
Abstract
The stingless bee genus Scaptotrigona is widely distributed across tropical Mexico and includes economically important species used in stingless beekeeping. As Scaptotrigona colonies are currently or potentially translocated across regions, it is important to analyze the extent of genetic diversity from different populations. Herein, every analyzed Scaptotrigona individual was correctly assigned through DNA barcoding to the three recognized species (Scaptotrigona mexicana, Scaptotrigona pectoralis, and Scaptotrigona hellwegeri). Intraspecific divergence showed a mean value of 0.70 %, whereas the interspecific value was 2.79 %. As predicted by traditional taxonomy, sequence analyses demonstrated the close affinity of S. mexicana with S. hellwegeri. However, this also suggested the existence of cryptic species within S. mexicana, one of the stingless bees exploited for honey production in Mesoamerica. These results confirm the hypothesis that the DNA barcoding technique may at least differentiate stingless bee taxa accepted by current taxonomy.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Stingless bees (tribe Meliponini, Michener 2007) are eusocial insects with an absent functional sting (Wille 1983) that are distributed in tropical regions with the highest concentration and diversity in the Amazon basin in South America. These bees have great importance as extensive pollinators in Neotropical ecosystems (Roubik 1989; Michener 2007; Freitas et al. 2009) and as an alternative to the domestic honey bee Apis mellifera Linnaeus for agricultural pollination purposes (Slaa et al. 2006; Quezada-Euán 2009). In contrast to A. mellifera, stingless bees have several advantages: they are less harmful to humans and domesticated animals, and are also effective pollinators in glasshouses (Kakutani et al. 1993; Heard 1999; Del Sarto et al. 2005). Despite this, they also have disadvantages, such as a poor level of domestication technologies and the low growth rate compared with A. mellifera (Quezada-Euán 2005).
The genus Scaptotrigona is composed of 24 species distributed from Mexico to Argentina (Michener 2007). Among them, only three Scaptotrigona species are currently reported for Mexico (Ayala 1999): Scaptotrigona hellwegeri Friese, Scaptotrigona mexicana Guérin, and Scaptotrigona pectoralis Dalla Torre (Ayala 1999). S. hellwegeri is endemic to Mexico across the Pacific coast between Oaxaca and Sinaloa, from the sea level to 1,500 m. S. mexicana is distributed from Chiapas to Tamaulipas across the Mexican Gulf coast from the sea level to 1,000 m. S. pectoralis is distributed throughout southeast Mexico in Chiapas, the Yucatan peninsula, and in the Gulf coast to Veracruz up to 1,200 m (Ayala 1999). The distribution of the latter two species continues through Guatemala. Both S. pectoralis and S. mexicana have a high degree of efficiency in pollinating crops such as avocado (Ish-Am et al. 1999). On the other hand, S. mexicana is one of the two stingless bees traditionally exploited for honey production in Mesoamerica and its use in stingless beekeeping is now increasing (Albores-González et al. 2011).
Due to the extensive deforestation of many regions, urban areas have become an alternative microhabitat for these bees. In fact, they have been encountered in wall cavities, although these bees usually nest in hollow trunks. The decline of the stingless bees' habitats is mainly due to anthropogenic factors such as habitat fragmentation and loss, parasites and pathogens, massive pesticide use, or invasive and emergent species like A. mellifera (Stout and Morales 2009). Managed colonies are also subject to translocation among different regions as stingless beekeeping gains interest across the country (Quezada-Euán 2005). The effect of such practices on the diversity of stingless bee species is unknown but could be potentially detrimental (Quezada-Euán et al. 2012). Given these threats, a rapid and accurate method for the identification of the bee species and their diversity is required to conserve native bee fauna (Gotelli 2004).
Barcoding is a useful technique for characterizing described and unknown biodiversity (Hebert et al. 2003). In animals, this method is based on the sequence data of a segment of 658 base pairs of the mitochondrial gene cytochrome oxidase I or cox1, and has been used as an accurate way to assess global diversity (Waugh 2007). In relation to bees, DNA barcoding has proven to be an essential tool in delimiting morphologically non-distinguishable species (Rehan and Sheffield 2011), to group individuals by sex in dimorphic species (Packer et al. 2008), to associate castes in species with a high sexual dimorphism between queens and workers, and to detect cryptic species (Sheffield et al. 2009). Focusing on stingless bees (tribe Meliponini), DNA barcoding has highlighted the existence of isolated reproductive units in S. hellwegeri (Quezada-Euán et al. 2012) as well as in other Melipona species such as Melipona yucatanica Camargo, Moure and Roubik (May-Itzá et al. 2010) and Melipona beecheii Bennett (May-Itzá et al. 2012).
