
Abstract
Molecular imprinting has shown significant advances in the recognition and separation of small molecules. This technology has been proposed for different applications, including solid-phase extraction, stationary phases in HPLC, chemical sensing, drug-delivery systems, passive sampling, among others. However, imprinting of biological macromolecules with increased structural complexity is still challenging. In this work, CaCO3 microparticles were synthesized using a precipitation method and employed as a novel support for the preparation of molecularly imprinted polymers (MIPs) towards a model protein (bovine serum albumin, BSA), through the polymerization of dopamine. Microparticles exhibited a rhombohedral morphology and a narrow size distribution (2.5 ± 0.4 µm). Reaction times showed to increase the polydopamine coating thickness, the MIP adsorption capacities, and the impression efficiency, reaching values of 5.1 nm, 50.2 ± 5.9 mg BSA/g sample, and 8.1 after 24 h, respectively. In addition, lower adsorption capacities were observed against proteins with similar physicochemical properties, such as ovalbumin (25.07 ± 2.5 mg/g) and casein (19.62 ± 7.01 mg/g). The adsorption kinetic assay indicated that MIPs present the highest BSA adsorption capacity after 1 h. In this regard, a methodology that offers a simple approach for the synthesis of materials designed for the specific recognition and separation of biological molecules is presented. The microparticles developed represent a potential use for protein separation in applications such as stationary phase in liquid chromatography.
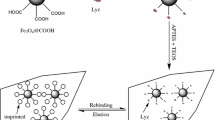
Graphical abstract

Similar content being viewed by others

Explore related subjects
Discover the latest articles, news and stories from top researchers in related subjects.Avoid common mistakes on your manuscript.
Introduction
Molecular imprinting technology allows the preparation of polymers with specific binding sites towards a molecule used as a template [1]. Molecularly imprinted polymers (MIPs) exhibit specific and selective recognition capacity towards the analyte of interest due to the presence of complementary binding sites in terms of shape, size, and functionality [2]. This is achieved by forming a cross-linked polymeric matrix around the template, which is then removed to generate complementary binding sites capable of subsequently recognizing the template molecule. Additionally, MIPs present unique features, such as high stability, easy preparation, low cost, and reusability [3], that have made them promising candidates in a wide variety of applications, including solid-phase extraction for different chemical families (proteins [4], antibiotics [5], non-steroidal anti-inflammatory drugs [6, 7], and contaminants [8]), stationary phases in HPLC for retention of proteins [9], enantiomers [10], hormones [11], and toxins [12], and removal of organic pollutants from wastewater [13, 14]. Additionally, MIP has been proposed for the development chemical sensors [15,16,17], drug-delivery systems [18,19,20], and passive sampling [21, 22], analysis of protein biomarkers [23], among others. However, the developed method still suffers from disadvantages such as sophisticated sensor fabrication, high chemical consumption, and expert operator for sensor construction as well.
In recent years, the molecular imprinting technology has made significant advances in the selective recognition of small molecules [24,25,26]. However, biological macromolecules and microorganisms (proteins, viruses, and bacteria) have a large size and increased structural complexity, making it difficult to develop MIPs for their specific recognition [27,28,29]. In particular, the size of the protein templates hampers their penetration in the MIPs to access the binding sites. In this regard, surface imprinting technology has been proposed as a promising alternative to overcome these drawbacks [30,31,32]. This technique is based on functional monomers’ polymerization on the surface of different substrates introduced into the polymerization system. In this way, proteins can easily access the specific recognition sites of the MIPs [33]. On the other hand, the preparation conditions of protein-imprinted polymers must be similar to the biological environment to ensure their conformational integrity [34]. Dopamine (DA) is a functional, water-soluble monomer with excellent biological compatibility. The self-polymerization of DA in mild conditions on different particulate substrates has been reported to prepare protein-selective molecularly imprinted nano- and microparticles preserving the target molecule conformational integrity during the synthesis [9, 34]. Wang et al. prepared poly(glycidyl methacrylate)/polystyrene microparticles imprinted with bovine serum albumin (BSA) exhibiting various morphologies ("raspberry", "golf ball", and porous-shaped microspheres) [34]. Although the imprinting efficiency (IE) was similar for all particle types, some differences in adsorption capacity (Q) were observed. This parameter was higher for porous microparticles (72.70 mg BSA/g particles) compared to "raspberry" and "golf ball"-shaped microparticles (53.35 and 49.15 mg BSA/g particles, respectively). On the other hand, Nematollahzadeh et al. developed a nanometer coating of human serum albumin (HSA)-imprinted polydopamine (PDA) by oxidative polymerization of DA on the pore surface of HSA-modified porous silica particles [9]. The thickness of the coating increased with reaction time, reaching 12 nm at 48 h. The adsorption capacity also improved with the reaction time (11.6 mg protein/g particles). However, the best performance of the MIPs was obtained at 24 h, where the Q was lower (6.2 mg protein/g particles), but the IE was higher than that achieved at 48 h (9.5 versus 2.8, respectively). The relatively low IE for the longer reaction times is attributed to the fact that the surface coating of the MIP hinders the protein removal or uptake.
