
Abstract
Purpose of the Review
The aim of this paper is to review the historical perspective for the need for new agents in the treatment of acute heart failure that may impact morbidity and mortality. Also, the purpose is to introduce serelaxin and discuss how it may play a role in the treatment for acute heart failure, in particular in the emergency department.
Recent Findings
Prospective, randomized trials in acute heart failure are limited. No single intervention or treatment in acute heart failure has improved mortality. However, registry data show that rapid treatment for acute heart failure produces better outcomes. Serelaxin is a pharmaceutical analog of relaxin which is a naturally occurring peptide hormone released in pregnancy that helps regulate hemodynamic function and renovascular blood flow through a number of effects including stimulation of nitric oxide production, vascular endothelial growth factor, and matrix metalloproteinases, and inhibition of endogenous vasoconstrictors. Serelaxin improved mortality at 180 days in RELAX-AHF when patients received 48 h of intravenous serelaxin within hours after presentation to the emergency department.
Summary
Serelaxin is a new agent with a different mechanism of action than previous agents studied in the treatment of acute heart failure. It produces vasodilation and improved renovascular blood flow and may prevent end organ damage to improve outcomes in patients with acute heart failure. Rapid treatment in the emergency department with intravenous diuretics and serelaxin in appropriate patients with normal to high blood pressure may have a beneficial effect on patients with acute heart failure.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Nearly 1 million people are admitted with acute heart failure (AHF) every year. About 80% are initially treated in the emergency department (ED) and of these 80% are ultimately admitted to the hospital [1]. Despite advances in the treatment of chronic heart failure (HF), patients who develop AHF face a worse prognosis with up to one third of patients dying or requiring rehospitalization within 90 days of their discharge. In-patient morbidity and mortality is also increased in AHF patients who have a delay in their diagnosis or treatment. Risk stratification of AHF patients may guide aggressive therapy and/or prompt admission to an intensive care unit for patients in higher risk groups. Despite the poor prognosis for delayed therapy or high-risk patients, there are no Class I guidelines from the American College of Cardiology (ACC) or the Heart Failure Society of America (HFSA) for pharmacologic therapy of AHF due to lack of clinical trial data. However, the medical community is aware of the gap of understanding in the care of this population as well as the lack of guidelines for the prompt care of this high-risk population. Although in 2015 recommendations on the early management of AHF were published by European Society of Cardiology and European Society of Emergency Medicine and the Society of Academic Emergency Medicine based on consensus from experienced practitioners, there remains a paucity of data to support treatment choice in addition to intravenous diuretics [2••]. This paper will discuss the approach to AHF using a case for illustration and will review the literature on specifically the use of vasodilators to date as well as a focus on the role of serelaxin, a potential game changer in the treatment of AHF.
Case
A 37-year-old man with no past medical history presented with cough, shortness of breath, and chest pain. He reports chest pain with inspiration but no fever/chills/sputum production or sick contacts. Exam revealed a blood pressure (BP) of 135/95 mmHg and heart rate (HR) of 126 bpm with tachypnea, no elevated jugular venous distension, but rales to the mid lung fields were noted in the absence of lower extremity edema. Labs were significant for only elevated troponin I at 0.151 (reference range 0.04 ng/ml). B-type natriuretic peptide (BNP) was not checked. Chest x-ray showed a “multifocal process, likely infectious.” The patient was admitted to a medicine team for the treatment of pneumonia. He received oxygen via nasal cannula, antibiotics, and cough suppressant. Later that day, a BNP level was checked and found to be over 500 pg/ml. Following this result, the patient received the first dose of intravenous (IV) furosemide 20 mg, 12 h after he presented to the ED. The cardiology team was consulted the next day and an echocardiogram was done which showed a left ventricular ejection fraction (LVEF) of <15%. The diagnosis of AHF was made. Antibiotics were recommended to be discontinued. Due to suboptimal diuresis and the need for inotropic support to improve diuresis, he was transferred to the cardiac care unit (CCU). He was discharged on day 14. In the subsequent 90 days, the patient was admitted three more times for AHF.