The aim of this study was to evaluate the DNA barcode technique in order to identify Mexican Scaptotrigona species and assign individuals classified by morphology (including non-classified individuals) to the barcode-defined species. The intra- and interspecific genetic variation within S. mexicana, S. hellwegeri, and S. pectoralis were also described to investigate the possible existence of cryptic species (Silveira et al. 2002) as suggested by Quezada-Euán et al. (2012) in S. hellwegeri. The opening hypotheses were that (1) DNA barcoding would at least differentiate stingless bee taxa accepted by current taxonomy, and that (2) according to classical taxonomy (Ayala 1999), S. mexicana would be evolutionarily closer to S. hellwegeri than to S. pectoralis.
2 Materials and methods
2.1 Samples
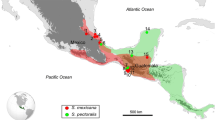
As part of an ongoing study of Mesoamerican stingless bee diversity, 88 Scaptotrigona colonies (38 of them identified as S. mexicana, 33 as S. pectoralis, 14 as S. hellwegeri, and 3 as Scaptotrigona sp) were sampled in different locations throughout their distribution range (Figure 1), from both managed and feral colonies (Table I). Each sample consisted of three to five worker bees collected from each colony (one to ten colonies per site) and preserved in absolute ethanol at −20 °C. Given the maternal inheritance of the mitochondrial DNA molecule, all of the individuals within each colony (workers and drones) share the same queen molecule; therefore, just one worker bee per colony was used to characterize the whole colony.

Location of the sampled Mesoamerican Scaptotrigona colonies. S. mexicana is marked with circles; S. hellwegeri is marked with squares, while S. pectoralis is outlined with triangles. Letters correspond to the locations in Table I.
2.2 DNA extraction and PCR amplification of the barcoding region
Genomic DNA was extracted using a non-destructive protocol, from two right legs dissected from each individual with the DNeasy tissue kit (QIAGEN) following the manufacturer's instructions. Total dilution volume was 100 μl. Vouchers from each colony preserved in ethanol were deposited in the stingless bee collection at the Zoology Laboratory at the Veterinary Faculty (University of Murcia, Spain).
Primers used for the amplification of the cox1 region were LepF (5′-ATTCAACCAATCATAAAGATATTGG-3′) and LepR (5′-TAAACTTCTGGATGTCCAAAAAATCA-3′) (Sheffield et al. 2009). PCR reactions were carried out in 12.5 μl volume with PureTaq TM Ready-To-Go TM PCR beads (GE Healthcare) in a PTC-200 Thermal Cycler (Biorad). PCR conditions involved an initial denaturation at 96 °C for 2 min, then 35 cycles of 96 °C for 30 s, 50 °C for 30 s, 72 °C for 1 min and a final extension of 72 °C for 10 min. Amplified PCR products were electrophoresed in 1.5 % agarose gels and then purified with isopropanol and ammonium acetate. Sequencing was performed in both directions using the standard protocol for ABI BigDye(r) Terminator v3.1 Cycle sequencing kit (Applied Biosystems).
2.3 Sequence analysis
DNA sequences of the cox1 region were unambiguously aligned with MEGA 4. Low-resolution ends were eliminated to get a final matrix of 629 bp. Sequences were deposited in GenBank. Nucleotide content was calculated with the MEGA 4 program. DNAsp v. 5.0 program (Librado and Rozas 2009) was used to evaluate genetic variability and calculate the number of haplotypes or identical sequences, their diversity, and the nucleotide diversity.