Despite the investigations, several issues related to DA polymerization on different substrates, as well as MIP–protein interactions, remain to be elucidated. In this sense, calcium carbonate (CaCO3) microparticles are promising substrates for the development of surface MIPs due to their controlled morphology and particle size, abundance, reduced cost, and low environmental impact. The synthesis of CaCO3 particles can be carried out by different methodologies, including wet precipitation or carbonation, the use of emulsion membranes, and novel technologies such as high gravity reactive precipitation [35]. The precipitation method, based on the mixture of saturated aqueous solutions of sodium carbonate and calcium nitrate, is relatively simple and allows obtaining nano- and microparticles of controlled size. Furthermore, this methodology is an environmentally friendly approach, since it does not involve the use of organic solvents. Commonly employed protein separation techniques include ion-exchange chromatography, affinity chromatography, dialysis, ultrafiltration, size-exclusion chromatography, SDS-PAGE, isoelectric focusing, and capillary electrophoresis. In this sense, MIPs present a facile and low-cost process for the preparation of materials, which eliminates the need of complex or expensive equipment. In light of these benefits, MIPs have gained significant interest and attention as a promising route for the separation and purification of proteins. The objective of this work is to study the synthesis of molecularly imprinted microparticles by surface coating of CaCO3 microparticles with PDA for the specific recognition of proteins. For this purpose, CaCO3 microparticles were synthesized and used to prepare MIPs in the presence of BSA as a model protein. The MIPs were characterized in terms of morphology, coating thickness, composition, thermal properties, and recognition capacity towards BSA and proteins with similar physicochemical characteristics. These types of materials present a potential application as stationary phase in liquid chromatography and could be applied for the selective recognition of proteins of biological interest.
Experimental
Materials
Sodium carbonate, sodium hydroxide, calcium nitrate tetrahydrate, sodium nitrate, sodium dodecyl sulfate (SDS), methanol, ethanol, hydrochloric acid, and phosphoric acid were all pro analysis (ACS) supplied by Cicarelli, Argentina. Dopamine hydrochloride, Tris–HCl, and Coomassie brilliant Blue G-250 were purchased from Sigma-Aldrich, USA. Polyanionic cellulose (PAC) and BSA were provided by MI Swaco Company, USA and Biorgen, Argentina, respectively. All reactants were used without further purification. Deionized water was used to prepare all solutions.
Methodology
Synthesis of CaCO3 microparticles
Microparticles were synthesized by the chemical precipitation method from saturated aqueous solutions of sodium carbonate and calcium nitrate employing the procedure developed by Babou-Kammoe et al. [35] with slight modifications. The sodium carbonate solution consisted of 0.53 g of sodium carbonate (NaCO3), 0.4 g of sodium hydroxide (NaOH), and 0.765 g of sodium nitrate (NaNO3) in 50 mL of 0.5% polyanionic cellulose (PAC) solution, and the calcium nitrate solution consisted of 0.82 g of calcium nitrate Ca(NO3)2 in 50 mL of 0.5% PAC aqueous solution. For particle preparation, 40 mL of calcium nitrate solution was added dropwise to 40 mL of sodium carbonate solution using a peristaltic pump with a flow rate of 1 mL/min, under continuous stirring at 9000 rpm using a homogenizer (Polytron 2500e, Kinematica, Switzerland) and in an ice bath at 4 °C. The resulting dispersion was kept under continuous stirring for 1 h. Subsequently, microparticles were separated by centrifugation at 7500 rpm for 5 min. Finally, the particles were washed three times with distilled water and dried in an oven at 90 °C.
Synthesis of PDA-based MIPs
A polymeric coating was generated on the surface of CaCO3 microparticles using DA as a functional monomer and BSA as a model protein following the methodology reported by Wang et al. [34]. Aqueous solutions of BSA (1.66 mg/mL) containing DA (6.25 mg/mL) were prepared and the solution was left under agitation for 30 min to allow the interaction between the functional monomer and the protein. Then, 6 mL of an aqueous dispersion of CaCO3 microparticles (8.3 mg/mL) and 3 mL of Tris–HCl buffer (0.01 M, pH 8.5) were added to 6 mL of DA and BSA solution to initiate the polymerization reaction. The mixture was left to react for different times: 1, 5, and 24 h (MIP-1 h, MIP-5 h, and MIP-24 h, respectively). The structures obtained were washed with ethanol and distilled water to remove the adsorbed oligomers. Also, BSA template was removed by successive washings with 0.5 M NaCl solution and allowed to dry in an oven at 40 °C. Non-imprinted polymeric structures (NIPs) were also synthesized in the absence of BSA.
