
Abstract
Early blight disease samples were collected from six different states of India. Seventeen different isolates of Alternaria spp were obtained in pure culture which were designated as PB-2, PB-4, PB-13, PB-17, PB-19, HR-12, HR-44, HR-62, MP-6, MP-7, JT-4, JT-11, UP-7, UP-10, UP-15, UP-20 and RJ-T-1. Characterization based on culture colour and conidial dimensions (conidial length and breadth, and beak length) indicated that no distinguished variation of the isolates was observed on the colour parameters. Although, a wide variation was observed in the length of conidia and beak, no significant difference was observed in conidial breadth except RJ-T-1. Among the isolates, conidial length of five isolates viz., HR-12, UP-7, UP-10, UP-15 and RJ-T-1 ranged from 80 to 97 µm whereas others were of significantly lesser size (30–51 µm). Similarly, significant difference was observed in beak length. The PCR amplification of the fungal DNA using universal primers ITS1 and ITS4 and sequencing indicated that, out of 17 isolates, 12 (JT-4, JT-11, HR-62, HR-44, PB-2, PB-4, PB-13, PB-17, PB-19, UP-20, MP-6 and MP-7) were closely matching with A. alternata (JX993756) and five isolates (HR-12, UP-7, UP-10, UP-15 and RJ-T-1) with A. solani (JF796068). Further, pathogenicity test on tomato revealed that both A. alternata and A. solani isolates were of virulent category indicating that former is also an incitant of early blight in northern India.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Alternaria belonging to sub-division Deutromycotina is a cosmopolitan fungus and is the causal organism of leaf blight diseases in Brassicaceous, Cucurbitaceous and Solanaceous vegetables. Early blight in tomato is the most destructive disease as it accounted for 78 % yield loss at 72 % disease intensity [1]. The pathogen of the disease has been documented as A. solani globally [2–4] as well as in India [5–7]. On the flip side, Bhatt et al. [8] collected early blight infected tomato samples from Almora, a hilly area in Uttarakhand, India and the pathogen responsible for the disease was identified through Indian Type Culture Collection, NewDelhi, India as A. alternata (IARI, Identification No: 3314.97). Moreover, certain tomato genotypes susceptible to A. solani under in vivo screening showed complete resistance under field conditions hinting the possibility of existence of other species of Alternaria in the field. The above two studies directed that documentation of Alternaria spp infecting tomato in India and characterization of pathogen based on pathogenic potentiality are mandatory to develop appropriate management practices. Identification of early blight pathogen is generally based on conidial morphology, but for conidial production, the pathogen needs specific media, temperature, relative humidity and light [6, 9]. Hence, it is a time consuming process apart from the fact, that species level identification of the pathogen is very difficult. Alternatively, genetic identification is progressively used to identify the pathogens [10, 11] and internal transcribed spacer (ITS) rRNA has been successfully employed to identify the fungal pathogens at species level [12–14]. In the present study, Alternaria early blight infected tomato samples collected from diverse locations especially from north and central India were used to characterize Alternaria spp., based on cultural characteristics, pathogenic potentiality and ITS region.
Material and Methods
Early blight infected tomato plants were collected from six states (Jammu and Kashmir, Haryana, Punjab, Uttar Pradesh, Madhya Pradesh and Rajasthan) of India (Table 1) and pure pathogen cultures were isolated on potato dextrose agar (PDA) and subsequently purified by single hyphal tip method. The cultures were transferred on PDA slants and incubated at 25 ± 2 °C under alternate light/darkness (12 h each) for 72 h. After that the cultures were stored at 4 °C for further use. All the cultures were tested for Koch’s postulates by standard procedure.