2.4 Species delimitation
Sequences were compared by the neighbor-joining (NJ) method by applying the correction of the model Kimura 2-parameter (K2P) (Kimura 1980) as recommended by the Consortium of Barcode of Life (CBOL, http://www.barcoding.si.edu/protocols.html). The analysis of confidence estimates of the relations in the NJ trees was performed with a bootstrap analysis of 2,000 replications with the program MEGA 4.0.2 (Tamura et al. 2007). The strict tree-based method (Ross et al. 2008) was followed for the identification of unclassified and misidentified individuals. This method assumed that query sequences belonged to a specific species if they were incorporated within a cluster (Pettengill and Maile 2010).
In order to compare the cluster delimitation of the tree-based method, the generalized mixed Yule coalescent (GMYC) method (Pons et al. 2006; Fontaneto et al. 2007) was applied. This method identified genetic clusters as independently evolving entities by using a maximum likelihood approach to optimize the shift in the branching patterns of the gene tree from interspecific branches (Yule model) to intraspecific branches (neutral coalescent). Sequences were collapsed to haplotypes with ALTER (González-Peña et al. 2010) and an ultrametric tree was generated with BEAST v. 1.5.4 (Drummond and Rambaut 2007) using a relaxed lognormal clock model, a GTR + I + α substitution model and a coalescence (constant size) tree (Monaghan et al. 2009). The cluster delimitation analysis was carried out using the R package SPLITS (SPecies Lmits by Threshold Statistics) available at http://r-forge.r-project.org/projects/splits/. Both single and multiple threshold optimizations (Monaghan et al. 2009) were analyzed.
TaxonDNA v. 1.5 (Meier et al. 2006) was used to obtain the distribution frequency of intra- and interspecific genetic variability and to evaluate the adequacy of barcoding in identifying stingless bee individuals at the species level. The proportion of correct matches followed three distance-based identification criteria: Best Match (BM), Best Close Match (BCM), and All Species Barcodes (ASB) as described by Meier et al. (2006). The distance below which 95 % of all intraspecific distances are found was used as the cutoff value.
3 Results
3.1 Nucleotide variation analysis
The final analyzed matrix included 629 positions with 592 conserved, 37 variable, and 35 phylogenetic informative positions. Average nucleotide composition showed an A + T bias (T = 46.4 % and A = 32.2 %, C = 11.3 %). No insertions or deletions were observed that could lead to a disruption of the reading frame in the translation, thereby confirming the absence of pseudogenes or NUMTs (López et al. 1994). In total, 15 haplotypes were found (eight in S. mexicana, three in S. hellwegeri, and four in S. pectoralis) with an overall haplotype diversity of 0.907 and nucleotide diversity of 0.019 (Table II). The sequences have been submitted to GenBank under the accession numbers JQ783136-JQ783157.
3.2 Species delimitation
The NJ tree allowed the identification of three undetermined individuals that corresponded to S. hellwegeri, and another three individuals previously misidentified by morphometry were reclassified. In the end, two individuals initially identified as S. mexicana were molecularly assigned one to S. hellwegeri and a second one to S. pectoralis. Likewise, an individual identified as S. pectoralis was molecularly assigned to the clade formed by S. mexicana individuals (Figure 2). These results were confirmed by observation of some diagnostic morphological characteristics such as the color of the tergites after sequence analyses. All of the analyzed sequences were properly assigned to their respective clade corresponding to one of the three species following the strict tree-based method (Ross et al. 2008).

Neighbor-joining tree for Mesoamerican Scaptotrigona individuals using Kimura-2-parameters distance. Bootstrap values (2,000 replicates) are shown above each branch. Black circles indicate the clades corresponding to each species, arrows indicate individuals assigned through DNA barcoding to its corresponding species and those with an asterisk correspond to misidentified individuals.
Genetic K2P distance analyses showed that every species formed a monophyletic group in the NJ tree (Figure 2). The three species formed well-supported clades with high bootstrap values: 99 (S. hellwegeri), 82 (S. mexicana), and 100 (S. pectoralis). S. hellwegeri and S. mexicana formed a supported clade (89). Furthermore, at least two clades were observed within each species, each of them corresponding to populations from separated localities within the distribution area of each species.
GMYC analyses showed that both single (L GMYC = 85.3911) and multiple threshold (L GMYC = 85.3911) models had a higher likelihood than the null model (L 0 = 83.3653). Both analyses yielded three clusters that agreed with the morphological species. It is noteworthy that the second single threshold model recovered with a higher likelihood two clusters within S. mexicana.