Diagnosis
This case illustrates the difficulty in diagnosing AHF in the ED and delay in treatment that can occur due to that difficulty. Thus, it is important to use tools to help aid in the diagnosis. In this case, the presentation, the exam, and even chest x-ray were confusing. Identification of AHF patients may be challenging as the presenting symptoms of dyspnea, edema, or abnormal lung findings (rales or effusion) may overlap with other conditions such as chronic obstructive pulmonary disease, pneumonia, liver disease, or pulmonary thromboembolic disease. Diagnostic testing can be used, including electrocardiogram (ECG), chest x-ray, and bedside echocardiography, to complement clinical judgment. Biomarkers have been particularly useful to improve clinical diagnosis among suspected AHF patients, most specifically natriuretic peptides. BNP and N-terminal proBNP have shown to improve diagnostic accuracy from ~75 to ~80% when used in conjunction with clinical judgment [3, 4]. As in the case above, the one lab that finally helped to confirm the diagnosis was BNP. The use of BNP or N-terminal-proBNP (NT-proBNP) is useful in the exclusion of AHF when a value is <50 pg/ml with a negative predictive value of 96% and >100 pg/nl with an accuracy of 83% [5]. When >400 pg/ml in a patient without a previous diagnosis of HF, the result is highly specific [6]. In addition, BNP >1000 pg/ml in the ED suggests a worse prognosis for AHF patients with a greater risk for inpatient mortality [7].
Treatment
In order to expedite the treatment of AHF, the diagnosis of AHF should frequently be considered for patients who present with shortness of breath. Once the diagnosis of AHF is suspected, an algorithm can be followed as outlined in a consensus paper from the European Society of Cardiology and Emergency Medicine [2••]. They suggest rapid identification of the unstable patient. These patients include those who are in shock, require ventilatory support with invasive or non-invasive mechanical ventilation, who have evidence of acute coronary syndrome, uncontrolled tachyarrhythmias, or clinically significant hypotension. Triaging this group of patients to an intensive care unit is important when invasive monitor is required.
Stable patients with AHF usually include those who have signs and symptoms of congestion in the setting of normal to high BP. Still, diagnostic tests need to be done to further confirm suspected AHF and evaluate for any comorbid conditions. In reality if there are no other extenuating conditions, the goal in many EDs is to decongest the patient and acutely optimize BP with discharge from the ED or to quickly determine if patient will need hospitalization. In general, if the patient quickly is diuresed and feels better with just a dose of IV diuretics, they may be a discharge candidate. When further management of volume or BP is required, the patient may require hospitalization. Regarding diuretic management, the ACCF/AHA guidelines provide the strongest recommendation of Class I, evidence level B, for AHF treatment to the use of IV loop diuretics in AHF patients with clinical evidence of volume overload. The initial IV dose generally exceeds the home dose, although in de novo HF a lower dose may be used, with close monitoring of effect on urine output and improvement in symptoms. If symptoms are inadequately relieved, addition of a synergistic diuretic can be added, typically a thiazide. Ultimately, if AHF patients do not respond to diuretics, and after ICU admission, the addition of dopamine may be considered to augment renal perfusion [8].
Diuretics, however, have not been shown to improve survival in of themselves. Chronic heart failure guidelines are based on abundant data supporting long-term therapy with angiotensin converting enzyme (ACE) inhibitor, angiotensin receptor blocker (ARB), β-blockers, and mineralocorticoid receptor antagonists. Pharmacologic therapies which address the underlying neurohormonal dysregulation have also been shown to improve LV function, decrease fibrosis and improve heart failure symptoms. These therapies are prescribed in the outpatient setting or in the hospital and do indeed improve long-term outcomes including reducing mortality and morbidity by 20–30%.
Rehospitalization and mortality have been shown to be reduced in HF patients in large randomized trials using ACE inhibitors [9,10,11,12], ARBs [13, 14], β-blockers [15,16,17,18], mineralocorticoid antagonists [19, 20], and the most recently validated angiotensin-neprylisin inhibitor (ARNI) and sacubitril/valsartan combination [21]. However, among the subset of patients who present with AHF and are hospitalized, their outcomes remain poor and rates of death and rehospitalization increase with age. Among patients that are >65 years of age, there is a 25% risk of rehospitalization at 30 days after discharge with an increase to 50% at 6 months [22]. Improving the outcomes of AHF patients is of prime interest for the HF community, but how to accomplish this remains elusive.
In AHF, there is some evidence that “time to therapy” may be important with early therapy improving outcomes [23,24,25]. Some suggest that like acute coronary syndromes, “time to therapy” is important in the treatment of AHF [2]. Early initiation of therapies in the ED for AHF has shown to improve outcomes for patients. In one large retrospective study, delays in IV diuretic administration of >4 h was associated with an increased mortality (OR of 1.004 per hour; CI 1.002 to 1.006, p = 0.001) and was more significant among AHF patients with a BNP level >856 pg/ml [26]. Similarly, other registries have reviewed timing of the administration of vasoactive substances such as dopamine, dobutamine, milrinone, nitroglycerin, and nitroprusside in AHF either in the ED (acute) or on an inpatient floor (delayed). Those who received vasoactive agents in the ED had significantly decreased length of hospital stay (4.5 versus 7 days, p < 0.0001) and in-hospital mortality (4.3 versus 10.9%, p < 0.0001) [27•, 28]. This evidence is important as it underscores the importance of initiating therapy early in the ED rather than waiting until the patient is admitted and on the unit.