Characterization Based on Culture and Spore Morphology
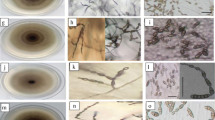
Pure cultures of 17 isolates (PB-2, PB-4, PB-13, PB-17, PB-19, HR-12, HR-44, HR-62, MP-6, MP-7, JT-4, JT-11, UP-7, UP-10, UP-15, UP-20 and RJ-T-1) were individually transferred onto PDA in petri dishes and incubated at 25 ± 2 °C. For each treatment (culture), three replications (two plates per replication) were maintained and followed completely randomized design (CRD). After 9 days, the cultures were observed for morphological characters viz., culture colour, pigmentation and growth pattern. Similarly, in another experiment, the cultures were transferred onto ready-made available V-8 juice agar medium containing asparagine for spore morphological studies. The culture plates were incubated at 25 ± 2 °C under alternate light/darkness (12 h each). After 9 days, for each culture, the spore morphologies were observed under microscope.
Molecular Characterization
Fungal DNA Extraction
Fungal isolates were grown on potato dextrose broth at 25 ± 2 °C for 10 days. DNA was extracted from the cultures by using Manicom et al. [15] protocol with minor modifications. Mycelium of the cultures was taken out by filtration through Whatman No 1 filter paper and washed thoroughly in distilled water and properly dried. Three gram of mycelium was macerated along with liquid nitrogen with the help of mortar and pestle to get a powder form. The powder was transferred into a centrifuge tube and added with 15 mL of CTAB buffer (2 % (w/v) CTAB, 1.4 M NaCl, 20 mM EDTA, 100 mM Tris–HCl, pH 8.0, 1 % PVP, 1 % (v/v) 2-mercaptoethanol). The mixture was incubated at 65 °C in a water bath for 30 min with intermittent shaking and the entire content was centrifuged at 13,000 rpm for 10 min at 4 °C. The supernatant was transferred into a fresh Oakridge tube and an equal volume of phenol:chloroform:isoamylalcohol (25:24:1) was slowly added to it. Again, the content was centrifuged at 13,000 rpm for 10 min at 4 °C. The supernatant was transferred into a fresh tube and added with 0.6 volume isopropanol and incubated overnight at −20 °C. On the following day, it was again centrifuged at 13,000 rpm for 10 min at 4 °C temperature. The pellet was retained and supernatant was discarded. The pellet was briefly washed with 75 % ethanol and dried at room temperature. Finally the pellet was dissolved in 100 µl of TE buffer and kept at −20 °C for further use.
PCR
The fungal DNA was amplified using universal internal transcribe spacer region (ITS) primers (ITS1-TCCGTAGGTGAACCTGCGG/ITS4-TCCTCCGCTTATTGATATGCA) as described by White et al. [16]. The PCR reaction was carried out in a Biorad Thermocycler (S 1000™) with 35 cycles of denaturation for 1 min at 94 °C, primer annealing for 45 s at 55 °C and primer extension for 1 min 30 s at 72 °C, with an initial denaturation at 94 °C for 3 min and a final extension for 15 min at 72 °C. The reaction was carried out in a volume of 25 μL containing 1 μL DNA template, 1.5 U pfu DNA polymerase, 25 mM MgCl2, 2 mM dNTPs and 25 pmol of each primer. PCR products were electrophoresed (1 h at 80 volts) in 0.8 % agarose gel in Tris–borate-EDTA buffer at pH 8. Gels were stained with ethidium bromide (10 µg/mL) and viewed in a Gel documentation system (Alpha Innotech, USA).
Cloning of PCR Product and Sequencing
The PCR amplified product was excised from gel and purified by Gel extraction kit (Qiagen) and cloned into the pTZ57R/T vector (Fermentas, Germany) according to the manufacturers’ instructions. The vector was transformed into Escherichia coli DH5α competent cells (Novagen) by following standard molecular biology procedures [17]. Recombinant clones were identified by restriction endonuclease digestion, and selected clones were sequenced with automated sequencer ABI PRISM 3730 (Applied Biosystems) from Eurofin Genomic India Pvt. Ltd DNA Sequencing facility, Bangalore, Karnataka, India.
Sequence Analysis
The sequences obtained from the ITS region of all Alternaria isolates were subjected to NCBI (www.ncbi.nlm.nih.gov) BLAST search and the sequences showing highest scores were retrieved. Multiple alignments were carried out with CLUSTAL-X program [18]. The phylogenetic tree was generated by MEGA 6.06 software [19] using the neighbour joining method with 1,000 bootstrapped replications to estimate evolutionary distances between all pairs of sequences simultaneously.