The intraspecific divergence values for these three species ranged from 0.00 to 1.90 %, whereas the interspecific divergence values ranged from 1.37 to 3.70 %. There was an overlap of the intra- and interspecific divergence values in the analyzed individuals between 1.37 and 1.90 %, corresponding to 12.14 % of the values of divergence (Figure 3). The limit or “cutoff” between intra and interspecific variation was 1.58 % of divergence with 95 % of probability. The mean value of intraspecific divergence in S. hellwegeri was 0.30 % (0.00–0.63 %), in S. pectoralis was 0.5 % (0.00–1.10 %) and in S. mexicana 0.8 % (0.00–1.90 %). S. mexicana individuals from Veracruz showed a higher divergence value than the cutoff when compared to the samples from Chiapas.

Mean intraspecific (a) and interspecific (b) divergence values for Scaptotrigona individuals.
The proportion of correct matches with Best Match, Best Close Match, and All Species Barcodes criteria reached 100 % of the samples.
4 Discussion
These findings on the use of DNA barcoding confirm that this technique provided reliable identification of the Mexican species of the stingless bee genus Scaptotrigona in congruence with the results obtained for other Hymenoptera taxa (Packer et al. 2008; Gibbs 2009; Sheffield et al. 2009). It is also an efficient tool for the unequivocal reassignment of individuals previously defined by current taxonomy. In this sense, the DNA obtained from a single leg allows the re-examination of morphological characteristics after molecular analysis. Thanks to the non-destructive DNA extraction method, the vouchers were perfectly conserved, which allowed the re-examination of undetermined or misidentified individuals.
The ability of DNA barcoding to distinguish among species is supported by low values of intraspecific variation and relatively high levels of interspecific variation (Packer et al. 2008). The results for the genus Scaptotrigona fulfilled this guideline, as the mean percentage of interspecific variation (2.79 %) was four times higher than the intraspecific value (0.70 %). These values are comparable to those obtained in a comprehensive study of bees (Hymenoptera: Apoidea) from Nova Scotia (Sheffield et al. 2009). In that study of 144 bee species, intraspecific cox1 divergences averaged 0.49 %, which was an expected value due to the elevated mitochondrial evolution rates observed in honey bees (Crozier et al. 1989) and other hymenopterans (Hebert et al. 2003). A ten times higher interspecific divergence than intraspecific divergence has been proposed as a criterion for barcode species identification (Hebert et al. 2004), which was not addressed in the present study. However, the proportion of correctly identified individuals under the Best Match, Best Close Match, and All Species Barcodes criteria reached 100 % of the samples, which shows that barcodes are useful in identifying species within this tribe.
The analysis of the cox1 sequence variation was also helpful in revealing species complexes within taxa with close morphologies, such as S. mexicana. In this study, TaxonDNA analysis showed that some intraspecific divergence values within S. mexicana (1.58–1.90 %) exceeded the interspecific cutoff (1.58 %), thus suggesting the existence of cryptic species or genetic lineages within this species. These values were found when comparing individuals sampled in Veracruz and Chiapas; these two regions are located in opposite extremes of the species distribution range. This finding is in congruence with genetic distance clustering and the second best solution of the single threshold model. The clustering obtained in the NJ tree also suggested some degree of differentiation within S. hellwegeri and S. pectoralis supported by high bootstrap values (95 and 98, respectively), although both species showed intraspecific divergence values that did not exceed the specific cutoff obtained with TaxonDNA. In a previous study performed with samples of the endemic S. hellwegeri, significant morphometric differences coupled with complementary analysis of microsatellite loci and the cox1 region resulted in a marked differentiation among populations, which is in accordance with the diversity of habitats occupied by these bees (Quezada-Euán et al. 2012). Given these results, they proposed the existence of genetic lineages (possibly resembling cryptic species) within S. hellwegeri. The finding of this hidden diversity requires the implementation of appropriate conservation measures to preserve the populations in their environments and avoid translocations of colonies between distant areas. These criteria should be specifically applied to S. mexicana, a species experiencing increasing management for honey production.
