
Abstract
Purpose of Review
Metabolic syndrome (MetS), also called the ‘deadly quartet’ comprising obesity, diabetes, dyslipidemia, and hypertension, has been ascertained to have a causal role in the pathogenesis of osteoarthritis (OA). This review is aimed at discussing the current knowledge on the contribution of metabolic syndrome and its various components to OA pathogenesis and progression.
Recent Findings
Lately, an increased association identified between the various components of metabolic syndrome (obesity, diabetes, dyslipidemia, and hypertension) with OA has led to the identification of the ‘metabolic phenotype’ of OA. These metabolic perturbations alongside low-grade systemic inflammation have been identified to inflict detrimental effects upon multiple tissues of the joint including cartilage, bone, and synovium leading to complete joint failure in OA. Recent epidemiological and clinical findings affirm that adipokines significantly contribute to inflammation, tissue degradation, and OA pathogenesis mediated through multiple signaling pathways. OA is no longer perceived as just a ‘wear and tear’ disease and the involvement of the metabolic components in OA pathogenesis adds up to the complexity of the disease.
Summary
Given the global surge in obesity and its allied metabolic perturbations, this review aims to throw light on the current knowledge on the pathophysiology of MetS-associated OA and the need to address MetS in the context of metabolic OA management. Better regulation of the constituent factors of MetS could be profitable in preventing MetS-associated OA. The identification of key roles for several metabolic regulators in OA pathogenesis has also opened up newer avenues in the recognition and development of novel therapeutic agents.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
Osteoarthritis (OA) is the most common type of arthritis affecting about 3.3 to 3.6% of the world’s population. It is the 11th most debilitating disease across the globe inflicting moderate to severe disability in about 43 million people [1]. According to the Global Burden of Diseases, Injuries, and Risk Factors Study 2017 (GBD) report, OA ranked among the highly prevalent rheumatic musculoskeletal disorder that had affected around 303 million people worldwide in 2017 with an overwhelming estimate of 9,604,000 years lost to OA-associated disability [2]. In the USA, it is estimated that 80% of the population above 65 years of age exhibit radiographic evidence of OA. Not to mention the intense physical and emotional ramifications manifested with the disease, OA is also affiliated with a huge personal, societal, and economic burden. OA was the second most expensive medical condition treated in US hospitals in 2013, contributing for 4.3% ($18.4 billion) of the $415 billion total cost of hospitalization [3]. OA patients are at a greater risk of all-cause mortality especially for cardiovascular diseases bearing direct relevance to the level of disability [4]. The enormous disease encumbrance associated with OA led to the submission of a White Paper by Osteoarthritis Research Society International (OARSI) in 2016, describing OA as a serious disease [5].
More than being just a disease of the articular cartilage, OA has been identified as a complex multi-factorial degenerative disease involving various components of the entire joint [6]. Advancing deterioration and destruction of articular cartilage accompanied by diverse structural and functional alterations in various tissues of the joint including subchondral bone remodeling, osteophyte formation, development of bone marrow lesions, synovial inflammation, weakening of the periarticular muscles, and modifications in the joint capsule, ligaments, and menisci have all been identified to be the hallmarks of OA [7,8,9,10]. OA may develop in any joint, but the knees, hips, hands, facet joints, and feet are the most commonly affected, with women having a higher prevalence rate compared to men [11]. It has been estimated that the global prevalence of knee OA was 16% in individuals aged 15 and over and 22.9% in individuals aged 40 and over [12•]. Various factors have been implicated to have a role in the disease pathogenesis of OA constituting discrete phenotypes including post-traumatic, ageing-related, genetic, and symptomatic [13], eventually resulting in clinical and radiographic manifestations. While it was originally thought that OA was a disease of the elderly, risk factors other than age have been identified to predispose an individual to OA. The prevalence of OA is on the rise and is attributed partly to a surge in the preponderance of OA risk factors, including obesity, lack of physical activity, and traumatic injuries of the joint. Compelling evidence from recent studies connote that OA could be a metabolic disease with several components of the metabolic syndrome (MetS) adding up to the disease pathogenesis and progression and that metabolic syndrome increased the risk for OA [14,15,16]. Metabolic syndrome, also called the syndrome X, is a clustering of closely associated clinical conditions comprising central obesity, glucose intolerance (type 2 diabetes, impaired glucose tolerance), insulin resistance (IR), dyslipidemia, and hypertension, all of which present a risk for cardiovascular diseases [17]. Of late, there has been a profound interest to decipher the plausible link between these metabolic perturbations and OA that has led to the identification of yet another phenotype of OA known as the metabolic OA [18, 19]. Evidence(s) from cohort studies have ascertained a strong positive association between metabolic syndrome and OA incidence and that there was a significant increase in the risk for developing OA with the addition of each component of metabolic syndrome [20]. This review is focused on discussing the contribution of each of the several components of the metabolic syndrome towards OA pathogenesis and progression.
Obesity and OA
Osteoarthritis (OA) is a complex disease having a multifactorial pathophysiology comprising biomechanical, metabolic, and inflammatory components to its etiology [21]. Obesity has been long established as a predominant and possibly avertable risk factor for OA, possessing multiple repercussions on the incidence, progression, and symptom severity associated with the disease. The role of obesity and overweight in contributing to OA progression is conceivably the most commonly researched topic in OA research. Several epidemiological studies have established the link between obesity and OA as listed in Table 1. A positive association between higher body mass and greater lower extremity joint loading has been established [22]. Coggon et al. [23] reported that subjects whose body mass index (BMI) exceeded 30 kg/m2 had 6.8 times greater risk of developing knee OA compared to subjects who recorded normal body weights. Excess body weight not only enhances the load on the weight-bearing joints [24] but also causes misalignment and unfavorable joint mechanics especially in the knees thereby increasing mechanical stress and cartilage degradation leading to OA [25]. Also, obesity is associated with a reduction in muscle strength highly essential for joint stabilization and hence consequently a decrease in the ability to withstand mechanical stress in the joints [26]. In addition to its direct detrimental effects on the cartilage matrix, mechanical loading can also alter the inflammatory state of chondrocytes. Application of high-magnitude cyclic tensile strain to chondrocytes significantly elevated the expression of pro-inflammatory mediators such as interleukin (IL)-1β, tumor necrosis factor (TNF)-α, cyclooxygenase (COX)-2, and matrix metalloproteinases (MMPs)-3, -13 mediated through FAK, ERK, JNK, p38, and NF-κB signaling [27,28,29]. Mechanical overloading has also been identified to promote chondrocyte senescence and OA development in human and mice chondrocytes [30]. Evidence(s) from in vivo studies indicate that compressive loading of the knee joints led to increased cartilage fibrillation and erosion, and osteophyte formation [31]. In addition to cartilage, obesity could also adversely affect the subchondral bone where the mechanical overburdening leads to thickening of subchondral cortical bone impairing the underlying cartilage [32, 33], and induce an inflammatory phenotype in both sclerotic and non-sclerotic osteoblasts identified by an increase in their expression of IL-6, IL-8, COX-2, Receptor activator of nuclear factor kappa-Β ligand (RANKL), MMP-3, MMP-9, and MMP-13 with a decrease in osteoprotegerin (OPG) expression resulting in an increased susceptibility to OA [34]. In obese subjects, malalignment and hyperextension in the knee joints also contributes to OA [35].
Although excessive joint loading contributes for an important etiological factor for obesity-mediated OA, altered biomechanics fully fail to justify the increased risk for OA in non-weight-bearing joints including the hands and the wrists in obese subjects, pointing to a systemic, non-mechanical influence on the risk for OA [36]. Indeed, perpetual inflammation leading to cartilage loss, osteophyte formation, and synovitis have been implicated as the main pathophysiological mechanism behind obesity-associated OA [37]. The adipose tissue (AT) is a complex and a highly metabolic organ comprising adipocytes, nerve tissue, stromovascular cells, and immune cells such as the macrophages, T cells, B cells and dendritic cell subsets, mast cells, neutrophils, and eosinophils [38]. Under obese conditions, the adipose tissue macrophages (ATMs) infiltrating and accumulating in the adipose tissue with increasing body weight undergo a phenotype switch leading to a shift in their activation state from an M2-polarized state (lean) that protect adipocytes from inflammation to an M1 pro-inflammatory state that leads to enhanced production of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-12, IL-15, IL-17, IL-18, IL-23), chemokines (monocyte chemoattractant protein (MCP)-1, C-X-C motif ligand (CXCL)-9, CXCL-10, CXCL-11, CXCL-13, C–C motif ligand (CCL)-8, CCL-15, CCL-19, CCL-20), interferon (IFN)-γ, and reactive oxygen species (ROS) such as nitric oxide (NO) resulting in chronic low-grade sterile inflammation and IR [39,40,41,42]. The role of IR in the pathophysiology of OA has been discussed elsewhere in this manuscript.
Higher levels of pro-inflammatory cytokines have been observed in overweight and obese adults [43]. Evidences from experimentally induced obesity in rats using a high-carbohydrate/high-fat diet also revealed a spontaneously induced infiltration of pro-inflammatory macrophages (M1) into the synovium of the joint tissue and also an activation of the M1 phenotype in the resident macrophages with a concomitant exacerbation of OA-like pathological changes [44]. The M1-associated cytokines IL-6, IL-1β, and TNF-α promote detrimental processes in chondrocytes such as decreased production of collagen II and aggrecan, and upregulation of several inflammatory molecules and matrix-degrading proteases mediated by the various signaling pathways including the transforming growth factor (TGF)-β, c-Jun N-terminal kinase (JNK), extracellular signal-regulated kinase (ERK), p38, AKT, NF-κB, and β-catenin signaling that result in cartilage degradation and bone resorption [45]. IL-1β plays a potential catabolic role in OA where it stimulates chondrocytes to release increased amounts of A Disintegrin and Metalloproteinase with Thrombospondin motifs (ADAMTS)-4, ADAMTS-5, MMP-1, MMP-3, MMP-13, and other intermediates including ROS, NO, cytosolic phospholipase A2 (cPLA2), COX-2, and prostaglandin E2 (PGE2) and also regulate Fas-mediated chondrocyte apoptosis. IL-1β also exerts a detrimental effect on osteoblasts by increasing the expression of MMP-2, MMP-3, MMP-9, MMP-13, ADAMTS-4, ADAMTS-5, and RANKL contributing to subchondral bone remodeling OA [37, 46]. TNF-α plays a critical role in OA by its ability to induce collagenases and aggrecanases including MMP-1, MMP-3, MMP-13, ADAMTS-4, IL-6, IL-8, RANTES, VEGF, iNOS, COX-2, and PGE2 synthase while also inhibiting the synthesis of proteoglycan components and collagen II [47]. TNF-α and IL-1β have also been demonstrated to significantly decrease the expression of SOX9, which is essential for chondrocyte differentiation [48, 49]. IL-6 has been identified to induce catabolic mediators in MMP-3, MMP-13, and ADAMTS which mediate cartilage degeneration, promote proteoglycan loss, reduce chondrocyte proliferation, and enhance ROS production. IL-6 has also been identified to affect other tissues of the joint including synovium, subchondral bone, and muscles in the context of OA [50]. IL-1 and IL-6 have also been identified to play decisive roles in driving Th17 signaling leading to the production of IL-17 [51, 52]. IL-17 could regulate several OA pathophysiology-related pathways in chondrocytes and synovial fibroblasts (SFs) observed in end-stage OA patients [53•]. In addition to solely effecting adverse effects on the cartilage structure and function, the pro-inflammatory cytokines such as IL-6, IL-1β, TNF-α, IL-15, IL-17, and IL-18 could also work in synergy with one another to maximize their potent adverse effects in OA including enhancing inflammation and upregulating the expression of proteases, aid cartilage ECM degradation, and ultimately resulting in total joint failure [37, 42].