Characterization of particulate systems
Size and morphology of microparticles
CaCO3 particulate suspensions were observed under an optical microscope (DM 2500 M, Leica, Germany) equipped with an imaging camera (DFC 290 HD, Leica). To determine the average size and size distribution of the particles, approximately 300 particles per sample were measured using an image processing software (ImageJ, National Institutes of Health, Bethesda, Maryland, USA).
The particle size and morphology were also studied by scanning electron microscopy (SEM). Samples were placed on a piece of aluminum and coated with gold under an argon atmosphere (SPI Supplies, 12,157-AX, USA) using mild conditions (two sputtering sprays of 40 s each with an intensity of 15 mA). The samples were examined using an accelerating voltage of 5 kV on a Phenom ProX microscope (Thermo Fisher Scientific, USA).
The morphology of CaCO3 microparticles coated with MIPs was investigated by transmission electron microscopy (TEM). The samples were placed on a carbon-coated copper grid, air-dried, and observed at an accelerating voltage of 200 kV using a JEOL-2100 Plus electron microscope (JEOL, Tokyo, Japan).
Thermogravimetric analysis (TGA)
Thermogravimetric analysis of microparticles were performed using a thermogravimetric analyzer (Q500 TA instrument, USA) under a nitrogen atmosphere at a flow rate of 80 mL/min. Samples of approximately 5 mg were prepared and heated from 10 to 800 °C at a heating rate of 10 °C/min.
The PDA content of the functionalized CaCO3 microparticles and the thickness of the PDA coating were estimated from the results obtained from TGA thermograms. The coating thickness was calculated from Eq. 1 [31]
where tp is the coating thickness (nm), dp (nm) is the mean particle diameter of CaCO3, \(\rho_{{{\text{CaCO}}_{{3}} }}\) is the density of CaCO3 (2.7 g/cm3), ρPDA is the density of PDA (1.2 g/cm3), and w is the weight percentage of PDA in the samples.
Fourier transform infrared spectroscopy (FTIR)
Microparticles were characterized by Fourier transform infrared spectroscopy (FTIR) using an FTIR-8201PC infrared spectrophotometer (Shimadzu, Japan) in the 400–4000 cm−1 frequency region with a resolution of 4 cm−1 and 40 scans per spectrum. Approximately 3 mg of samples and 100 mg of potassium bromide (KBr) were dried for 24 h at 40 and 105 °C, respectively. The samples were mixed in a mortar and the resulting mixture was compacted into discs using a hydraulic press at a pressure of 4–6 tons.
X-ray diffraction (XRD) analysis
X-ray diffraction analysis (XRD) was conducted to investigate the mineralogical composition of calcium carbonate and PDA-coated particles. It was performed using a Malvern PANAlytical EMPYREAN X-ray diffractometer employing 2 theta ranges between 5 and 90°, Cu-Kα radiation, a scan speed of 2°/min, and an acceleration of voltage and current of 40 kV and 45 mA, respectively. The HighScore Plus software was utilized to analyze the XRD data obtained from the scans.
Protein adsorption assay
To study the recognition ability of MIPs, adsorption experiments were performed by incubating 8 mg of MIPs and NIPs in 5 mL of an aqueous solution of BSA.
(0.5 mg/mL) until the equilibrium was reached (5 h) at room temperature. The microparticles were then centrifuged at 5000 rpm for 5 min and the remaining concentration of BSA in the supernatant was determined following the Bradford method [34]. For this purpose, samples were prepared containing 0.1 mL of supernatant, 0.9 mL of H2O, and 5 mL of Coomassie blue solution. The Coomassie blue solution (0.1 mg/ mL) was prepared using a mixture of ethanol, phosphoric acid (85%), and distilled water in a 5/10/85 volumetric ratio; the solution was filtered before use. For BSA quantification, standard solutions with different concentrations were prepared: 0.1, 0.2, 0.4, 0.6, 0.8, and 1 mg/mL. Quantification of BSA in the samples was performed using a UV–Vis spectrometer (Lambda 25, Perkin Elmer, USA) at a wavelength of 595 nm. The adsorption capacity of MIPs and NIPs (Q) was calculated according to Eq. 2
where Q (mg/g) is the mass of protein adsorbed per gram of dried microparticles, C0 (mg/mL) is the initial concentration of the protein solution, C1 (mg/mL) is the equilibrium concentration of the supernatant after the adsorption process, V (mL) is the volume of the protein solution, and w (g) is the weight of dried microparticles. The IE was calculated from Eq. 3
where QMIP (mg/g) and QNIP (mg/g) are the adsorption capacities of MIPs and NIPs, respectively.