Analysis of Pathogenic Variability
In order to test aggressiveness, the cultures were inoculated on tomato seedlings grown in pots filled with sterile potting mixture (Soil: Sand: well decomposed farm yard manure at 2:1:1). For this study two highly susceptible tomato cultivars CO3 and DVRT-2 seedlings were raised in pots. After 25 days, seedlings were transplanted in 30 cm diameter pots. Three seedlings were planted at equal distance in each pot. After 20 days of transplanting (45 days old), the plants were spray inoculated with spore suspension of fungal cultures. For preparation of spore suspension, the cultures were established on readymade V-8 agar medium containing asparagine as described in spore characterization study. After 10 days, culture mat was harvested by applying 10-15 ml of sterile water and scraping the mycelial mat with spores. To harvest the spores, the content was mixed well and filtered through three layers of sterile muslin cloth. The filtrate containing spores was adjusted to 105 spores/ml by diluting with sterile distilled water. The spore suspension was spray inoculated on seedlings and for each culture, three pots (total nine seedlings) were maintained. A humidifier was set to create sufficient humidity (>80 %). After 15 days of spray inoculation, the plants were observed for early blight development and scored for per cent disease index as described by Pandey et al. [20].
Statistical Analysis
The percentage (%) values were transformed in arcsine values before analysis and the data were analysed using IRRISTAT (v. 92) developed by the International Rice Research Institute (IRRI).
Results and Discussion
Cultural Characterization
A total of 17 isolates were collected from major tomato growing states (Jammu and Kashmir, Haryana, Punjab, UP, Rajasthan and MP) of India covering northern and central India (Table 1), and all the cultures proved Koch’s postulates. Characterization of the isolates indicated that most of the cultures were gray or brown in colour with light or dark brown pigmentation and regular or irregular growth pattern (Table 2). Hence no distinguished variation was observed on culture colour of isolates. Regarding conidial morphologies viz., conidial length, breadth and beak length, a wide variation was observed in length of conidia and beak but no significant difference was observed in conidial breadth except that of RJ-T-1. Hence, conidial length and beak length can be considered for characterization of early blight pathogen. Among the isolates, conidial length of five isolates viz., HR-12, UP-7, UP-10, UP-15 and RJ-T-1 ranged from 80 to 97 µm whereas others were having significantly smaller size (30–51 µm). Similarly, significant difference was observed in beak length. This indicated the existence of two categories of isolates, one group with five isolates (HR-12, UP-7, UP-10, UP-15 and RJ-T-1) and the other with 12 isolates (JT-4, JT-11, HR-62, HR-44, PB-2, PB-4, PB-13, PB-17, PB-19, UP-20, MP-6 and MP-7) and probably they might be belonging to A. solani and A. alternata respectively. Most of the reports available in India indicated that the early blight of tomato is caused by A. solani [6, 21] but the present study showed two species of Alternaria infecting tomato and hence to further confirm, molecular based identification of Alternaria isolates was included.
PCR Amplification and Phylogenetic Analysis
The 17 early blight pathogen isolates collected from different geographical locations were amplified by PCR using universal primer pairs ITS1/ITS4. The process resulted in amplification of ~580 bp in all infected tomato samples which corresponds to ITS rRNA of the fungal species. The PCR products were sequenced and nucleotide sequences of ITS region of all 17 isolates of Alternaria spp were compared with the representative ITS region of selected fungal species from the GeneBank database. BLAST searches using ITS sequences obtained from 12 isolates produced 96.7–98.0 % identities to A. alternata sequences existing in the GeneBank (JX993756), while five others produced 71.1–85.7 % identities to A. solani (JF796068) (Table 3).
The study clearly indicated that majority of Alternaria spp infecting tomato are A. alternata followed by A. solani. Similar identification of fungal pathogens through phylogenetic relationship was successfully documented [22–25].