Furthermore, the factors secreted by M1 synovial macrophages have been demonstrated to impede the chondrogenic differentiation of the resident mesenchymal stem cells in the OA synovium suggestive of the fact that the M1-polarized macrophage subsets orchestrate an anti-chondrogenic effect within the OA joint [54]. Recently, Liu et al. [55] reported a markedly higher ratio of M1 to M2 macrophages in the synovial fluid (SF) and peripheral blood of knee OA patients compared to controls with a significant positive correlation with the level of Kellgren–Lawrence grade in knee OA, strongly suggestive of the involvement of macrophages in knee OA pathogenesis. The shift in macrophage phenotype from M2 to M1 with increasing adiposity significantly augments the M1 cytokine-induced cartilage deterioration and diminishes the efficacy for tissue repair and angiogenesis mediated by the M2 macrophage-derived factors. Therefore, prospective therapeutic approaches directed at the synovial macrophage phenotype could be decisive in breaking the bond between obesity and OA and also promote the efficiency of MSC-based cartilage regeneration approaches [56, 57].
In obesity, the adipose tissue (AT) has been recognized to function as the largest endocrine metabolic organ secreting a battery of pro-inflammatory cytokines, chemokines, and adipokines. Adipokines comprise a range of pleiotrophic molecules including bioactive peptides and immune and inflammatory mediators secreted by the adipose tissue, and exhibit their effects in an autocrine/paracrine and endocrine manner [58]. The infrapatellar fat pad (IPFP) which is in close proximity with the synovium is the major source of adipokines in the SF of the knee. Despite the fact the adipokines are majorly secreted by adipocytes, other joint tissue resident cells including chondrocytes, osteoblasts, synoviocytes, stromal cells, macrophages, and immune cells have also been identified to produce some adipokines [59]. The presence of adipokine receptors in many joint cell types is indicative of the complex regulatory network of adipokine signaling within the joint.
Leptin was the first identified adipokine that is predominantly produced by the adipose tissue and executes its functions mediated by the Ob receptor [60]. Owing to the wide expression of leptin receptors in peripheral tissues and the involvement of leptin in several physiological processes including insulin secretion, bone metabolism, and immune responses, it is contemplated to be a potential link between obesity and OA. Higher levels of systemic leptin have been identified in OA patients in comparison to healthy subjects. Several studies have identified that significantly elevated leptin levels in OA patients were positively correlated with disease severity and pain [61,62,63,64], making it a potential biomarker for OA. One of the earlier studies showed that leptin could have an anabolic role on chondrocytes by inducing insulin-like growth factor 1 (IGF-1) and TGF-β expression [65]. However, in advanced OA cartilage and SF, the leptin and leptin’s receptor (Ob-Rb) are expressed in significantly increased levels. Leptin exerts a pro-inflammatory and catabolic function in cartilage metabolism by its inherent ability to function alone or in association with other pro-inflammatory factors to target chondrocytes, synoviocytes, and osteoblasts in exerting crucial functions of OA pathogenesis [66]. Inflammatory and catabolic factors such as IL-1β, MMP-9, and MMP-13 can induce the expression of leptin and in turn increase the production of T helper 1 (TH1) type cytokines by immune cells, and suppress TH2 type cytokines which corroborate a catabolic and pro-inflammatory role for leptin in OA pathophysiology [67, 68]. Adiposity in the absence of leptin signaling was found to be inadequate in inducing systemic inflammation and knee OA which underscores leptin’s noteworthy role in OA pathogenesis as well as its utility as a potential biomarker in OA [69].
The adipokine adiponectin, also known as AdipoQ, exerts its effects mediated through AdipoR1 and AdipoR2 receptors. While evidences from clinical and experimental studies point to a role for adiponectin in OA pathophysiology, it is yet not clear whether adiponectin exerts a protective role in OA or not. Adiponectin has been found to be expressed by synoviocytes, IPFP, osteophytes, cartilage, and bone tissues of the joint [70]. Two recent meta-analyses indicated that circulating adiponectin levels were elevated in OA patients compared to healthy controls [71, 72] whereas earlier studies showed decreased circulating levels of adiponectin in OA patients [73]. However, a positive association has been reported between increased serum adiponectin levels with a higher radiographic score in knee OA but not in hand OA suggestive of different pathological mechanisms in OA development of the two joints [74]. It has also been elucidated that adiponectin is upregulated in OA cartilage and the full-length form of adiponectin, but not the globular form, has a stimulatory effect on PGE2 and MMP-13 activity. It was also identified that AdipoR1 mRNA levels are strongly associated with the mRNA expression of cartilage-specific components, suggesting that adiponectin could be involved in matrix remodeling [75]. Recently, it was shown that there was a negative association between serum levels of adiponectin and bone mineral density (BMD) in symptomatic knee OA patients suggestive of its adverse effect on BMD [76]. Also, a recent cross-sectional study indicated that there was a greater association for synovial adiponectin with clinical severity of knee OA in women than for synovial leptin underscoring its clinical relevance in OA pathogenesis [77].
Visfatin, also known as the pre-B cell colony-enhancing factor (PBEF) or nicotinamide phosphoribosyltransferase (NAMPT), is another key adipokine widely expressed in white adipose tissue (WAT) and implicated in OA [78, 79]. Studies have revealed that both circulating as well as SF visfatin levels were significantly higher in OA patients compared to healthy controls with the cartilage and synovial tissues of OA patients shown to exhibit higher secretion of visfatin compared to healthy subjects [80]. Besides, the IPFP tissue expression of visfatin in OA patients was found to be higher than that in matched subcutaneous WAT [81]. OA patients with greater radiographic evidence of joint damage and disease severity reportedly had higher levels of SF visfatin compared to those with less disease severity [82]. Visfatin has been found to be expressed in osteophytes by the osteoblasts, osteoclasts, and chondrocytes in OA patients indicating its destructive role in OA especially by unfavorably altering the extracellular matrix homeostasis resulting in cartilage destruction [83]. Visfatin has also been demonstrated to contribute to OA progression by its ability to exert a pro-inflammatory effect by inducing the production of IL-1β, IL-6, and TNF-α in lymphocytes [84].
The adipokine resistin is a cysteine-rich polypeptide hormone predominantly secreted by macrophages and adipocytes in humans and mice, respectively [85]. Resistin has been identified to have an association with radiographic knee OA [86]. Epidemiological and clinical studies have indicated that serum and SF resistin levels positively correlated with OA severity, synovitis, and structural abnormalities in OA patients [87,88,89]. With a role identified for resistin in enhancing the pro-inflammatory milieu by aiding the synthesis of MMPs and release of pro-inflammatory cytokines in chondrocytes, resistin is believed to promote OA progression [85]. Chemerin is another adipokine mainly expressed in the WAT, which exerts its functions by binding the ChemR23 receptor. Chemerin is a chemoattractant adipokine which stimulates chemotaxis of immune cells to the inflammatory site as in the case of OA where macrophages and other immune cells are recruited to the synovium as a part of the inflammatory cascade [90]. Obese patients have been reported to have increased levels of serum chemerin which is correlated to OA disease severity. The SF chemerin levels have been found to be positively correlated with BMI and OA severity [91]. With an inherent ability for the human articular chondrocytes to produce chemerin, express ChemR23, and also to promote inflammatory signaling [92], it is speculated that chemerin could play a role in OA pathogenesis.
Lipocalin -2 is another adipokine whose production within the joint tissues is triggered by both mechanical as well as inflammatory stimuli [93], serving as a sensor of mechanical load and joint inflammation in OA pathophysiology. Lipocalin-2 levels were found to be elevated in the synovial fluid and cartilage of OA patients, and proposed to be involved in matrix degradation by its ability to reduce chondrocyte proliferation and form a covalent complex with MMP-9 thereby preventing its auto-degradation [94]. Nesfatin-1 (nesfatin) is an N-terminal 82 amino acid peptide nucleobindin-2-derived adipokine implicated to have a role in OA. Serum nesfatin levels have been elevated in OA incidences [95], with the serum and SF concentrations found to be associated with radiographic severity in OA [96]. Moreover, the increased serum and chondrocyte expression levels of nesfatin-1 in OA subjects were found to be positively correlated with high-sensitivity C-reactive protein (hsCRP) and IL-18 levels [97], making it a prospective biomarker for OA progression. However, a recent study has reported that nesfatin-1 could protect against IL-1β induced OA progression in rats [98].
Apelin is another member of the adipokine superfamily highlighted to be associated with increased bone marrow lesions in OA [64]. A positive correlation was observed between SF apelin levels and disease severity in OA subjects with significantly elevated apelin levels identified in OA serum compared to the normal subjects. The expression of apelin and its receptor APJ was also relatively higher in OA cartilage compared to healthy controls suggestive of its role in contributing to OA pathophysiology [99]. Reports have identified a catabolic role for apelin in OA by virtue of its ability to induce the expression of inflammatory and matrix degrading proteases in vivo and in vitro [100, 101]. Apelin has also been reportedly identified to mediate the synovial VEGF-mediated angiogenesis in OA progression, making it an ideal pharmaceutical and therapeutic target in OA [102].
Owing to advancements in clinical research and the pursuit to gain newer knowledge concerning adipokine contribution to OA pathophysiology, novel adipokines are being identified lately. The novel adipokines such as Serpin Peptidase Inhibitor, Clade E, Member 2 (SERPINE2), WNT1 Inducible Signaling Pathway Protein 2 (WISP2), Glycoprotein Nmb (GPNMB), and Inter-Alpha-Trypsin Inhibitor Heavy Chain family, member 5 (ITIH5) are upregulated in obesity and have been identified to be expressed in the OA synovium, IPFP, and chondrocytes exhibiting differential expression patterns with a potential for involvement in OA onset and progression [103]. Another adipokine serum amyloid A (SAA) has been found to be significantly elevated in circulation as well as SF of OA patients and proven to contribute to the inflammatory process in OA [104]. Metrnl is another newly identified adipokine which has been discussed to have a connection with obesity–OA interplay [105]. The Retinol binding protein 4 (RBP4) is another novel adipokine that is a member of the lipocalin family that is produced within the OA joints and positively correlated with increased levels of adipokines and MMPs [106]. The fatty acid-binding protein 4 (FABP4) is a novel adipokine that is found in elevated levels in the circulation as well as the SF of OA patients with a positive correlation between the IPFP and SF levels, and perceived to be a potential biomarker for OA [107]. Adipsin is an adipokine recently identified to bear clinical relevance as a biomarker as well as potential therapeutic target for OA. Adipsin levels were significantly higher in human OA serum, SF, synovial membrane, and cartilage compared with controls. Higher serum adipsin levels have been reported in OA patients which was strongly associated with greater cartilage loss [108]. Also, adipsin deficiency in transgenic mice rendered protection against cartilage degradation when subjected to anterior cruciate ligament (ACL) injury thereby accentuating its role in OA [109]. In addition, a few adipokines such as omentin-1, vaspin, progranulin, and SERPINE2 have also been ascertained to play a protective role in OA progression [110,111,112,113]. The various roles of different adipokines in the context of OA have been discussed in Table 2.
OA and Dyslipidemia
Obesity is characterized not only by an abnormal loading of the weight-bearing joints, but also by an aberrant lipid metabolism leading to dyslipidemia identified by low levels of systemic high-density lipoproteins (HDLs) and higher levels of free fatty acids (FFAs), triglycerides (TGs), oxidized low-density lipoproteins (ox-LDLs), and cholesterol [114].
