, Marklund T. Neuroendocrine response in acute heart failure and the influence of treatment. Eur Heart J. 1989;10:1075–83.
Danielson LA, Sherwood OD, Conrad KP. Relaxin is a potent renal vasodilator in conscious rats. J Clin Invest. 1999;103:525–33.
Sasser JM, Molnar M, Baylis C. Relaxin ameliorates hypertension and increases nitric oxide metabolite excretion in angiotensin II but not N(ω)-nitro-l-arginine methyl ester hypertensive rats. Hypertension. 2011;58:197–204.
Debrah DO, Conrad KP, Jeyabalan A, Danielson LA, Shroff SG. Relaxin increases cardiac output and reduces systemic arterial load in hypertensive rats. Hypertension. 2005;46:745–50.
Jelinic M, Leo CH, Uiterweer ED, et al. Localization of relaxin receptors in arteries and veins, and region-specific increases in compliance and bradykinin-mediated relaxation after in vivo serelaxin treatment. FASEB J. 2014;28:275–87.
Fallick C, Sobotka PA, Dunlap ME. Sympathetically mediated changes in capacitance: redistribution of the venous reservoir as a cause of decompensation. Circ Heart Fail. 2011;4:669–75.
Perna AM, Masini E, Nistri S, et al. Novel drug development opportunity for relaxin in acute myocardial infarction: evidences from a swine model. FASEB J. 2005;19:1525–7.
Nistri S, Pini A, Sassoli C, et al. Relaxin promotes growth and maturation of mouse neonatal cardiomyocytes in vitro: clues for cardiac regeneration. J Cell Mol Med. 2012;16:507–19.
Perna AM, Masini E, Nistri S, Bani Sacchi T, Bigazzi M, Bani D. Human recombinant relaxin reduces heart injury and improves ventricular performance in a swine model of acute myocardial infarction. Ann N Y Acad Sci. 2005;1041:431–3.
Málek F, Ošťádal P, Pařenica J, et al. Uric acid, allopurinol therapy, and mortality in patients with acute heart failure—results of the Acute HEart FAilure Database registry. J Crit Care. 2012;27:737.e11–24.
Alimonda AL, Núñez J, Núñez E, et al. Hyperuricemia in acute heart failure. More than a simple spectator? Eur J Intern Med. 2009;20:74–9.
Collino M, Rogazzo M, Pini A, et al. Acute treatment with relaxin protects the kidney against ischaemia/reperfusion injury. J Cell Mol Med. 2013;17:1494–505.
Boehnert MU, Armbruster FP, Hilbig H. Relaxin as a protective substance in the preserving solution for liver transplantation: spectrophotometric in vivo imaging of local oxygen supply in an isolated perfused rat liver model. Ann N Y Acad Sci. 2009;1160:320–1.
Boehnert MU, Hilbig H, Armbruster FP. Relaxin as an additional protective substance in preserving and reperfusion solution for liver transplantation, shown in a model of isolated perfused rat liver. Ann N Y Acad Sci. 2005;1041:434–40.
Samuel CS, Unemori EN, Mookerjee I, et al. Relaxin modulates cardiac fibroblast proliferation, differentiation, and collagen production and reverses cardiac fibrosis in vivo. Endocrinology. 2004;145:4125–33.
Conrad KP, Schroff SG. Effects of relaxin on arterial dilation, remodeling, and mechanical properties. Curr Hypertens Rep. 2011;13:409–20.
Mookerjee I, Unemori EN, Du XJ, Tregear GW, Samuel CS. Relaxin modulates fibroblast function, collagen production, and matrix metalloproteinase-2 expression by cardiac fibroblasts. Ann N Y Acad Sci. 2005;1041:190–3.
Samuel CS, Zhao C, Bathgate RA, et al. The relaxin gene-knockout mouse: a model of progressive fibrosis. Ann N Y Acad Sci. 2005;1041:173–81.
Samuel CS, Cendrawan S, Gao XM, et al. Relaxin remodels fibrotic healing following myocardial infarction. Lab Invest. 2011;91:675–90.
Du XJ, Xu Q, Lekgabe E, et al. Reversal of cardiac fibrosis and related dysfunction by relaxin. Ann N Y Acad Sci. 2009;1160:278–84.
Sassoli C, Chellini F, Pini A, et al. Relaxin prevents cardiac fibroblast-myofibroblast transition via notch-1-mediated inhibition of TGF-β/Smad3 signaling. PLoS One. 2013;8:e63896.
Chow BS, Chew EG, Zhao C, Bathgate RA, Hewitson TD, Samuel CS. Relaxin signals through a RXFP1-pERK-nNOS-NO-cGMP-dependent pathway to up-regulate matrix metalloproteinases: the additional involvement of iNOS. PLoS One. 2012;7:e42714.
Parikh A, Patel D, McTiernan CF, et al. Relaxin suppresses atrial fibrillation by reversing fibrosis and myocyte hypertrophy and increasing conduction velocity and sodium current in spontaneously hypertensive rat hearts. Circ Res. 2013;113:313–21.
Fallowfield J, Hayden A, Snowdon V, et al. Relaxin modulates human and rat hepatic myofibroblast function and ameliorates portal hypertension in vivo. Hepatology. 2013 (Epub ahead of print).
Bennett RG, Kharbanda KK, Tuma DJ. Inhibition of markers of hepatic stellate cell activation by the hormone relaxin. Biochem Pharmacol. 2003;66:867–74.
Bennett RG, Heimann DG, Tuma DJ. Relaxin reduces fibrosis in models of progressive and established hepatic fibrosis. Ann N Y Acad Sci. 2009;1160:348–9.