BP is of particular importance in assigning risk as it is also useful in guiding treatment among AHF patients. Most patients can be stratified into three groups of hypertensive (systolic BP >140 mmHg), normotensive (SBP 100–140 mmHg), and hypotensive (SBP <100 mmHg). Patients in the hypertensive category with pulmonary edema may represent a distinct group of patients whose symptoms of dyspnea are driven predominantly due to increased vascular tone and filling pressures, rather than increased intravascular volume. These patients may benefit from aggressive vasodilator therapy rather or in addition to IV diuretics.
In AHF patients who present with significant hypertension (SBP >160 mmHg or mean arterial pressure >120 mmHg), early initiation of vasodilators has been suggested to decrease the need for endotracheal intubation (ETI), positive-pressure ventilation (PPV), and intensive care admission. In a single-arm, open-label study of high-dose nitroglycerin, a decrease in the risk of intubation was found in comparison to a retrospectively reviewed group of similar AHF patients. Between the two groups, a 12.9% lower risk of ETI (13.8 versus 26.7%), 13.1% lower risk of PPV use (6.9 vs. 20.0%), and 42.1% lower risk of ICU admission (37.9 vs. 80.0%) was observed [29]. High-dose isosorbide dinitrate plus low-dose furosemide has also been compared to therapy with high-dose diuretics and low-dose isosorbide dinitrate in patients presenting with signs of AHF. In patients who received high-dose isosorbide dinitrite, there was 27% lower use of PPV when compared to the high-dose diuretic group (p = 0.0041) [30]. Mean arterial BP in the two groups was 132 and 124 mmHg, respectively. However, outcome data do not demonstrate that use of nitrates in the ED for AHF produce any acute or near-term survival benefit [31].
The success of vasodilators in the treatment of AHF has demonstrated the importance of elevated systemic vascular resistance to the development of pulmonary edema and symptoms. Research has focused on weight loss (fluid loss/diuresis) and its prediction of improvement in symptoms as well as outcomes after discharge for AHF. Among discharged patients, there has been a higher risk of rehospitalization suggested due to inadequate diuresis. In one analysis, over 50% of patients admitted for AHF were found to lose <5 lbs., or even gain weight during their hospitalization [32]. When followed after discharge from the hospital, AHF patients with residual symptoms [33], pulmonary capillary wedge pressure (PCWP) >16 mmHg [34], or BNP >350 pg/ml [35] face greater risk of rehospitalization and mortality. The focus of many studies has been to augment diuresis in patients with AHF admitted to the hospital using continuous administration of diuretics, diuretic combinations, and even ultrafiltration. None of these has been shown to affect long-term outcomes among AHF patients.
The premise that greater volume removal is associated with superior outcomes is controversial. Analysis in the IMPACT-HF registry found that weight loss above and below the median did not predict improvement in fatigue, dyspnea, or paroxysmal nocturnal dyspnea. There was also no improvement in recurrent AHF or mortality at 60 days among those that lost more weight [36]. This refutes the idea that heart failure symptoms and increased pulmonary pressures are due solely to increased intravascular volume. Importantly, this underscores the fact that decongestion alone does not impact morbidity and mortality.
Thus, in the setting of AHF with normal or elevated BP, one can speculate that the use of vasodilators may be helpful and have a larger role in the management of AHF than previously thought, and maybe even more helpful if used expeditiously in the ED or early during hospitalization. Diuretics relieve congestion, but do not afford any mortality benefit. Vasodilators improve hemodynamics with venodilation and arterial dilation producing both preload and afterload reduction. Together, they both produce relief of congestive signs and symptoms and importantly for the patient marked improvement in dyspnea. However, AHF is a syndrome with a pathophysiology that really is poorly understood, likely due to the lack of experimental models and due the heterogeneity of presentation. However, it is known that it involves impaired hemodynamic function as well as neurohormonal activation, inflammation, and oxidative stress which can result in myocardial, renal, and hepatic injury, and remodeling [36,37,38]. These different mechanisms, and downstream organ damage, may result in further disease progression and worse short- and long-term prognosis despite decongestion and improved hemodynamics. Markers such as troponin and others confirm this likelihood [39,40,41,42,43,44,45,46,47,48,49,50,51,52,53,54,55].
