
Abstract
Background
Breast cancer (BC) is a highly prevalent solid cancer with a high-rise infiltration of immune cells, turning it into a significant candidate for tumor-specific immunotherapies. Chimeric antigen receptor (CAR)-T cells are emerging as immunotherapeutic tools with genetically engineered receptors to efficiently recognize and attack tumor cells that express specific target antigens. Technological advancements in CAR design have provided five generations of CAR-T cells applicable to a wide range of cancer patients while boosting CAR-T cell therapy safety. However, CAR-T cell therapy is ineffective against breast cancer because of the loss of specified antigens, the immunosuppressive nature of the tumor and CAR-T cell-induced toxicities. Next-generation CAR-T cells actively pass through the tumor vascular barriers, persist for extended periods and disrupt the tumor microenvironment (TME) to block immune escape.
Conclusion
CAR-T cell therapy embodies advanced immunotherapy for BC, but further pre-clinical and clinical assessments are recommended to achieve maximized efficiency and safety.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
Breast cancer (BC) is the most common female malignancy worldwide that even exceeds lung cancer as the leading cause of cancer, with an estimated 2.3 million new cases each year, accounting for 11.7% of all cancers. Around 25% of newly diagnosed and 15% of cancer death cases in the female population are attributed to BC [1]. BC is a complex disease encompassing four subtypes: Luminal A (ER+/PR+/HER2−, grade 1/2), Luminal B (ER+/PR+/HER2− grade 3, ER+/PR+/HER2+), HER2 overexpressed (ER−/PR−/HER2+) and triple-negative (TNBC, ER−/PR−/HER2−) BC [2]. Non-specific therapies such as surgery, radiotherapy, endocrine therapy, chemotherapy, and even herbal remedies do not fulfill expectations of a better tumor prognosis, decreased metastasis and/or beneficial overall and disease-free survival rates [3]. Thus, precise tumor-targeting strategies with low toxicity and adverse effects are urgently needed [4].
HER2+ and TNBC subtypes of BC exhibit various mutations, molecular heterogeneity and genetic instability. Also, immune checkpoint overexpression and tumor-infiltrated lymphocytes (TILs) enhance tumor immunogenicity. Lifestyle-related factors such as obesity, diet, pregnancy, lactation and anti-cancer therapies also affect BC immune cell infiltration and immunological responses [5]. Immunogenic tumors may define responsive candidates to specific immunotherapy treatments. The common immunotherapeutic strategies for targeting BC are shown in Fig. 1.

Immunotherapeutic strategies for targeting breast cancer. Monoclonal antibodies target surface antigens and disrupt the following signaling pathways relevant to cancer cell survival. Antibody–drug conjugates are monoclonal antibodies combined with cytotoxic anti-tumor agents to deliver tumor-specific drugs and reduce systemic toxicity. Tyrosine kinase inhibitors exhibit structural homology to ATP, competing for binding to the intracellular domains of the HER family to prevent tyrosine kinase phosphorylation (activation) and to block downstream signaling cascades. Cancer vaccines, whether administered as tumor-derived antigens or cell and virus-based, activate host T cells and provoke immune responses against tumor cells. Immune checkpoint inhibitors block ligand-receptor immunomodulatory interactions between T cells and tumor cells to prevent cancer immune escape. Adoptive cell therapy includes stimulating and activating innate and adaptive immune cells against target antigens and genetically engineering immune cells to express specialized receptors to overcome tumor progression
Adoptive cell therapy (ACT) strategies represent a step forward in the design of efficient immunotherapeutics by which immune cells get artificially manipulated to overcome BC progression. Cytokine-induced killer (CIK) cells are adopted from peripheral mononuclear cells, expanded in vitro and stimulated with IFN-γ and IL-2 cytokines. CIK cells acquire phenotypic and functional characteristics of both natural killer (NK) and T cells. The combined therapy of CIK cells and cetuximab remarkably impeded the growth of patient- and cell line-derived TNBC xenografts and tumors [6]. In a clinical trial, T cells were collected from peripheral blood activated with anti-CD3, grew in IL-2, and armored with bispecific anti-CD3 and anti- HER2 antibodies. Armored activated T cells stimulated anti-tumor responses and that IL-2 production was associated with stable disease in 59.1% of metastatic BC patients, which enhanced the overall survival rate [7]. In the latter case, isolated T cells were genetically modified by cloning T-cell receptors (TCRs) or engineered CARs to obtain a specialized generation of anti-tumor T cells with more potent effects than the former.
To evaluate the anti-tumor effect of TCR-engineered T cells, TCRα- and β-chains were transduced into CD8+ T lymphocytes to allow placenta-specific 1 (PLAC1) recognition in BC cells. Upon co-culture with BC cells, PLAC1-specific TCR T cells produced IFN-γ and TNFα, indicative of TCR activation and delayed tumor progression [8]. γδ TCRs derived from TILs residing in BC showed potent anti-tumor activity, independent of additional costimulatory signals [9]. However, tumor cells may downregulate MHC molecules, resulting in abortive MHC-based immunotherapy. Conversely, TCR-modified T cells may misrecognize tumor antigen-activated host T cells as tumor cells, thereby acting against and destroying them through an MHC-dependent pathway. These obstacles stimulated researchers to manufacture engineered CAR-T cell populations working in a non-MHC-restricted manner.
2 Evolution of general CAR constructs
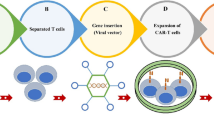
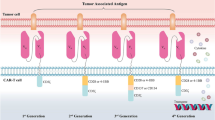
The first successful clinical result of CAR-T cancer therapy was released by the National Cancer Institute in 2010 when post-chemotherapy autologous CD19 CAR-T cells were re-infused into a case of advanced follicular lymphoma. This treatment was associated with long-term depletion of CD19+ lymphoma cells and repopulation of the non-cancerous B lineage [10]. Due to CD19 expression on tumor cells originating from the B lineage, CD19 CAR constructs were subsequently employed to elicit anti-tumor responses in various B cell cancers, ranging from B-acute lymphoblastic leukemias (B-ALL) to B-lymphomas [11]. The main parts of a CAR construct include an extracellular antigen-binding domain and a hinge region, a transmembrane linker domain, and an intracellular signaling domain. The antigen-binding domain comprises a single-chain variable fragment (scFv) from monoclonal antibodies that arrange as VH-linker-VL or VL-linker-VH [12]. This fragment recognizes a broad spectrum of protein, glycoprotein and glycolipid membrane antigens on cancer cells and directly binds to them. Thus, these antigens are not processed by antigen-presenting cells (APCs) and are not exposed to T cells in an MHC-dependent manner. The antigen-binding part and other CAR elements are cloned into plasmids, separately or together, and transfected into target T cells using retroviral or lentiviral vectors. By doing so, genetically engineered T cells (CAR-T cells) acquire the unique potential of antigen-specific recognition [13]. The hinge (stalk) represents an extracellular moiety with variable length obtained from CH2-CH3 (Fc) domains of the constant region of immunoglobulins G (IgG) or spacer sequences of CD4 or CD8. The hinge region confers CAR flexibility and proper location of scFv for efficient ligand binding and links scFv to the transmembrane part. It provides a practical function to CAR marked by upgraded cytokine production, anti-tumor immune responses and cytotoxicity [12, 14]. The transmembrane (TM) domain mediates the connection of extra and intracellular parts and transduces signals from CD3z, CD4, CD7, CD8, CD28, FcεRIγ and H2-Kb, each with different effects on the surface expression of CARs [15]. The intracellular signaling domains are the most prominent constituents reflecting engineered T cell survival, proliferation and immunoreactivity, making new terms for "four generations" of CAR-T cells (Fig. 2). In first-generation CARs, the intracellular domain is derived from the CD3z domain of TCR, composed of three immunoreceptor tyrosine-based activation motifs (ITAMs), with the potential to initiate adaptive immune signal transduction independent of other CD3 domains. First-generation CAR-T cells targeted NKG2D ligands in TNBC cells and induced tumor cytotoxicity and IFN-γ production. However, antigen-specific signals were insufficient to direct T cell proliferation and activation [16]. Full activation of T cells requires two main signs: antigen-binding and co-stimulation. Costimulatory molecules expressed by T cells interact with counterparts on the surface of APCs to elicit T cell immunity and to prevent T cell anergy and immunotolerance. Therefore, costimulatory endodomains from CD27, CD28 family (CD28, inducible costimulator (ICOS, CD278)), 4-1BB (CD134) and OX40 (CD137) were added to second-generation CARs to obtain long-lasting immune activities [17]. CD28 co-stimulation yielded a higher rate of effector T cell activation, regulatory T cell (Treg) suppression and cytokine (IL-2, IFNγ, and TNFα) secretion. At the same time, 4-1BB supports short-term T cell toxicity and IFN-γ production, and long-term in vivo T cell persistence [18]. Third-generation CARs were manufactured by fusing two costimulatory domains of CD28 with either 4-1BB or OX40. Dual co-stimulation prolonged CAR-T cell intratumor persistence and upgraded pro-inflammatory and cytokine release [19, 20]. Fourth-generation CAR-T cells own the most sophisticated structure, with additional costimulatory domains or transgenes producing soluble anti-tumor factors. Fourth-generation anti-folate receptor alpha antigen (FRα) CAR-T cells containing CD28, 4-1BB and CD27 costimulatory domains showed effective cytolytic activity against BC cells and in vitro spheroids [21]. A category of fourth-generation CAR-T cells called T cells redirected for universal cytokine-mediated killing (TRUCKs) possesses a framework similar to that of second-generation CARs accompanied by inducible transgenic cytokines such as IL-2, IL-7, IL-12, IL-15 and IL-18, chemokines, or suicide genes to robust CAR-T cell therapy safety and efficacy for solid tumors. The rationale for designing fourth-generation CARs is that effective T cell responses can be achieved by the co-existence of three main signals of antigen recognition, co-stimulation and cytokine production [22].

Classical CAR-T cells. First-generation CAR-T cells express a single-chain variable fragment (scFv) derived from a monoclonal antibody to recognize a tumor antigen, a hinge domain, a transmembrane domain and the signal transduction domain. Second and third-generation CARs contain one and two extra costimulatory domains to prompt full T cell activation. Fourth-generation CARs resemble second-generation CARs, carrying additive costimulatory domains or cytokine transgenes to elicit robust anti-tumor responses
3 Next-generation CAR-T cells: improved fighters for universal cancer immunotherapy
Upon introducing CAR-T cells as a breakthrough in cancer treatment, efforts have focused on designing universal CARs applicable to many cancer patients (Fig. 3). Generating patient-derived CAR-T cells is advantageous for preventing immune reactions such as graft-versus-host disease (GVHD). Still, it requires a large population of T cells that might get lost in cancer patients who undergo severe radiotherapy and chemotherapy interventions. Moreover, CAR-T cell production is time-consuming, a negative point for patients with limited time to make therapeutic decisions. Accordingly, to dispel the limitations of autologous sources, "off-the-shelf" CAR-T cells from allogeneic donors have been a development focus. Gene-modifying technologies like zinc finger nucleases and CRISPR/Cas9 have been used to target TCR and beta-2 microglobulin (B2M) and avert HLA class I (HLA-I) and PD-1 expression in allogeneic CAR-T cells, thus decreasing alloreactivity and GVHD complications [23, 24]. The efficacy of anti-mesothelin CAR-T cells with CRISPR-Cas9-mediated knockout of TCRαβ and PD-1 is currently being evaluated in patients with mesothelin-expressing solid tumors (NCT03545815).

Next-generation CAR-T cells. (A) Off-the-shelf universal CAR-T cells are allogenic sources of T cells that undergo genetic manipulation to prevent alloreactivity and GVHD. (B) Split, universal and programmable (SUPRA) CARs harbor an intermediate extracellular leucine zipper domain between the extracellular and transmembrane domains to add various antigen recognition domains for a broad range of anti-tumor effects. (C) Anti-αvβ6 integrin CAR-T cells are constructed by co-expressing αvβ6-binding CARs and a 4αβ chimeric cytokine receptor comprising an ectodomain of IL-4 receptor α, fused to transmembrane and endodomain regions of IL-2/IL-15 receptor β. (D) Anti-FITC CARs are manufactured by adding an intermediate fluorescein-labeled bispecific adapter that links to tumor antigens and anti-FITC CAR-T cells. (E) Tandem CAR-T cells harbor two individual extracellular antigen recognition domains for simultaneously detecting two antigens and eliciting specific responses. (F) Armored CAR-T cells have an extra transgene for expressing cytokines, chemokines and immune checkpoints targeting tumor immunosuppressive elements. (G) Conditional CAR-T cells use tumor niche conditions to activate responses in a spatially controllable manner. Oxygen-responsive CAR-T cells equipped with a hypoxia detection domain get activated in response to a hypoxic tumor niche, followed by inactivation upon removal of the hypoxic signal. (H) Switchable CAR-T cells are constructed by incorporating FITC or grafting a peptide neo-epitope (PNE) into the antigen-binding domain at specific sites as switch molecules. (I) Self-destructing CAR-T cells manufactured by co-transducing suicide genes and specific biomolecules govern their expression to provide controllable CAR-T cell destruction
Split, universal and programmable (SUPRA) CAR systems are engineered by adding an intermediate extracellular leucine zipper domain between the antigen recognition and transmembrane domains. This manipulation has provided CAR-T cells with multiple simultaneous features for targeting various antigens in a switchable specific mode with fine-tuned control over CAR-T cell function against leukemia and BC cells [25]. Several strategies are applied in CAR design to strengthen the tumor infiltration, persistence and anti-tumor responses of engineered T cells, as well as better safety to minimize possible consequences. Incorporating signaling elements upstream to pathways relevant to CAR-T cell expansion and anti-tumor immunity ensures higher success in CAR-T cell therapy. Anti-αvβ6 integrin CARs have been designed by transducing a 4αβ chimeric cytokine receptor comprising an ectodomain of IL-4 receptor α, fused to transmembrane and endodomain regions of IL-2/IL-15 receptor β. Binding IL-4 to 4αβ chimeric cytokine receptors induces a potent and selective growth signal in 4αβ + T cells, associated with robust anti-tumor immunity in TNBC cells and breast tumor xenografts [26].
Multiple antigen-targeting CAR-T cells potentiate the targeting of patient-to-patient variations in tumor-specific antigens and enhance the curative potential of CAR-T cells for drug-resistance solid tumors [27]. An extracellular biotin-binding immune receptor (BBIR), comprising a dimeric avidin linker, enables the binding of various biotinylated antigen-specific domains such as full-length antibodies and scFV segments to assemble universal CAR-T cells directed against lymphoma cells [28]. In a similar effort, a bispecific adapter of fluorescein linked to a tumor-specific ligand has been designed (anti-FITC CARs) to recognize multiple antigens and form functional immunologic synapses between CAR-T cells and BC cells, leading to CAR-T cell activation and BC cell killing [29]. Tandem CARs (TanCARs) harbor two individual extracellular antigen recognition domains, connecting a Gly-Ser linker for simultaneous binding to MUC1 or ErbB2 antigens and making a more robust cytolytic response and cytokine secretion against BC cells [30]. The immunosuppressive nature of solid tumors and its prominence in tumor resistance to CAR-T cell therapy entails targeting relevant signaling cues such as immune checkpoints to get better clinical outcomes. Armored CAR-T cells targeting PD-L1 have been found to block PD-1/PD-L1 signaling within the breast tumor milieu and to show more extended cancer cell killing. PD-L1 CAR-T cells stimulated PD-L1 expression on target BC cells with low PD-L1 expression, with a unique self-amplifying effect on effector CAR-T cells [31].