The intraspecific divergence found for S. mexicana and S. hellwegeri might be reflecting unrecognized isolation-by-distance phenomena within morphological species. In addition, it is expected that the genetic intraspecific divergence would also be reflected in phenotypic differences. In other stingless bee species, morphometric analyses have supported the existence of genetic lineages (M. yucatanica, May-Itzá et al. 2010; M. beecheii Francoy et al. 2011, May-Itzá et al. unp. data). The morphometric study of S. mexicana and S. hellwegeri is currently in progress to corroborate this hypothesis. Complementarily, microsatellite analysis of a wider sampling covering the whole distribution range will provide new insights in relation to the extent of the gene flow. This is an important aspect to study given the low dispersal rate in Meliponini due to the reduced dispersion of swarms and short flight distances (Engels and Imperatriz-Fonseca 1990).
Although the main aim of the barcode analysis was to delineate species boundaries, a phylogenetic signal from the cox1 sequence data could be observed. In this sense, the NJ phylogram and the General mixed Yule Coalescent (GMYC) results correctly delineated the three species described with traditional morphological analysis and support the hypothesis stated by Ayala (1999) concerning the close evolutionary affinity of S. mexicana to S. hellwegeri, with S. pectoralis as a more distantly related taxon.
In conclusion, Meliponini represents another tribe for which DNA barcoding is a tool for species identification and that provides new insights into the diversity of this group of bees. The main problem in Mesoamerica related to bee conservation is the scarce studies or registers about biodiversity, richness, and the impact of human activities. At present, describing the genetic diversity of organisms spread throughout important biodiversity hotspots like Mesoamerica plays a pivotal role in the scenario of dramatic bee declines and pollination-dependent systems of both wild and managed bees.
References
Albores-González, M.L., García Guerra, T.G., Durán Olguín, L., Aguliar Ayón, A. (2011) Experiencia de la Unión de Cooperativas Tosepan en el fomento a la cría de las abejas nativas Pitsilnejmej (Scaptotrigona mexicana) in Memorias del VII Seminario Mesaomerciano sobre abejas nativas. pp 95–99
Ayala, R. (1999) Revisión de las abejas sin aguijón de México (Hymenoptera, Apoidea). Folia Entomol. Mex. 106, 1–123. Xalapa, Veracruz México
Crozier, R.H., Crozier, Y.C., Mackinley, A.G. (1989) The CO-I and CO-II region of the honeybee mitochondrial DNA: evidence of variation in insect mitochondrial evolutionary rates. Mol. Biol. Evol. 6, 399–411
Del Sarto, M.C.L., Peruquetti, R.C., Campos, L.A.O. (2005) Evaluation of the neotropical stingless bee Melipona quadrifasciata (Hymenoptera: Apidae) as pollinator of greenhouse tomatoes. J. Econ. Entomol. 98, 260–266
Drummond, A.J., Rambaut, A. (2007) BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol. Biol. 7, 214
Engels, W., Imperatriz-Fonseca, V.L. (1990) Caste development, reproductive strategies and control of fertility in honeybees and stingless bees. In: Engels, W. (ed.) Social insects: an evolutionary approach to castes and reproduction, pp. 166–230. Springer, Berlin
Fontaneto, D., Herniou, E.A., Boschetti, C., Caprioli, M., Melone, G., Ricci, C., Barraclough, T.G. (2007) Independently evolving species in asexual bdelloid rotifers. PLoS Biol. 5, 914–921
Francoy, T.M., Grassi, M.L., Imperatriz-Fonseca, V.L., May-Itzá, W.D.J., Quezada-Euán, J.J.G. (2011) Geometric morphometrics of the wing as a tool for assigning genetic lineages and geographic origin to Melipona beecheii (Hymenoptera: Meliponini). Apidologie 44, 499–507