Bennett RG, Heimann DG, Singh S, Simpson RL, Tuma DJ. Relaxin decreases the severity of established hepatic fibrosis in mice. Liver Int. 2013 (Epub ahead of print).
Gukathasan N, Mehran R. Acute coronary syndromes: advances in antithrombotics. Curr Atheroscler Rep. 2013;15:318.
Chen SA, Perlman AJ, Spanski N, et al. The pharmacokinetics of recombinant human relaxin in nonpregnant women after intravenous, intravaginal, and intracervical administration. Pharm Res. 1993;10:834–8.
Stough WG, O’Connor CM, Gheorghiade M. Overview of current noninodilator therapies for acute heart failure syndromes. Am J Cardiol. 2005;96(6A):41G–6G.
Metra M, Teerlink JR, Voors AA, et al. Vasodilators in the treatment of acute heart failure: what we know, what we don’t. Heart Fail Rev. 2009;14:299–307.
Moore XL, Tan SL, Lo CY, et al. Relaxin antagonizes hypertrophy and apoptosis in neonatal rat cardiomyocytes. Endocrinology. 2007;148:1582–9.
Mookerjee I, Hewitson TD, Halls ML, et al. Relaxin inhibits renal myofibroblast differentiation via RXFP1, the nitric oxide pathway, and Smad2. FASEB J. 2009;23:1219–29.
Wilson BC, Rappaport R. An in vitro study of the protective effect of relaxin on brain tissue under ischemic stress. Ann N Y Acad Sci. 2009;1160:265–8.
Heeg MH, Koziolek MJ, Vasko R, et al. The antifibrotic effects of relaxin in human renal fibroblasts are mediated in part by inhibition of the Smad2 pathway. Kidney Int. 2005;68:96–109.
Brecht A, Bartsch C, Baumann G, Stangl K, Dschietzig T. Relaxin inhibits early steps in vascular inflammation. Regul Pept. 2011;166:76–82.
Armbruster FP, Grön HJ, Maier I, et al. A sensitive homologous radioimmunoassay for human relaxin-2 (h-RLX-2) based on antibodies characterized by epitope mapping studies. Eur J Med Res. 2001;6:1–9.
Fisher C, Berry C, Blue L, Morton JJ, McMurray J. N-terminal pro B type natriuretic peptide, but not the new putative cardiac hormone relaxin, predicts prognosis in patients with chronic heart failure. Heart. 2003;89:879–81.
Dschietzig T, Richter C, Bartsch C, et al. The pregnancy hormone relaxin is a player in human heart failure. FASEB J. 2001;15:2187–95.
Kupari M, Mikkola TS, Turto H, Lommi J. Is the pregnancy hormone relaxin an important player in human heart failure? Eur J Heart Fail. 2005;7:195–8.
Callander GE, Thomas WG, Bathgate RA. Prolonged RXFP1 and RXFP2 signaling can be explained by poor internalization and a lack of beta-arrestin recruitment. Am J Physiol Cell Physiol. 2009;296:C1058–66.
SOLVD Investigators. Effect of enalapril on survival in patients with reduced left ventricular ejection fractions and congestive heart failure. N Engl J Med. 1991;325:293–302.
Hjalmarson A, Goldstein S, Fagerberg B, et al. Effects of controlled-release metoprolol on total mortality, hospitalizations, and well-being in patients with heart failure: the Metoprolol CR/XL Randomized Intervention Trial in congestive heart failure (MERIT-HF). MERIT-HF Study Group. JAMA. 2000;283:1295–302.
Granger CB, McMurray JJ, Yusuf S, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function intolerant to angiotensin-converting-enzyme inhibitors: the CHARM-Alternative trial. Lancet. 2003;362:772–6.
Zannad F, McMurray JJ, Krum H, et al. Eplerenone in patients with systolic heart failure and mild symptoms. N Engl J Med. 2011;364:11–21.
Krumholz HM, Lin Z, Keenan PS, et al. Relationship between hospital readmission and mortality rates for patients hospitalized with acute myocardial infarction, heart failure, or pneumonia. JAMA. 2013;309:587–93.
Vaduganathan M, Bonow RO, Gheorghiade M. Thirty-day readmissions: the clock is ticking. JAMA. 2013;309:345–6.
Gorodeski EZ, Starling RC, Blackstone EH. Are all readmissions bad readmissions? N Engl J Med. 2010;363:297–8.
Gheorghiade M, Vaduganathan M, Fonarow GC, Bonow RO. Rehospitalization for heart failure: problems and perspectives. J Am Coll Cardiol. 2013;61:391–403.
Clinicaltrials.gov identifier NCT01870778. http://clinicaltrials.gov/ct2/show/NCT01870778. Accessed January 2014.
Acknowledgments
The author would like to thank Graham Allcock and Marianne Wells of CircleScience for providing writing assistance, which was funded by Novartis Pharma AG, Basel, Switzerland.
Financial disclosure
The author has received adviser and/or lecturer fees from Novartis, Merck Sharp and Dohme, and Abbvie.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Open Access This article is distributed under the terms of the Creative Commons Attribution Noncommercial License which permits any noncommercial use, distribution, and reproduction in any medium, provided the original author(s) and the source are credited.
About this article
Cite this article
Díez, J. Serelaxin: A Novel Therapy for Acute Heart Failure with a Range of Hemodynamic and Non-Hemodynamic Actions. Am J Cardiovasc Drugs 14, 275–285 (2014). https://doi.org/10.1007/s40256-014-0069-0
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40256-014-0069-0