Other vasodilator type therapies have been studied including nesiritide and ularitide. Different from nitrates, the natriuretic peptides have venous, arterial, and coronary vasodilatory properties. These effects decrease preload and afterload, increase cardiac output without direct inotropic effects, improve echocardiographic indices of diastolic function, and improve symptoms in patients with AHF. Nesiritide, a recombinant BNP, was approved in 2001 for use in patients with AHF to improve dyspnea and PCWP [56]. While there were high expectations for this drug in treating AHF, concern for increased mortality and renal toxicity, based on meta-analyses, prevented its use. In 2011, the ASCEND-HF trial reported on the use of nesiritide within 24 h of treatment for AHF in addition to standard care. It found no effect on dyspnea, no difference in rate of death or hospitalization, and no change in renal function; however, it was associated with more hypotension [56].
What was the reason for the failure of nesiritide? Was it used too late in the cascade of AHF? Was its impact on the kidneys not beneficial? Answers to these questions remain unclear. However, since then another natriuretic peptide, ularitide, has shown promise in early trials. Ularitide is the chemically synthesized form of urodilatin, a human natriuretic peptide, produced by differential processing of pro-atrial natriuretic peptide in distal renal tubule cells. It produces vasodilation, diuresis, and natriuresis through the natriuretic peptide receptor [57]. In phase 1 and 2 trials, SIRIUS I and II, the drug was found to improve dyspnea at 6 h and reduce PCWP by 10–12 mmHg compared to placebo at 4 mmHg. There was no worsening renal function. An additional and surprising result was a marked reduction in mortality, although the study was not powered to assess this outcome in only about 75 patients [58, 59]. This led to the most recent study, TRUE-AHF in a phase III efficacy study involving over 2100 patients. Although results have not been published, they were presented at AHA meeting in the fall of 2016. This trial sought to study efficacy of ularitide in patients with AHF treated with ularitide vs. placebo within 12 h of presentation. In addition, it included a sicker cohort of patients with dyspnea at rest, BNP >500 pg/ml and SBP ≥116 mmHg, but SBP ≤180 mmHg. Patients had mild to moderate renal impairment, but not severe. No LVEF cut-off was used to exclude participants. Importantly, about 50% of patients began treatment within 6 h of presentation with AHF. Follow-up over 15 months revealed no difference in the primary endpoint of mortality which showed 22% for ularitide group and 21% for placebo group. There were no differences in length of hospital stay, rehospitalization at 30 days or combined endpoint of all-cause mortality or cardiovascular hospitalization within 6 months [60]. Thus, with this trial and past trials with nesiritide, it seems that natriuretic peptides are unlikely to have a place in the contemporary treatment of AHF.
This strategy of targeting AHF early in its presentation with vasodilators has thus far been unsuccessful. Yet, we are still left with another drug in the vasodilator category, serelaxin. How might this drug be different? Relaxin is a naturally occurring peptide hormone released in pregnancy that helps regulate hemodynamic function and renovascular blood flow through a number of effects including stimulation of nitric oxide production, vascular endothelial growth factor, and matrix metalloproteinases and inhibition of endogenous vasoconstrictors. Serelaxin is a pharmaceutical analog of relaxin. Completed in 2012, RELAX-AHF trial enrolled over 1100 patients with mild to moderate renal insufficiency and SBP >125 mmHg. Overall, the mean SBP of enrolled patients was 142 mmHg. Dyspnea scores were better in the serelaxin group than placebo. There was a reduction in BP with serelaxin but a built-in protocol for medication reduction resulted in no difference in hypotension related adverse events (5 vs. 4%, serelaxin vs. placebo). There were significant decreases in worsening heart failure, clinical signs of vascular congestion, adverse events related to renal impairment, length of initial hospital stay, and total dose of IV diuretic through day 5 in the serelaxin group. However, treatment with serelaxin did not improve acute dyspnea in a clinically relevant fashion, did not increase days alive out of hospital, and there was no difference in cardiovascular death or hospital readmission for heart failure or renal failure up to day 60. Notably, serelaxin was associated with a significant reduction in both all-cause (6.1 vs. 9.6%; p = 0.028) and cardiovascular (7.3 vs. 11.3%; p = 0.02) mortality when follow-up was increased to 180 days, a pre-specified safety endpoint [61••].
So how should we view this data? The FDA has not approved the use of serelaxin and the results of RELAX-AHF-2 [62] will be announced in the next year. In the meantime, however, it is interesting to speculate how one might explain the results of RELAX-AHF and postulate how to approach use of this medication in the setting of AHF.