Altered lipid metabolism could well play a causal role in the pathobiology of OA as identified by several study findings. Epidemiological studies have also reported a positive correlation between hypercholesterolemia and OA [115], implying that cholesterol might be a systemic risk factor for OA. Studies carried out in rodents using ApoE−/− mice and diet-induced hypercholesterolemia (DIHC) rats showed that hypercholesterolemia was able to induce OA-like changes in these animals characterized by cartilage degradation, osteophyte formation, and alterations to the subchondral bone tissue architecture, accentuating the role of cholesterol in the pathogenesis of OA [116]. Impairment of cholesterol efflux genes accompanied by an increase in intracellular lipid accumulation in osteoarthritic chondrocytes has been established with a positive correlation to disease severity [117]. The downstream adverse effect of cholesterol accumulation in chondrocytes is manifested as an impairment of mitochondrial functions which could further exacerbate other downstream pathways critically involved in cartilage degradation such as ROS production, amplification of cytokine-induced chondrocyte inflammation, matrix catabolism, and increased chondrocyte apoptosis [118, 119]. Also, a recent study discovered that retinoic acid-related orphan receptor alpha in chondrocytes is directly activated by cholesterol and its metabolites, upregulating matrix-degrading enzymes and raising the risk of OA [120]. High levels of cholesterol also inhibited LRP3 gene in chondrocytes adversely affecting cartilage ECM metabolism and eventually resulting in OA cartilage degeneration [121]. Higher circulating levels of cholesterol and TG levels, and dysfunctional HDL have also been identified to accelerate joint pathology and induce cartilage loss in knee OA by synovial activation, ectopic bone formation [122], and an increased occurrence of bone marrow lesions which are a source of intense pain in OA [123].
In addition, reduced serum levels of HDL-c observed in the serum of OA patients could possibly have a propensity in OA pathogenesis. Studies using LCAT−/− and ApoA-1−/− (both are necessary to form mature HDL-c particles) mice have proven that these KO mice had greatly reduced levels of functional HDL-c and also exhibited cartilage fibrillation, vertical clefts, chondrocyte clustering, and reduced PG content with increased MMP-2, MMP-9, and MMP-13 expression compared to their controls [124]. Alterations in HDL-c metabolic pathway could adversely tinker cartilage homeostasis in an untoward direction leading to OA.
Higher circulating levels of ox-LDL are another feature of obesity-related dyslipidemia. In OA, inflammation accelerates vascular porosity expediting infusion of biological factors into the synovial fluid [125] including ox-LDL which has been oxidatively altered extra-articularly. In addition, activated endothelial cells in the inflamed synovium and chondrocytes in the degrading cartilage release ROS which could further oxidatively modify the native LDL to ox-LDL [126]. Binding of ox-LDL to its scavenger receptor—lectin-like ox-LDL receptor-1 (LOX-1)—reduces cell viability and PG synthesis in cartilage matrix, and increases intracellular ROS production leading to activation of NF-κB [127]. This further activates expression of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6), chemokines (IL-8, macrophage inflammatory protein-1b), enzymes (COX-2, iNOS, cPLA2, metalloproteinases), and adhesion molecules (intercellular adhesion molecule-1 and vascular cell adhesion molecule-1) [128]. Also, ox-LDL/LOX-1 stimulates VEGF release in chondrocytes which increases the expression of proteinases like MMP-1, MMP-3, and MMP-13 and pro-inflammatory cytokines like IL-1β, TNF-α, and IL-6 leading to cartilage degradation [129]. Besides, chondrocytes in OA cartilage exhibit increased LOX-1 expression and localization in comparison to normal controls [130]. Chondrocytes express membrane receptors both for FAs and lipoproteins, including G-protein-coupled receptor 40 (GPR40) and GPR120, TLR4, and CD36, as well as some members of the low-density lipoprotein receptor (LDLR) and LDL receptor‐related protein (LRP) families [131]. The LRP5-mediated Wnt/β-catenin signaling pathway is involved in downregulating type II collagen production, while upregulating MMP-3 and MMP-13 synthesis thereby inducing cartilage degradation [132]. Ox-LDL could also trigger the release of MMPs, pro-inflammatory cytokines, and other growth factors by their inherent ability to also activate synovial macrophages, endothelial cells, and synovial fibroblasts [133]. These findings underline the significance of hypercholesterolemia and ox-LDL in the initiation and progression of OA by their inherent ability to impair the joint tissue homeostasis and induce inflammation and cartilage degradation.
Higher systemic FFA levels are also a characteristic of obesity-related dyslipidemia. Impairment in the inhibition of lipolysis in adipocytes is chiefly responsible for enhanced FFA release from adipose tissue into the circulation [134]. These FFAs induce a macrophage inflammatory response by triggering toll-like receptors (TLRs) and activating downstream signaling by phosphorylating TAK1, JNK, p-38, c-Jun, and NF-κB leading to the production of cytokines, iNOS, and COX-2 [135]. Resident macrophages of the synovial tissue lining can also be activated by FFAs to induce local joint inflammation. In addition, FFAs coming from a diet with imbalanced fat composition have also been shown to affect the various cells of the joint leading to inflammation and OA. Saturated fatty acids (SFAs) such as palmitic and stearic acid have been shown to promote chondrocyte matrix remodeling by activating autophagy [136]. Dietary saturated fatty acid palmitate promoted chondrocyte apoptosis and cartilage lesions in knee joint of mice mediated through the promotion of unfolding protein response (UPR)/endoplasmic reticulum (ER) stress in a mouse model of diet-induced OA [137]. In addition to chondrocytes, FAs and their derivatives function as signaling molecules to bind to receptors on the other joint tissue cells including osteoblasts, osteoclasts, and synoviocytes [131]. They activate various downstream signaling pathways to trigger detrimental effects on the joint including apoptosis of cells, altered tissue homeostasis, remodeling, and inflammation.
Diabetes and OA
Evidence from epidemiological and experimental data not only suggest a conceivable association between OA and diabetes but also endorse the proposition that diabetes could be in itself an important independent risk factor for OA [138]. OA and type 2 diabetes mellitus (T2DM) intermittently co-exist due to the common risk factors they share—obesity and aging and also due to their higher prevalence. Epidemiological studies reveal that the overall risk of OA in patients with T2DM is 1.46 while that of T2DM in patients with OA is 1.41. The prevalence of OA among T2DM patients and that of T2DM in OA patients was 29.5% and 14.4%, respectively [139]. A meta-analysis study carried out to assess the prevalence of OA in patients with DM revealed a high association between the two, even suggestive of identification of a T2DM-related OA within a metabolic phenotype [140].
Evidences suggest that T2DM elicits a pathological role on OA effected via two important pathways: (1) chronic hyperglycemia, which promotes oxidative stress, bolsters pro-inflammatory cytokines and AGEs production in joint tissues but also decreases the chondrogenic differentiation potential of the various stem cells thereby further decreasing the already impaired cartilage repair in OA; and (2) insulin resistance, which executes its effects both locally and also through low-grade inflammation systemically [138]. Articular chondrocytes are highly glycolytic cells expressing glucose transporters (GLUT 1, 3, and 9) that need a stable glucose supply for maintenance of cellular energy homeostasis [134]. Normal human chondrocytes sense fluctuations in the extracellular glucose levels and accordingly adapt themselves by regulating GLUT-1 synthesis and its lysosomal mediated degradation [141]. This ability is compromised in OA chondrocytes which become incapacitated to adapt to higher extracellular concentrations of glucose vis-à-vis impaired GLUT-1 downregulation leading to accumulation of glucose within the cells. This has a noxious effect on chondrocyte homeostasis and function manifested as increased and prolonged ROS production, advanced glycation end products (AGEs) accumulation, and expression of inflammatory and catabolic mediators including pro-inflammatory cytokines and matrix metalloproteinases [142, 143]. At diabetic glucose concentrations, chondrocytes also become non-responsive to IGF-1 leading to a condition of IGF-1 resistance in chondrocytes [144]. This could also constitute a pathogenic mechanism for cartilage degeneration as IGF-1 exerts anabolic effects in articular cartilage by inducing production of PGs, collagen type II, and other ECM components by the chondrocytes.
ROS conduce to OA pathogenesis by their ability to induce IL-1β, diminish the production and stimulate the degradation of cartilage matrix proteins [145], enhance chondrocyte apoptosis [146], and activate transcription factors like activator protein-1 and NF-κB that play pivotal roles in joint inflammation and cartilage degradation [147]. Accumulation of AGEs in cartilage primarily modifies its mechano-chemical functioning by making the cartilage brittle, promoting matrix stiffness, and making the cartilage more sensitive to mechanical stress resulting in degradation [148]. The accumulated AGEs are also recognized by pattern recognition receptors (PRRs) expressed by the chondrocytes, namely, Receptor for Advanced Glycation Endproducts (RAGE) and TLR-4 which trigger downstream signaling pathways including the MAP kinases and NF-κB pathways leading to a pro-inflammatory and pro-catabolic state of the chondrocytes [149]. AGEs also reduce the AMPKα/SIRT1/PGC-1α signaling in chondrocytes, leading to mitochondrial dysfunction as a result of increased oxidative stress, inflammation, and apoptosis [150]. Accumulation of AGEs is also higher in the subchondral bone of diabetic patients compared to healthy subjects which may impair the mechanical resistance of subchondral bone and also portray pro-inflammatory effects [138]. Hyperglycemia-induced AGE accumulation in fibroblast-like synoviocytes increased the release of inflammatory factors which in turn induce chondrocyte degradation and promote OA progression [151].
Diabetes also accelerates OA by damaging and deteriorating the functions of the subchondral bone by adversely altering its microarchitecture, chemical composition, and biomechanical properties [152]. In women, higher fasting serum glucose levels were shown to have a positive association with two key predictors of structural OA damage—tibial cartilage volume loss and the occurrence of bone marrow lesions [153]. Together, these evidences indicate that higher levels of glucose adversely affect chondrocytes not only by aiding catabolic responses, but also by modifying their response to anabolic elements ultimately leading to cartilage destruction.
IR and T2DM develop as a consequence of visceral adiposity which presents itself with chronic low-grade systemic inflammation leading to dysregulated joint metabolism precipitating as OA [154]. Insulin receptors are expressed by the chondrocytes which make these cells sensitive to insulin. Insulin has also been identified to induce anabolic effects in a variety of musculoskeletal tissues including cartilage, bone, and tendon promoting cell differentiation, proliferation, and extracellular matrix production [155]. Regardless of the fact that insulin negatively regulates synovial inflammation and catabolism, obese subjects with T2DM develop synovial IR which abates the ability of higher insulin levels to curtail the production of OA-promoting inflammatory and catabolic mediators [138, 156]. In OA, higher insulin levels could facilitate macrophage infiltration and production of chemokines, inhibit autophagy in fibroblast-like synoviocytes, and intensify the inflammatory response by the activation of PI3K/AKT/mTOR/NF-ĸB signaling and a positive feedback loop with the pro-inflammatory cytokines (IL-1β, IL-6, and TNF-α) [157]. Also, at supraphysiological insulin concentrations, chondrocytes exhibit impaired autophagy mediated by the increased activity of AKT/mTOR signaling pathway, loss of PG, and increased IL-1β and MMP-13 expression contributing to inflammatory OA-like changes [158]. Impaired autophagy could be one of the mechanisms responsible for accelerated cartilage degradation in diabetes-associated OA patients. Pharmacological intervention to address impaired autophagy may prove effective in preventing T2DM-induced cartilage damage.