Conditional CAR-T cells with spatially controllable cytolytic activities respond to local conditions exclusive to the tumor mass. An oxygen-responsive multichain CAR construct has been engineered by adding oxygen-sensitive subdomains of HIF1α to the conventional CAR framework (HIF-CAR), acting as an oxygen sensor responding to oxygen variations. Hypoxia actively stimulated CAR expression and later tumor-killing, followed by rapid CAR down switching after removing the hypoxic signal [32]. Switchable CARs were constructed by incorporating FITC or grafting a peptide neo-epitope (PNE) into the anti-Her2 antibody trastuzumab at specific sites as switch molecules. These switch entities vary the distance and orientation between the CAR and tumor antigens and, thus, optimize immunological synapse activities [33].
Self-destructing CARs have been developed by co-transducing suicide genes such as inducible caspase 9 (iC9), truncated EGFR (tEGFR or EGFRt) and herpes simplex virus thymidine kinase (HSV-TK) into the CAR structure. The expression of suicide genes responding to specific biomolecules induces apoptosis in CAR-T cells by programmed destruction of DNA and Fas-mediated apoptotic signals to halt further anti-tumor immune functions [34].
4 CAR- delivery strategies
4.1 Viral vectors
Viral vectors are popular in genetic engineering applications due to their inherent potential to load genetic elements and easy integration within the host genome [35]. Retroviruses, gammaretroviruses and lentiviruses are common CAR vectors, among which the last one is noticed most. Lentivirus (LV) vectors are cost–benefit gene delivery vehicles with simple manufacturing procedures and reduced risk of malignant transformation and random integration. Moreover, LV vectors are well arranged to transduce many dividing and non-dividing, slowly growing, and difficult-transducable cells such as hematopoietic progenitors and lymphoid cells [36].
LV vectors developed from the first to the third generation, lacking unnecessary viral components, and the remaining main elements were broken into parts, each carried by different plasmids. Delivery of viral components in separate plasmids permits LV vector self-inactivation and improved biosafety [35]. The cloning of EGFR-targeted CARs into the LV vector pCDH-CMV yielded approximately 30% transfection efficiency and a 40-fold increased CAR expression. EGFR-targeted CAR-T cells showed strong cytotoxic effects against TNBC cells in vitro and in an in vivo xenograft mouse model associated with the expansion of naïve‐associated CAR‐T cells [37]. The clinical feasibility, safety and efficacy of lentiviral-transduced anti-mesothelin CAR-T cell therapies have been investigated in some solid tumors [38]. Integrative viral vectors are advantageous for prolonging CAR transgene expression. Yet, they are somewhat expensive, limit the size of transduced DNA and may integrate transgenes at cancer-related sites, causing insertional mutagenesis. Therefore, several non-viral vectors and delivery vehicles have been investigated as proper alternatives.
4.2 Transposon vectors
Transposons are mobile elements of genetic material that can move through the genome thanks to the cut-and-paste features of transposase. Transposon-based systems present safer and more cost-effective non-viral headways of CAR transduction, which may translate into large-scale settings. The Sleeping Beauty (SB) transposon is a fascinating CAR gene transduction system, and clinical observations have illustrated post-infusion CAR-T cell proliferation and satisfactory quality control outcomes [39]. SB11 and SB100X are advanced versions of the primary SB system with more effective transposition rates. SB systems exhibit superior integration profiles accompanied by minimized epigenetic mutations in the host genome, but they hold low rates of gene transfer and nearly random integration profiles [40]. SB-generated CAR-T cells specific for anti-human endogenous retrovirus-K (HERV-K) envelope (env) protein significantly decreased BC cell growth and increased IFNγ, TNF-α and Granzyme B release in vitro. Anti-HERV-K CAR-T cells repressed tumor growth, burden and metastasis in BC xenograft mouse models, mechanistically controlled by p53, MDM2 and ERK signaling regulation [41].
PiggyBac (PB) transposons possess an integration profile resembling LV vectors but non-random by inserting transgenes at TTAA sequences. In addition, PB transposons carry more significant segments of transgenes (≥ 200 kb) than SB and TOL2 counterparts, turning them into favorable candidates to deliver multiple cargos [40]. PB transposon-based gene transduction has been found to avoid the pre-activation of T cells and to preserve their memory phenotype without early T cell exhaustion. PB-transduced anti-HER2 CAR-T cells have shown potent and sustained cytolytic activity against invasive BC cells with low HER-2 expression and HER2 monoclonal antibody-based therapy resistance [42]. Despite various advantages in applying transposon-based systems, constructing transposon vectors is relatively sophisticated and time-consuming and necessities co-transfection with two or more plasmids and other purification processes.
4.3 Minicircle DNA vectors
Due to the obstacles mentioned above with transposon systems to generate CARs, non-viral and non-integrative alternatives such as minicircle DNA (mcDNA) have been proposed. mcDNA vectors are double-stranded, circular, supercoiled DNA molecules derived from bacterial plasmid DNA. Comparatively, mcDNA vectors lack some antibiotic resistance and immunogenic sequences, and the bacterial backbone lacks sequences that silence transgene transcription. Short sequence mcDNA vectors enhance the time and level of gene expression. Also, because of its non-integrating nature, mcDNA reduces the possibility of insertional mutagenesis [43]. Many CARs engineered against BC cells have been successfully transfected into T lymphocytes by DNA vector platforms based on scaffold/matrix attachment region (S/MAR) motifs. In xenograft mouse models, CAR-T cells showed long-term persistence and function in cytokine release, tumor infiltration and anti-tumor immunity [44]. Non-bacterial mcDNA represents a next-generation, rapidly established system without the risk of bacterial contamination. However, non-integrative CAR transgene manifests shorter maintenance of CAR-T cells in vitro and in vivo, requiring multiple rounds of infusion [45].
4.4 Nanoparticle delivery systems
Nanoparticles (NPs) link nanotechnology with other medical and biological fields of science, imparting stable nano-scale techniques to deliver a varied spectrum of cargos, including CAR-T cells [46]. Lipid NPs (LNPs) are easily internalized in target cells via membrane-mediated endocytosis and harbor an ionizable core that remains stable at physiological pH while getting charged under acidic situations, like in endosomes. Both mechanisms allow LNPs to escape endosomal degradation and are suitable for delivering fast-degrading elements such as RNA. Furthermore, the easy change in LNP composition beneficially improves cellular uptake and delivery efficacy [47]. LNPs have been demonstrated to successfully deliver anti-CD19-CAR mRNA into primary human T cells with minimal toxicity compared to electroporation [48].
The delivery of PB-transfected EGFRvIII CARs with reduction-sensitive polymer nanomicelles permits cellular uptake beyond endocytic pathways and gives site-specific DNA release. The packaging system provides efficient transfection (30%), continuous CAR expression, and T cell memory formation [49]. In vitro transcribed mRNA (IVT-mRNA) generates temporary CAR expression to avoid severe therapeutic side effects arising from CAR overexpression. Due to time-limited expression, structural instability and rapid degradation, IVT- mRNA CAR-T cells unveil poor tumor infiltration [50].
5 Antigen targets
Selecting appropriate tumor-determined antigens is critical to bringing about desired clinical outcomes on CAR-T cell therapy with minimum adverse side effects. Immunotherapy-targeted antigens classify into various types: 1) tumor-specific antigens (TSAs) that are exclusively expressed in tumor cells, 2) tumor-associated antigens (TAAs)/tumor-selective antigens that overexpress in tumor cells, and are also narrowly distributed in normal cells, 3) oncofetal antigens that are expressed during embryogenesis but not in adult tissues, and may be detected in adult tumor cells [51].
Hepatocyte growth factor receptor, or c- mesenchymal-epithelial transition factor (c-Met), is a surface protein tyrosine kinase that primarily functions during embryonic organ development and differentiation [52]. It also acts as a protooncogene to control cancer cell motility, propagation and invasion, linked with a reduced survival rate of 47% in invasive ductal breast carcinoma [53]. In a clinical trial conducted in metastatic BC cases, intratumor injection of mRNA-transfected anti-c-Met CAR-T cells yielded inflammatory responses within tumors, loss of c-Met expression and tumor necrosis [54].
Human epidermal growth factor receptor 1 (HER1)/EGFR belongs to the HER family of receptor tyrosine kinases with various ligands, each governing signaling cascades such as ERK1/2-MAPK, PKC, PI3K/Akt and Jak/STAT, regulating cell survival and proliferation [55]. Abnormal HER1 expression in several epithelial cancers indicates a poor prognosis, therapy resistance, enhanced tumor inflammation and a weak immune response due to its down-regulatory effect on MHC-1 and its stimulation of PD-L1 expression [56].
Anti-tumor activity of anti-HER1-CAR-T cells was found to be coupled with cell lysis, tumor shrinkage and higher levels of IFN-γ, IL-2 and IL-4 in both TNBC cells and cell line-derived and xenograft breast tumor models [57]. However, Xia et al. reported that approximately 30% of TNBC xenograft tumors resisted third-generation anti-EGFR CAR-T cell regimens. Transcriptomic analysis of resistant tumors revealed the activation of a large set of immune suppression-associated genes, possibly activated by CAR-T cell-released IFNγ. CAR-T cell-mediated upregulation of immunosuppressive genes was sensitive to THZ1, a CDK7 inhibitor, which inhibited tumor immune resistance [58].
Human epidermal growth factor receptor 2 (HER2) is a member of the HER family showing overexpression in breast, pancreatic, colon and lung cancers and has been found to be related to cancer progression, metastasis, recurrence and reduced patient survival [59]. HER2 serves as a potent target for solid tumor immunotherapy. However, CAR-T cell functionality assessments remain restricted to pre-clinical reports in BC. CAR-T cells are powered to penetrate HER2+ breast tumor spheroids and extracellular matrix to reach cancer cells located out of access to monoclonal antibodies. CAR-T cells whose antigen-binding domains are derived from trastuzumab stimulate active cell lysis, tumor regression, and IL-2 and IFN-γ production in trastuzumab-resistant BC xenografts [60, 61].
Mesothelin (meso) is a 40 kDa glycosyl-phosphatidyl inositol (GPI)-linked glycoprotein expressed to a limited extent in mesothelial cells, pleura, pericardium, tunica vaginalis, ovaries, fallopian tube and rete testis. Abnormal meso overexpression has been observed in mesothelioma, pancreatic and ovarian cancer and TNBC [62]. Negligible expression in healthy tissues and immunogenicity make meso promising for BC monoclonal antibody, antibody–drug conjugate, vaccine and CAR-T cell therapies [63, 64]. Meso mediates cancer cell viability, propagation, migration, invasion and therapy resistance via activating Wnt/NF-κB/PI3K/Akt signaling pathways [65].
Anti-meso CAR-T cells infiltrated into TNBC xenograft models and released IL-6, IL-12 and IFN-γ, producing repressed tumor masses and liver metastases [66]. CARs designed against different epitopes of meso and those targeting proximal region epitopes evoked a vigorous immune response characterized by higher levels of cell killing, CD107α, IL-2, TNF-α and IFN-γ production in vitro and tumor shrinkage in vivo [67].
Epithelial cell adhesion molecule (EpCAM, CD326) is a 40 kDa transmembrane glycoprotein found in normal epithelial cells and to be overexpressed in breast, prostate, pancreatic and liver cancers, mediating cell adhesion, proliferation, invasion, as well as a poor prognosis and therapy resistance [68, 69]. Pre-treatment with catumaxomab followed by adding activated T cells abolished chemoresistant EpCAM+ TNBC cells via the upregulation of the T cell activating receptor NKG2D [70]. Anti-EpCAM CAR-NK and CAR-T cells have shown increased cytotoxicity and cytokine release (IFN-γ, perforin and granzyme B) against colorectal and ovarian cancer cells, respectively [71, 72]. Anti-EpCAM CAR-T cells have been found to diminish tumor burden and improve host survival in a breast tumor model. However, accumulation of CAR-T cells in the lung and cytokine release may have dose-dependent fatal consequences [73].
L1 cell adhesion molecule (L1CAM, CD171) is a 200–220 kDa transmembrane glycoprotein consisting of six extracellular Ig-like domains and five fibronectin III repeats bound to transmembrane and cytoplasmic compartments [74]. It is typically expressed in the nervous system and developing neurons, hematopoietic and endothelial cells, renal epithelium, intestinal crypts, as well as in relevant malignancies [75]. Clinicopathological observations revealed associations of L1CAM expression with high breast tumor stage, advanced grade, lymph node and vasculature-dependent lung metastases and recurrences [76, 77].
Mucin1 glycoprotein (MUC1) is a transmembrane glycoprotein expressed by most epithelial cells in low amounts but exists remarkably on the surface of epithelial tumors including BC. MUC1 bears an extracellular domain containing a variable number of tandem repeats (VNTR of 20 amino acids) with different glycosylation patterns in normal and cancerous tissues. Due to varied glycosylation, protein residues of tumor MUC1 are not entirely covered by glycans, thus exposed to immune cells. This expression pattern turns MUC1 into a tumor neoantigen for immunotherapy and vaccine design [78]. Anti-MUC1 CAR-T cells have shown strong TNBC cell killing in an antigen dose-dependent manner to produce higher IL-2, IFN-γ, TNF-α and Granzyme B and to repress tumor growth in an orthotopic in vivo model. However, remaining MUC1-expressing tumor cells were evident in long-term follow-up, necessitating further CAR-T cell injections for complete tumor elimination [79].
Carcinoembryonic antigen (CEA) is a 180 kDa GPI-linked membrane glycoprotein expressed during embryonic development and in adult gastrointestinal epithelial cells and has been found to overexpress in gastrointestinal, colorectal, pancreatic, non-small cell lung, and breast cancers [51]. High expression by tumor cells and immunogenicity without autoimmune reactions make CEA a suitable tumor antigen for pre-clinical investigations and clinical trials [80, 81]. Co-treatment of second-generation anti-CEA-CAR-T cells with IL-2 has provided a timely dose-dependent cytotoxic immune response, an enhanced IFN-γ secretion, a retarded tumor growth and a significantly diminished tumor infiltration of FoxP3+Treg cells in murine models of breast and colon cancer [82].