Selectivity assays
This assay aims to determine the recognition capacity of BSA-imprinted MIPs against other proteins with similar physicochemical characteristics: ovalbumin (OVA) and casein (CAS). The OVA adsorption assays were performed following the methodology previously described for BSA. For CAS evaluation, an alkaline medium (NaOH 0.25 M) was used due to the low solubility of the protein in aqueous media. Measurements were performed in triplicate.
Gel electrophoresis
The stacking and separating gels used were 5% and 15% polyacrylamide, respectively. Samples were loaded into the individual wells, and the electrophoretic separation was carried out at 200 V for 75 min. Gel staining was performed with Coomassie blue and destaining was achieved by incubation with a solution containing 15% (v/v) methanol and 10% (v/v) acetic acid. In case desalination was required, samples were ultra-centrifugated into a 10,000 MWCO centrifugal concentrator (Vivaspin, Sartorius, US).
Results and discussion
CaCO3 microparticles were prepared by the precipitation method. Several additives (including surfactants, synthetic polymers, and biomolecules) have been employed to obtain CaCO3 particles with different sizes, morphologies, and polymorphs [36]. The additives can interact with Ca2+ ions and provide active sites for CaCO3 nucleation. In turn, the additives can bind to preferential crystal surfaces to prevent further growth on specific planes. CaCO3 exists mainly in four polymorphs: calcite, barite, aragonite, and amorphous calcium carbonate, being calcite the most thermodynamically stable phase. Figures 1 and 2 show optical and SEM micrographs at different magnifications of the synthesized microparticles. From the optical microscopy observations (Fig. 1a), microparticles exhibit a rhombohedral morphology, typical of calcite, and a narrow size distribution in the range of 1–4 µm. The particle-size distribution is shown in Fig. 1b. The mean particle diameter was 2.5 ± 0.4 µm.

a Optical microscopy micrograph of CaCO3 microparticles, and b particle-size distribution

SEM micrographs of CaCO3 microparticles
Figure 2 shows SEM micrographs of CaCO3 microparticles. They exhibited a homogeneous rhombohedral morphology and a smooth surface. The mean particle diameter determined by SEM was similar to that obtained by optical microscopy.
The morphology of CaCO3 microparticles before and after MIP coating (24 h reaction time) was studied by TEM (Fig. 3). Difference in particle geometries can be observed in the coated microparticles samples. Uncoated macroparticles presented defined edges and angles, while those synthesized with MIPs exhibited a nanometer-thick polymeric coating on their surface. From TEM image analysis, the thickness of the MIP coating was estimated to be approximately 10 nm

a, b TEM micrographs of CaCO3 microparticles, and c, d microparticles coated with MIPs
The thermal degradation properties of the particulate materials were studied by TGA. From these results, it was possible to estimate the PDA content in the polymer-coated particles and the coating thickness. The TGA thermograms of the samples are presented in Fig. 4. The results revealed a first decomposition process starting at 110 °C related to the loss of particles adsorbed water. The CaCO3 particles present a high thermal stability in the temperature range from 25 to 600 °C with a weight loss (between 600 and 720 °C) corresponding to its thermal decomposition into calcium oxide and carbon dioxide. Analysis of the MIP-coated samples indicated that the polymer coating exhibits complete thermal decomposition at 500 °C. This finding is in agreement with the data reported by Mirzayi et al. [37]. From these results, it is possible to conclude that the thickness of the polymer coating increases at longer reaction times.

TGA and DTGA thermograms: a, c CaCO3 microparticles coated with MIPs, and b, d CaCO3 microparticles coated with NIPs
The microparticles’ PDA content was estimated from the TGA analyses employing Eq. 1 and the results are presented in Table 1. The PDA content after 1 h of reaction is considerably lower than those obtained after 5 and 24 h of reaction, as well as the resulting PDA thickness. On the other hand, a difference between the coating thickness of MIPs and NIPs was observed. This variation is attributable to the interaction between the BSA and the monomer during the synthesis of the MIPs, which may decrease the polymerization rate of DA.
Crystalline forms of CaCO3 microparticles and PDA-coated microparticles were analyzed by employing XRD diffractogram (Fig. 5). Peaks related to CaCO3 structures at 23.05, 29.35, 35.95, 39.35, 43.05, 47.05, and 56.45° were observed in both samples, which were are also reported in the previous works [38, 39]. Additionally, MIP particles displayed a broad and low-intensity peak between 20 and 25° that was attributed to diffraction from the amorphous PDA structures [40].