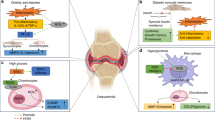
Herrero-Beaumont et al. [159] have proposed an additional pathogenic pathway called O-GlcNAcylation to explain the link between OA and diabetes. O-GlcNAcylation is a dynamic post-translational modification where a single O-N-Acetyl-glucosamine residue is incorporated to nucleocytoplasmic and mitochondrial proteins. O-GlcNAcylation is a glucose-dependent process and is involved in regulating cellular activities and the stress response. UDP-GlcNAc serves as the donor for protein GlcNAc, the synthesis of which increases under hyperglycemic conditions mediated by the hexosamine biosynthetic pathway [160]. Findings from studies indicate that the O-GlcNAcylated proteins accumulate in human OA cartilage which could partly induce hypertrophic-like phenotype changes in OA chondrocytes [161], thereby delineating a possible link between diabetes and OA. An integrative view of the pathophysiology of the metabolic-syndrome associated OA is depicted in Fig. 1.

An integrative view of the pathophysiology of metabolic-syndrome associated OA
Hypertension and OA
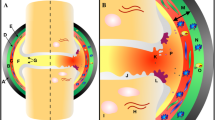
Hypertension is an important component of MetS and an independent risk factor for cardiovascular and cerebrovascular disease [162]. However, epidemiological studies have now established that OA is more common in subjects with hypertension [163] and is highly likely a key factor in the pathogenesis of MetS-associated OA. In the latest Framingham Osteoarthritis study, the investigators observed that even after adjusting for BMI or body weight, there was a significant association between hypertension and the occurrence of OA [164]. Studies attempting to understand the mechanism behind the role of hypertension in the pathogenesis of OA have centered on vascular pathology leading to subchondral ischemia [165]. Hypertension-induced vasoconstriction over a period of time could reduce flow of blood through the small vessels in the subchondral bone. Also, venous occlusion or microemboli development in subchondral blood vessels can narrow the vessel lumen leading to blockage and reduced blood flow ultimately resulting in subchondral ischemia [166]. The impending pernicious effects of subchondral ischemia are (1) a debilitated nutrient and oxygen exchange across cartilage and bone triggering cartilage degradation and (2) apoptosis of osteocytes in the ischemic regions of subchondral bone which might elicit osteoclastic resorption rendering deprivation of bony support for the above lying cartilage [19, 167]. Joint loading also results in subchondral trabecular loss that leads to cartilage breakdown by favoring cartilage deformation. Subchondral bone remodeling plays an important role in hypertension-mediated joint deterioration in OA. Evidences also show that there could be an involvement of multiple genes in OA and hypertension such as the OPG/RANKL, OPG, and LDRP 6, gene polymorphisms of vitamin D receptor and IL-6 [168]. Recent epidemiological evidence have also ascertained a positive association between hypertension and knee OA of both radiological and symptomatic disease and pain severity, accentuating the significant relationship between hypertension and OA [167, 169]. In addition to a plethora of clinical and epidemiological evidence(s), several in vivo models portraying features of MetS such as UC-Davis-T2DM rats [170], WNIN/Gr-Ob obese rats [32•], Zucker Diabetic Fatty (ZDF) rats [171], obese Spontaneously Hypertensive Heart Failure (SHHFcp/cp) rats [172], diet-induced obese mice, rat, and guinea pig models [173, 174], and T2DM db/db mice [175] have also helped better decipher and establish the association between MetS and OA.
Even as multiple components of metabolic syndrome predispose to OA, optimal management of OA must encompass modification of risk factors through targeted interventions. Obesity/overweight, physical activity, and diet are among the chief modifiable risk factors that could affect the course of OA [176]. Survival analysis from a recent Osteoarthritis Initiative data has concluded that every 1% weight loss was associated with a 2% reduced risk of knee replacement in subjects with clinical knee OA and that public health strategies which include weight loss interventions have the potential to lessen the burden of knee and hip replacement surgery [177]. Furthermore, findings from a recent systematic review carried out to assess the effects of exercise on knee OA revealed that strengthening and aerobic exercises had positive effects on OA patients, and both aquatic and land-based programs improved pain, physical function, and quality of life [178]. Data from the recent Osteoarthritis Initiative also revealed that knee OA progression was inversely associated with a prudent dietary pattern comprising high intake of vegetables, fruits, fish, whole grain, and legumes, while a Western dietary pattern characterized by a high intake of processed/red meats, refined grains, high-fat dairy products, sugar-containing beverages, desserts, and sweets increased the radiographic and symptomatic progress of knee OA [179]. Of late, pharmacological agents such as metformin conventionally used for treating type II diabetes have also been shown to be beneficial in treating OA by inhibiting inflammation, modulating autophagy, countering oxidative stress and reducing pain levels [180, 181], and reducing leptin secretion from adipose tissue [182]. Given the multifactorial etiology of MetS-associated OA, current evidence supports lifestyle modifications as a safe and effective means to alter the parameters of MetS, and also yield promising results for decreasing symptomatic and radiographic knee OA [183].
Conclusion
With an abundance of novel evidence arising out of advancements in preclinical, clinical, and epidemiological studies, there has been a paradigm shift in the way OA pathogenesis is perceived. There is undeniable confirmation that OA is not merely a ‘wear and tear’ disease of the elderly as it has been commonly thought of. Given the alarming rate at which obesity and its allied metabolic perturbations are on the rise globally, the need to address metabolic syndrome and its modifiable risk factors gains preeminence in the holistic approach of metabolic OA management. Chronic low-grade inflammation orchestrated by several adipokines and pro-inflammatory cytokines associated with obesity, dysregulated lipid and glucose homeostasis have been among the chief factors that drive the pathogenesis of OA associated with MetS. The identification of key roles for several metabolic regulators in OA pathogenesis has opened up newer avenues in the recognition of therapeutic targets and the development of novel treatments in addressing metabolic OA.
References
Jaggard M, et al. Can metabolic profiling provide a new description of osteoarthritis and enable a personalised medicine approach? 2020;39:3875–3882.
Wang H, et al. Metabolic syndrome increases the risk for knee osteoarthritis: a meta-analysis. Evidence-Based Complementary and Alternative Medicine. 2016;2016:7242478.
Liu S-Y, et al. Bidirectional association between metabolic syndrome and osteoarthritis: a meta-analysis of observational studies. Diabetol Metab Syndr. 2020;12(1):38.
Pan F, et al. Association between metabolic syndrome and knee structural change on MRI. Rheumatology. 2019;59(1):185–93.
Jha BK, et al. Progress in understanding metabolic syndrome and knowledge of its complex pathophysiology. 2023;4(2):134–59.
Courties A, Berenbaum F, Sellam JJJBS. The phenotypic approach to osteoarthritis: a look at metabolic syndrome-associated osteoarthritis. 2019;86(6):725–730.
Zhuo Q, et al. Metabolic syndrome meets osteoarthritis. Nat Rev Rheumatol. 2012;8(12):729–37.
Askari A, et al. Relationship between metabolic syndrome and osteoarthritis: the Fasa Osteoarthritis Study. Diabetes Metab Syndr. 2017;11:S827–32.
He Y, et al. Pathogenesis of osteoarthritis: risk factors, regulatory pathways in chondrocytes, and experimental models. 2020;9(8):194.
Al Khatib F, et al. Biomechanical characteristics of the knee joint during gait in obese versus normal subjects. 2022;19(2):989.
Coggon D, et al. Knee osteoarthritis and obesity. Int J Obes Relat Metab Disord. 2001;25(5):622–7.
Sun AR, et al. Cartilage tissue engineering for obesity-induced osteoarthritis: physiology, challenges, and future prospects. Journal of Orthopaedic Translation. 2021;26:3–15.
Chen L, et al. Pathogenesis and clinical management of obesity-related knee osteoarthritis: impact of mechanical loading. Journal of Orthopaedic Translation. 2020;24:66–75.
Zhang X, et al. Relationship between knee muscle strength and fat/muscle mass in elderly women with knee osteoarthritis based on dual-energy X-ray absorptiometry. 2020;17(2):573.
Yanoshita M, et al. Cyclic tensile strain upregulates pro-inflammatory cytokine expression via FAK-MAPK signaling in chondrocytes. Inflammation. 2018;41(5):1621–30.
Hirose N, et al. Protective effects of cilengitide on inflammation in chondrocytes under excessive mechanical stress. 2020;44(4):966–74.
Hikida M, Nakajima M, Nakata K. Cyclic compressive mechanical loading on threedimensional cultured tissue of human chondrocytes synergistically upregulates MMP3 gene expression with IL-1β. J Osaka Dent Univ. 2021;55(1):91–8.
Zhang H, et al. Mechanical overloading promotes chondrocyte senescence and osteoarthritis development through downregulating FBXW7. 2022;81(5):676–686.
Roemhildt ML, et al. Chronic in vivo load alteration induces degenerative changes in the rat tibiofemoral joint. Osteoarthr Cartil. 2013;21(2):346–57.
• Pragasam SSJ, Venkatesan V. Metabolic syndrome predisposes to osteoarthritis: lessons from model system. CARTILAGE. 2020;1947603520980161. The incidence of knee OA associated with metabolic syndrome in an obese mutant rodent model confirms the role played by various components of MetS in promoting the incidence of OA
Zhu, J., et al., Instability and excessive mechanical loading mediate subchondral bone changes to induce osteoarthritis. 2020, 2020;8(6):350.
Kovács B, Vajda E, Nagy EE. Regulatory effects and interactions of the Wnt and OPG-RANKL-RANK signaling at the bone-cartilage interface in osteoarthritis. 2019;20(18):4653.
Karimi MT. and F. Hemati, Knee joint osteoarthritis in obese subjects, effects of diet and exercise on knee-joint loading: a review of literature. 2022;33(4):376–83.
Gløersen M, et al. Associations of body mass index with pain and the mediating role of inflammatory biomarkers in people with hand osteoarthritis. 2022;74(5):810–7.
Wang T, He C. Pro-inflammatory cytokines: the link between obesity and osteoarthritis. Cytokine Growth Factor Rev. 2018;44:38–50.
Lu J, et al. Adipose tissue-resident immune cells in obesity and type 2 diabetes. 2019;10(1173).
Castoldi A, et al. The macrophage switch in obesity development. Front Immunol. 2015;6:637.
Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab. 2004;89(6):2548–56.
Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Investig. 2007;117(1):175–84.
Liu S, et al. Cartilage tissue engineering: from proinflammatory and anti-inflammatory cytokines to osteoarthritis treatments (review). Mol Med Rep. 2022;25(3):99.
Purdy JC, Shatzel JJ. The hematologic consequences of obesity. 2021;106(3):306–19.
Sun AR, et al. Obesity-associated metabolic syndrome spontaneously induces infiltration of pro-inflammatory macrophage in synovium and promotes osteoarthritis. PLoS ONE. 2017;12(8): e0183693.
Zhang H, Cai D, Bai X. Macrophages regulate the progression of osteoarthritis. Osteoarthritis Cartilage. 2020;28(5):555–61. Activated macrophages generate pro-inflammatory mediators, as well as multiple tissue-degrading enzymes that escalate the inflammatory milieu and contribute to the destruction of cartilage and bone.
Jenei-Lanzl Z, Meurer A, Zaucke F. Interleukin-1β signaling in osteoarthritis – chondrocytes in focus. Cell Signal. 2019;53:212–23.
Molnar V, et al. Cytokines and chemokines involved in osteoarthritis pathogenesis. 2021;22(17):9208.
Zhang X, et al. Telmisartan mitigates TNF-α-induced type II collagen reduction by upregulating SOX-9. ACS Omega. 2021;6(17):11756–61.