Fibroblast activation protein (FAP) is a transmembrane serine protease generally expressed during embryonic development and pathologically overexpressed during wound healing, fibrosis formation and cancer by stromal fibroblasts of damaged tissues or the tumor matrix. Most epithelial malignancies, including breast, pancreatic, lung and brain cancers, are rich in FAP-reactive fibroblasts, rendering them suitable for immune-targeting coupled with tumor growth suppression and enhanced chemotherapy efficacy [83]. Targeting FAP has been found to destroy the tumor-supportive niche in BC allografts by activating T cells, thereby inducing apoptosis in cancer-associated fibroblasts and reducing type I collagen and CD31 expression [84].
Receptor tyrosine kinase orphan receptor 1 (ROR1) is a Wnt receptor expressed during embryogenesis and in hematological and solid malignancies, including BC. ROR1 mediates crucial signaling pathways of cAMP-response-element-binding protein (CREB) and Wnt to advance EMT, tumor growth and aggression [85]. Several extracellular epitopes make ROR-1 a candidate for immunotherapeutic strategies based on mono- and polyclonal antibodies, TKIs, miRNAs, siRNAs and CAR-T cells [86]. ROR-1-CAR-T cells can spike into 3D tumor models of non-small cell lung cancer and TNBC in vitro, migrate through the tumor matrix, and propagate and actively eliminate high percentages of cancer cells [87]. EpCAM-synthetic Notch receptor CARs enable engineered T cells to specifically quarry ROR1+ tumor cells but not those co-localized with non-malignant counterparts [88].
6 Challenges
6.1 Biomarker identification
Challenges in advance of CAR-T cell therapy for solid tumors such as BC negatively influence its treatment efficacy and unfavorable clinical consequences. CAR-T cells recognize surface antigens independent from MHC pathways, but they fail to identify intracellular tumor antigens presented through the MHC-dependent route. Moreover, most antigens that CAR-T cells target are not exclusive to tumor cells and express in various normal tissues to a lesser extent. Thus, if the antigen of interest is not tumor-specific, the occurrence of severe clinical consequences, which may ultimately lead to death, may be inevitable [89].
One strategy to impart precise T cell-mediated tumor recognition is generating dual receptor CARs that work based on the co-existence of two antigens in tumor cells. For example, a synthetic notch (synNotch) receptor designed for a primary antigen may be embedded into the CAR, activating CAR-T cells against a second antigen [90]. Han et al. developed tumor-specific anti-EGFR CAR-T cells comprising a masking peptide blocking the CAR antigen-binding site and a protease-sensitive linker. Exposure to proteases within the TME cleaved the linker domain and separated the masking peptide, activating CAR-T cells against BC cells [91].
6.2 Vasculature barrier, CAR-T cell trafficking and infiltration
CAR-T cells follow the same route as host T cells to reach the tumor. A successful anti-cancer immunity depends on the extravasation of cytotoxic T cell populations to the tumor site and subsequent passing through the vasculature barrier of the malignant mass (Fig. 4). Pericytes and endothelial cells (ECs) represent first-line physical barriers to infiltrating immune cells, and their unique features enhance tumor invasion and immunomodulation. Tumor ECs are distinguished from typical equivalents by non-uniform and fenestrated membranes, chromosomal instability, gene mutations, and higher glycolytic and proliferative activities [92]. The tumor vasculature consists of multiple layers of ECs laid irregularly on an interrupted basement membrane. Inconstant smooth muscles and pericytes surrounding ECs enable higher fluid flow into the tumor, forming a physical barrier to the penetration of anti-cancer drugs [93]. Leukocytes exclusively attach to inflammation-activated ECs in a multistep process mediated by selectins, integrin ligands and adhesion molecules. Tumor ECs remain unresponsive to activating signals (TNFα and IFNγ), thereby downregulating the expression of chemoattractants (CCL2, CCL18, CXCL10 and CXCL11) and adhesion molecules (ICAM1, VCAM1 and E-selectin), thus preventing immune cell chemotaxis, attachment and trafficking into TME [94]. In contrast, P-selectin expression in BC cells boosted the infiltration of regulatory T cells secreting high levels of IL-4 and IL-10 [95]. ECs express both co-stimulatory and co-inhibitory molecules such that their balance can determine T cell infiltration and intratumoral function. ECs from solid tumors like BC subjected to TME cytokines such as VEGFA, IL10 and prostaglandin E2 (PGE2) outperform infiltrating Treg cells more than cytotoxic ones in a Fas ligand (FasL)-dependent mode. FasL arbitrates an immune tolerance by inducing selective apoptosis in CD8+ effector T cells with a lower c-FLIP expression [96].

Vasculature barrier, CAR-T cell trafficking and infiltration. Tumor blood vessels exhibit an abnormal architecture of multiple irregular layers of endothelial cells laid on an interrupted basement membrane. The breast tumor vasculature prevents cytotoxic T cell infiltration and instigates apoptosis while facilitating Treg trafficking
6.3 CAR-T cell exhaustion
The remaining cytotoxic T cells that overcome the vasculature barrier and infiltrate into the tumor encounter a complex physico/chemical environment that dictates that they lose function. T cell exhaustion or dysfunction occurs following long-term exposure to tumor antigens, inflammation, hypoxia and nutrient deprivation. This process can be identified by the upregulation of T cells' inhibitory receptors such as CTLA-4, PD-1, T cell immunoglobulin and mucin domain 3 (Tim-3). Exhausted T lymphocytes meet depleted cytolytic function, memory formation, shortened cytokines (IL-2, TNF-α, IFN-γ, and granzyme B) and undergo apoptosis [97]. CAR-T cell exhaustion has increasingly been studied to reach appropriate solutions. Trogocytosis followed by CAR-T cell fratricide and tonic signaling caused by the antigen-independent activity of CARs have been proposed as mechanisms that exhaust CAR-T cells in hematological malignancies and some solid tumors [98, 99]. In the case of solid cancers including BC, however, interactions among CAR-T cells and immunosuppressive elements within the TME have been noted (Fig. 5).

The tumor microenvironment (TME). The TME represents a complex collection of resident and infiltrating cells on a supportive solid matrix exposed to various soluble and physicochemical factors
7 Immunosuppressive TME
The TME is a complex niche consisting of cell types, matrix and soluble factors that support tumor progression, metastasis and immune system escape that may influence T cell dysfunction and CAR-T cell therapy inefficacy (Fig. 6). Complex interactions among TME components involving resident and infiltrating cell populations, cytokines and chemokines produced by cells, matrix components (epitope masking), vascular barriers, inflammatory state, oxygen and nutrient tension contribute to an immunosuppressive milieu restricting CAR-T cell trafficking, infiltration, persistence and effector function [100]. The main components of the TME controlling immunosuppressive responses and CAR-T cell interactions will be discussed in more detail below (Fig. 7).

Cellular components of the TME. (A) Cancer cells overexpress (shown in blue) immunomodulatory molecules, which block CAR-T cell infiltration. BC cells downregulate NK cell-activating molecules (shown in red), and tumor cell-released autophagosomes (TRAPs) stimulate regulatory phenotypes in tumor-associated macrophages and cytotoxic T cells. KAT6A and ALDH1A1 overexpression result in the recruitment and expansion of MDSCs. (B) Tumor-associated macrophages (TAMs) secrete matrix metalloproteinase (MMP) and chitinase-3-like-1 (Chi3L1), which upregulate (shown in blue color) signaling pathways affecting BC survival and metastasis and produce chemokines to recruit Treg cells. They release immunosuppressive molecules, suppress NK and cytotoxic T cells, and bring them to arginine-starving conditions (red). (C) Myeloid-derived suppressor cells (MDSCs) enhance the recruitment of large populations of MDSCs and Treg cells and induce nutrient deficiency (shown in red color), inhibition, regulatory phenotype switching and apoptosis in cytotoxic T cells. (D) Cancer-associated fibroblasts (CAFs) accelerate MDSC, TAM and Treg cell recruitment while inhibiting cytotoxic T cell infiltration. Also, CAFs express immune checkpoint molecules and induce a regulatory phenotype in T cells. (E) Mesenchymal stem cells (MSCs) interact with cytotoxic T cells, inhibit their proliferation, and drive them into immunoregulatory phenotypes and IL-10 production. They secret various immunomodulatory molecules to polarize TAMs into the M2 phenotype

ECM components of the TME. (A) Desmoplastic collagen creates a physical barrier hampering CAR-T cell infiltration and availability to cancer cells. Collagen interaction with immune cells leads to T cell dysfunction, regulatory phenotyping and M2 macrophage polarization. (B) Binding CXCL-2 to the fibronectin III domain of tenascin C (TNC) forms a robust adhesive substrate trapping T cells into the matrix. (C) Thrombospondin-1 (TSP1) is an H2S antagonist and T cell inhibitor that restricts endogenous H2S via downregulating cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH). TSP1-CD47 interaction blocks the MEK/ERK and PKA pathways, which prevents T cell activity by inducing a regulatory phenotype and apoptosis. (D) Secreted protein acidic and rich in cysteine (SPARC) overexpression in breast tumors is associated with collagen deposition, cytokine production, MDSC recruitment and Treg cell phenotyping. (E) Versican overexpression correlates with TAM accumulation, CCL2 and TGF-β1 production. Versican-TLR2 interactions induce DCs to secrete IL-6 and IL-10 cytokines that restrict immune cell functions
7.1 Cellular components
7.1.1 Cancer cells
Cancer cells are central residents that orchestrate tumor metabolism and its biological behavior. They express higher immune checkpoint PD-L1 molecules, co-inhibitory elements such as TIGIT ligands (CD112, CD155) and B7-class molecules. BC cells and the TGF-β content of tumor stroma impair NK cell function by downregulating NK-activating receptors (NKp30, NKG2D, DNAM-1 and CD16). On the contrary, they upregulate the expression of inhibitory receptors (such as NKG2A) to intensify self-tolerance and escape from NK cell-induced toxicity [101]. Overexpression of receptor activator of nuclear factor-κB (RANK) in BC cells has been found to restrain the infiltration of cytotoxic T cells while increasing the recruitment of immunosuppressive immune cells [102]. TNBC cells express high levels of Annexin-A1 acting through both endogenous and exogenous paracrine pathways to prompt M2 macrophage phenotype and inflammation induction [103]. BC cells stimulate lysosome-associated membrane protein type 2A (LAMP2a) overexpression in tumor-associated macrophages. LAMP2a activates downstream peroxiredoxin 1, CREB-regulated transcription coactivator 1 and immune-responsive gene 1 protein to sustain immunosuppressive characteristics of breast tumors [104]. Tumor cell-released autophagosomes (TRAPs) impel an immunomodulatory phenotype in CD4+ T cells and polarize CD163+ TAMs by initiating the TLR4-mediated MyD88/p38/STAT3 pathway [105]. Breast tumor-initiating cells exhibit high aldehyde dehydrogenase 1A1 (ALDH1A1) activity, reducing intratumor pH. Acidic conditions activate the NFκB pathway and raise granulocyte–macrophage colony-stimulating factor (GM-CSF) production, leading to the expansion of myeloid-derived suppressor cells (MDSCs) and immunosuppression [106]. Lysine acetyltransferase 6A (KAT6A) overexpression in metastatic TNBC has been linked to the acetylation and activation of SMAD3, a pathway that mediates IL-6, IL-22 and TNFα expression and MDSC recruitment [107]. The application of a positron emission tomography (PET) technology to track the infiltration of CAR-T cells into TNBC models uncovered a different retention pattern inversely correlated with the expression of immunomodulatory molecules. Immune checkpoint elements hamper CAR-T cells from infiltrating the breast tumor. Thus, they exclusively kill cancer cells at the tumor borders, but not the bulk [108].
7.1.2 Tumor-associated macrophages (TAMs)
TAMs predominantly exhibit M2 phenotypes derived from resident intratumor macrophages or infiltrated circulating monocytes induced by IL-4, IL-10 and IL-13 released from type 2 T helper (Th2) cells. Transcriptional analyses showed that TAM transcriptomes in BCs are distinct from TAMs found in other types of cancers or their monocytic precursors. Sialic acid-binding Ig-like lectin 1 (SIGLEC1) and CCL8 upregulated in BC-TAMs, mediating cancer-TAM crosstalk and monocyte recruitment [109]. Interaction of SIGLEC10 with CD24 molecules on BC cells triggers an immune evasion signal through which BC cells can not undergo macrophage-mediated phagocytosis [110]. Macrophage cyclooxygenase-2 (COX-2) induces IL-10 and indoleamine 2,3-dioxygenase (IDO) to inhibit CD8+ cytotoxic T cell growth and interferonγ (IFN-γ) production. TAMs catabolize arginine sources by arginase-1 activity and bring CD8+ T cells into an arginine-deprived state, making them metabolically and functionally disabled. The release of IL-10, TGF-β, PGE2 and prostanoids from TAMs restrains the effector function of cytotoxic T and NK cells and enhances Treg recruitment [111]. TAMs secrete CCL-18, thereby attracting circulating naive CD4+ T cells via CCL-18-PITPNM3 receptor interaction which, in turn, induces regulatory phenotypes [112].
7.1.3 Myeloid-derived suppressor cells (MDSCs)
MDSCs derive from monocytic (Mo-MDSCs) and polymorphonuclear granulocytic (PMN-MDSCs) precursors arising from the abundance of immunosuppressors granulocyte-colony stimulating factor (G-CSF), GM-CSF and IL-6 in BC stroma [113]. Highly frequent CD14+ HLA−DR−/low MDSCs have been found to suppress autologous T cell proliferation in breast tumors via ROS-dependent mechanisms [114]. MDSCs possess less significant functions than TAMs for nutrient-starving and inhibiting effector T cells, Treg development and immunosuppression. They reside in less perfused and highly hypoxic regions in breast tumors and play a more prominent role in generating metastatic and angiogenic niches [115]. Tumor-infiltrating MDSCs secrete chemokines (CCL3, CCL4 and CCL5) to attract infiltrating Treg cells, repress T cell proliferation and the Th1 phenotype, and intensify T cell apoptosis through STAT3 activation [116].
7.1.4 Cancer-associated fibroblasts (CAFs)
CAFs inhabit the tumor stroma and regulate cancer cell proliferation, invasion, inflammation, angiogenesis, immune resistance and ECM remodeling. Paracrine factors like exosomal miR-9 and osteopontin produced by BC cells may transform normal fibroblasts into pro-inflammatory CAFs [117, 118]. Also, epigenetic mediators such as histone deacetylase 6 controlled by the prostaglandin E2/cyclooxygenase-2 /STAT3 axis overexpressed in CAFs, has been found to maintain BC immunosuppressive niches [119]. CAFs release high Chi3L1 contents that activate MAPK and PI3K pathways in BC cells, accompanied by high levels of pro-inflammatory and metastatic factors. Chi3L1 instigates angiogenesis, macrophage recruitment and M2 polarization while suppressing T cell infiltration [120]. Four CAF subsets (CAFS1-S4) have been detected in BC, especially TNBC showing a higher number of CAF-S1 and CAF-S4 subsets. The CAF-S1 population induces immune suppression by amplifying CXCL-12-dependent recruitment of Treg cells and upregulation of PD-1 and CTLA4 protein levels, supporting their persistence through the upregulation of JAM2, PD-L2 and OX40L, and directing the differentiation and survival of CD25HighFOXP3High T lymphocytes [121, 122]. IL6/STAT3 upregulation in CAFs results in adenosine overproduction, inducing CD73+ γδ Treg cells to positively affect IL-6 levels in CAFs via the adenosine/A2BR/p38MAPK pathway [123]. CAFs secrete CXCL16 and develop infiltrated primary monocytes into S100A9+ myeloid-derived suppressor cells [124].