XRD patterns of CaCO3 microparticles and MIP particles
Figure 6a shows the FTIR spectra of CaCO3 microparticles, DA, and CaCO3 microparticles with PDA coating after 24 h of reaction. In the FTIR spectrum corresponding to CaCO3, the following signals are distinguished: the bending of the C–O bond of the carbonate group at 865 cm−1, the stretching of the C–O bond of the carbonate at 1425 cm−1 and the band associated with water adsorption at 3722 cm−1 [41]. In the DA spectrum, the bands located at 878 and 1600 cm−1 are related to bending of the N–H bond of the amino group (H–N–H), and those located at 1190 and 1290 cm−1 are attributed to bending of the C–C–H bond [9]. These characteristic peaks are absent or reduced in the spectra obtained for MIP and NIP samples. Therefore, the results support the effective polymerization of DA in the experimental conditions studied. It can be observed that the spectra of microparticles coated with MIPs and NIPs (Fig. 6a) exhibit several structural features common to PDA and CaCO3. For example, the band centered at 1600 cm−1 corresponding to the N–H bending of the aromatic regions of PDA and the bands of the carbonate group of CaCO3 are observed in the MIP and NIP samples. Figure 6b shows the FTIR spectra of BSA, MIP-BSA, and MIP. The MIP-BSA sample corresponds to MIPs that were incubated in a BSA solution for 5 h. In the BSA spectrum, characteristic peaks arise from the amide bonds linking the amino acids, in particular, the primary amide and secondary amide bands. The characteristic primary amide peak of BSA, corresponding to the stretching of the C–O bond of the amide, can be observed at 1650 cm−1; the peak at 1545 cm−1 is attributed to the bending mode of the N–H bond of the secondary amide, while the band centered at 3435 cm−1 can be attributed to the primary amines. The hydroxyl stretching peaks are dominant in the region between 2500 and 3500 cm−1 [42]. The spectrum obtained from MIP-BSA presents characteristic peaks from BSA, indicating that the specific binding sites of the MIPs are able to recognize the protein. In Fig. 6c, the FTIR spectra of MIPs prepared at different reaction times are displayed. It is worth to be noted that an increase in the peak corresponding to the N–H bond of PDA (1640 cm−1) is observed with the evolution of the polymerization reaction.

FTIR spectra: a DA, PDA-coated and uncoated CaCO3 microparticles, b MIPs in the presence of BSA, and c MIPs synthesized at different reaction times
Adsorption experiments were performed to determine the binding properties of MIPs and NIPs towards BSA. Figure 7 compares the adsorption capacities for the MIPs and NIPs’ samples prepared at different reaction times. The results show that the adsorption capacities of MIPs and NIPs obtained after 1 h of polymerization (MIP-1 h and NIP-1 h, respectively) are very low, possibly due to the low formation of specific binding sites for BSA related to incomplete polymerization of DA. The binding capacities of MIPs obtained after 5 and 24 h of polymerization were notably higher compared to NIPs, indicating a higher interaction with the protein due to the presence of specific binding. In the case of MIPs and NIPs synthesized during 24 h of reaction, they exhibit a slight decrease in adsorption capacity possibly due to the generation of a denser polymeric matrix that affects the protein removal or uptake. Table 2 presents the values of Q and IE for the different systems. The IE values obtained after 24 h of reaction were higher than those reported by Wang et al. for particles with different morphologies (around 4.55). Due to its high Q and IE values, MIP-24 h is going to be employed to perform selectivity and adsorption kinetics studies.

Adsorption capacity of MIPs and NIPs prepared at different reaction times
To investigate the BSA adsorption capacity of the developed MIP, an adsorption isotherm experiment was performed by incubating a fixed mass of MIP in BSA aqueous solutions of different concentrations (0.125, 0.25, 0.5, and 0.75 mg/mL) (Fig. 8a). The data showed a significant increase in the Q value from protein concentrations of 0.125 to 0.5 mg/mL, after which the Q value remained essentially stable evidencing that MIP-binding sites are completely occupied under this condition. BSA adsorption as a function of time was also evaluated to determine the kinetic behavior of MIPs towards BSA (Fig. 8b). It is observed that the MIPs present the highest adsorption capacity after 1 h, implying that BSA has completely occupied the binding sites of the MIPs under this condition. During the first 60 min of experiment, the adsorption capacity increases rapidly, reaching adsorption capacity values of about 50 mg/g approximately at equilibrium. Two stages can be distinguished in the adsorption kinetics: in the first stage, BSA diffuses rapidly from the solution towards the surface of the MIPs generating non-covalent type interactions, such as hydrogen bonding. In a second stage, the adsorption rate of BSA decreases due to the resulting steric hindrance between the polymeric matrix of the imprinting site and BSA.