We can look at the patients studied in both nesiritide and serelaxin studies (we do not have ularitide details available). Both studies included AHF with heterogeneous population with regards to sex, race, and etiology of heart failure. The breakdown of patient characteristics did show that for both groups there was a similar rate of ischemic heart disease and male sex. However, the serelaxin study group was predominantly white (94–95%) and older (mean age 72) vs. the nesiritide study cohort (56% white, mean age 67). The LVEF in both groups included reduced and preserved left ventricular systolic function with the nesiritide group having 80% with LVEF <40% and serelaxin study having only 55% LVEF <40%. However, both groups had similar NT-pro BNP levels. In addition, because of pre-specified entry criteria, the creatinine was mildly to moderately elevated in the serelaxin study, but and close to normal in the nesiritide study. Finally, two key items that may have impacted study results are BP which was higher in the serelaxin group than the nesiritide (SBP 142 vs. 123 mmHg) and the time to drug administration which was almost 16 h for nesiritide and about 8 h for serelaxin. Thus, a broad conclusion may be that vasodilator type drugs are more effective in patients with higher BPs and when the drug is administered expeditiously after AHF presentation in the ED [56, 61••]. Although serelaxin did not reduce readmissions, it did have an impact on both all-cause mortality as well as cardiovascular mortality. This may be due to its beneficial effect on markers of injury, such as troponin T, cystatin C, AST, and ALT [63]. Thus, if myocardial, renal, and hepatic injuries are lessened, long-term mortality may be impacted even though readmissions for hospitalization in the short-term are not affected.
Conclusion
In the ED, when evaluating a patient with AHF, time may be of the essence. What if AHF is similar to acute coronary syndrome where time wasted and therapy delayed may be lives lost due to myocardial and other organ injury? Thus, in the ED the algorithm for assessing someone with AHF should include ruling out any immediate comorbid conditions that may need addressing, such as ACS, pneumonia, DKA, etc. [2••, 64]. This also includes considering clinical severity, heart rate and rhythm, BP, AHF precipitants, comorbidities, and if the patient has de novo or chronic HF. Although all of these issues need careful evaluation, stability of the patient and BP are most important for appropriate triaging. Pang et al. describe three BP phenotypes: hypertensive (SBP > 140 mmHg), normotensive (90–140 mmHg), and hypotensive (<90 mmHg). If serelaxin is FDA cleared and we are going to use it in the ED, different thresholds may need to be defined as per the RELAX-AHF entry criteria of SBP ≥125 and SBP <125 mmHg. Importantly, knowledge of the LVEF is not needed, but BNP should be >350 pg/ml or pro NT-BNP > 1400 pg/ml. Also, renal function needs to be assessed and GFR should be >25 cm3/min/1.73 m^2. If a patient is volume overloaded, then attempt at decongestion should be made with intravenous loop diuretics and if SBP remains ≥125 mmHg after initial diuresis, then serelaxin may be considered at 30 mcg/kg/day, as long as renal function is not severely impaired. Vasoactive drugs should be avoided, except IV nitroglycerin, which has been demonstrated to be safe if used within 2 h prior to initiation of IV serelaxin. As per protocol, if SBP remains stable, then serelaxin can be continued for 48 h. If SBP decreases by 40 mmHg or is <100 mmHg, then the dose should be reduced by half and if SBP is persistently <100 mmHg, the drug should be stopped. It is important to also exclude pregnancy as this drug has not been assessed in patients who were pregnant. Real-life experience with serelaxin should be considered. The drug can produce profound hypotension if the SBP is not elevated at presentation. This can be problematic if patients are or were treated with not just IV nitroglycerin but also ACEI, ARBs, BB, or other anti-hypertensives. Caution should be used when considering serelaxin for this reason. Administration requires close monitoring in an ICU setting with one-on-one observation for the first 12 h to ensure that if hypotension occurs, the dose can be adjusted or the drug can be discontinued if necessary.
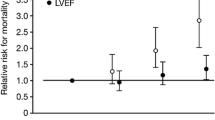
Our 37-year-old patient may have benefited from serelaxin given that he was hemodynamically stable with SBP of >125 mmHg. He had markers of increased risk including elevated troponin I 0.175 ng/ml and BNP > 500 pg/ml. Renal function was preserved at 88 ml/mi/1.73 m^2. According to data from RELAX-AHF, with improved indices of myocardial, renal, and liver injury as shown in Table 1, our patient would be expected to have improved mortality at 180 days. This protection may be the explanation for mortality benefit among those who received serelaxin vs. placebo (Fig. 1). Our recommendations for the use of serelaxin in AHF are only a proposed algorithm, given it has not been approved for use by the FDA. The results of RELAX-AHF2 are anxiously awaited so that we may confirm the beneficial effect on mortality that was shown in RELAX-AHF and add to the ED armamentarium of drugs with which to treat AHF and impact long-term outcomes of this high-risk patient population.