7.1.5 Mesenchymal stem cells (MSCs)
MSCs are characterized by multipotent fibroblast-like cells in adult tissues such as skin, adipose tissue and bone marrow [125]. Although naive classic MSCs (MSC1) repress cancer cell growth, invasion and angiogenesis, those that inhabit the TME (MSC2) excrete CXCR4, CCL2, CCL5, IDO and adhesion molecules (ICAM-1, VCAM-1) to promote tumorigenesis and recruit immunosuppressor cells [126]. Physical interactions of adhesion molecules with LFA-11, VLA-4 and HLA-G1 instigate CTLA-4 expression, T cell proliferation inhibition, the Treg phenotype and IL-10 production [127]. Tumor-educated MSCs that secrete various immunomodulatory molecules (CCL5, TGF-β, C1q and semaphorins) induce PD-1 overexpression and the M2 macrophage phenotyping in immature MDSCs and differentiated macrophages. IL-4, IL-10 and TGF-β1 secretomes from BC adipose tissues have been found to increase the quantity of Treg cells within the ME [128,129,130].
7.2 Extracellular matrix (ECM) components
7.2.1 Collagen
Collagen is the most frequent component of the ECM, mainly synthesized by CAFs and TAMs in which type I (in stroma) and IV (in basement membrane) are more abundant and provide structural support and tissue polarization. Physical tension, inflammation and carcinogenic factors such as TGF-β induce collagen overproduction resulting in a severe pathological condition known as "desmoplasia." Desmoplastic tissue is characterized by a linear re-arrangement of collagen fibers that influence BC prognosis, invasion and metastasis [131]. ECM constituents like tenascin C and versican intensify the tumor stroma's desmoplastic status by recruiting inflammatory cells and producing MMP and TGF-β. The highly dense and parallel organization of collagen networks around tumor clusters reduce the influx of cytotoxic T cells into the tumor, their motility and physical access to cancer cells. High-density collagen matrices have diminished the proliferation of cytotoxic T cells and CIP2A expression involved in cytolytic behavior. Furthermore, collagen densities have been found to induce Treg cells by overexpressing CD101, SMAD4, FOXO1, PML and FOXM1 [132]. Collagen-dense breast tumors exhibit a higher cyclooxygenase-2 content, which is correlated with IL-2, IL-4 and IL-17A overexpression and macrophage and neutrophil recruitment [133]. Although the exact mechanism is not well understood, it is believed that the higher stiffness and collagen-binding receptors available may stimulate M2 macrophage polarization and YAP/TAZ mechanotransduction pathways [134]. Collagen type IV has been shown to stimulate the production of CCL-2, CXCL-3, CXCL-10 and TGF-β in BC cells, intensifying immunosuppressive functions and driving T cell dysfunction [135].
7.2.2 Tenascin-C (TNC)
TNC is a 180–300 kDa ECM glycoprotein highly distributed in embryonic tissues while restricted to tendons and stem cell niches in adult tissues [136]. The pathological abundance of TNC appears in the ECM of most solid tumors, including breast, ovarian, prostate, lung, glioblastoma, gastrointestinal and liver cancers, mediating tumor growth, metastasis, angiogenesis and relapse [137]. Its hexameric multimodular architecture enables TNC to interact with various ECM compounds like fibronectin and integrin to regulate cell behavior [136]. TNC-enriched breast tumors contain higher numbers of M2 TAMs, contributing to reduced cytotoxic T-cell proliferation and induced IL17-secreting T cells [138]. TNC has been found to affect the localization of immune cells in BC stroma, by which cytotoxic T cells were entrapped into the stroma without invading tumor cell nests. Mechanistically, TNC upregulates CXCL12 in a TLR4-dependent pathway and binds it through fibronectin type III (FNIII) domains. TNC-CXCL-12 forms a robust adhesive substrate to retain T cells in the BC stroma. Also, TNC negatively impacts killer T cell action by targeting granzyme B and perforin [139]. Accumulation of TNC in TNBC cells harboring an irregular autophagy-lysosome degradation system inhibits T cell infiltration and cytotoxic activity [140].
7.2.3 Thrombospondin-1 (TSP1)
TSP1 is a 450 kDa matricellular glycoprotein highly expressed in BC and is known to contribute to different biological processes via interactions with other ECM components and receptors, including CD36, CD47, CD148 and LRP1 [141]. TSP1 is a H2S antagonist and T cell inhibitor that restricts the synthesis of endogenous H2S via downregulating cystathionine β-synthase (CBS) and cystathionine γ-lyase (CTH). It binds to CD47 and suppresses the H2S-activated MEK/ERK pathway and T cell activation [142]. Also, TSP1-CD47 interaction dictates T cell apoptosis through inhibiting protein kinase A and intracellular cAMP [143] and induces naive or memory T cells into regulatory phenotypes [144]. Furthermore, TGF-β2-expressing APCs in mucosal tissues have been found to use the TSP1-CD36 signaling axis to enhance the Treg phenotype in an integrin-independent manner by TGF-β1 production [145].
7.2.4 Secreted protein acidic and rich in cysteine (SPARC)
SPARC (osteonectin) is a non-structural matricellular glycoprotein distributed in normal and malignant tissues orchestrating cell–cell and cell-ECM interactions, as well as cellular growth, migration and differentiation. SPARC is detected in high-grade breast tumors and reflects a higher EMT and therapy resistance [146]. SPARC-rich breast tumors overexpress cyclooxygenase-2 (COX-2), GM-CSF and IL-6, permitting higher recruitment of MDSCs and collagen deposition, and CCL-2 and IL-10 to induce Treg differentiation [147, 148].
7.2.5 Versican
Versican is a > 1000 kDa chondroitin/dermatan sulfate proteoglycan from the hyalectan family exhibiting various interactions with hyaluronan, fibronectin, tenascin and CD44 [149]. Versican enrichment halts T cell-hyaluronan interaction and prevents T cell binding and migration into the matrix [150]. Versican overexpression at early BC stages directly correlates with TAM accumulation, CCL2, VEGF, TGF-β1, collagen production, and metastatic tumor development [151]. Versican is a central ligand for TLR2 through which dendritic cells (DCs) release high amounts of autocrine IL-10 and IL-6 that restrain DC function, T cell expansion and anti-tumor immunity [152].
7.3 Soluble factors
7.3.1 TGF-β1
In addition to insoluble components, various soluble factors such as cytokines, chemokines and metabolites released from cellular entities generate an immunomodulatory TME (Table 1). TGF-β family members play contrasting roles in cancer, among which TGF-β1 acts as a cancer-developing isoform produced by tumor and immune cells. Autocrine and paracrine TGF-β1 induces immune checkpoint elements and anti-inflammatory cytokines bringing down the immune responses and generating immune escape [153]. TGF-β1 raises EMT in HER2+ colorectal, ovarian and BC cells via upregulation of PD-L1 depending on the MEK/ERK and PI3K/Akt pathways. EMT impairs CAR-T cell anti-tumor and cytokine release functions. Blockade of the TGF-β signaling pathway refines the proliferation, anti-tumor activity and cytokine production of anti-ROR1-CAR-T cells in 3D models of TNBC [154].
7.3.2 IL-1β
IL-1β produced during advanced BC stages is supposed to play converse roles in cancer progression and anti-tumor immunity. IL-1β production and maturation into the active form are mediated through TLR signaling and inflammasome-dependent cleavage pathways. Inflammasome activation results in higher IL-1β followed by the penetration of MDSCs and TAMs into BC mouse models and activating and expanding MDSCs via the IL-1RI/NF-κB pathway in gastric cancer [155, 156]. IL-1β-bearing breast tumors exhibit higher levels of CCL-2, which outperforms recruiting and inducing inflammatory CCR-2+ immune cells compared to cytotoxic T cells [157]. Breast tumor-derived IL-1β induces IL-22 production in memory T cells via aryl hydrocarbon receptor (AhR) and RORγ signaling pathways [158].
7.3.3 IL-10
IL-10 is an anti-inflammatory cytokine with pro- and anti-tumorigenic activities released by infiltrated immune cells and it modulates tumor immune status by upregulating TNF-α, IL-1 and IL-12. At the same time, IL-10 has a reverse effect on costimulatory molecules (CD80 and CD86) and obstructs APCs' access to tumor antigens [159]. IL-10 upregulates heparanase expression in inflammatory BC, which positively correlates with TAMs infiltration [160]. It also inhibits CD4+ T cell proliferation by reducing IL-2 and stabilizes Treg cell function through the STAT3 signaling pathway [161, 162]. IL-12/IL-10 ratios with higher contents of IL-10 control the immunosuppressive character of orthotopic breast tumor models [157].
7.3.4 IL-18
IL-18 is a pro-inflammatory cytokine from the IL-1 family acting through the NLRP/inflammasome pathway. TNBC-derived IL-18 signifies immature and non-toxic NK cell fractions and induces PD-1 expression in them, correlating with a poor disease prognosis [163].
7.3.5 IL-35
IL-35 is a cytokine from the IL-12 family responsible for recruiting MDSCs and TME immunotolerance. IL-35 expressed by BC cells hinders the expansion of conventional T cells and incites STAT1/STAT3 signaling to convert them into Treg cells producing IFN-γ, IL-10, IL-17 and IL-35 [164]. IL-35 expressing Treg cells overexpress BLIMP-1 in intratumoral CD8+ T cells and exhaust them [165]. T cell loss of function caused by IL-35 is associated with CD28 downregulation, inhibitory receptor (CTLA4, PD-1, Tim-3, LAG-3) expression and Th1 cytokine release [166, 167].
7.3.6 Prostaglandin-E2 (PGE2)
PGE2 is a lipid metabolite deriving from arachidonic acid as a function of COX-2 activity targeting downstream mitogenic signaling pathways to enhance cancer cell proliferation. Tumor cells, TAMs and MSCs produce high amounts of PGE2 with a negative effect on propagation, antigen presentation, tumor killing and cytokine release of dendritic and T cells [168].
7.3.7 Immunosuppressive metabolites
IDO is a heme-bearing immunotolerance-inducing enzyme that gets L-tryptophan from access to T cells by catalyzing it to the toxic metabolite kynurenine (Kyn), thereby reducing T cell activity and survival and inciting apoptosis in them [169]. IDO expression is coupled with escalated PD-L1 content within the TME, and STAT3-dependent IDO overexpression in MDSCs blocks T cell function, proliferation and Th1 polarization, and prompts apoptosis [116, 170]. Tryptophan-2, 3-dioxygenase (TDO2) catalyzes the rate-limiting step of tryptophan catabolism in TNBC cells. The final metabolite of TDO2 activity, Kyn, binds to the AhR in cytotoxic T cells and drives them toward a Treg phenotype and apoptosis [171, 172]. Upregulation of Kynurenine 3-monooxygenase (KMO) and L-kynureninase (KYNU) directs the Kyn pathway, alters the metabolomic profile in BC cells, and causes the generation of immunosuppressive anthranilic acid derivatives [173].
BC cellular components produce and secrete different soluble factors and govern various aspects of tumor immunosuppression. PGE2: Prostaglandin-E2, IDO: Indoleamine 2,3-dioxygenase, TDO2: Tryptophan-2, 3-dioxygenase, KMO: Kynurenine 3-monooxygenase, KYNU: L-kynureninase.
7.4 Tumor environmental factors
7.4.1 Hypoxia
Oxygen tension (hypoxia) is indicative of the invasive nature of a tumor, associated with disease progression and therapy resistance. Upon tumor penetration, T cells metabolically reprogram from oxidative phosphorylation to glycolytic metabolism and undergo structural remodeling of mitochondria. Nutrient withdrawal induced by hypoxia may hamper this transformation and impede T cell activation [174]. Glycolytic activities in human TNBC orchestrate a molecular network of the AMPK-ULK1, autophagy and CCAAT/enhancer-binding protein beta (CEBPB) pathways, collectively supporting MDSC maintenance and immunosuppression [175].
Hypoxia mediates HIF1α overexpression to direct tumor angiogenesis/vasculature remodeling. HIF1α restricts innate and adaptive immune cell survival, cytotoxicity and cytokine production by forcing glucose metabolism into lactate formation and forwarding immunosuppressive activities [176]. Low oxygen pressure increases the population and function of Tregs, MDSCs and M2 macrophages and induces the production of immunomodulatory cytokines and immune checkpoint elements [177]. Mild photothermal ablation has been shown to oxygenate the melanoma mass, imbalance its hypoxic status, and enhance tumor perfusion.
Under hypoxic conditions, carbonic anhydrase IX (CAIX) enhances and promotes a cascade of events to produce G-CSF and reduce the tumor pH. G-CSF signaling plays a significant role in recruiting immunosuppressor cells in various cancers, including BC. Hypoxia stimulates the expression of CD39 and CD73 on tumor-resident cells that mediate the conversion of ATP into ADP and ADP into adenosine, respectively. Elevated extracellular adenosine imposes strong immunomodulation on T cells, macrophages, DCs and MDSCs [178]. Binding adenosine to the A2aR receptor on the surface of T cells triggers intracellular cAMP-dependent PKA/CREB signaling. The activated pathway interrupts TCR-induced proliferation, tumor killing and IL-2, TNF and IFNγ cytokine secretion of TILs [178, 179].
7.4.2 Acidic pH
Due to oxygen tension and HIF1α expression, cancer cells undergo metabolic reprogramming from oxidative phosphorylation to glycolysis. Anaerobic glycolysis is accompanied by intracellular accumulation of lactate and protons. Besides, ALDH1A1 activity leads to an intracellular acidic pH in BC cells, which enhances TAK1 phosphorylation, NFκB activation and GMCSF secretion, which leads to MDSC infiltration [106]. Cancer cells govern the acidic balance via a series of transporters and pumps through which they efflux acidic metabolites into the TME and reduce its pH. Lactic acid also prevents the infiltration, homing and migration of T cells, DCs and NK cells into the tumor and induces tolerogenic phenotypes [180]. Lactate abundance and an acidic pH of TME block MCT transporters (MCT-1, MCT-2, MCT-4) responsible for effluxing acidic metabolites from cytotoxic T cells in a gradient-dependent manner. Blockade of MCT transporters has been found to impede T cell function and to impose apoptosis [181]. A low pH inhibits IL-2 receptor (CD25) expression while instigating upregulation of the immunoinhibitory molecules CTLA-4 and IFNγ-R2 in TILs [182].