a Adsorption isotherm and b adsorption kinetics of BSA on MIPs
Selectivity tests were performed to study the recognition capacity of MIPs towards other proteins with similar physicochemical properties: OVA and CAS. The molecular weights of BSA, OVA, and CAS are: 66.4, 45, and 24 kDa, respectively; and the isoelectric points are 4.7, 4.5, and 4.6, respectively. The results presented in Fig. 9a showed that the adsorption capacity of OVA and CAS is lower than that of BSA. In particular, the lower CAS adsorption capacity is possibly due to greater differences in chemical structure and molecular weights, and to the alkaline environment of the adsorption medium. A similar behavior was observed for MIPs obtained after 5 and 24 h of reaction. It should be noted that the materials presented a remarkable differential adsorption capacity towards BSA despite the structural complexity of the proteins. To study the selectivity of MIPs, adsorption experiments of BSA and OVA mixed solutions (0.5 mg/mL each) were carried out. After the assay, MIP particles were recovered by centrifugation and the proteins were desorbed from MIP using a 0.5 M NaCl solution or a 150 mM NH4OH solution. The protein solution after the adsorption assay and the MIPs desorption solution with NaCl or NH4OH were studied by SDS-PAGE gel electrophoresis, and the results are shown in Fig. 9b. In both desorption conditions, the bands related to BSA (about 66 kDa) showed a higher intensity than those related to OVA (about 40 kDa). To sum up, the adsorption studies showed not only a higher selectivity of the MIP towards BSA but also demonstrated the potential application of the developed material for protein purification.

a Selectivity studies of MIPs towards OVA and CAS, and b SDS-PAGE analysis [molecular weight markers (lane 1), protein solution after MIP elution with NH4OH solution (lane 2), NaCl solution (lane 3), and protein solution after the adsorption assay (lane 4)]
Conclusion
CaCO3 microparticles were prepared as support for the synthesis of BSA-imprinted polymeric coatings by the self-polymerization of DA in alkaline solution. DA as a water-soluble functional monomer did not affect the protein structure, and thus, the imprinted cavity structures on the MIPs allowed the selective binding of the template protein. The PDA-based molecular imprinted polymers exhibited excellent adsorption capacity towards BSA, and lower adsorption capacities were observed against other proteins with similar characteristics. The presented method is a simple and promising alternative for the development of materials for the selective recognition and separation of biological molecules, with potential application as stationary phase in liquid chromatography.
Data availability
Should any data files be needed they are available from the corresponding author upon reasonable request.
References
Rico Yuste A, Carrasco S (2019) Molecularly imprinted polymer-based hybrid materials for the development of optical sensors. Polymers 11:1173. https://doi.org/10.3390/POLYM11071173
Belbruno JJ (2019) Molecularly imprinted polymers. Chem Rev 119:94–119. https://doi.org/10.1021/acs.chemrev.8b00171
Liu G, Huang X, Li L, Xu X, Zhang Y, Lv J, Xu D (2019) Recent advances and perspectives of molecularly imprinted polymer-based fluorescent sensors in food and environment analysis. Nanomaterials 9:1030. https://doi.org/10.3390/NANO9071030
Mostafa AM, Barton SJ, Wren SP, Barker J (2022) Development of magnetic molecularly imprinted polymers for the extraction of salivary pepsin prior to analysis by a novel HPLC-SEC method. Polymer 261:125417. https://doi.org/10.1016/j.polymer.2022.125417
Lu W, Liu J, Li J, Wang X, Lv M, Cui R, Chen L (2019) Dual-template molecularly imprinted polymers for dispersive solid-phase extraction of fluoroquinolones in water samples coupled with high performance liquid chromatography. Analyst 144:1292–1302. https://doi.org/10.1039/C8AN02133C
Olcer YA, Demirkurt M, Demir MM, Eroglu AE (2017) Development of molecularly imprinted polymers (MIPs) as a solid phase extraction (SPE) sorbent for the determination of ibuprofen in water. RSC Adv 7:31441–31447. https://doi.org/10.1039/C7RA05254E
Madikizela LM, Tavengwa NT, Chimuka L (2018) Applications of molecularly imprinted polymers for solid-phase extraction of non-steroidal anti-inflammatory drugs and analgesics from environmental waters and biological samples. J Pharm Biomed Anal 147:624–633. https://doi.org/10.1016/j.jpba.2017.04.010