A significant improvement in mortality at 180 days was noted among patients who received serelaxin in RELAX in acute heart failure (RELAX-AHF) compared to those who received placebo [61••]
References
Papers of particular interest, published recently, have been highlighted as: • Of importance •• Of major importance
Go A, Mozaffarian D, Roger VL, et al. Heart disease and stroke statistics—2014 update: a report from the American Heart Association. Circulation. 2014;129:e28–292.
•• Mebazaa A, Yilmaz MB, Levy P, et al. Recommendations on pre-hospital and early Hospital Management of Acute Heart Failure: a consensus paper from the Heart Failure Association of the European Society of Cardiology, the European Society of Emergency Medicine and the Society of Academic Emergency Medicine. Eur J of Heart Failure. 2015;17:544–58. This aticle discusses expert opinion on the management and treatment of acute heart failure
Januzzi J, van Kimmenade R, Lainchbury J, et al. NT-proBNP testing for diagnosis and short-term prognosis in acute destabilized heart Failure: an international pooled analysis of 1256 patients: the International Collaborative of NT-proBNP Study. Eur Heart J. 2006;27:330–7.
McCullough P, Nowak RM, McCord J, et al. B-type natriuretic peptide and clinical judgement in emergency diagnosis of heart failure: analysis from Breathing Not Properly (BNP) Multinational Study. Circulation. 2002;106:416–22.
Maisel A, Krishnaswamy P, Nowak RM, et al. Rapid measurement of B-type natriuretic peptide in the emergency diagnosis of heart failure. NEJM. 2002;347:161–7.
Maisel A, Daniels LB. Breathing not properly 10 years later: what we have learned and what we still need to learn. JACC. 2012;60:277–82.
Fonarow G, Peacock WF, Horwich TB, et al. Usefulness of B-type natriuretic peptide and cardiac troponin levels to predict in-hospital mortality from ADHERE. Am J Cardiol. 2008;101:231–7.
Yancy CW, Jessup M, Bozkurt B, et al. ACCF/AHA guideline for the Management of Heart Failure—a report of the American College of Cardiology Foundation/American Heart Association Task Force on practice guidelines. Circulation. 2013;2013:138.
Lewis G. Comparison of Lisinopril versus placebo for congestive heart failure. Am J Cardiol. 1989;63:12D–6D.
Lechat P, Garnham SP, Desche P, et al. Efficacy and acceptability of perindopril in mild to moderate chronic congestive heart failure. Am Heart J. 1993;136:798–806.
Magnani B, Magelli C. Captopril in mild heart failure: preliminary observations of a long-term, double-blind, placebo-controlled multicentre trial. Postgrad Med J. 1986;61:153–8.
Swedberg K, Kjekshus J. Effects of Enalapril on mortality in severe congestive heart failure: results of the Cooperative North Scandinavian Enalapril Survival Study. Am J Cardiol. 1988;62:60A–6A.
Maggioni A, Anand I, Gottlieb SO, et al. Effects of valsartan on morbidity and mortality in patients with heart failure not receiving angiotensin-converting enzyme inhibitors. Journal of American College of Cardiology. 2002;40:1414–21.
McMurray J, Ostergren J, Swedberg K, et al. Effects of candesartan in patients with chronic heart failure and reduced left-ventricular systolic function taking angiogtensin-converting-enzyme inhibitors: the CHARM-added trial. Lancet. 2003;362:767–71.
CIBIS II Authors. The cardiac Insuffiency bisoprolol study II (CIBIS-II): a randomised trial. Lancet. 1999;353:9–13.
MERIT-HF-Investigators. Effect of metoprolol CR/XL in chronic heart failure (MERIT-HF). Lancet. 1999;353:2001–7.
Packer M, Bristow MR, Cohn JN, et al. The effect of carvedilol on morbidity and mortality in patients with chronic heart failure. NEJM. 1996;334:1349–55.
Packer M, Coats AJS, Fowler M, et al. Effect of carvedilol on survival in severe chronic heart failure. NEJM. 2001;344:1651–8.
Pitt B, Remme W, Zannad F, et al. Eplerenone, a selective aldosterone blocker, in patients with left ventricular dysfunction after myocardial infarction. NEJM. 2003;348:1309–21.
Zannad F, McMurray JJV, Krum H, et al. Eplerenone in patient with systolic heart failure and mild symptoms. NEJM. 2011;364:11–21.
McMurray J, Packer M, Desai AS, et al. Angiotensin-neprilysin inhibition versus enalapril in heart failure. NEJM. 2014;371:993–1004.
Chen J, Ross JS, Carlson MD, et al. Skilled nursing referral and hospital readmission rates after heart failure or myocardial infarction. Am J Med. 2012;125:e1–9.