7.4.3 Mechanical force
Increased cancer cell proliferation and matrix synthesis during tumor growth in a limited area generate mechanical forces decisive to cancer progression and metastasis. Exposure to oscillatory strain drives TNBC cells to release PD-L1-rich exosomes. Exosomes engulfed by MDSCs, M2 macrophages and T cells are responsible for immunosuppressive effects [183]. Mechanical stimuli transduced in myeloid cells through mechanosensitive ion channel Piezo1 upregulate histone deacetylase 2, thereby silencing the retinoblastoma gene Rb1. This signal transduction enhances the expansion of MDSCs and strengthens their immunomodulatory status [184]. BC cells express higher CTLA-4 interacting with CD80 molecules on APCs. High levels of CTLA-4 generate strong forces that deplete bounded CD80 molecules from APCs and trans-endocytose them into BC cells in a myosin-dependent manner leading to T cell suppression [185].
7.4.4 CAR-T cell-induced toxicity
A range of mild to severe side effects observed in patients infused with CAR-T cells is unavoidable and challenging for the therapeutic procedure. The adverse effects encompass CAR-T cell anti-tumor activities. For example, eradicating cancer cells to a large extent and their lysates results in electrolytic and metabolic harassment. This imbalanced state may lead to tumor lysis syndrome (TRS), manifested by hyperuricemia, hyperphosphatemia, hyperkalemia and hypocalcemia [186]. Cytokine release syndrome (CRS) is a common consequence of CAR-T cell therapy and results from a systemic inflammatory reaction due to the overproduction of inflammatory cytokines (IL-6, IL-10, TNF-α and IFN-γ) from infused CAR-T cells and host immune cells. CRS symptoms include high fever, sinus tachycardia, blood pressure, oxygen tension and host organ dysfunction. CAR-T cell dose and disease burden may determine the severity of CRS symptoms [187].
Another undesired side effect is that most tumor-associated antigens known as CAR-T cell therapy targets are also expressed in healthy tissues, which CAR-T cells may also recognize. In a case report, the infusion of ERBB2-targeting CAR-T cells to a patient with metastatic colon cancer gave rise to a rapid respiratory distress reaction followed by death five days post-infusion [89]. Clinical manifestations of CAR-T cell-induced toxicity involve various reversible and irreversible symptoms depending on the affected organ. The most common adverse effects are toxicities involving cardiovascular, respiratory, renal, hepatic, gastrointestinal, musculoskeletal, immunologic/hematologic and nervous systems (reviewed by Brudno et al. [188]).
8 Discussion
CAR-T cell therapy represents a milestone in cancer immunotherapy, and promising clinical outcomes on hematological malignancies have resulted in more consideration into administering CAR-T cell therapy to solid tumors, including BC. Customized CAR design provides antigen-specific and safe responses in engineered T cells. High-affinity scFV entities, short hinges and extracellular spacer domains afford higher tumor lysis potential of anti-ROR CAR-T cells [189]. The more specific a tumor antigen target is selected for CAR-T cell therapy, the more satisfactory clinical outcomes may be expected. Considerable advancements in genomics and proteomics, RNA, DNA, whole-genome and exome sequencing have enabled the identification of new neoantigens and neoepitopes as potentially targetable TAAs in some solid tumors.
Despite favorable in vitro results on tumor-killing and cytokine release, in vivo proof pointed out impaired anti-tumor function and delicate infiltration of IL-7/IL-15 co-expressing anti-Neu-CAR-T cells. Replacing T cell signaling endodomains with IL-17A+ helper and cytotoxic T cells (Th17 and Tc17) has led to modestly higher CAR-T cell infiltration and persistence within the TME and short-term anti-tumor immunity [190]. Local and regional administration of CAR-T cells is preferred over systemic infusion, which ensues long-lasting survival and anti-tumor activity [191]. Intravenous injection of anti-IL13Rα2 CAR-T cells has not been found to potentiate enough trafficking into glioblastoma tumors, while local intracranial administration supported T cell traffic, persistence and effector function [192].
A beneficial strategy to prevent possible CAR-T cell-mediated on-target/off-tumor toxicity is using suicide genes to initiate switch-on/switch-off mechanisms of CAR-T cell activity regulation [193]. However, suicide genes derived from viral sources enhance immunogenicity, are time-consuming to synthesize interfering DNA, and abolish CAR-T cells [194]. In vitro transcribed mRNA-based approaches generate transiently expressed CARs with lower on-target/off-tumor side effects. However, short-term CAR expression, poor infiltration and impaired anti-tumor function remain challenging [195].
The development of synNotch CAR-T cells introduced an intelligent mechanism to reduce off-tumor toxicity and to amplify T cells' anti-tumor immunity. SynNotch receptors specific for EpCAM or B7-H3 activate anti-ROR1 CAR-T cells directed against ROR1+ expressing BC cells and obstruct CAR-T cell-mediated death in sub-lethal radiation preconditioned tumors. Although synNotch CAR-T cells discriminate primary breast tumors from normal tissues and eradicate them selectively, they fail to do the same with metastasized secondary tumors, bringing about toxicities to adjacent non-cancer cells [88]. The solid TME is a fundamental immunosuppressive barrier, and conjugation of TME targeting methods with CAR-T cells may overcome tumor immune escape. Abnormal angiogenesis and deformed, not fully developed vasculature are prominent features of solid tumors that significantly determine tumor survival, growth and malignancy and prevent T cell infiltration [196]. Combination therapy with VEGF, CD276 or endothelin B receptors has been found to restore tumor vessels' regular organization and to facilitate CAR-T cell infiltration into the tumor mass [197].
Genetic engineering strategies prevent cancer cells from staying hidden from immune effector cells. EpCAM aptamer-linked small-interfering RNA chimeras (AsiCs) warrant the knockdown of genes related to the immunity of EpCAM expressing HER2+ and TNBC cells in vivo. Selective gene knockdown increases neoantigen expression and presentation of tumor-infiltrating immune cell toxicity while decreasing immune checkpoint expression and tumor growth [198]. Cytokine-producing CAR-T cells lack short-time tumor regression potential, but their combination with stimulator of IFN gene (STING) antagonists results in effective recruiting and trafficking of CAR-T cells with long-term maintenance and proliferation [190]. The combination of CAR-T cell and PD-1/PD-L1 checkpoint blockade overwhelms CAR-T cell loss of functions and upgrades tumor eradication [199].
Targeting the SDF1α/CXCR4 axis hinders MDSC migration into breast tumors and boosts CAR-T cell anti-tumor activity [200]. Genetic modification of anti-MUC1 CAR-T cells to co-express TR2.41BB costimulatory receptors targets MUC1-expressing BC cells and MDSCs expressing tumor necrosis factor-related apoptosis-inducing apoptosis ligand receptor 2 (TR2) [113]. Anti-Meso CAR-T cells with TGF-β targeting adenoviruses yield more robust anti-tumor responses. Oncolytic adenoviruses have an inherent potential for tumor cell lysis, reduce tumor burden at earlier stages and interfere with the TME by TGF-β suppression-mediated IL-6 and IL-12 production [66].
Targeting extrinsic and intrinsic factors impacting CAR-T cell exhaustion ameliorates the CAR-T cell therapeutic success. Bajgain et al. fused IL-4/IL-7 domains to second-generation anti-MUC1 CARs to support CAR-T cells against the inhibitory effect of tumor-associated IL-4. The IL-4/IL-7 fragment reversed the IL-4 inhibitory signaling into a stimulatory IL-7, resulting in T cell persistence, survival, expansion and effective anti-tumor responses [22]. Knocking down the transcription factor IKZF3 from anti-HER2 CAR-T cells using CRISPR technology augmented the killing potential of CAR-T cells against BC cells through influencing cytokine signaling, chemotaxis and cytotoxicity [201]. Determining the effective dose of CAR-T cells is a principal matter of the clinical implication of CAR-T cells to reach high efficiency and minimum toxicity. A small proportion of anti-HER2 CAR-T cells (175,000 or 17,500 cells) in combination with mouse T cells (2.5 million or 250,000 cells respectively) has been found to result in a complete immune response in xenograft breast tumor models [202].
9 Conclusion
In short, CAR-T cell therapy and developing next-generation CAR constructs with refined antigen recognition and effector function properties while ensuring minimized toxicities hold great promise.
References
H. Sung, J. Ferlay, R.L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal et al., Global cancer statistics 2020: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 71, 209–249 (2021)
L.-Y. Yu, J. Tang, C.-M. Zhang, W.-J. Zeng, H. Yan, M.-P. Li et al., New immunotherapy strategies in breast cancer. Int. J. Environ. Res. Public Health 14, 68 (2017)
A. Bozorgi, S. Khazaei, A. Khademi, M. Khazaei, Natural and herbal compounds targeting breast cancer, a review based on cancer stem cells. Iran J. Basic Med. Sci. 23, 970–983 (2020)
V. Seledtsov, A. Goncharov, G. Seledtsova, Multiple-purpose immunotherapy for cancer. Biomed. Pharmacother. 76, 24–29 (2015)
G. Kroemer, L. Senovilla, L. Galluzzi, F. André, L. Zitvogel, Natural and therapy-induced immunosurveillance in breast cancer. Nat. Med. 21, 1128–1138 (2015)
R. Sommaggio, E. Cappuzzello, A. Dalla Pietà, A. Tosi, P. Palmerini, D. Carpanese et al., Adoptive cell therapy of triple negative breast cancer with redirected cytokine-induced killer cells. Oncoimmunology 9, 1777046 (2020)
L.G. Lum, A. Thakur, Z. Al-Kadhimi, G.A. Colvin, F.J. Cummings, R.D. Legare et al., Targeted T-cell therapy in stage IV breast cancer: A phase I clinical trial. Clin. Cancer Res. 21, 2305–2314 (2015)
Q. Li, M. Liu, M. Wu, X. Zhou, S. Wang, Y. Hu et al., PLAC1-specific TCR-engineered T cells mediate antigen-specific antitumor effects in breast cancer. Oncol. Lett. 15, 5924–5932 (2018)
A. Janssen, J. Villacorta Hidalgo, D.X. Beringer, S. van Dooremalen, F. Fernando, E. van Diest et al., γδ T-cell receptors derived from breast cancer-infiltrating T lymphocytes mediate antitumor reactivity. Cancer Immunol. Res. 8, 530–543 (2020)
J.N. Kochenderfer, W.H. Wilson, J.E. Janik, M.E. Dudley, M. Stetler-Stevenson, S.A. Feldman et al., Eradication of B-lineage cells and regression of lymphoma in a patient treated with autologous T cells genetically engineered to recognize CD19. Blood 116, 4099–4102 (2010)
J.N. Kochenderfer, S.A. Rosenberg, Treating B-cell cancer with T cells expressing anti-CD19 chimeric antigen receptors. Nat. Rev. Clin. Oncol. 10, 267–276 (2013)
D.T. Harris, D.M. Kranz, Adoptive T cell therapies: A comparison of T cell receptors and chimeric antigen receptors. Trends. Pharmacol. Sci. 37, 220–230 (2016)
B. Heyman, Y. Yang, New developments in immunotherapy for lymphoma. Cancer Biol. Med. 15, 189–209 (2018)
R.D. Guest, R.E. Hawkins, N. Kirillova, E.J. Cheadle, J. Arnold, A. O’Neill et al., The role of extracellular spacer regions in the optimal design of chimeric immune receptors: evaluation of four different scFvs and antigens. J. Immunother. 28, 203–211 (2005)
H. Shi, L. Liu, Z. Wang, Improving the efficacy and safety of engineered T cell therapy for cancer. Cancer Lett. 328, 191–197 (2013)
Han Y, Xie W, Song D-G, Powell DJ, Jr. Control of triple-negative breast cancer using ex vivo self-enriched, costimulated NKG2D CAR T cells 11,92. J. Hematol. Oncol. (2018).