Kalogiouri NP, Tsalbouris A, Kabir A, Furton KG, Samanidou VF (2020) Synthesis and application of molecularly imprinted polymers using sol–gel matrix imprinting technology for the efficient solid-phase extraction of BPA from water. Microchem J 157:104965. https://doi.org/10.1016/j.microc.2020.104965
Nematollahzadeh A, Shojaei A, Abdekhodaie MJ, Sellergren B (2013) Molecularly imprinted polydopamine nano-layer on the pore surface of porous particles for protein capture in HPLC column. J Colloid Interface Sci 404:117–126. https://doi.org/10.1016/j.jcis.2013.04.004
Hroboňová K, Lomenova A (2018) Molecularly imprinted polymer as stationary phase for HPLC separation of phenylalanine enantiomers. Monatshefte für Chemie Chem Mon 149:939–946. https://doi.org/10.1007/s00706-018-2155-5
Podjava A, Šilaks A (2021) Study of chromatographic properties of catecholamines and their acidic metabolites using novel molecularly imprinted polymers as stationary phases. Key Eng Mater 903:15–21. https://doi.org/10.4028/www.scientific.net/KEM.903.15
Yang F, Wang R, Na G, Yan Q, Lin Z, Zhang Z (2018) Preparation and application of a molecularly imprinted monolith for specific recognition of domoic acid. Anal Bioanal Chem 410:1845–1854. https://doi.org/10.1007/s00216-017-0843-3
Zare EN, Fallah Z, Le VT, Doan VD, Mudhoo A, Joo SW, Vasseghian Y, Tajbakhsh M, Moradi O, Sillanpää M, Varma RS (2022) Remediation of pharmaceuticals from contaminated water by molecularly imprinted polymers: a review. Environ Chem Lett 20:2629–2664
Heidari G, Afruzi FH, Zare EN (2023) Molecularly imprinted magnetic nanocomposite based on carboxymethyl dextrin for removal of ciprofloxacin antibiotic from contaminated water. Nanomaterials 13:489. https://doi.org/10.3390/nano13030489
Casis N, Busatto C, Fidalgo de Cortalezzi MM, Ravaine S, Estenoz DA (2015) Molecularly imprinted hydrogels from colloidal crystals for the detection of progesterone. Polym Int 64:773–779. https://doi.org/10.1002/pi.4851
Kadhem A, Xiang S, Nagel S, Lin CH, Fidalgo de Cortalezzi M (2018) Photonic molecularly imprinted polymer film for the detection of testosterone in aqueous samples. Polymers 10:349. https://doi.org/10.3390/polym10040349
Gao M, Gao Y, Chen G, Huang X, Xu X, Lv J, Wang J, Xu D, Liu G (2020) Recent advances and future trends in the detection of contaminants by molecularly imprinted polymers in food samples. Front Chem 8:616326. https://doi.org/10.3389/fchem.2020.616326
Zaidi SA (2020) Molecular imprinting: a useful approach for drug delivery. Mater Sci Energy Technol 3:72–77. https://doi.org/10.1016/j.mset.2019.10.012
Liu R, Poma A (2021) Advances in molecularly imprinted polymers as drug delivery systems. Molecules 26:3589. https://doi.org/10.3390/molecules26123589
Sanadgol N, Wackerlig J (2020) Developments of smart drug-delivery systems based on magnetic molecularly imprinted polymers for targeted cancer therapy: a short review. Pharmaceutics 12:831. https://doi.org/10.3390/pharmaceutics12090831
Khulu S, Ncube S, Nuapia Y, Madikizela LM, Mavhunga E, Chimuka L (2022) Development and application of a membrane assisted solvent extraction-molecularly imprinted polymer based passive sampler for monitoring of selected pharmaceuticals in surface water. Water Res 225:119145. https://doi.org/10.1016/j.watres.2022.119145
Garnier A, Montigny C, Causse L, Spinelli S, Avezac M, Otazaghine B, Gonzalez C (2022) Synthesis of an organotin specific molecularly imprinted polymer for organotin passive sampling in seawater. Water 14:1786. https://doi.org/10.3390/w14111786
Mostafa AM, Barton SJ, Wren SP, Barker J (2021) Review on molecularly imprinted polymers with a focus on their application to the analysis of protein biomarkers. Trend Anal Chem 144:116431. https://doi.org/10.1016/J.TRAC.2021.116431
Pupin RR, Foguel MV, Gonçalves LM, del Sotomayor M, PT, (2020) Magnetic molecularly imprinted polymers obtained by photopolymerization for selective recognition of penicillin G. J Appl Polym Sci 137:48496. https://doi.org/10.1002/app.48496
Wei X, Yu M, Guo J (2019) A core-shell spherical silica molecularly imprinted polymer for efficient selective recognition and adsorption of dichlorophen. Fiber Polym 20:459–465. https://doi.org/10.1007/s12221-019-8822-2