Maisel A, Peacock WF, McMullin N, et al. Timing of immunoreactive B-type natriuretic peptide levels and treatment delay in acute decompensated heart failure: an ADHERE (Acute Decompensated Heart Failure National Registry) analysis. Journal of American College of Cardiology. 2008;52:534–40.
Peacock W, Emerman CL, Costanzo MR, et al. Early vasoactive drugs improve heart failure outcomes. Congst Heart Fail. 2009;15:256–64.
Wuerz R, Meador SA. Effects of prehospital medications on mortality and length of stay in congestive heart failure. Ann Emerg Med. 1992;21:669–74.
Maisel AS, Peacock W, McMullin N. Timing of Immunoreactive B-type natriuretic peptide levels and treatment delay in acute decompensated heart failure. Journal of American College of Cardiology. 2008;52:534–40.
• Peacock W, Emerman CL, Costanzo MR, et al. Early initiation of intravenous therapy improve heart failure outcomes: an analysis from the ADHERE registry database. Ann Emerg Med. 2003;42:S26. Article provides registry data for early initiation of treatment for acute heart failure for better outcomes
Peacock W, Emerman CL, Costanzo MR, et al. Acute heart failure mortality is dependent on time to intravenous vasoactive administration. J Card Fail. 2006;12(Suppl 1):S117.
Levy P, Compton S, Welch R, et al. Treatment of severe decompensated heart failure with high-dose intravenous nitroglycerin: a feasibility and outcome analysis. Ann Emerg Med. 50:144–52.
Cotter G, Metzkor E, Kaluski E, et al. Randomised trial of high-dose isosorbide dinitrate plus low-dose furosemide versus high-dose furosemide plus low-dose isosorbide dinitrate in severe pulmonary oedema. Lancet. 1998;351:389–93.
Ho E, Parker JD, Austin PC, et al. Impact of nitrate use on survival in acute heart failure: a propensity-matched analysis. J of the American Heart Association. 2016;5:e002531.
Gheorgihiade M, Filippatos G. Reassessing treatment of acute heart failure syndromes: the ADHERE registry. European Heart Journal Supplement. 2005;7:B13–9.
Lucas C, Johnson W, Hamilton MA. Freedom from congestion predicts good survival despite previous Class IV symptoms of heart failure. Am Heart J. 2000;140:840–7.
Fonarow G, Stevenson LW, Steimle AE, et al. Persistently high left ventricular filling pressures predict mortality despite angiotensin converting enzyme inhibition in advanced heart failure. Circulation. 1994;90(pt 2):1–488.
Logeart D, Thabut G, Jourdain P, et al. Predischarge B-type natriuretic peptide assay for identifying patients at high risk of re-admission after decompensated heart failure. JACC. 2004;43:635–41.
Cotter G, Felker GM, Adams KF, et al. The pathophysiology of acute heart failure-is it all about fluid accumulation? Am Heart J. 2008;155:9–18.
Cotter G, Milo O, Davison BA. Increased mortality after an acute heart failure episode: new pathophysiological insights from RELAX-AHF study and beyond. Curr Heart Failure Rep. 2014;11:19–30.
Cotter G, Metra M, Milo-Cotter O, et al. Fluid overload in acute heart failure-re-distribution and other mechanisms beyond fluid accumulation. Eur J of Heart Failure. 2008;10:165–9.
You J, Austin PC, Alter DA, et al. Relation between cardiac troponin I and mortality in acute decompensated heart failure. Am Heart J. 2007;153:462–70.
Perna E, Aspromonte N, Cimbaro Canella JP, et al. Minor myocardial damage in a prevalent condition in patients with acute heart failure syndromes and preserved systolic function with long-term prognostic implications: a report from the CIAST-HF (Collaborative Italo-Argentinean Study on Cardiac Troponin T in Heart Failure) study. J Cardiac Fail. 2012;18:822–30.
Perna E, Macin SM, Cimbaro Canella JP, et al. Minor myocardial damage dectected by troponin T is a powerful predictor of long-term pronosis in patients with acute decompensated heart failure. Int J Cardiol. 2005;99:253–61.
Peacock WF, De Marco T, Fonarow GC, et al. Cardiac troponin and outcome in acute heart failure. NEJM. 2008;358:2117–26.
Parenti N, Bartolacci S, Carle F, et al. Cardiac troponin I as prognostic marker in heart failure patients discharged from emergency department. Intern Emerg Med. 2008;3:43–7.
Metra M, Cotter G, Davison BA, et al. Effect of serelaxin on cardiac, renal, and hepatic biomarkers in the relaxin in acute heart failure (RELAX-AHF) development program: correlation with outcomes. Journal of American College of Cardiology. 2013;61:196–206.
Metra M, Cotter G, Gheorghiade M, et al. The role of the kideny in heart failure. Eur Heart Journal. 2012;33:2135–42.