X. Meng, R. Jing, L. Qian, C. Zhou, J. Sun, Engineering cytoplasmic signaling of CD28ζ CARs for improved therapeutic functions. Front. Immunol. 11, 1046 (2020)
V. Date, S. Nair, Emerging vistas in CAR T-cell therapy: challenges and opportunities in solid tumors. Expert. Opin. Biol. Ther. 21, 145–160 (2021)
A.A. Hombach, H. Abken, Costimulation by chimeric antigen receptors revisited the T cell antitumor response benefits from combined CD28-OX40 signalling. Int. J. Cancer 129, 2935–2944 (2011)
P. Li, L. Yang, T. Li, S. Bin, B. Sun, Y. Huang et al., The third generation anti-HER2 chimeric antigen receptor mouse T cells alone or together with anti-PD1 antibody inhibits the growth of mouse breast tumor cells expressing HER2 in vitro and in immune competent mice. Front. Oncol. 10, 1143 (2020)
P. Luangwattananun, M. Junking, J. Sujjitjoon, Y. Wutti-In, N. Poungvarin, C. Thuwajit et al., Fourth-generation chimeric antigen receptor T cells targeting folate receptor alpha antigen expressed on breast cancer cells for adoptive T cell therapy. Breast Cancer Res. Treat. 186, 25–36 (2021)
P. Bajgain, S. Tawinwung, L. D’Elia, S. Sukumaran, N. Watanabe, V. Hoyos et al., CAR T cell therapy for breast cancer: harnessing the tumor milieu to drive T cell activation. J. Immunother. Cancer 6, 34 (2018)
J. Ren, X. Liu, C. Fang, S. Jiang, C.H. June, Y. Zhao, Multiplex Genome Editing to Generate Universal CAR T Cells Resistant to PD1 Inhibition. Clin. Cancer Res. 23, 2255–2266 (2017)
H. Torikai, A. Reik, P.Q. Liu, Y. Zhou, L. Zhang, S. Maiti et al., A foundation for universal T-cell based immunotherapy: T cells engineered to express a CD19-specific chimeric-antigen-receptor and eliminate expression of endogenous TCR. Blood 119, 5697–5705 (2012)
J.H. Cho, J.J. Collins, W.W. Wong, Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell 173, 1426-1438.e11 (2018)
L.M. Whilding, A.C. Parente-Pereira, T. Zabinski, D.M. Davies, R.M.G. Petrovic, Y.V. Kao et al., Targeting of aberrant αvβ6 integrin expression in solid tumors using chimeric antigen receptor-engineered T cells. Mol. Ther. 25, 259–273 (2017)
K. Bielamowicz, K. Fousek, T.T. Byrd, H. Samaha, M. Mukherjee, N. Aware et al., Trivalent CAR T cells overcome interpatient antigenic variability in glioblastoma. Neuro. Oncol. 20, 506–518 (2018)
K. Urbanska, E. Lanitis, M. Poussin, R.C. Lynn, B.P. Gavin, S. Kelderman et al., A universal strategy for adoptive immunotherapy of cancer through use of a novel T-cell antigen receptor. Cancer Res. 72, 1844–1852 (2012)
Y.G. Lee, I. Marks, M. Srinivasarao, A.K. Kanduluru, S.M. Mahalingam, X. Liu et al., Use of a single CAR T cell and several bispecific adapters facilitates eradication of multiple antigenically different solid tumors. Cancer Res. 79, 387–396 (2019)
S. Wilkie, M.C. van Schalkwyk, S. Hobbs, D.M. Davies, S.J. van der Stegen, A.C. Pereira et al., Dual targeting of ErbB2 and MUC1 in breast cancer using chimeric antigen receptors engineered to provide complementary signaling. J. Clin. Immunol. 32, 1059–1070 (2012)
M. Bajor, A. Graczyk-Jarzynka, K. Marhelava, A. Burdzinska, A. Muchowicz, A. Goral et al., PD-L1 CAR effector cells induce self-amplifying cytotoxic effects against target cells. J. Immunother. Cancer 10, e002500 (2022)
A. Juillerat, A. Marechal, J.M. Filhol, Y. Valogne, J. Valton, A. Duclert et al., An oxygen sensitive self-decision making engineered CAR T-cell. Sci. Rep. 7, 39833 (2017)
Y. Cao, D.T. Rodgers, J. Du, I. Ahmad, E.N. Hampton, J.S. Ma et al., Design of switchable chimeric antigen receptor T cells targeting breast cancer. Angew Chem. Int. Ed. Engl. 55, 7520–7524 (2016)
X. Wang, W.C. Chang, C.W. Wong, D. Colcher, M. Sherman, J.R. Ostberg et al., A transgene-encoded cell surface polypeptide for selection, in vivo tracking, and ablation of engineered cells. Blood 118, 1255–1263 (2011)
M.C. Milone, U. O’Doherty, Clinical use of lentiviral vectors. Leukemia 32, 1529–1541 (2018)
M. Poorebrahim, S. Sadeghi, E. Fakhr, M.F. Abazari, V. Poortahmasebi, A. Kheirollahi et al., Production of CAR T-cells by GMP-grade lentiviral vectors: latest advances and future prospects. Crit. Rev. Clin. Lab. Sci. 56, 393–419 (2019)
L. Xia, Z.Z. Zheng, J.Y. Liu, Y.J. Chen, J.C. Ding, N.S. Xia et al., EGFR-targeted CAR-T cells are potent and specific in suppressing triple-negative breast cancer both in vitro and in vivo. Clin. Transl. Immunol. 9, e01135 (2020)
A.R. Haas, J.L. Tanyi, M.H. O’Hara, W.L. Gladney, S.F. Lacey, D.A. Torigian et al., Phase I study of lentiviral-transduced chimeric antigen receptor-modified T cells recognizing mesothelin in advanced solid cancers. Mol. Ther. 27, 1919–1929 (2019)
H. Singh, M.J. Figliola, M.J. Dawson, S. Olivares, L. Zhang, G. Yang et al., Manufacture of clinical-grade CD19-specific T cells stably expressing chimeric antigen receptor using Sleeping Beauty system and artificial antigen presenting cells. PLoS One 8, e64138 (2013)
V. Lukjanov, I. Koutná, P. Šimara, CAR T-Cell production using nonviral approaches. J. Immunol. Res. 2021, 6644685 (2021)
F. Zhou, J. Krishnamurthy, Y. Wei, M. Li, K. Hunt, G.L. Johanning et al., Chimeric antigen receptor T cells targeting HERV-K inhibit breast cancer and its metastasis through downregulation of Ras. Oncoimmunology 4, e1047582 (2015)
K. Nakamura, S. Yagyu, S. Hirota, A. Tomida, M. Kondo, T. Shigeura et al., Autologous antigen-presenting cells efficiently expand piggyBac transposon CAR-T cells with predominant memory phenotype. Mol. Ther. - Methods Clin. Dev. 21, 315–324 (2021)
L.M. Arévalo-Soliz, C.L. Hardee, J.M. Fogg, N.R. Corman, C. Noorbakhsh, L. Zechiedrich, Improving therapeutic potential of non-viral minimized DNA vectors. Cell. Gene Ther. Insights. 6, 1489–1505 (2020)
M. Bozza, A.D. Roia, M.P. Correia, A. Berger, A. Tuch, A. Schmidt et al., A nonviral, nonintegrating DNA nanovector platform for the safe, rapid, and persistent manufacture of recombinant T cells. Sci. Adv. 7, eab1333 (2021)
C. Cheng, N. Tang, J. Li, S. Cao, T. Zhang, X. Wei et al., Bacteria-free minicircle DNA system to generate integration-free CAR-T cells. J. Med. Genet. 56, 10–17 (2019)
A. Bozorgi, M. Khazaei, M. Soleimani, Z. Jamalpoor, Application of nanoparticles in bone tissue engineering; a review on the molecular mechanisms driving osteogenesis. Biomater. Sci. 9, 4541–4567 (2021)
K.A. Hajj, K.A. Whitehead, Tools for translation: non-viral materials for therapeutic mRNA delivery. Nat. Rev. Mater. 2, 17056 (2017)
M.M. Billingsley, N. Singh, P. Ravikumar, R. Zhang, C.H. June, M.J. Mitchell, Ionizable lipid nanoparticle-mediated mRNA delivery for human CAR T cell engineering. Nano. Lett. 20, 1578–1589 (2020)
Y. Zheng, Z.R. Li, R. Yue, Y.L. Fu, Z.Y. Liu, H.Y. Feng et al., PiggyBac transposon system with polymeric gene carrier transfected into human T cells. Am. J. Transl. Res. 11, 7126–7136 (2019)
N.N. Parayath, S.B. Stephan, A.L. Koehne, P.S. Nelson, M.T. Stephan, In vitro-transcribed antigen receptor mRNA nanocarriers for transient expression in circulating T cells in vivo. Nat. Commun. 11, 6080 (2020)
P. Sarobe, E. Huarte, J.J. Lasarte, F. Borrás-Cuesta, Carcinoembryonic antigen as a target to induce anti-tumor immune responses. Curr. Cancer Drug Targets 4, 443–454 (2004)
J.I. Mori, K. Adachi, Y. Sakoda, T. Sasaki, S. Goto, H. Matsumoto et al., Anti-tumor efficacy of human anti-c-met CAR-T cells against papillary renal cell carcinoma in an orthotopic model. Cancer Sci. 112, 1417–1428 (2021)
R.A. Ghoussoub, D.A. Dillon, T. D’Aquila, E.B. Rimm, E.R. Fearon, D.L. Rimm, Expression of c-met is a strong independent prognostic factor in breast carcinoma. Cancer 82, 1513–1520 (1998)
J. Tchou, Y. Zhao, B.L. Levine, P.J. Zhang, M.M. Davis, J.J. Melenhorst et al., Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res. 5, 1152–1161 (2017)
J.A. Krall, E.M. Beyer, G. MacBeath, High- and low-affinity epidermal growth factor receptor-ligand interactions activate distinct signaling pathways. PLoS One 6, e15945 (2011)
G. Bergado Báez, D.R. Hernández Fernández, Z. Mazorra Herrera, R.B. Sánchez, HER1-based vaccine: Simultaneous activation of humoral and cellular immune response. Semin. Oncol. 45, 75–83 (2018)
Y. Liu, Y. Zhou, K.-H. Huang, Y. Li, X. Fang, L. An et al., EGFR-specific CAR-T cells trigger cell lysis in EGFR-positive TNBC. Aging 11, 11054–11072 (2019)
L. Xia, Z. Zheng, J.Y. Liu, Y.J. Chen, J. Ding, G.S. Hu et al., Targeting triple-negative breast cancer with combination therapy of EGFR CAR T cells and CDK7 inhibition. Cancer Immunol. Res. 9, 707–722 (2021)
C. Jackisch, P. Lammers, I. Jacobs, Evolving landscape of human epidermal growth factor receptor 2-positive breast cancer treatment and the future of biosimilars. Breast 32, 199–216 (2017)
Á. Szöőr, G. Tóth, B. Zsebik, V. Szabó, Z. Eshhar, H. Abken et al., Trastuzumab derived HER2-specific CARs for the treatment of trastuzumab-resistant breast cancer: CAR T cells penetrate and eradicate tumors that are not accessible to antibodies. Cancer Lett. 484, 1–8 (2020)
H. Li, W. Yuan, S. Bin, G. Wu, P. Li, M. Liu et al., Overcome trastuzumab resistance of breast cancer using anti-HER2 chimeric antigen receptor T cells and PD1 blockade. Am. J. Cancer Res. 10, 688–703 (2020)
G. Tozbikian, E. Brogi, K. Kadota, J. Catalano, M. Akram, S. Patil et al., Mesothelin expression in triple negative breast carcinomas correlates significantly with basal-like phenotype, distant metastases and decreased survival. PLoS ONE 9, e114900 (2014)
J. Tchou, L.-C. Wang, B. Selven, H. Zhang, J. Conejo-Garcia, H. Borghaei et al., Mesothelin, a novel immunotherapy target for triple negative breast cancer. Breast Cancer Res. Treat. 133, 799–804 (2012)
R. Hassan, A. Thomas, C. Alewine, D.T. Le, E.M. Jaffee, I. Pastan, Mesothelin immunotherapy for cancer: ready for prime time? J. Clin. Oncol. 34, 4171–4179 (2016)
Z. Tang, M. Qian, M. Ho, The role of mesothelin in tumor progression and targeted therapy. Anticancer Agents Med. Chem. 13, 276–280 (2013)
Y. Li, F. Xiao, A. Zhang, D. Zhang, W. Nie, T. Xu et al., Oncolytic adenovirus targeting TGF-β enhances anti-tumor responses of mesothelin-targeted chimeric antigen receptor T cell therapy against breast cancer. Cell Immunol. 348, 104041 (2020)
Z. Zhang, D. Jiang, H. Yang, Z. He, X. Liu, W. Qin et al., Modified CAR T cells targeting membrane-proximal epitope of mesothelin enhances the antitumor function against large solid tumor. Cell Death Dis. 10, 476 (2019)
M. Jäger, A. Schoberth, P. Ruf, J. Hess, M. Hennig, B. Schmalfeldt et al., Immunomonitoring results of a phase II/III study of malignant ascites patients treated with the trifunctional antibody catumaxomab (anti-EpCAM× anti-CD3). Cancer Res. 72, 24–32 (2012)
A. Martowicz, G. Spizzo, G. Gastl, G. Untergasser, Phenotype-dependent effects of EpCAM expression on growth and invasion of human breast cancer cell lines. BMC Cancer 12, 501 (2012)
M. Kubo, M. Umebayashi, K. Kurata, H. Mori, M. Kai, H. Onishi et al., Catumaxomab with activated T-cells efficiently lyses chemoresistant EpCAM-positive triple-negative breast cancer cell lines. Anticancer Res. 38, 4273–4279 (2018)
Q. Zhang, H. Zhang, J. Ding, H. Liu, H. Li, H. Li et al., Combination Therapy with EpCAM-CAR-NK-92 Cells and Regorafenib against Human Colorectal Cancer Models. J. Immunol. Res. 2018, 4263520 (2018)
J. Fu, Y. Shang, Z. Qian, J. Hou, F. Yan, G. Liu et al., Chimeric Antigen receptor-T (CAR-T) cells targeting Epithelial cell adhesion molecule (EpCAM) can inhibit tumor growth in ovarian cancer mouse model. J. Vet. Med. Sci. 83, 241–247 (2021)
D. Qin, D. Li, B. Zhang, Y. Chen, X. Liao, X. Li et al., Potential lung attack and lethality generated by EpCAM-specific CAR-T cells in immunocompetent mouse models. Oncoimmunology 9, 1806009 (2020)
K. Lund, J.L. Dembinski, N. Solberg, A. Urbanucci, I.G. Mills, S. Krauss, Slug-dependent upregulation of L1CAM is responsible for the increased invasion potential of pancreatic cancer cells following long-term 5-FU treatment. PLoS One 10, e0123684 (2015)
K. Doberstein, K. Milde-Langosch, N.P. Bretz, U. Schirmer, A. Harari, I. Witzel et al., L1CAM is expressed in triple-negative breast cancers and is inversely correlated with androgen receptor. BMC Cancer 14, 958 (2014)
J. Zhang, F. Yang, Y. Ding, L. Zhen, X. Han, F. Jiao et al., Overexpression of L1 cell adhesion molecule correlates with aggressive tumor progression of patients with breast cancer and promotes motility of breast cancer cells. Int. J. Clin. Exp. Pathol. 8, 9240–9247 (2015)
H. Zhang, C.C. Wong, H. Wei, D.M. Gilkes, P. Korangath, P. Chaturvedi et al., HIF-1-dependent expression of angiopoietin-like 4 and L1CAM mediates vascular metastasis of hypoxic breast cancer cells to the lungs. Oncogene 31, 1757–1770 (2012)
X. Wu, Z. Yin, C. McKay, C. Pett, J. Yu, M. Schorlemer et al., Protective epitope discovery and design of MUC1-based vaccine for effective tumor protections in immunotolerant mice. J. Am. Chem. Soci. 140, 16596–16609 (2018)
R. Zhou, M. Yazdanifar, L.D. Roy, L.M. Whilding, A. Gavrill, J. Maher et al., CAR T cells targeting the tumor MUC1 glycoprotein reduce triple-negative breast cancer growth. Front. Immunol. 10, 1149 (2019)