Aggressiveness of A. solani and A. alternata on Tomato
Though two species A. solani and A. alternata were observed in association of early blight of tomato, it is essential to study their potentiality to cause disease (virulence) because in many instances these species exists as saprophytes or weak parasite or opportunistic pathogen [26]. Observations on virulence of different isolates of A. solani and A. alternata revealed that the species could produce similar kind of reaction on susceptible genotypes CO3 and DVRT-2 (Table 4). However, the isolates belonging to same species manifested observable variation in pathogenicity. Among 12 A. alternata isolates, seven isolates of JT-4, HR-62, PB-4, PB-13, UP-20, MP-6 and MP-7 were categorised into virulent group as they produced 80–90 % PDI and remaining five (JT-11, HR-44, PB-2, PB-17 and PB-19) were grouped as weak virulent (10–20 % PDI). In case of A. solani, all five A. solani (HR-12, UP-7, UP-10, UP-15 and RJ-T-1) were able to produce 80–100 % PDI and hence categorised as virulent group. The study indicated that A. solani exist as a virulent form to cause early blight disease in tomato as documented earlier [5–7, 21]. In the present experiment, contrary to the earlier studies, majority of isolates were A. alternata and also proved that many of them are in virulent form. Although, Bhatt et al. [8] have reported the association of A. alternata with early blight disease of tomato in Uttarakhand, they did not prove the extent of damage caused by it or categorisation of the pathogen as virulent or weak virulent. Hence the present study clearly indicated the existence of A. alternata as virulent form to cause early blight in tomato and this species is widely distributed in northern India emphasizing the fact that in future breeding for resistance against early blight in tomato should be done against both A. solani and A. alternata.
Future Perspective
In general, A. solani is known as causal agent of early blight of tomato but the present study indicates that A. alternata also exists in virulent form to cause the disease. Hence in future, screening for resistance to early blight disease is to be performed using A. solani and A. alternata. It also paves the way to look for predisposing factors responsible for A. alternata to cause infection, if any.
Conclusion
Molecular and pathogenic characterization of Alternaria spp infecting tomato in northern India revealed the presence of A. solani and A. alternata in virulent form and emphasised that apart from A. solani, A. alternata also needs to be used for early blight resistance screening.
References
Datar VV, Mayee CD (1981) Assessment of loss in tomato yield due to early blight. Indian Phytopathol 34:191–195
Gomes SMDTP, Carneiro EDB, Romano EP, Teixeira MZ, da Costa ME, Vasconcelos JCG (2010) Effect of biotherapic of Alternaria solani on the early blight of tomato-plant and the in vitro development of the fungus. Int J High Dilution Res 9:147–155
Derbalah AS, El-Mahrouk MS, El-Sayed AB (2011) Efficacy and safety of some plant extracts against tomato early blight disease caused by Alternaria solani. Plant Pathol J 10(3):115–121
Alhussan KM (2012) Morphological and physiological characterization of Alternaria solani isolated from tomato in Jordan Valley. Res J Biol Sci 7:316–319
Varma KP, Singh S, Gandhi SK (2007) Variability among Alternaria solani isolates causing early blight of tomato. Indian Phytopathol 60:180–186
Kumar V, Haldar S, Pandey KK, Singh RP, Singh AK, Singh PC (2008) Cultural, morphological, pathogenic and molecular variability amongst tomato isolates of Alternaria solani in india. World J Microbiol Biotechnol 24:1003–1009
Radhajeyalakshmi R, Velazhahan R, Samiyappan R, Doraiswamy S (2009) Systemic induction of pathogenesis related proteins (PRs) in Alternaria solani elicitor sensitized tomato cells as resistance response. Sci Res Essay 4:685–689
Bhatt JC, Gahlain A, Pant SK (2000) Record of Alternaria alternata on tomato, capsicum and spinach in Kumaon hills. Indian Phytopathol 53:495–496