Freitas, B.M., Imperatriz-Fonseca, V.L., Medina, L.M., Kleinert, A.M.P., Galetto, L., Nates-Parra, G., Quezada-Euán, J.J.G. (2009) Diversity, threats and conservation of native bees in the Neotropics. Apidologie 40, 332–346
Gibbs, J. (2009) Integrative taxonomy identifies new (and old) species in the Lasioglossum (Dialictus) tegulare (Robertson) species group (Hymenoptera, Halictidae). Zootaxa 2032, 1–38
González-Peña, D., Gómez-Blanco, D., Reboiro-Jato, M., Fdez-Riverola, F., Posada, D. (2010) ALTER: program-oriented format conversion of DNA and protein alignments. Nucleic Acids Res. 38(suppl 2), W14–W18. doi:10.1093/nar/gkq321
Gotelli, N.J. (2004) A taxonomic wish-list for community ecology. Philos. Trans. R. Soc. Lond. [Biol] 359, 585–597
Heard, T.A. (1999) The role of stingless bees in crop pollination. Annu. Rev. Entomol. 44, 183–206
Hebert, P.D.N., Cywinska, A., Ball, S.L., de Waard, J.R. (2003) Biological identifications through DNA barcodes. Proc. R. Soc. Lond. [Biol] 270, 313–322
Hebert, P.D.N., Penton, E.H., Burns, J.M., Janzen, D.H., Hallwachs, W. (2004) Ten species in one, DNA barcoding reveals cryptic species in the neotropical skipper butterfly Astraptes fulgerator. Proc. Natl. Acad. Sci. U. S. A. 101, 14812–14817
Ish-Am, G., Barrientos-Priego, F., Castañeda-Vildozola, A., Gazit, S. (1999) Avocado (Persea americana Mill) pollinators in its region of origin. Rev. Chapingo Ser. Hortic. 5, 137–143
Kakutani, T., Inoue, T., Tezuka, T., Maeta, Y. (1993) Pollination of strawberry by the stingless bee, Trigona minangkabau, and the honey bee, Apis mellifera an experimental study of fertilization efficiency. Res. Popul. Ecol. 35, 95–111
Kimura, M. (1980) A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 15, 111–120
Librado, P., Rozas, J. (2009) DnaSP v5:A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 25, 1451–1452
López, J.V., Yuhki, N., Masuda, R., Modi, W., Obrien, S.J. (1994) Numt, a recent transfer and tandem amplification of mitochondrial DNA to the nuclear genome of the domestic cat. J. Mol. Evol. 39, 174–190
May-Itzá, W. de J., Quezada-Euán, J.J.G., Ayala, R., De la Rúa, P. (2012) Morphometric and genetic analyses reveal two taxonomic units within Melipona beecheii (Hymenoptera: Meliponidae), a Mesoamerican endangered stingless bee. J. Insect Conerv. doi:10.1007/s10841-012-9457-4
May-Itzá, W.D.J., Quezada-Euán, J.J.G., Medina Medina, L.A., Enríquez, E., De la Rúa, P. (2010) Morphometric and genetic differentiation in isolated populations of the endangered Mesoamerican stingless bee Melipona yucatanica (Hymenoptera, Apoidea) suggest the existence of a two species complex. Conserv. Genet. 11, 2079–2084
Meier, R., Kwong, S., Vaidya, G., Ng, P.K.L. (2006) DNA Barcoding and taxonomy in Diptera, a tale of high intraspecific variability and low identification success. Syst. Biol. 55, 715–728
Michener, C.D. (2007) The bees of the world, 2nd edn. The Johns Hopkins University Press, Baltimore
Monaghan, M.T., Wild, R., Elliot, M., Fujisawa, T., Balke, M., Inward, D.J.G., Lees, D.C., Ranaivosolo, R., Eggleton, P., Barraclough, T.G., Vogler, A.P. (2009) Accelerated species inventory on Madagascar using coalescent-based models of species delineation. Syst. Biol. 58, 298–311
Packer, L., Gibbs, J., Sheffield, C., Kevan, P. (2008) Barcoding the Bees of the World. Anais do VIII Encontro sobre Abelhas Ribeirão Preto-SP, Brasil
Pettengill, J.B., Maile, C.N. (2010) An evaluation of candidate plant DNA barcodes and assignment methods in diagnosing 29 species in the genus Agalinis (Orobanchaceae). Am. J. Bot. 97, 1391–1406
Pons, J., Barraclough, T.G., Gomez-Zurita, J., Cardoso, A., Duran, D.P., et al. (2006) Sequence-based species delimitation for the DNA taxonomy of undescribed insects. Syst. Biol. 55, 595–609