M. Antonilli, H. Rahimi, V. Visconti, C. Napoletano, I. Ruscito, I.G. Zizzari et al., Triple peptide vaccination as consolidation treatment in women affected by ovarian and breast cancer: Clinical and immunological data of a phase I/II clinical trial. Int. J. Oncol. 48, 1369–1378 (2016)
R. Bei, J. Kantor, S.V. Kashmiri, S. Abrams, J. Schlom, Enhanced immune responses and anti-tumor activity by baculovirus recombinant carcinoembryonic antigen (CEA) in mice primed with the recombinant vaccinia CEA. J. Immunother. Emphasis Tumor Immunol. 16, 275–282 (1994)
S.E. Cha, M. Kujawski, J.P. Yazaki, C. Brown, J.E. Shively, Tumor regression and immunity in combination therapy with anti-CEA chimeric antigen receptor T cells and anti-CEA-IL2 immunocytokine. Oncoimmunology 10, 1899469 (2021)
M. Loeffler, J.A. Krüger, A.G. Niethammer, R.A. Reisfeld, Targeting tumor-associated fibroblasts improves cancer chemotherapy by increasing intratumoral drug uptake. J. Clin. Invest. 116, 1955–1962 (2006)
M. Meng, W. Wang, J. Yan, J. Tan, L. Liao, J. Shi et al., Immunization of stromal cell targeting fibroblast activation protein providing immunotherapy to breast cancer mouse model. Tumour Biol. 37, 10317–10327 (2016)
S. Zhang, L. Chen, B. Cui, H.Y. Chuang, J. Yu, J. Wang-Rodriguez et al., ROR1 is expressed in human breast cancer and associated with enhanced tumor-cell growth. PLoS One 7, e31127 (2012)
A. Kamrani, A. Mehdizadeh, M. Ahmadi, L. Aghebati-Maleki, M. Yousefi, Therapeutic approaches for targeting receptor tyrosine kinase like orphan receptor-1 in cancer cells. Expert. Opin. Ther. Targets 23, 447–456 (2019)
L. Wallstabe, C. Göttlich, L.C. Nelke, J. Kühnemundt, T. Schwarz, T. Nerreter et al., ROR1-CAR T cells are effective against lung and breast cancer in advanced microphysiologic 3D tumor models. JCI Insight 4, e126345 (2019)
S. Srivastava, A.I. Salter, D. Liggitt, S. Yechan-Gunja, M. Sarvothama, K. Cooper et al., Logic-gated ROR1 chimeric antigen receptor expression rescues T cell-mediated toxicity to normal tissues and enables selective tumor targeting. Cancer Cell 35, 489–503 (2019)
R.A. Morgan, J.C. Yang, M. Kitano, M.E. Dudley, C.M. Laurencot, S.A. Rosenberg, Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol. Ther. 18, 843–851 (2010)
K.T. Roybal, L.J. Rupp, L. Morsut, W.J. Walker, K.A. McNally, J.S. Park et al., Precision tumor recognition by T cells with combinatorial antigen-sensing Circuits. Cell 164, 770–779 (2016)
X. Han, P.D. Bryson, Y. Zhao, G.E. Cinay, S. Li, Y. Guo et al., Masked chimeric antigen receptor for tumor-specific activation. Mol. Ther. 25, 274–284 (2017)
L. Nagl, L. Horvath, A. Pircher, D. Wolf, Tumor endothelial cells (TECs) as potential immune directors of the tumor microenvironment - new findings and future perspectives. Front. Cell. Dev. Biol. 8, 766 (2020)
D. Klein, The tumor vascular endothelium as decision maker in cancer therapy. Front. Oncol. 8, 367 (2018)
D. Lambrechts, E. Wauters, B. Boeckx, S. Aibar, D. Nittner, O. Burton et al., Phenotype molding of stromal cells in the lung tumor microenvironment. Nat. Med. 24, 1277–1289 (2018)
T.H. Nasti, D.C. Bullard, N. Yusuf, P-selectin enhances growth and metastasis of mouse mammary tumors by promoting regulatory T cell infiltration into the tumors. Life Sci. 131, 11–18 (2015)
G.T. Motz, S.P. Santoro, L.-P. Wang, T. Garrabrant, R.R. Lastra, I.S. Hagemann et al., Tumor endothelium FasL establishes a selective immune barrier promoting tolerance in tumors. Nat. Med. 20, 607–615 (2014)
Y. Jiang, Y. Li, B. Zhu, T-cell exhaustion in the tumor microenvironment. Cell Death Dis. 6, e1792 (2015)
Y. Liu, G. Liu, J. Wang, Z.-y Zheng, L. Jia, W. Rui et al., Chimeric STAR receptors using TCR machinery mediate robust responses against solid tumors. Sci Transl Med 13, eabb5191 (2021)
M. Hamieh, A. Dobrin, A. Cabriolu, S.J.C. van der Stegen, T. Giavridis, J. Mansilla-Soto et al., CAR T cell trogocytosis and cooperative killing regulate tumour antigen escape. Nature 568, 112–116 (2019)
M. Castellarin, K. Watanabe, C.H. June, C.C. Kloss, A.D. Posey Jr., Driving cars to the clinic for solid tumors. Gene Ther. 25, 165–175 (2018)
E. Mamessier, A. Sylvain, M.-L. Thibult, G. Houvenaeghel, J. Jacquemier, R. Castellano et al., Human breast cancer cells enhance self tolerance by promoting evasion from NK cell antitumor immunity. J. Clin. Invest. 121, 3609–3622 (2011)
C. Gómez-Aleza, B. Nguyen, G. Yoldi, M. Ciscar, A. Barranco, E. Hernández-Jiménez et al., Inhibition of RANK signaling in breast cancer induces an anti-tumor immune response orchestrated by CD8+ T cells. Nat. Commun. 11, 6335 (2020)
L.A. Moraes, S. Kar, S.L. Foo, T. Gu, Y.Q. Toh, P.B. Ampomah et al., Annexin-A1 enhances breast cancer growth and migration by promoting alternative macrophage polarization in the tumour microenvironment. Sci. Rep. 7, 17925 (2017)
R. Wang, Y. Liu, L. Liu, M. Chen, X. Wang, J. Yang et al., Tumor cells induce LAMP2a expression in tumor-associated macrophage for cancer progression. EBioMedicine 40, 118–134 (2019)
Z.F. Wen, H. Liu, R. Gao, M. Zhou, J. Ma, Y. Zhang et al., Tumor cell-released autophagosomes (TRAPs) promote immunosuppression through induction of M2-like macrophages with increased expression of PD-L1. J. Immunother. Cancer 6, 151 (2018)
C. Liu, J. Qiang, Q. Deng, J. Xia, L. Deng, L. Zhou et al., ALDH1A1 activity in tumor-initiating cells remodels myeloid-derived suppressor cells to promote breast cancer progression. Cancer Res. 81, 5919–5934 (2021)
B. Yu, F. Luo, B. Sun, W. Liu, Q. Shi, S.Y. Cheng et al., KAT6A acetylation of SMAD3 regulates myeloid-derived suppressor cell recruitment, metastasis, and immunotherapy in triple-negative breast cancer. Adv Sci (Weinh) 8, e2100014 (2021)
A. Volpe, C. Lang, L. Lim, F. Man, E. Kurtys, C. Ashmore-Harris et al., Spatiotemporal PET imaging rveals differences in CAR-T tumor retention in triple-negative breast cancer models. Mol. Ther. 28, 2271–2285 (2020)
L. Cassetta, S. Fragkogianni, A.H. Sims, A. Swierczak, L.M. Forrester, H. Zhang et al., Human tumor-associated macrophage and monocyte transcriptional landscapes reveal cancer-specific reprogramming, biomarkers, and therapeutic Targets. Cancer Cell 35, 588-602.e10 (2019)
A.A. Barkal, R.E. Brewer, M. Markovic, M. Kowarsky, S.A. Barkal, B.W. Zaro et al., CD24 signalling through macrophage Siglec-10 is a target for cancer immunotherapy. Nature 572, 392–396 (2019)
A.K. Mehta, S. Kadel, M.G. Townsend, M. Oliwa, J.L. Guerriero, Macrophage biology and mechanisms of immune suppression in breast cancer. Front. Immunol. 12, 643771 (2021)
S. Su, J. Liao, J. Liu, D. Huang, C. He, F. Chen et al., Blocking the recruitment of naive CD4(+) T cells reverses immunosuppression in breast cancer. Cell Res. 27, 461–482 (2017)
S.A. Nalawade, P. Shafer, P. Bajgain, M.K. McKenna, A. Ali, L. Kelly et al., Selectively targeting myeloid-derived suppressor cells through TRAIL receptor 2 to enhance the efficacy of CAR T cell therapy for treatment of breast cancer. J. Immunother. Cancer 9, e003237 (2021)
L. Speigl, H. Burow, J.K. Bailur, N. Janssen, C.B. Walter, G. Pawelec et al., CD14+ HLA-DR-/low MDSCs are elevated in the periphery of early-stage breast cancer patients and suppress autologous T cell proliferation. Breast Cancer Res. Treat. 168, 401–411 (2018)
Z. Fang, C. Wen, X. Chen, R. Yin, C. Zhang, X. Wang et al., Myeloid-derived suppressor cell and macrophage exert distinct angiogenic and immunosuppressive effects in breast cancer. Oncotarget 8, 54173–54186 (2017)
J. Yu, W. Du, F. Yan, Y. Wang, H. Li, S. Cao et al., Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J. Immunol. 190, 3783–3797 (2013)
Y. Sharon, Y. Raz, N. Cohen, A. Ben-Shmuel, H. Schwartz, T. Geiger et al., Tumor-derived osteopontin reprograms normal mammary fibroblasts to promote inflammation and tumor growth in breast cancer. Cancer Res. 75, 963–973 (2015)
S. Baroni, S. Romero-Cordoba, I. Plantamura, M. Dugo, E. D’Ippolito, A. Cataldo et al., Exosome-mediated delivery of miR-9 induces cancer-associated fibroblast-like properties in human breast fibroblasts. Cell Death Dis. 7, e2312 (2016)
A. Li, P. Chen, Y. Leng, J. Kang, Histone deacetylase 6 regulates the immunosuppressive properties of cancer-associated fibroblasts in breast cancer through the STAT3-COX2-dependent pathway. Oncogene 37, 5952–5966 (2018)
N. Cohen, O. Shani, Y. Raz, Y. Sharon, D. Hoffman, L. Abramovitz et al., Fibroblasts drive an immunosuppressive and growth-promoting microenvironment in breast cancer via secretion of Chitinase 3-like 1. Oncogene 36, 4457–4468 (2017)
A. Costa, Y. Kieffer, A. Scholer-Dahirel, F. Pelon, B. Bourachot, M. Cardon et al., Fibroblast heterogeneity and immunosuppressive environment in human breast cancer. Cancer Cell 33, 463-479.e10 (2018)
Y. Kieffer, H.R. Hocine, G. Gentric, F. Pelon, C. Bernard, B. Bourachot et al., Single-cell analysis reveals fibroblast clusters linked to immunotherapy resistance in cancer. Cancer Discov. 10, 1330–1351 (2020)
G. Hu, P. Cheng, J. Pan, S. Wang, Q. Ding, Z. Jiang et al., An IL6-adenosine positive feedback loop between CD73(+) γδTregs and CAFs promotes tumor progression in human breast cancer. Cancer Immunol. Res. 8, 1273–1286 (2020)
R. Allaoui, C. Bergenfelz, S. Mohlin, C. Hagerling, K. Salari, Z. Werb et al., Cancer-associated fibroblast-secreted CXCL16 attracts monocytes to promote stroma activation in triple-negative breast cancers. Nat. Commun. 7, 13050 (2016)
S. Khazaei, G. Keshavarz, A. Bozorgi, H. Nazari, M. Khazaei, Adipose tissue-derived stem cells: a comparative review on isolation, culture, and differentiation methods. Cell Tissue Bank 23, 1–16 (2021)
S. Aravindhan, S.S. Ejam, M.H. Lafta, A. Markov, A.V. Yumashev, M. Ahmadi, Mesenchymal stem cells and cancer therapy: insights into targeting the tumour vasculature. Cancer Cell Int. 21, 158 (2021)
M.E. Castro-Manrreza, Participation of mesenchymal stem cells in the regulation of immune response and cancer development. Bol. Med. Hosp. Infant Mex. 73, 380–387 (2016)
S. Biswas, G. Mandal, S. Roy Chowdhury, S. Purohit, K.K. Payne, C. Anadon et al., Exosomes produced by mesenchymal stem cells drive differentiation of myeloid cells into immunosuppressive M2-polarized macrophages in breast cancer. J. Immunol. 203, 3447–3460 (2019)
H. Aboulkheyr Es, B. Bigdeli, S. Zhand, A.R. Aref, J.P. Thiery, M.E. Warkiani, Mesenchymal stem cells induce PD-L1 expression through the secretion of CCL5 in breast cancer cells. J. Cell Physiol. 236, 3918–3928 (2021)
M. Razmkhah, M. Jaberipour, N. Erfani, M. Habibagahi, A.R. Talei, A. Ghaderi, Adipose derived stem cells (ASCs) isolated from breast cancer tissue express IL-4, IL-10 and TGF-β1 and upregulate expression of regulatory molecules on T cells: do they protect breast cancer cells from the immune response? Cell Immunol. 266, 116–122 (2011)
M. Egeblad, M.G. Rasch, V.M. Weaver, Dynamic interplay between the collagen scaffold and tumor evolution. Curr. Opin. Cell Biol. 22, 697–706 (2010)
D.E. Kuczek, A.M.H. Larsen, M.L. Thorseth, M. Carretta, A. Kalvisa, M.S. Siersbæk et al., Collagen density regulates the activity of tumor-infiltrating T cells. J. Immunother. Cancer 7, 68 (2019)
K. Esbona, D. Inman, S. Saha, J. Jeffery, P. Schedin, L. Wilke et al., COX-2 modulates mammary tumor progression in response to collagen density. Breast Cancer Res. 18, 35 (2016)
A.M.H. Larsen, D.E. Kuczek, A. Kalvisa, M.S. Siersbæk, M.L. Thorseth, A.Z. Johansen et al., Collagen density modulates the immunosuppressive functions of macrophages. J. Immunol. 205, 1461–1472 (2020)
C. Robertson, A. Sebastian, A. Hinckley, N.D. Rios-Arce, W.F. Hynes, S.A. Edwards et al., Extracellular matrix modulates T cell clearance of malignant cells in vitro. Biomaterials 282, 121378 (2022)
K.S. Midwood, M. Chiquet, R.P. Tucker, G. Orend, Tenascin-C at a glance. J. Cell Sci. 129, 4321–4327 (2016)
R. Chiquet-Ehrismann, M. Chiquet, Tenascins: regulation and putative functions during pathological stress. J. Pathol. 200, 488–499 (2003)
C. Deligne, D. Murdamoothoo, A.N. Gammage, M. Gschwandtner, W. Erne, T. Loustau et al., Matrix-targeting immunotherapy controls tumor growth and spread by switching macrophage phenotype. Cancer Immunol. Res. 8, 368–382 (2020)
D. Murdamoothoo, Z. Sun, A. Yilmaz, G. Riegel, C. Abou-Faycal, C. Deligne et al., Tenascin-C immobilizes infiltrating T lymphocytes through CXCL12 promoting breast cancer progression. EMBO Mol. Med. 13, e13270 (2021)
Z.L. Li, H.L. Zhang, Y. Huang, J.H. Huang, P. Sun, N.N. Zhou et al., Autophagy deficiency promotes triple-negative breast cancer resistance to T cell-mediated cytotoxicity by blocking tenascin-C degradation. Nat. Commun. 11, 3806 (2020)
J. Cen, L. Feng, H. Ke, L. Bao, L.Z. Li, Y. Tanaka et al., Exosomal thrombospondin-1 disrupts the integrity of endothelial intercellular junctions to facilitate breast cancer cell metastasis. Cancers (Basel). 11, 1946 (2019)
T.W. Miller, S. Kaur, K. Ivins-O’Keefe, D.D. Roberts, Thrombospondin-1 is a CD47-dependent endogenous inhibitor of hydrogen sulfide signaling in T cell activation. Matrix Biol. 32, 316–324 (2013)