Xu Z, et al. Agonism of GPR120 prevented IL-1β-induced reduction of extracellular matrix through SOX-9. Aging (Albany NY). 2020;12(12):12074–85.
Wiegertjes R, van de Loo FAJ, Blaney Davidson ENA. roadmap to target interleukin-6 in osteoarthritis. Rheumatology (Oxford). 2020;59(10):2681–2694.
Na HS, et al. Interleukin-1-interleukin-17 signaling axis induces cartilage destruction and promotes experimental osteoarthritis. 2020;11.
Kang S, et al. Targeting interleukin-6 signaling in clinic. Immunity. 2019;50(4):1007–23.
• Mimpen JY, et al. Interleukin-17A causes osteoarthritis-like transcriptional changes in human osteoarthritis-derived chondrocytes and synovial fibroblasts in vitro. 2021;12. IL-17 cytokines activated multiple catabolic pathways in knee OA patients making it a potential biomarker and a clincal target in OA
Zhu X, et al. Phenotypic alteration of macrophages during osteoarthritis: a systematic review. Arthritis Res Ther. 2021;23(1):110.
Liu B, et al. Imbalance of M1/M2 macrophages is linked to severity level of knee osteoarthritis. Exp Ther Med. 2018;16(6):5009–14.
Xie J, et al. Clinical implications of macrophage dysfunction in the development of osteoarthritis of the knee. 2019;46:36–44.
Wang W, et al. Targeting macrophage polarization as a promising therapeutic strategy for the treatment of osteoarthritis. Int Immunopharmacol. 2023;116: 109790.
Gu X, et al. Adipose tissue adipokines and lipokines: functions and regulatory mechanism in skeletal muscle development and homeostasis. Metabolism. 2023;139: 155379.
Xie C, Chen Q. Adipokines: new therapeutic target for osteoarthritis? Curr Rheumatol Rep. 2019;21(12):71.
Yan M, et al. The role of leptin in osteoarthritis. Medicine (Baltimore). 2018;97(14): e0257.
Lambova SN, et al. Serum leptin and resistin levels in knee osteoarthritis—clinical and radiologic links: towards precise definition of metabolic type knee osteoarthritis. 2021;9(8):1019.
Min S, et al. Serum levels of leptin, osteopontin, and sclerostin in patients with and without knee osteoarthritis. Clin Rheumatol. 2021;40(1):287–94.
Kroon FPB, et al. The role of leptin and adiponectin as mediators in the relationship between adiposity and hand and knee osteoarthritis. Osteoarthritis Cartilage. 2019;27(12):1761–7.
Zhu J, et al. Association of serum levels of inflammatory markers and adipokines with joint symptoms and structures in participants with knee osteoarthritis. Rheumatology. 2021;61(3):1044–52. A complex interplay between the metabolic components and inflammatory markers have been found to be associated with joint symptoms and strucutral deformities in knee OA.
Dumond H, et al. Evidence for a key role of leptin in osteoarthritis. 2003;48(11):3118–29.
Gao Y-H, et al. An update on the association between metabolic syndrome and osteoarthritis and on the potential role of leptin in osteoarthritis. Cytokine. 2020;129: 155043.
Simopoulou T, et al. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. 2007;15(8):872–883.
Ouchi N, et al. Adipokines in inflammation and metabolic disease. Nat Rev Immunol. 2011;11(2):85–97.
Griffin TM, et al. Extreme obesity due to impaired leptin signaling in mice does not cause knee osteoarthritis. Arthritis Rheum. 2009;60(10):2935–44.
Presle N, et al. Differential distribution of adipokines between serum and synovial fluid in patients with osteoarthritis. Contribution of joint tissues to their articular production. Osteoarthr Cartil. 2006;14(7):690–5.
Lee YH, Song GG. Association between circulating adiponectin levels and osteoarthritis: a meta-analysis. jrd. 2018;25(4):231–238.
Tang Q, et al. Association of osteoarthritis and circulating adiponectin levels: a systematic review and meta-analysis. Lipids Health Dis. 2018;17(1):189.
Laurberg TB, et al. Plasma adiponectin in patients with active, early, and chronic rheumatoid arthritis who are steroid- and disease-modifying antirheumatic drug-naive compared with patients with osteoarthritis and controls. 2009;36(9):1885–1891.
Xu H, et al. Increased adiponectin levels are associated with higher radiographic scores in the knee joint, but not in the hand joint. Sci Rep. 2021;11(1):1842.
Francin PJ, et al. Association between adiponectin and cartilage degradation in human osteoarthritis. Osteoarthritis Cartilage. 2014;22(3):519–26.
Wu J, et al. Associations between circulating adipokines and bone mineral density in patients with knee osteoarthritis: a cross-sectional study. BMC Musculoskelet Disord. 2018;19(1):16.
Orellana C, et al. Synovial adiponectin was more associated with clinical severity than synovial leptin in women with knee osteoarthritis. Cartilage. 2021;13(1_suppl):1675s-1683s.
Samal B, et al. Cloning and characterization of the cDNA encoding a novel human pre-B-cell colony-enhancing factor. Mol Cell Biol. 1994;14(2):1431–7.
Azamar-Llamas D, et al. Adipokine contribution to the pathogenesis of osteoarthritis. 2017;2017.
Han DF, et al. An update on the emerging role of visfatin in the pathogenesis of osteoarthritis and pharmacological intervention. Evid Based Complement Alternat Med. 2020;2020:8303570.
Conde J, et al. Differential expression of adipokines in infrapatellar fat pad (IPFP) and synovium of osteoarthritis patients and healthy individuals. Ann Rheum Dis. 2014;73(3):631–3.
Duan Y, et al. Increased synovial fluid visfatin is positively linked to cartilage degradation biomarkers in osteoarthritis. Rheumatol Int. 2012;32(4):985–90.
Junker S, et al. Expression of adipokines in osteoarthritis osteophytes and their effect on osteoblasts. Matrix Biol. 2017;62:75–91.
Moschen AR, et al. Visfatin, an adipocytokine with proinflammatory and immunomodulating properties. J Immunol. 2007;178(3):1748–58.
Zhao C-W, et al. An update on the emerging role of resistin on the pathogenesis of osteoarthritis. Mediators Inflamm. 2019;2019:1532164.
Lambova SN, et al. Serum leptin and resistin levels in knee osteoarthritis-clinical and radiologic links: towards precise definition of metabolic type knee osteoarthritis. Biomedicines. 2021;9(8).
Alissa EM, Alzughaibi LS, Marzouki ZM. Relationship between serum resistin, body fat and inflammatory markers in females with clinical knee osteoarthritis. Knee. 2020;27(1):45–50.
Lim DH, Choi S. High synovial fluid resistin levels are associated with radiographic severity in female patients with knee osteoarthritis. Osteoarthritis Cartilage. 2020;28:S195–6.
Han W, et al. Higher serum levels of resistin are associated with knee synovitis and structural abnormalities in patients with symptomatic knee osteoarthritis. J Am Med Dir Assoc. 2019;20(10):1242–6.
Iannone F, Lapadula G. Chemerin/ChemR23 pathway: a system beyond chemokines. Arthritis Res Ther. 2011;13(2):104.
Huang K, et al. Association of chemerin levels in synovial fluid with the severity of knee osteoarthritis. Biomarkers. 2012;17(1):16–20.
Berg V, et al. Human articular chondrocytes express ChemR23 and chemerin; ChemR23 promotes inflammatory signalling upon binding the ligand chemerin. Arthritis Res Ther. 2010;12:R228.
Carrión M, et al. The adipokine network in rheumatic joint diseases. 2019;20(17):4091.
Gupta K, et al. Neutrophil gelatinase-associated lipocalin is expressed in osteoarthritis and forms a complex with matrix metalloproteinase 9. Arthritis Rheum. 2007;56(10):3326–35.
Puzio I, et al. Role of nesfatin-1 in the metabolism of skeletal tissues. 2018;74(5):290–94.
Zhang Y, et al. Serum and synovial fluid nesfatin-1 concentration is associated with radiographic severity of knee osteoarthritis. Medical science monitor : international medical journal of experimental and clinical research. 2015;21:1078–82.
Jiang L, et al. Increased serum levels and chondrocyte expression of nesfatin-1 in patients with osteoarthritis and its relation with BMI, hsCRP, and IL-18. Mediators Inflamm. 2013;2013: 631251.
Jiang L, et al. Nesfatin-1 suppresses interleukin-1β-induced inflammation, apoptosis, and cartilage matrix destruction in chondrocytes and ameliorates osteoarthritis in rats. Aging (Albany NY). 2020;12(2):1760–77.
Hu P-F, et al. Increased apelin serum levels and expression in human chondrocytes in osteoarthritic patients. Int Orthop. 2011;35(9):1421–6.
Chang TK, et al. Apelin enhances IL-1β expression in human synovial fibroblasts by inhibiting miR-144-3p through the PI3K and ERK pathways. Aging (Albany NY). 2020;12(10):9224–39.
Hu P-F, et al. Apelin plays a catabolic role on articular cartilage: in vivo and in vitro studies. Int J Mol Med. 2010;26(3):357–63.
Wang Y-H, et al. Apelin affects the progression of osteoarthritis by regulating VEGF-dependent angiogenesis and miR-150–5p expression in human synovial fibroblasts. 2020;9(3):594.
Conde J, et al. Identification of novel adipokines in the joint. Differential expression in healthy and osteoarthritis tissues. PLOS ONE. 2015;10(4):e0123601.
Geurts J, et al. A novel Saa3-promoter reporter distinguishes inflammatory subtypes in experimental arthritis and human synovial fibroblasts. 2011;70(7):1311–1319.
Sobieh BH, et al. Potential emerging roles of the novel adipokines adipolin/CTRP12 and meteorin-like/METRNL in obesity-osteoarthritis interplay. Cytokine. 2021;138: 155368.
Scotece M, et al. Novel adipokine associated with OA: retinol binding protein 4 (RBP4) is produced by cartilage and is correlated with MMPs in osteoarthritis patients. Inflamm Res. 2020;69(4):415–21.
Zhang C, et al. FABP4 as a biomarker for knee osteoarthritis. Biomark Med. 2018;12(2):107–18.
Valverde-Franco G, et al. High in vivo levels of adipsin lead to increased knee tissue degradation in osteoarthritis: data from humans and animal models. Rheumatology. 2018;57(10):1851–60.
Paré F, et al. In vivo protective effect of adipsin-deficiency on spontaneous knee osteoarthritis in aging mice. Aging (Albany NY). 2020;12(3):2880–96.
Feng D, et al. Progranulin modulates cartilage-specific gene expression via sirtuin 1-mediated deacetylation of the transcription factors SOX9 and P65. J Biol Chem. 2020;295(39):13640–50.
He H, et al. Vaspin regulated cartilage cholesterol metabolism through miR155/LXRα and participated in the occurrence of osteoarthritis in rats. Life Sci. 2021;269: 119096.
Santoro A, et al. SERPINE2 inhibits IL-1α-induced MMP-13 expression in human chondrocytes: involvement of ERK/NF-κB/AP-1 pathways. PLoS ONE. 2015;10(8): e0135979.
Li Z, et al. Omentin-1 promotes mitochondrial biogenesis via PGC1α-AMPK pathway in chondrocytes. Arch Physiol Biochem. 2020;1–7.
Sanja Klobučar M, et al. Dyslipidemia: current perspectives and implications for clinical practice. In: Management of dyslipidemia, Wilbert SA. Editor. 2021;IntechOpen: Rijeka.Ch.1.
Cho BW, et al. Cross-sectional association between hypercholesterolemia and knee pain in the elderly with radiographic knee osteoarthritis: data from the Korean National Health and Nutritional Examination Survey. 2021;10(5):933.