Qasim S, Hsu SY, Rossi E, Salahshoor Z, Lin CH, Parada LP, Fidalgo M (2022) Detection of progesterone in aqueous samples by molecularly imprinted photonic polymers. Microchim Acta 189:174. https://doi.org/10.1007/s00604-022-05290-w
Malik AA, Nantasenamat C, Piacham T (2017) Molecularly imprinted polymer for human viral pathogen detection. Mater Sci Eng C 77:1341–1348. https://doi.org/10.1016/j.msec.2017.03.209
Nahhas AF, Webster TJ (2021) The promising use of nano-molecular imprinted templates for improved SARS-CoV-2 detection, drug delivery and research. J Nanobiotechnol 19:305. https://doi.org/10.1186/s12951-021-01032-x
Dar KK, Shao S, Tan T, Lv Y (2020) Molecularly imprinted polymers for the selective recognition of microorganisms. Biotechnol Adv 45:107640. https://doi.org/10.1016/j.biotechadv.2020.107640
El-Schich Z, Zhang Y, Feith M, Beyer S, Sternbæk L, Ohlsson L, Stollenwerk M, Wingren AG (2020) Molecularly imprinted polymers in biological applications. Biotechniques 69:406–419. https://doi.org/10.2144/btn-2020-0091
Zhang Z, Niu D, Li Y, Shi J (2018) Magnetic, core-shell structured and surface molecularly imprinted polymers for the rapid and selective recognition of salicylic acid from aqueous solutions. Appl Surf Sci 435:178–186. https://doi.org/10.1016/j.apsusc.2017.11.033
Cui F, Zhou Z, Zhou HS (2020) Molecularly imprinted polymers and surface imprinted polymers based electrochemical biosensor for infectious diseases. Sensors 20:996. https://doi.org/10.3390/s20040996
Zhang W, Zhang Y, Wang R, Zhang P, Zhang Y, Randell E, Zhang M, Jia Q (2022) A review: development and application of surface molecularly imprinted polymers towards amino acids, peptides, and proteins. Anal Chem Acta 1234:340319. https://doi.org/10.1016/J.ACA.2022.340319
Wang Y, Zhou J, Wu C, Tian L, Zhang B, Zhang Q (2018) Fabrication of micron-sized BSA-imprinted polymers with outstanding adsorption capacity based on poly(glycidyl methacrylate)/polystyrene (PGMA/PS) anisotropic microspheres. J Mater Chem B 6:5860–5866. https://doi.org/10.1039/C8TB01423J
Babou-Kammoe R, Hamoudi S, Larachi F, Belkacemi K (2012) Synthesis of CaCO3 nanoparticles by controlled precipitation of saturated carbonate and calcium nitrate aqueous solutions. Can J Chem Eng 90:26–33. https://doi.org/10.1002/cjce.20673
Boyjoo Y, Pareek VK, Liu J (2014) Synthesis of micro and nano-sized calcium carbonate particles and their applications. J Mater Chem A 2:14270–14288. https://doi.org/10.1039/C4TA02070G
Mirzayi B, Nematollahzadeh A, Seraj S (2015) Synthesis and characterization of magnetic maghemite/catecholamine core/shell nanoparticles. Powder Technol 270:185–191. https://doi.org/10.1016/j.powtec.2014.10.022
Kato T, Sugawara A, Hosoda N (2002) Calcium carbonate-organic hybrid materials. Adv Mater 14:869. https://doi.org/10.1002/1521-4095(20020618)14:12%3c869::AID-ADMA869%3e3.0.CO;2-E
Islam KN, Bakar MZBA, Ali ME, Bin HMZ, Noordin MM, Loqman MY, Miah G, Wahid H, Hashim U (2013) A novel method for the synthesis of calcium carbonate (aragonite) nanoparticles from cockle shells. Powder Technol 235:70–75. https://doi.org/10.1016/j.powtec.2012.09.041
Luo H, Gu C, Zheng W, Dai F, Wang X, Zheng Z (2015) Facile synthesis of novel size-controlled antibacterial hybrid spheres using silver nanoparticles loaded with polydopamine spheres. RSC Adv 5:13470–13477. https://doi.org/10.1039/c4ra16469e
Kusuktham B (2011) Spinning of PET fibres mixed with calcium carbonate. Asian J Text 1:106–113. https://doi.org/10.3923/ajt.2011.106.113
Lazarevic A, Pokrajac D, Marcano A, Melikechi N (2009) Support vector machine based classification of fast Fourier transform spectroscopy of proteins. Adv Biomed Clin Diag Syst 7169:63–70. https://doi.org/10.1117/12.809964
Acknowledgements
We acknowledge the financial support from CONICET, MINCyT, and UNL. The authors would also like to express their sincere gratitude to Lic. Malen Menegon for her valuable support in the gel electrophoresis assays.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Citta, M.d., Fookes, F., Busatto, C. et al. Molecularly imprinted CaCO3/polydopamine hybrid composite for selective protein recognition. Iran Polym J 32, 1111–1122 (2023). https://doi.org/10.1007/s13726-023-01189-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13726-023-01189-2