P.P. Manna, W.A. Frazier, The mechanism of CD47-dependent killing of T cells: heterotrimeric Gi-dependent inhibition of protein kinase A. J. Immunol. 170, 3544–3553 (2003)
P. Grimbert, S. Bouguermouh, N. Baba, T. Nakajima, Z. Allakhverdi, D. Braun et al., Thrombospondin/CD47 interaction: a pathway to generate regulatory T cells from human CD4+ CD25- T cells in response to inflammation. J. Immunol. 177, 3534–3541 (2006)
F.A. Mir, L. Contreras-Ruiz, S. Masli, Thrombospondin-1-dependent immune regulation by transforming growth factor-β2-exposed antigen-presenting cells. Immunology 146, 547–556 (2015)
T. Triulzi, P. Casalini, M. Sandri, M. Ratti, M.L. Carcangiu, M.P. Colombo et al., Neoplastic and stromal cells contribute to an extracellular matrix gene expression profile defining a breast cancer subtype likely to progress. PLoS One 8, e56761 (2013)
S. Sangaletti, C. Tripodo, A. Santangelo, N. Castioni, P. Portararo, A. Gulino et al., Mesenchymal transition of high-grade breast carcinomas depends on extracellular matrix control of myeloid suppressor cell activity. Cell Rep. 17, 233–248 (2016)
S. Sangaletti, L. Gioiosa, C. Guiducci, G. Rotta, M. Rescigno, A. Stoppacciaro et al., Accelerated dendritic-cell migration and T-cell priming in SPARC-deficient mice. J. Cell Sci. 118, 3685–3694 (2005)
A. Papadas, G. Arauz, A. Cicala, J. Wiesner, F. Asimakopoulos, Versican and versican-matrikines in cancer progression, inflammation, and immunity. J. Histochem. Cytochem. 68, 871–885 (2020)
S.P. Evanko, S. Potter-Perigo, P.L. Bollyky, G.T. Nepom, T.N. Wight, Hyaluronan and versican in the control of human T-lymphocyte adhesion and migration. Matrix Biol. 31, 90–100 (2012)
D.C. Dos Reis, K.A. Damasceno, C.B. de Campos, E.S. Veloso, G.R.A. Pêgas, L.R. Kraemer et al., Versican and tumor-associated macrophages promotes tumor progression and metastasis in canine and murine models of breast carcinoma. Front. Oncol. 9, 577 (2019)
M. Tang, J. Diao, M.S. Cattral, Molecular mechanisms involved in dendritic cell dysfunction in cancer. Cell Mol. Life Sci. 74, 761–776 (2017)
V.W. Xue, J.Y.-F. Chung, C.A.G. Córdoba, A.H.-K. Cheung, W. Kang, E.W.F. Lam et al., Transforming growth factor-β: A multifunctional regulator of cancer immunity. Cancers 12, 3099 (2020)
T. Stüber, R. Monjezi, L. Wallstabe, J. Kühnemundt, S.L. Nietzer, G. Dandekar et al., Inhibition of TGF-β-receptor signaling augments the antitumor function of ROR1-specific CAR T-cells against triple-negative breast cancer. J Immunother Cancer 8, e000676 (2020)
B. Guo, S. Fu, J. Zhang, B. Liu, Z. Li, Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep 6, 36107 (2016)
S. Tu, G. Bhagat, G. Cui, S. Takaishi, E.A. Kurt-Jones, B. Rickman et al., Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419 (2008)
I. Kaplanov, Y. Carmi, R. Kornetsky, A. Shemesh, G.V. Shurin, M.R. Shurin et al., Blocking IL-1β reverses the immunosuppression in mouse breast cancer and synergizes with anti-PD-1 for tumor abrogation. Proc. Natl. Acad. Sci. U. S A. 116, 1361–1369 (2019)
C. Voigt, P. May, A. Gottschlich, A. Markota, D. Wenk, I. Gerlach et al., Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc. Natl. Acad. Sci. U. S. A. 114, 12994–12999 (2017)
E. Sheikhpour, P. Noorbakhsh, E. Foroughi, S. Farahnak, R. Nasiri, H. Neamatzadeh, A survey on the role of interleukin-10 in breast cancer: A narrative. Rep. Biochem. Mol. Biol. 7, 30–37 (2018)
M. El-Nadi, H. Hassan, M.E. Saleh, E. Nassar, Y.M. Ismail, M. Amer et al., Induction of heparanase via IL-10 correlates with a high infiltration of CD163+ M2-type tumor-associated macrophages in inflammatory breast carcinomas. Matrix Biol. Plus. 6–7, 100030 (2020)
Malefyt R. de Waal, H. Yssel, J.E. de Vries, Direct effects of IL-10 on subsets of human CD4+ T cell clones and resting T cells Specific inhibition of IL-2 production and proliferation. J. Immunol. 150, 4754–65 (1993)
A. Chaudhry, R.M. Samstein, P. Treuting, Y. Liang, M.C. Pils, J.M. Heinrich et al., Interleukin-10 signaling in regulatory T cells is required for suppression of Th17 cell-mediated inflammation. Immunity 34, 566–578 (2011)
I.H. Park, H.N. Yang, K.J. Lee, T.S. Kim, E.S. Lee, S.Y. Jung et al., Tumor-derived IL-18 induces PD-1 expression on immunosuppressive NK cells in triple-negative breast cancer. Oncotarget 8, 32722–32730 (2017)
S. Hao, X. Chen, F. Wang, Q. Shao, J. Liu, H. Zhao et al., Breast cancer cell-derived IL-35 promotes tumor progression via induction of IL-35-producing induced regulatory T cells. Carcinogenesis 39, 1488–1496 (2018)
M. Damo, N.S. Joshi, T(reg) cell IL-10 and IL-35 exhaust CD8(+) T cells in tumors. Nat. Immunol. 20, 674–675 (2019)
H. Jiang, T. Zhang, M.-X. Yan, W. Wu, IL-35 inhibits CD8 + T cells activity by suppressing expression of costimulatory molecule CD28 and Th1 cytokine production. Transl. Cancer Res. 8, 1319–1325 (2019)
Q.Q. Ding, J.M. Chauvin, H.M. Zarour, Targeting novel inhibitory receptors in cancer immunotherapy. Semin. Immunol. 49, 101436 (2020)
B.A. Pockaj, G.D. Basu, L.B. Pathangey, R.J. Gray, J.L. Hernandez, S.J. Gendler et al., Reduced T-cell and dendritic cell function is related to cyclooxygenase-2 overexpression and prostaglandin E2 secretion in patients with breast cancer. Ann. Surg. Oncol. 11, 328–339 (2004)
J. Sun, J. Yu, H. Li, L. Yang, F. Wei, W. Yu et al., Upregulated expression of indoleamine 2, 3-dioxygenase in CHO cells induces apoptosis of competent T cells and increases proportion of Treg cells. J. Exp. Clin. Cancer Res. 30, 82 (2011)
E.A. Dill, P.M. Dillon, T.N. Bullock, A.M. Mills, IDO expression in breast cancer: an assessment of 281 primary and metastatic cases with comparison to PD-L1. Mod. Pathol. 31, 1513–1522 (2018)
J.D. Mezrich, J.H. Fechner, X. Zhang, B.P. Johnson, W.J. Burlingham, C.A. Bradfield, An interaction between kynurenine and the aryl hydrocarbon receptor can generate regulatory T cells. J. Immunol. 185, 3190–3198 (2010)
F. Fallarino, U. Grohmann, C. Vacca, R. Bianchi, C. Orabona, A. Spreca et al., T cell apoptosis by tryptophan catabolism. Cell Death Differ. 9, 1069–1077 (2002)
B. Heng, A.A. Bilgin, D.B. Lovejoy, V.X. Tan, H.H. Milioli, L. Gluch et al., Differential kynurenine pathway metabolism in highly metastatic aggressive breast cancer subtypes: beyond IDO1-induced immunosuppression. Breast Cancer Res. 22, 113 (2020)
A.N. Macintyre, V.A. Gerriets, A.G. Nichols, R.D. Michalek, M.C. Rudolph, D. Deoliveira et al., The glucose transporter Glut1 is selectively essential for CD4 T cell activation and effector function. Cell Metab. 20, 61–72 (2014)
W. Li, T. Tanikawa, I. Kryczek, H. Xia, G. Li, K. Wu et al., Aerobic glycolysis controls myeloid-derived suppressor cells and tumor immunity via a specific CEBPB isoform in triple-negative breast cancer. Cell Metab. 28, 87-103.e6 (2018)
A. Schurich, I. Magalhaes, J. Mattsson, Metabolic regulation of CAR T cell function by the hypoxic microenvironment in solid tumors. Immunotherapy 11, 335–345 (2019)
G. Multhoff, P. Vaupel, Hypoxia compromises anti-cancer immune responses. Adv. Exp. Med. Biol. 1232, 131–143 (2020)
K. Sek, L.M. Kats, P.K. Darcy, P.A. Beavis, Pharmacological and genetic strategies for targeting adenosine to enhance adoptive T cell therapy of cancer. Curr. Opin. Pharmacol. 53, 91–97 (2020)
D. Jin, J. Fan, L. Wang, L.F. Thompson, A. Liu, B.J. Daniel et al., CD73 on tumor cells impairs antitumor T-cell responses: a novel mechanism of tumor-induced immune suppression. Cancer Res. 70, 2245–2255 (2010)
V. Huber, C. Camisaschi, A. Berzi, S. Ferro, L. Lugini, T. Triulzi et al., Cancer acidity: An ultimate frontier of tumor immune escape and a novel target of immunomodulation. Semin. Cancer Biol. 43, 74–89 (2017)
K. Fischer, P. Hoffmann, S. Voelkl, N. Meidenbauer, J. Ammer, M. Edinger et al., Inhibitory effect of tumor cell-derived lactic acid on human T cells. Blood 109, 3812–3819 (2007)
M. Bosticardo, S. Ariotti, G. Losana, P. Bernabei, G. Forni, F. Novelli, Biased activation of human T lymphocytes due to low extracellular pH is antagonized by B7/CD28 costimulation. Eur. J. Immunol. 31, 2829–2838 (2001)
Y. Wang, K.F. Goliwas, P.E. Severino, K.P. Hough, D. Van Vessem, H. Wang et al., Mechanical strain induces phenotypic changes in breast cancer cells and promotes immunosuppression in the tumor microenvironment. Lab. Invest. 100, 1503–1516 (2020)
B. Aykut, R. Chen, J.I. Kim, D. Wu, S.A.A. Shadaloey, R. Abengozar et al., Targeting Piezo1 unleashes innate immunity against cancer and infectious disease. Sci Immunol 5, eabb5168 (2020)
S. Park, Y. Shi, B.C. Kim, M.H. Jo, L.O. Cruz, Z. Gou et al., Force-dependent trans-endocytosis by breast cancer cells depletes costimulatory receptor CD80 and attenuates T cell activation. Biosens Bioelectron. 165, 112389 (2020)
S.C. Howard, D.P. Jones, C.H. Pui, The tumor lysis syndrome. N Engl. J. Med. 364, 1844–1854 (2011)
K.A. Hay, L.A. Hanafi, D. Li, J. Gust, W.C. Liles, M.M. Wurfel et al., Kinetics and biomarkers of severe cytokine release syndrome after CD19 chimeric antigen receptor-modified T-cell therapy. Blood 130, 2295–2306 (2017)
J.N. Brudno, J.N. Kochenderfer, Recent advances in CAR T-cell toxicity: Mechanisms, manifestations and management. Blood Rev. 34, 45–55 (2019)
M. Hudecek, M.T. Lupo-Stanghellini, P.L. Kosasih, D. Sommermeyer, M.C. Jensen, C. Rader et al., Receptor affinity and extracellular domain modifications affect tumor recognition by ROR1-specific chimeric antigen receptor T cells. Clin. Cancer Res. 19, 3153–3164 (2013)
N. Xu, D.C. Palmer, A.C. Robeson, P. Shou, H. Bommiasamy, S.J. Laurie et al., STING agonist promotes CAR T cell trafficking and persistence in breast cancer. J. Exp Med. 218, e20200844 (2021)
P.S. Adusumilli, L. Cherkassky, J. Villena-Vargas, C. Colovos, E. Servais, J. Plotkin et al., Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 6, 261ra151 (2014)
C.E. Brown, B. Aguilar, R. Starr, X. Yang, W.C. Chang, L. Weng et al., Optimization of IL13Rα2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 26, 31–44 (2018)
A. Klampatsa, V. Dimou, S.M. Albelda, Mesothelin-targeted CAR-T cell therapy for solid tumors. Expert. Opin. Biol. Ther. 21, 473–486 (2020)
S. Yu, M. Yi, S. Qin, K. Wu, Next generation chimeric antigen receptor T cells: safety strategies to overcome toxicity. Mol. Cancer 18, 125 (2019)
T. Soundara Rajan, A. Gugliandolo, P. Bramanti, E. Mazzon, In vitro-transcribed mRNA chimeric antigen receptor T cell (IVT mRNA CAR T) therapy in hematologic and solid tumor management: A preclinical update. Int. J. Mol. Sci. 21, 6514 (2020)
A. Ager, High endothelial venules and other blood vessels: critical regulators of lymphoid organ development and function. Front. Immunol. 8, 45 (2017)
M. Martinez, E.K. Moon, CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 10, 128 (2019)
Y. Zhang, X. Xie, P.N. Yeganeh, D.J. Lee, D. Valle-Garcia, K.F. Meza-Sosa et al., Immunotherapy for breast cancer using EpCAM aptamer tumor-targeted gene knockdown. Proc. Natl. Acad. Sci. U. S. A. 118, e2022830118 (2021)
L. Cherkassky, A. Morello, J. Villena-Vargas, Y. Feng, D.S. Dimitrov, D.R. Jones et al., Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 126, 3130–3144 (2016)
R. Sun, H. Luo, J. Su, S. Di, M. Zhou, B. Shi et al., Olaparib suppresses MDSC recruitment via SDF1α/CXCR4 axis to improve the anti-tumor efficacy of CAR-T cells on breast cancer in mice. Mol. Ther. 29, 60–74 (2021)
Y. Zou, B. Liu, L. Li, Q. Yin, J. Tang, Z. Jing et al., IKZF3 deficiency potentiates chimeric antigen receptor T cells targeting solid tumors. Cancer Lett. 524, 121–130 (2022)
G. Tóth, J. Szöllősi, H. Abken, G. Vereb, Á. Szöőr, A small number of HER2 redirected CAR T cells significantly improves immune response of adoptively transferred mouse lymphocytes against human breast cancer xenografts. Int. J. Mol. Sci. 21, 1039 (2020)
Acknowledgements
The authors would like to thank the Fertility and Infertility Research Center, Health Technology Institute, Kermanshah University of Medical Sciences for supporting the present manuscript ideas.
Funding
The authors did not receive support from any organization for the submitted work.
Author information
Authors and Affiliations
Contributions
AB collected data from databases, wrote and revised the manuscript, and designed the figures. MB participated in writing and revising the manuscript. MKh conceptualized and supervised the project and revised the manuscript.
Corresponding author
Ethics declarations
Ethical approval and Consent to participate
Not applicable.
Consent for publication
Not applicable.
Data availability
Not applicable.
Competing interests
The authors have no competing interests to declare relevant to this article's content.
Additional information
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Bozorgi, A., Bozorgi, M. & Khazaei, M. Immunotherapy and immunoengineering for breast cancer; a comprehensive insight into CAR-T cell therapy advancements, challenges and prospects. Cell Oncol. 45, 755–777 (2022). https://doi.org/10.1007/s13402-022-00700-w
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13402-022-00700-w