Farnaghi S, et al. Protective effects of mitochondria-targeted antioxidants and statins on cholesterol-induced osteoarthritis. Faseb j. 2017;31(1):356–67.
Tsezou A, et al. Impaired expression of genes regulating cholesterol efflux in human osteoarthritic chondrocytes. J Orthop Res. 2010;28(8):1033–9.
Papathanasiou I, Anastasopoulou L, Tsezou A. Cholesterol metabolism related genes in osteoarthritis. Bone. 2021;152: 116076.
Zheng L, et al. The role of metabolism in chondrocyte dysfunction and the progression of osteoarthritis. Ageing Res Rev. 2021;66: 101249.
Choi W-S, et al. The CH25H–CYP7B1–RORα axis of cholesterol metabolism regulates osteoarthritis. Nature. 2019;566(7743):254–8.
Cao C, et al. Cholesterol-induced LRP3 downregulation promotes cartilage degeneration in osteoarthritis by targeting Syndecan-4. Nat Commun. 2022;13(1):7139.
de Munter W, et al. Cholesterol accumulation caused by low density lipoprotein receptor deficiency or a cholesterol-rich diet results in ectopic bone formation during experimental osteoarthritis. Arthritis Res Ther. 2013;15(6):R178.
Davies-Tuck ML, et al. Total cholesterol and triglycerides are associated with the development of new bone marrow lesions in asymptomatic middle-aged women - a prospective cohort study. Arthritis Res Ther. 2009;11(6):R181.
Triantaphyllidou IE, et al. Perturbations in the HDL metabolic pathway predispose to the development of osteoarthritis in mice following long-term exposure to western-type diet. Osteoarthritis Cartilage. 2013;21(2):322–30.
Sokolove J, Lepus CM. Role of inflammation in the pathogenesis of osteoarthritis: latest findings and interpretations. Ther Adv Musculoskelet Dis. 2013;5(2):77–94.
Bostan M, et al. Effects of synovial fluid on the respiratory burst of granulocytes in rheumatoid arthritis. J Cell Mol Med. 2001;5(2):188–94.
Nishimura S, et al. Oxidized low-density lipoprotein (ox-LDL) binding to lectin-like ox-LDL receptor-1 (LOX-1) in cultured bovine articular chondrocytes increases production of intracellular reactive oxygen species (ROS) resulting in the activation of NF-kappaB. Osteoarthritis Cartilage. 2004;12(7):568–76.
Pahl HL. Activators and target genes of Rel/NF-kappaB transcription factors. Oncogene. 1999;18(49):6853–66.
Kanata S, et al. Oxidized LDL binding to LOX-1 upregulates VEGF expression in cultured bovine chondrocytes through activation of PPAR-gamma. Biochem Biophys Res Commun. 2006;348(3):1003–10.
Simopoulou T, Malizos KN, Tsezou A. Lectin-like oxidized low density lipoprotein receptor 1 (LOX-1) expression in human articular chondrocytes. Clin Exp Rheumatol. 2007;25(4):605–12.
Harasymowicz NS, et al. Physiologic and pathologic effects of dietary free fatty acids on cells of the joint. Ann N Y Acad Sci. 2019;1440(1):36–53.
Shin Y, et al. Low-density lipoprotein receptor-related protein 5 governs Wnt-mediated osteoarthritic cartilage destruction. Arthritis Res Ther. 2014;16(1):R37.
Hashimoto K, Akagi M. The role of oxidation of low-density lipids in pathogenesis of osteoarthritis: a narrative review. J Int Med Res. 2020;48(6):300060520931609.
Wu X, et al. The metabolic landscape in osteoarthritis. Aging Dis. 2022;13(4):1166–82.
Tsai Y-W, et al. Palmitoleic acid ameliorates palmitic acid-induced proinflammation in J774A.1 macrophages via TLR4-dependent and TNF-α-independent signallings. Prostaglandins, Leukotrienes and Essential Fatty Acids. 2021;169:102270.
Sekar S, et al. Saturated fatty acids promote chondrocyte matrix remodeling through reprogramming of autophagy pathways. Nutrition. 2018;54:144–52.
Tan L, et al. Dietary saturated fatty acid palmitate promotes cartilage lesions and activates the unfolded protein response pathway in mouse knee joints. PLoS ONE. 2021;16(2): e0247237.
Veronese N, et al. Type 2 diabetes mellitus and osteoarthritis. Semin Arthritis Rheum. 2019;49(1):9–19.
Sellam J, Berenbaum F. Is osteoarthritis a metabolic disease? Joint Bone Spine. 2013;80(6):568–73.
Louati K, et al. Association between diabetes mellitus and osteoarthritis: systematic literature review and meta-analysis. RMD Open. 2015;1(1): e000077.
Ashrafizadeh H, Ashrafizadeh M, Oroojan AA. Type 2 diabetes mellitus and osteoarthritis: the role of glucose transporters. Clinical Reviews in Bone and Mineral Metabolism. 2020;18(1):1–17.
Juybari KB, Hosseinzadeh A, Sharifi AM. Protective effects of atorvastatin against high glucose-induced nuclear factor-κB activation in cultured C28I2 chondrocytes. J Recept Signal Transduction. 2019;39(1):1–8.
Njoto I, et al. Chondrocyte intracellular matrix strain fields of articular cartilage surface in hyperglycemia model of rat: cellular morphological study. Med Arch. 2018;72(5):348–51.
Doré S, et al. Human osteoarthritic chondrocytes possess an increased number of insulin-like growth factor 1 binding sites but are unresponsive to its stimulation. Possible role of IGF-1-binding proteins. Arthritis Rheum. 1994;37(2):253–63.
Bolduc JA, et al. Reactive oxygen species, aging and articular cartilage homeostasis. 2019;132:73–82.
Zhuang C, et al. Oxidative stress induces chondrocyte apoptosis through caspase-dependent and caspase-independent mitochondrial pathways and the antioxidant mechanism of Angelica sinensis polysaccharide. 2020;2020.
Kan S, et al. Role of mitochondria in physiology of chondrocytes and diseases of osteoarthritis and rheumatoid arthritis. 2021;13(2_suppl):1102S-1121S.
Moshtagh PR, et al. Effects of non-enzymatic glycation on the micro- and nano-mechanics of articular cartilage. J Mech Behav Biomed Mater. 2018;77:551–6.
Suzuki A, Yabu A, Nakamura H. Advanced glycation end products in musculoskeletal system and disorders. Methods. 2022;203:179–86.
Yang Q, et al. Advanced glycation end products induced mitochondrial dysfunction of chondrocytes through repression of AMPKα-SIRT1-PGC-1α pathway. Pharmacology. 2022;107(5–6):298–307.
Li Q, et al. Hyperglycemia-induced accumulation of advanced glycosylation end products in fibroblast-like synoviocytes promotes knee osteoarthritis. Exp Mol Med. 2021;53(11):1735–47.
Wang HJ, et al. Diabetes mellitus accelerates the progression of osteoarthritis in streptozotocin-induced diabetic mice by deteriorating bone microarchitecture, bone mineral composition, and bone strength of subchondral bone. Ann Transl Med. 2021;9(9):768.
Davies-Tuck ML, et al. Increased fasting serum glucose concentration is associated with adverse knee structural changes in adults with no knee symptoms and diabetes. Maturitas. 2012;72(4):373–8.
Bradley D. The intriguing intersection of type 2 diabetes, obesity-related insulin resistance, and osteoarthritis. J Clin Endocrinol Metab. 2021;106(5):e2370–2.
Sakhrani N, et al. Toward development of a diabetic synovium culture model. Front Bioeng Biotechnol. 2022;10: 825046.
Griffin TM, Huffman KM. Editorial: Insulin resistance: releasing the brakes on synovial inflammation and osteoarthritis? Arthritis Rheumatol. 2016;68(6):1330–3.
Qiao L, Li Y, Sun S. Insulin exacerbates inflammation in fibroblast-like synoviocytes. Inflammation. 2020;43(3):916–36.
Ribeiro M, et al. Insulin decreases autophagy and leads to cartilage degradation. Osteoarthritis Cartilage. 2016;24(4):731–9.
Herrero-Beaumont G, et al. Targeting chronic innate inflammatory pathways, the main road to prevention of osteoarthritis progression. Biochem Pharmacol. 2019;165:24–32.
Hart GW, Housley MP, Slawson C. Cycling of O-linked β-N-acetylglucosamine on nucleocytoplasmic proteins. Nature. 2007;446(7139):1017–22.
Tardio L, et al. O-linked N-acetylglucosamine (O-GlcNAc) protein modification is increased in the cartilage of patients with knee osteoarthritis. Osteoarthritis Cartilage. 2014;22(2):259–63.
Kokubo Y, Iwashima Y. Higher blood pressure as a risk factor for diseases other than stroke and ischemic heart disease. Hypertension. 2015;66(2):254–9.
Ching K, et al. Hypertension meets osteoarthritis — revisiting the vascular aetiology hypothesis. Nat Rev Rheumatol. 2021;17(9):533–49.
Niu J, et al. Metabolic syndrome, its components, and knee osteoarthritis: the Framingham Osteoarthritis Study. Arthritis Rheumatol. 2017;69(6):1194–203.
Khamidov O, et al. The role of vascular pathology in the development and progression of deforming osteoarthritis of the joints of the lower extremities (Literature review). 2021:214–225.
Findlay DM. Vascular pathology and osteoarthritis. Rheumatology (Oxford). 2007;46(12):1763–8.
Shi X, Schlenk EAJPMN. Association of hypertension with knee pain severity among people with knee osteoarthritis. 2022;23(2):135–141.
Zhang YM, Wang J, Liu XG. Association between hypertension and risk of knee osteoarthritis: a meta-analysis of observational studies. Medicine (Baltimore). 2017;96(32): e7584.
Lo K, et al. Association between hypertension and osteoarthritis: a systematic review and meta-analysis of observational studies. Journal of Orthopaedic Translation. 2022;32:12–20. A significant relationship between hypertension and structural damages of knee OA points to a plausible vascular etiology of OA.
Onur T, et al. Characterisation of osteoarthritis in a small animal model of type 2 diabetes mellitus. Bone Joint Res. 2014;3(6):203–11.
Fu Y, et al. Effects of leptin and body weight on inflammation and knee osteoarthritis phenotypes in female rats. n/a;(n/a):e10754.
Deng C, et al. Eplerenone treatment alleviates the development of joint lesions in a new rat model of spontaneous metabolic-associated osteoarthritis. Ann Rheum Dis. 2018;77(2):315–6.
Siriarchavatana P, et al. The preventive effects of greenshell mussel (Perna canaliculus) on early-stage metabolic osteoarthritis in rats with diet-induced obesity. 2019;11(7):1601.
Radakovich LB, et al. Calorie restriction with regular chow, but not a high-fat diet, delays onset of spontaneous osteoarthritis in the Hartley guinea pig model. Arthritis Res Ther. 2019;21(1):145.
Rui F, et al. Undenatured type II collagen prevents and treats osteoarthritis and motor function degradation in T2DM patients and db/db mice. Food Funct. 2021;12(10):4373–91.
Georgiev T, Angelov AK. Modifiable risk factors in knee osteoarthritis: treatment implications. Rheumatol Int. 2019;39(7):1145–57.
Salis Z, et al. Weight loss is associated with reduced risk of knee and hip replacement: a survival analysis using Osteoarthritis Initiative data. Int J Obes. 2022;46(4):874–84.
Raposo F, Ramos M. and A. Lúcia Cruz, Effects of exercise on knee osteoarthritis: a systematic review. 2021;19(4):399–435.