Benlioglu S, Delen N (1996) Studies on the sporulation of the early blight agent of tomatoes. J Turk Phytopathol 25:23–28
Vunch RA, Rosner A, Stein A (1999) The use of the polymerase chain reaction (PCR) for detection of bean yellow mosaic virus in gladiolus. Ann Appl Biol 117:561–569
Bridge PD, Singh T, Arora DK (2004) The application of molecular markers in the epidemiology of plant pathogenic fungi. In: Arora DK, Bridge PD, Bhatnagar D (eds) Fungal biotechnology in agricultural, food, and environmental applications. Marcel Dekker Inc, New York, p 475
Abd-Esalem KA (2003) Non-gel based techniques for plant pathogen genotyping. Acta Microbiol Pol 52:329–341
Abd-Esalem KA, Asran-Amel A, Schneider F, Migheli Q, Verreet JA (2006) Molecular detection of Fusarium oxysporum. f. sp. vasinfectum in cotton roots by PCR and real-time PCR assay. J Plant Dis Prot 113:14–19
Bowmann KD, Albrecht U, Graham JH, Bright DB (2007) Detection of Phytophtora nicotianae and P. palmivora in citrus roots using PCR-RFLP in comparison with other methods. Eur J Plant Pathol 119:143–158
Manicom BQ, Bar-Joseph M, Rosner A, Vigodsky-Haas H, Kotze JM (1987) Potential applications of random DNA probes, and restriction fragment length polymorphisms in the taxonomy of Fusaria. Phytopathology 77:669–672
White TJ, Bruns T, Lee S, Taylor JW (1990) Amplification and direct sequencing of fungal ribosomal RNA genes for phylogenetics. In: Innis MA, Gelfand DH, Sninsky JJ, White TJ (eds) PCR Protocols: a Guide to methods and applications. Academic Press Inc, New York, pp 315–322
Sambrook J, Russell DW (2001) Molecular cloning: a laboratory manual. Cold Spring Harbor Laboratory Press, New York
Thompson JD, Higgins DG, Gibson TJ (1994) CLUSTALW: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties and weight matrix choice. Nucleic Acids Res 22:4673–4680
Tamura K, Stecher G, Peterson D, Filipski A, Kumar S (2013) MEGA6: molecular evolutionary genetics analysis version 6.0. Mol Biol Evol 30:2725–2729
Pandey KK, Pandey PK, Kallo G, Banerjee MK (2003) Resistance to early blight of tomato with respect to various parameters of disease epidemics. J Gen Plant Pathol 69:364–371
Naik MK, Prasad Y, Bhat KV, Devika Rani GS (2010) Morphological, physiological, pathogenic and molecular variability among isolates of Alternaria solani from tomato. Indian Phytopathol 63:168–173
Kusaba M, Tsuge T (1995) Phylogeny of Alternaria fungi known to produce host-specific toxins on the basis of variation in internal transcribed spacers of ribosomal DNA. Curr Genet 28:491–498
Pryor BM, Gilbertson RL (2000) Molecular phylogenetic relationships amongst Alternaria species and related fungi based upon analysis of nuclear ITS and mt SSU rDNA sequences. Mycol Res 104:1312–1321
Wang HK, Zhang TY, Zhang M (2001) Application of sequencing of 5.8S rDNA, ITS1 and ITS2 on identification and classification of Alternaria at species level. Mycosystema 20:168–173
Pryor BM, Michailides TJ (2002) Morphological, pathogenic, and molecular characterization of Alternaria isolates associated with Alternaria late blight of pistachio. Phytopathology 92:406–416
Guo LD, Xu L, Zheng WH, Hyde KD (2004) Genetic variation of Alternaria alternata, an endophytic fungus isolated from Pinus tabulaeformis as determined by random amplified microsatellites (RAMS). Fungal Divers 16:53–65
Acknowledgments
The present work was carried-out with the financial support of ICAR-Out Reach Project on Diagnosis and Management of Leaf Spot Diseases of Field and Horticultural Crops under EFC of ICAR - Indian Institute of Horticultural Research, Bangalore, India. There is no conflict of interest regarding the research work done.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article

Cite this article
Loganathan, M., Venkataravanappa, V., Saha, S. et al. Morphological, Pathogenic and Molecular Characterizations of Alternaria Species Causing Early Blight of Tomato in Northern India. Proc. Natl. Acad. Sci., India, Sect. B Biol. Sci. 86, 325–330 (2016). https://doi.org/10.1007/s40011-014-0446-0
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s40011-014-0446-0