Metra M, Bettari L, Pagani F, et al. Troponin T levels in patients with acute heart failure: clinical and prognostic significance of their detection and release during hospitalisation. Clin Res Cardiol. 2012;101:663–72.
Maisel A, Choudhary R. Biomarkers in acute heart failure-state of the art. Nature Reviews-Cardiology. 2012;9:478–90.
Maisel A, Mueller C, Fitzgerald R, et al. Prognostic utility of plasma neutrophil gelatinase-associated lipocalin in patients with acute heart failure: the NGAL evaLuation along with B-type NaTriuretic peptide in acutely decompensated heart failure (GALLANT) trial. Eur J of Heart Failure. 2011;13:846–51.
La Vecchia L, Mezzena G, Zanolla L, et al. Cardiac troponin I as diagnostic and prognostic marker in severe heart failure. Journal of Heart Lung Transplant. 2000;19:644–52.
Kociol R, Pang PS, Gheorghiade MD, et al. Troponin elevation in heart failure: prevalence, mechanisms, and clinical implications. Journal of American College of Cardiology. 2010;56:1071–8.
Felker G, Hasselblad V, Tang WH, et al. Troponin in acute decompensated heart failure: insights from the ASCEND-HF study. Eur J of Heart Failure. 2012;14:1257–64.
Del Carlo C, Pereira-Barretto AC, Cassaro-Strunz C, et al. Serial measure of cardiac troponin T levels for prediction of clinical events in decompensated heart failure. J Cardiac Fail. 2004;10:43–8.
Damman K, Van Veldhuisen DJ, Navis G, et al. Tubular damage in chronic systolic heart failure is associated with reduced survival independent of glomerular filtration rate. Heart. 2010;96:1297–302.
Braga, JR, Tu, JV, Austin, PC, et al. Outcomes and care of patients with acute heart failure syndromes and cardiac troponin elevation. Circulation Heart Failure 2013;6:193–202.
Ambrosy A, Vaduganathan M, Huffman MD, et al. Clinical course and predictive value of liver function tests in patients hospitalized for worsening heart failure with reduced ejection fraction: an analysis of the EVEREST trial. Eur J of Heart Failure. 2012;14:302–11.
O’Connor C, Starling RC, Hernandez AF, et al. Effect of nesiritde in patients with acute decompensated heart failure. NEJM. 2011;365:32–43.
Anker S, Ponikowski P, Mitrovic V, et al. Ularitide for the treatment of acute decompensated heart failure: from preclinical to clinical studies. Eur Heart Journal. 2015;36:715–23.
Mitrovic V, Seferovic P, Simeunovic D, et al. Hemodynamic and clinical effects of ularitide in decompensated heart failure. Euro Heart J. 2006;27:2823–32.
Mitrovic V, Luss H, Nitsche K, et al. Effects of the renal natriuretic peptide urodilatin (ularitide) in patients with decompensated chronic heart failure: a double-blind, placebo-controlled, ascending-dose trial. Am Heart J. 2005;150:1239.e1–8.
Cardiorentis. Efficacy and safety of ularitide for the Treatment of Acute Decompensated Heart Failure (TRUE-AHF). ClinicalTrialsgov. 2016;https://clinicaltrials.gov/ct2/show/NCT01661634.
•• Teerlink J, Cotter G, Davison BA, et al. Serelaxin, recombinant human relaxin-2, for treatment of acute heart failure (RELAX-AHF): a randomized, placebo-controlled trial. Lancet. 2013;381:29–39. This is a seminal article on the RELAX AHF study discussing the results and endpoints in the use of intravenous serelaxin in the setting of acute heart
Novartis Pharmaceuticals. Efficacy, safety and tolerability of serelaxin when added to standard therapy in AHF (RELAX-AHF-2). ClinicalTrialsgov (Bethesda, MD). 2017;https://clinicaltrials.gov/ct2/show/NCT01870778.NCT01870778.
Diez J, Ruilope LM. Serelaxin for the treatment of acute heart failure: a review with a focus on end-organ protection. Eur Heart Journal. 2016;2:119–30.
Gheorghiade M, Braunwald E. A proposed model for initial assessment and management of acute heart failure syndromes. Journal of American Medical Association. 2011;305:1702–3.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
Drs Cunningham and Misra declare no conflict of interest.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
This article is part of the Topical Collection on Heart Failure
Rights and permissions
About this article

Cite this article
Cunningham, L., Misra, A. Serelaxin in the Treatment of Acute Heart Failure in the Emergency Department. Curr Emerg Hosp Med Rep 5, 68–75 (2017). https://doi.org/10.1007/s40138-017-0136-3
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40138-017-0136-3