Xu C, et al. Dietary patterns and risk of developing knee osteoarthritis: data from the osteoarthritis initiative. Osteoarthritis Cartilage. 2021;29(6):834–40.
Song, Y, Wu Z, Zhao P. The effects of metformin in the treatment of osteoarthritis: Current perspectives. 2022;13.
Liu X, et al. Metformin attenuates high-fat diet induced metabolic syndrome related osteoarthritis through inhibition of prostaglandins. Front Cell Dev Biol. 2023;11:1184524.
Li D, et al. Metformin attenuates osteoarthritis by targeting chondrocytes, synovial macrophages and adipocytes. Rheumatology. 2022;62(4):1652–61.
Maderitz RLJLJoMS. Easing the burden: the value of lifestyle modifications for management of knee osteoarthritis in patients with metabolic syndrome. 2023;5(1):102.
Park D, et al. Association of general and central obesity, and their changes with risk of knee osteoarthritis: a nationwide population-based cohort study. Sci Rep. 2023;13(1):3796. General obesity and central obesity are proven risk factors for OA.
Chen L, et al. The burden of end-stage osteoarthritis in Australia: a population-based study on the incidence of total knee replacement attributable to overweight/obesity. Osteoarthritis Cartilage. 2022;30(9):1254–62.
Raud B, et al. Level of obesity is directly associated with the clinical and functional consequences of knee osteoarthritis. Sci Rep. 2020;10(1):3601.
Misra D, et al. Risk of knee osteoarthritis with obesity, sarcopenic obesity, and sarcopenia. Arthritis Rheumatol. 2019;71(2):232–7.
Hussain SM, et al. Relationship of weight and obesity with the risk of knee and hip arthroplasty for osteoarthritis across different levels of physical performance: a prospective cohort study. Scand J Rheumatol. 2019;48(1):64–71.
Reyes C, et al. Association between overweight and obesity and risk of clinically diagnosed knee, hip, and hand osteoarthritis: a population-based cohort study. Arthritis Rheumatol. 2016;68(8):1869–75.
Lee S, et al. Obesity, metabolic abnormality, and knee osteoarthritis: a cross-sectional study in Korean women. Mod Rheumatol. 2015;25(2):292–7.
Apold H, et al. Weight gain and the risk of knee replacement due to primary osteoarthritis: a population based, prospective cohort study of 225,908 individuals. Osteoarthritis Cartilage. 2014;22(5):652–8.
Mork PJ, Holtermann A, Nilsen TI. Effect of body mass index and physical exercise on risk of knee and hip osteoarthritis: longitudinal data from the Norwegian HUNT Study. J Epidemiol Community Health. 2012;66(8):678–83.
Holliday KL, et al. Lifetime body mass index, other anthropometric measures of obesity and risk of knee or hip osteoarthritis in the GOAL case-control study. Osteoarthritis Cartilage. 2011;19(1):37–43.
Yoshimura N. Epidemiology of osteoarthritis in Japan: the ROAD study. Clin Calcium. 2011;21(6):821–5.
Toivanen AT, et al. Obesity, physically demanding work and traumatic knee injury are major risk factors for knee osteoarthritis—a population-based study with a follow-up of 22 years. Rheumatology (Oxford). 2010;49(2):308–14.
Lohmander LS, et al. Incidence of severe knee and hip osteoarthritis in relation to different measures of body mass: a population-based prospective cohort study. Ann Rheum Dis. 2009;68(4):490–6.
Grotle M, et al. Obesity and osteoarthritis in knee, hip and/or hand: an epidemiological study in the general population with 10 years follow-up. BMC Musculoskelet Disord. 2008;9:132.
Reijman M, et al. Body mass index associated with onset and progression of osteoarthritis of the knee but not of the hip: the Rotterdam Study. Ann Rheum Dis. 2007;66(2):158–62.
Karlson EW, et al. Total hip replacement due to osteoarthritis: the importance of age, obesity, and other modifiable risk factors. Am J Med. 2003;114(2):93–8.
Gelber AC, et al. Body mass index in young men and the risk of subsequent knee and hip osteoarthritis. Access the “Journal Club” discussion of this paper at http://www.elsevier.com/locate/ajmselect. Am J Med. 1999;107(6):542–548.
Shiozaki H, et al. Obesity and osteoarthritis of the knee in women: results from the Matsudai Knee Osteoarthritis survey. Knee. 1999;6(3):189–92.
Felson DT, et al. Risk factors for incident radiographic knee osteoarthritis in the elderly: the Framingham Study. Arthritis Rheum. 1997;40(4):728–33.
Hart DJ, Spector TD. The relationship of obesity, fat distribution and osteoarthritis in women in the general population: the Chingford Study. J Rheumatol. 1993;20(2):331–5.
Felson DT, et al. Obesity and knee osteoarthritis. The Framingham Study Ann Intern Med. 1988;109(1):18–24.
Hartz AJ, et al. The association of obesity with joint pain and osteoarthritis in the HANES data. J Chronic Dis. 1986;39(4):311–9.
Otero M, et al. Signalling pathway involved in nitric oxide synthase type II activation in chondrocytes: synergistic effect of leptin with interleukin-1. Arthritis Res Ther. 2005;7(3):R581.
Simopoulou T, et al. Differential expression of leptin and leptin’s receptor isoform (Ob-Rb) mRNA between advanced and minimally affected osteoarthritic cartilage; effect on cartilage metabolism. Osteoarthritis Cartilage. 2007;15(8):872–83.
Lago R, et al. A new player in cartilage homeostasis: adiponectin induces nitric oxide synthase type II and pro-inflammatory cytokines in chondrocytes. Osteoarthritis Cartilage. 2008;16(9):1101–9.
Vuolteenaho K, et al. Leptin enhances synthesis of proinflammatory mediators in human osteoarthritic cartilage—mediator role of NO in leptin-induced PGE2, IL-6, and IL-8 production. Mediators Inflamm. 2009;2009:345838–345838.
Bao J-P, et al. Leptin plays a catabolic role on articular cartilage. Mol Biol Rep. 2010;37(7):3265–72.
Mutabaruka M-S, et al. Local leptin production in osteoarthritis subchondral osteoblasts may be responsible for their abnormal phenotypic expression. Arthritis Res Ther. 2010;12(1):R20.
Conde J, et al. Adiponectin and leptin induce VCAM-1 expression in human and murine chondrocytes. PLoS ONE. 2012;7(12): e52533.
Hui W, et al. Leptin produced by joint white adipose tissue induces cartilage degradation via upregulation and activation of matrix metalloproteinases. 2012;71(3):455–62.
Yang W-H, et al. Leptin induces IL-6 expression through OBRl receptor signaling pathway in human synovial fibroblasts. PLoS ONE. 2013;8(9): e75551.
Yaykasli KO, et al, Leptin induces ADAMTS-4, ADAMTS-5, and ADAMTS-9 genes expression by mitogen-activated protein kinases and NF-ĸB signaling pathways in human chondrocytes. 2015;39(1):104–112.
Koskinen A, et al. Leptin enhances MMP-1, MMP-3 and MMP-13 production in human osteoarthritic cartilage and correlates with MMP-1 and MMP-3 in synovial fluid from OA patients. Clin Exp Rheumatol. 2011;29(1):57–64.
Scotece M, et al. Adipokines induce pro-inflammatory factors in activated Cd4+ T cells from osteoarthritis patient. 2017;35(6):1299–1303.
Zhang ZM, et al. Leptin induces the apoptosis of chondrocytes in an in vitro model of osteoarthritis via the JAK2-STAT3 signaling pathway. Mol Med Rep. 2016;13(4):3684–90.
Huang ZM, et al. Leptin promotes apoptosis and inhibits autophagy of chondrocytes through upregulating lysyl oxidase-like 3 during osteoarthritis pathogenesis. Osteoarthritis Cartilage. 2016;24(7):1246–53.
Wang W, et al. Effects of the leptin-mediated MAPK/ERK signaling pathway on collagen II expression in knee cartilage of newborn male mice from obese maternal offspring. 2022;12(3):477.
Zhang S, et al. Effects of leptin on differentiation and proliferation of chondrocytes. Journal of Hard Tissue Biology. 2019;28(1):51–6.
Conde J, et al. E74-like factor (ELF3) and leptin, a novel loop between obesity and inflammation perpetuating a pro-catabolic state in cartilage. Cell Physiol Biochem. 2018;45(6):2401–10.
Su Y-P, et al. Leptin induces MMP1/13 and ADAMTS 4 expressions through bone morphogenetic protein-2 autocrine effect in human chondrocytes. J Cell Biochem. 2018;119(4):3716–24.
Xiong H, et al. Elevated leptin levels in temporomandibular joint osteoarthritis promote proinflammatory cytokine IL-6 expression in synovial fibroblasts. 2019;48(3):251–259.
Zhao X, et al. Activation of the leptin pathway by high expression of the long form of the leptin receptor (Ob-Rb) accelerates chondrocyte senescence in osteoarthritis. 2019;8(9):425–436.
Mourmoura E, et al. Leptin-depended NLRP3 inflammasome activation in osteoarthritic chondrocytes is mediated by ROS. Mech Ageing Dev. 2022;208: 111730.
Primrose JGB, et al. Concentration-dependent effects of leptin on osteoarthritis-associated changes in phenotype of human chondrocytes. Connect Tissue Res. 2023;1–12.
Liang J, et al. Leptin-mediated cytoskeletal remodeling in chondrocytes occurs via the RhoA/ROCK pathway. J Orthop Res. 2011;29(3):369–74.
Gómez R, et al. Adiponectin and leptin increase IL-8 production in human chondrocytes. 2011;70(11):2052–2054.
Jiang M, et al. Leptin induced TLR4 expression via the JAK2-STAT3 pathway in obesity-related osteoarthritis. Oxid Med Cell Longev. 2021;2021:7385160.
Strebkova E, et al. The role of leptin in the metabolic phenotype of osteoarthritis. Osteoarthritis Cartilage. 2021;29:S158. Leptin is an aggravating predcitor of metabolic syndrome associated OA with higher leptin levels found in more advanced stages of OA making it a potential therapeutic target.
Tang CH, et al. Adiponectin enhances IL-6 production in human synovial fibroblast via an AdipoR1 receptor, AMPK, p38, and NF-kappa B pathway. J Immunol. 2007;179(8):5483–92.
Kang EH, et al. Adiponectin is a potential catabolic mediator in osteoarthritis cartilage. Arthritis Res Ther. 2010;12(6):R231.
Tong K-M, et al. Adiponectin increases MMP-3 expression in human chondrocytes through adipor1 signaling pathway. 2011;112(5):1431–1440.
Zuo W, et al. Adiponectin receptor 1 mediates the difference in adiponectin-induced prostaglandin E2 production in rheumatoid arthritis and osteoarthritis synovial fibroblasts. 2011 Chinese Medical Journals Publishing House Co., Ltd. 42 Dongsi Xidajie. 3919–3924.
Chen H-T, et al. Adiponectin enhances intercellular adhesion molecule-1 expression and promotes monocyte adhesion in human synovial fibroblasts. PLoS ONE. 2014;9(3): e92741.
Udomsinprasert W, et al. Decreased serum adiponectin reflects low vitamin D, high interleukin 6, and poor physical performance in knee osteoarthritis. Arch Immunol Ther Exp. 2020;68(3):16.
Ali SA, et al. Adiponectin is a potential mediator of synovial fibrosis from early to late knee osteoarthritis. Osteoarthritis Cartilage. 2019;27:S479–80.