Quezada-Euán, J.J.G. (2005) Biología y uso de las abejas sin aguijón de la península de Yucatán, México (Hymenoptera: Meliponini). Ediciones de la Universidad Autónoma de Yucatán. México. pp.112
Quezada-Euán, J.J.G. (2009) Potencial de las abejas nativas en la polinización de cultivos. Acta Biol. Colomb. 14, 169–172
Quezada-Euán, J.J.G., May-Itzá, W.D.J., Rincón, M., De la Rúa, P., Paxton, R.J. (2012) Genetic and phenotypic differentiation in endemic Scaptotrigona hellwegeri (Apidae: Meliponini): implications for the conservation of stingless bee populations in contrasting environments. Insect Conserv. Divers.. doi:10.1111/j.1752-4598.2011.00179.x
Rehan, S.M., Sheffield, C.S. (2011) Morphological and molecular delineation of a new species in the Ceratina dupla species-group (Hymenoptera: Apidae: Xylocopinae) of eastern North America. Zootaxa 2873, 35–50
Ross, H.A., Murugan, S., Sibon Li, W.L. (2008) Testing the reliability of genetic methods of species identification via simulation. Syst. Biol. 57, 216–230
Roubik, D.W. (1989) Ecology and natural history of tropical bees. Cambridge University Press
Sheffield, C.S., Hebert, P.D.N., Kevan, P., Packer, L. (2009) DNA barcoding a regional bee (Hymenoptera, Apoidea) fauna and its potential for ecological studies. Mol. Ecol. Res. 9, 196–207
Silveira, F.A., Melo, G.A.R., Almeida, E.A.B. (2002) Abelhas do Brasil: Sistemática e Identificação. 1ª edição, Belo Horizonte
Slaa, E.J., Sanchez Chaves, L.A., Malagodi-Braga, K.S., Hofstede, F.E. (2006) Stingless bees in applied pollination: practice and perspectives. Apidologie 37, 293–315
Stout, J.C., Morales, C.L. (2009) Ecological impacts of invasive alien species on bees. Apidologie 40, 388–409
Tamura, K., Dudley, J., Nei, M., Kumar, S. (2007) MEGA4, Molecular Evolutionary Genetics Analysis (MEGA) software version 4,0. Mol. Biol. Evol. 24, 1596–1599
Waugh, J. (2007) DNA barcoding in animal species, progress, potential and pitfalls. BioEssays 29, 188–197
Wille, A. (1983) Biology of stingless bees. Annu. Rev. Entomol. 28, 41–64
Acknowledgments
We very much appreciate the helpful comments of Prof. José Serrano and two anonymous reviewers that greatly improved the manuscript. We also acknowledge the help of the stingless beekeepers and Manuel Rincón Rabanales for providing the analyzed samples. This work has been supported by the projects MUTUAL “Mutualismos y abejas en paisajes tropicales: riesgos y rescate para la biodiversidad y la agricultura”, 4293 “Fondo de Cooperación Internacional en Ciencia y Tecnología UE—México” (FONCICYT), and “Conservación de las abejas sin aguijón de México (Hymenoptera: Meliponini): identificación de especies crípticas e indicadores de diversidad genética” (SEP CONACYT 103341-Z).
Author information
Authors and Affiliations
Corresponding author
Additional information
Manuscript editor: James Nieh
Barcoding des abeilles sans aiguillon: diversité génétique du genre Scaptotrigona, d'importance économique en Amérique centrale.
abeille sans aiguillon / Scaptotrigona / barcoding / espèce cryptique / Amérique centrale
Barcoding von Stachellosen Bienen: Genetische Diversität von Bienen der wirtschaftlich bedeutsamen Gattung Scaptotrigona in Mittelamerika
stachellose bienen / Scaptotrigona / barcoding / kryptische Arten / Mittelamerika
Rights and permissions
About this article
Cite this article
Hurtado-Burillo, M., Ruiz, C., de Jesús May-Itzá, W. et al. Barcoding stingless bees: genetic diversity of the economically important genus Scaptotrigona in Mesoamerica. Apidologie 44, 1–10 (2013). https://doi.org/10.1007/s13592-012-0146-9
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13592-012-0146-9