Harasymowicz NS, et al. Chondrocytes from osteoarthritic cartilage of obese patients show altered adiponectin receptors expression and response to adiponectin. 2021;39(11):2333–9.
Gosset M, et al. Crucial role of visfatin/pre-B cell colony-enhancing factor in matrix degradation and prostaglandin E2 synthesis in chondrocytes: possible influence on osteoarthritis. Arthritis Rheum. 2008;58(5):1399–409.
Dvir-Ginzberg M, et al. Regulation of cartilage-specific gene expression in human chondrocytes by SirT1 and nicotinamide phosphoribosyltransferase*. J Biol Chem. 2008;283(52):36300–10.
McNulty AL, et al. The effects of adipokines on cartilage and meniscus catabolism. Connect Tissue Res. 2011;52(6):523–33.
Hong EH, et al. Nicotinamide phosphoribosyltransferase is essential for interleukin-1beta-mediated dedifferentiation of articular chondrocytes via SIRT1 and extracellular signal-regulated kinase (ERK) complex signaling. J Biol Chem. 2011;286(32):28619–31.
Yammani RR, Loeser RF. Extracellular nicotinamide phosphoribosyltransferase (NAMPT/visfatin) inhibits insulin-like growth factor-1 signaling and proteoglycan synthesis in human articular chondrocytes. Arthritis Res Ther. 2012;14(1):R23.
Chauffier K, et al. Induction of the chemokine IL-8/Kc by the articular cartilage: possible influence on osteoarthritis. Joint Bone Spine. 2012;79(6):604–9.
Pecchi E, et al. Induction of nerve growth factor expression and release by mechanical and inflammatory stimuli in chondrocytes: possible involvement in osteoarthritis pain. Arthritis Res Ther. 2014;16(1):R16.
Laiguillon M-C, et al. Expression and function of visfatin (Nampt), an adipokine-enzyme involved in inflammatory pathways of osteoarthritis. Arthritis Res Ther. 2014;16(1):R38.
Yang S, et al. NAMPT (visfatin), a direct target of hypoxia-inducible factor-2α, is an essential catabolic regulator of osteoarthritis. Ann Rheum Dis. 2015;74(3):595–602.
Oh H, et al. Reciprocal regulation by hypoxia-inducible factor-2α and the NAMPT-NAD+-SIRT axis in articular chondrocytes is involved in osteoarthritis. Osteoarthritis Cartilage. 2015;23(12):2288–96.
Won Y, et al. Pleiotropic roles of metallothioneins as regulators of chondrocyte apoptosis and catabolic and anabolic pathways during osteoarthritis pathogenesis. 2016;75(11):2045–52.
Cheleschi S, et al. Exploring the crosstalk between hydrostatic pressure and adipokines: an in vitro study on human osteoarthritic chondrocytes. 2021;22(5):2745.
Wu M-H, et al. Visfatin promotes IL-6 and TNF-α production in human synovial fibroblasts by repressing miR-199a-5p through ERK, p38 and JNK signaling pathways. 2018;19(1):190.
Cheleschi S, et al. MicroRNA mediate visfatin and resistin induction of oxidative stress in human osteoarthritic synovial fibroblasts via NF-κB pathway. 2019;20(20):5200.
Law Y-Y, et al. Visfatin increases ICAM-1 expression and monocyte adhesion in human osteoarthritis synovial fibroblasts by reducing miR-320a expression. Aging. 2020;12(18):18635–48.
Chang, S-F, et al. Effects of visfatin on intracellular mechanics and catabolism in human primary chondrocytes through glycogen synthase kinase 3β inactivation. 2021;22(15):8107.
Cheleschi S, et al. MicroRNA-34a and microRNA-181a mediate visfatin-induced apoptosis and oxidative stress via NF-κB pathway in human osteoarthritic chondrocytes. 2019;8(8):874.
Philp AM, et al. eNAMPT is localised to areas of cartilage damage in patients with hip osteoarthritis and promotes cartilage catabolism and inflammation. 2021;22(13):6719.
Tsai C-H, et al. Visfatin increases VEGF-dependent angiogenesis of endothelial progenitor cells during osteoarthritis progression. 2020;9(5):1315.
Silswal N, et al. Human resistin stimulates the pro-inflammatory cytokines TNF-α and IL-12 in macrophages by NF-κB-dependent pathway. Biochem Biophys Res Commun. 2005;334(4):1092–101.
Lee JH, et al. Resistin is elevated following traumatic joint injury and causes matrix degradation and release of inflammatory cytokines from articular cartilage in vitro. Osteoarthritis Cartilage. 2009;17(5):613–20.
Zhang Z, et al. Resistin induces expression of proinflammatory cytokines and chemokines in human articular chondrocytes via transcription and messenger RNA stabilization. Arthritis Rheum. 2010;62(7):1993–2003.
Koskinen A, et al. Resistin as a factor in osteoarthritis: synovial fluid resistin concentrations correlate positively with interleukin 6 and matrix metalloproteinases MMP-1 and MMP-3. Scand J Rheumatol. 2014;43(3):249–53.
Zhang Z, et al. Resistin stimulates expression of chemokine genes in chondrocytes via combinatorial regulation of C/EBPβ and NF-κB. 2014;15(10):17242–55.
Chen W-C, et al. Resistin enhances VCAM-1 expression and monocyte adhesion in human osteoarthritis synovial fibroblasts by inhibiting MiR-381 expression through the PKC, p38, and JNK signaling pathways. 2020;9(6):1369.
Chen WC, et al. Resistin enhances IL-1β and TNF-α expression in human osteoarthritis synovial fibroblasts by inhibiting miR-149 expression via the MEK and ERK pathways. Faseb j. 2020;34(10):13671–84.
Eisinger K, et al. Chemerin induces CCL2 and TLR4 in synovial fibroblasts of patients with rheumatoid arthritis and osteoarthritis. Exp Mol Pathol. 2012;92(1):90–6.
Wang C, et al. Chemerin promotes MAPK/ERK activation to induce inflammatory factor production in rat synoviocytes. Exp Ther Med. 2022;24(5):684.
Conde J, et al. Expanding the adipokine network in cartilage: identification and regulation of novel factors in human and murine chondrocytes. 2011;70(3):551–9.
Yu Chengshuai DG. Pang Shenning. Lao Shan, Chemerin, a pro-inflammatory adipokine, regulates chondrocyte proliferation and metabolism by increasing production of nitric oxide. 2021;25(2):258–63.
Villalvilla A, et al. The adipokine lipocalin-2 in the context of the osteoarthritic osteochondral junction. Sci Rep. 2016;6(1):29243.
Francisco V, et al. Adipokines and inflammation: is it a question of weight? 2018;175(10):1569–1579.
Jiang Z, et al. Whole-transcriptome sequence of degenerative meniscus cells unveiling diagnostic markers and therapeutic targets for osteoarthritis. Front Genet. 2021;12: 754421.
Shen F, et al. Cytokines link osteoblasts and inflammation: microarray analysis of interleukin-17- and TNF-alpha-induced genes in bone cells. J Leukoc Biol. 2005;77(3):388–99.
Scotece M, et al. NUCB2/nesfatin-1: a new adipokine expressed in human and murine chondrocytes with pro-inflammatory properties, an in vitro study. 2014;32(5):653–60.
Lee KT, et al. Nesfatin-1 facilitates IL-1β production in osteoarthritis synovial fibroblasts by suppressing miR-204-5p synthesis through the AP-1 and NF-κB pathways. Aging (Albany NY). 2021;13(18):22490–501.
Lu D, et al. Apelin alleviates meniscus endothelial cell apoptosis in osteoarthritis. Dis Markers. 2022;2022:3556372.
Takano S, et al. Vascular endothelial growth factor expression and their action in the synovial membranes of patients with painful knee osteoarthritis. BMC Musculoskelet Disord. 2018;19(1):204.
de Seny D, et al. Acute-phase serum amyloid A in osteoarthritis: regulatory mechanism and proinflammatory properties. PLoS ONE. 2013;8(6): e66769.
Zhang C, et al. FABP4 as a biomarker for knee osteoarthritis. 2018;12(2):107–118.
Zhao Y-P, et al. Progranulin protects against osteoarthritis through interacting with TNF-α and β-catenin signalling. 2015;74(12):2244–53.
Pan Y, et al. Progranulin regulation of autophagy contributes to its chondroprotective effect in osteoarthritis. Genes & Diseases. 2023;10(4):1582–95.
Guo F, et al. Granulin-epithelin precursor binds directly to ADAMTS-7 and ADAMTS-12 and inhibits their degradation of cartilage oligomeric matrix protein. Arthritis Rheum. 2010;62(7):2023–36.
Papers of particular interest, published recently, have been highlighted as: • Of importance
Bortoluzzi A, Furini F, Scirè CA. Osteoarthritis and its management - epidemiology, nutritional aspects and environmental factors. Autoimmun Rev. 2018;17(11):1097–104.
Global, regional, and national incidence, prevalence, and years lived with disability for 354 diseases and injuries for 195 countries and territories, 1990–2017: a systematic analysis for the Global Burden of Disease Study 2017. Lancet, 2018;392(10159):1789–1858.
Leifer VP, Katz JN, Losina E. The burden of OA-health services and economics. Osteoarthritis Cartilage. 2022;30(1):10–6.
Wang Y, et al. Knee osteoarthritis, potential mediators, and risk of all-cause mortality: data from the osteoarthritis initiative. Arthritis Care Res (Hoboken). 2021;73(4):566–73.
March L, et al. Osteoarthritis: A Serious Disease: Submitted to the US Food and Drug Administration. 2016.
Coaccioli S, et al. Osteoarthritis: new insight on its pathophysiology. 2022;11(20):6013.
Donell SJEOR. Subchondral bone remodelling in osteoarthritis. 2019;4(6):221.
Lee JY, et al. Association of leg muscle symmetry with knee osteoarthritis. Clin Rheumatol. 2019;38(12):3549–56.
Ozeki N, Koga H, Sekiya IJL. Degenerative meniscus in knee osteoarthritis: from pathology to treatment. 2022;12(4):603.
Schulze-Tanzil G. Intraarticular ligament degeneration is interrelated with cartilage and bone destruction in osteoarthritis. 2019;8(9):990.
Allen KD, Thoma LM, Golightly YM. Epidemiology of osteoarthritis. Osteoarthritis Cartilage. 2022;30(2):184–95.
• Cui A, et al. Global, regional prevalence, incidence and risk factors of knee osteoarthritis in population-based studies. EClinicalMedicine. 2020;29. The global prevalence and incidence of knee OA increased with age and peaked at 70–79 years while increasing education attainment was negatively associated with the prevalence of knee OA.
Acknowledgements
The authors thank the Department of Health Research, Ministry of Health and Family Welfare, New Delhi for financial support to Samuel Joshua Pragasam Sampath through the DHR – Young Scientist Research Fellowship (DHR – YSS Grant No. YSS/2020/000185/PRCYSS).
Funding
Department of Health Research,Ministry of Health and Family Welfare,New Delhi,India,YSS/2020/000185/PRCYSS,Samuel Joshua Pragasam Sampath
Author information
Authors and Affiliations
Contributions
SJPS completed the review; KN, SG, and VV reviewed the content and approach of the manuscript, and critically evaluated the review.
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare no competing interests.
Human and Animal Rights and Informed Consent
This article does not contain any studies with human or animal subjects performed by any of the authors.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Sampath, S.J.P., Venkatesan, V., Ghosh, S. et al. Obesity, Metabolic Syndrome, and Osteoarthritis—An Updated Review. Curr Obes Rep 12, 308–331 (2023). https://doi.org/10.1007/s13679-023-00520-5
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13679-023-00520-5