
Abstract
Streptomyces albus J1074 is one of the most popular heterologous expression platforms among streptomycetes. Identification of new genes and mutations that influence specialized metabolism in this species is therefore of great applied interest. Here, we describe S. albus KO-1304 that was isolated as a spontaneous lincomycin-resistant variant of double rpsLR94G rsmGR15SG40E mutant KO-1295. Besides altered antibiotic resistance profile, KO-1304 exhibited increased antibiotic activity as compared to its parental strains. KO-1304 genome sequencing revealed mutations within gene XNR_2147 encoding putative TetR-like protein. Gene XNR_2146 for efflux protein is the most likely target of repressing action of Xnr_2147. Our data agree with the scenario where lincomycin resistance phenotype of KO-1304 arose from inability of mutated Xnr_2147 protein to repress XNR_2146. Introduction of additional copy of XNR_2146 into wild type strain increased antibiotic activity of the latter, attesting to the practical value of transporter genes for strain improvement.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The actinobacterium Streptomyces albus (= albidoflavus (Shashkov et al. 2016)) J1074 was isolated in 1980 as a S. albus G derivative deficient in the SalGI restriction-modification system (Chater and Wilde 1980). Later, J1074 served as a model to study phage restriction phenomena in streptomycetes, and in the 1990s, it has gained prominence as a host for heterologous expression of antibiotic biosynthetic gene clusters (BGCs) (Myronovskyi and Luzhetskyy 2019). It is expected that simple-to-use expression platforms will accelerate the access to novel natural products encrypted in environmental DNA and dormant gene clusters of actinobacteria. The need for bioactive small molecules is felt by all relevant fields of medicine, agriculture, and animal healthcare, but is especially pronounced in the area of chemotherapy of infectious diseases (Wright 2017; Bergeijk et al. 2020).
Fast, highly dispersed growth and genetic amenability make J1074 a strain of first choice for expression of BGCs from actinobacteria (Iqbal et al. 2016; Yuzawa et al. 2018). Recent rigorous comparative studies (Tan et al. 2017) have further strengthened the rather intuitive notion of outstanding properties of J1074. A variety of genetic approaches have been applied to J1074 to optimize it for high-level expression of BGCs. These include the development of synthetic promoters (Siegl et al. 2013), new inducible expression systems (Horbal et al. 2014), introduction of beneficial regulatory genes (Kallifidas et al. 2018), and genome reduction (Myronovskyi et al. 2018). We recently described a method of genetic engineering of J1074 derivatives carrying point mutations within gene rpsL for ribosomal protein S12 (Lopatniuk et al. 2019). The rpsL mutants of Streptomyces are often characterized by increased or activated specialized metabolism, and identification of many beneficial rpsL mutations is facilitated by the fact that they also cause resistance to streptomycin (Tamehiro et al. 2003; Gromyko et al. 2004; Tanaka et al. 2009). It has to be noted that rpsL mutations are not only ones known to boost specialized metabolism. We showed previously that antibiotic overproducers can be found among spontaneous mutants resistant to rifampicin, gentamicin, paramomycin, and other drugs (Wang et al. 2008; Tanaka et al. 2013); these mutations are not located in rpsL and in many cases were not mapped at all. It is attractive therefore to combine several antibiotic resistance-conferring mutations in one strain in order to further improve its antibiotic production (Ochi 2017). We set out to apply this approach to strain S. albus R94G, one of the most promising rpsL mutants that we previously described (Lopatniuk et al. 2019). In this report, we focus on characterization of triple mutant KO-1304 which has resulted from sequential introduction of mutations leading to low-level resistance to streptomycin and then to lincomycin (Fig. 1). We provide evidence that both increased lincomycin resistance and antibiotic productivity of KO-1304 stem from deregulated expression of XNR_2146 gene for transport protein of major facilitator superfamily. To our knowledge, this is the first report that genetic perturbations within efflux-related genes may lead to increased lincomycin resistance and antibiotic activity of a streptomycete.

Pedigree of the strains mentioned in this work. S. albus SAM2 is the initial (wild type) strain for the mutants R94G, KO-1295, and KO-1304. Genotypes are shown in the brackets next to the strain’s name. Strr and Linr stand for phenotypes of streptomycin and lincomycin resistance, respectively. Please see the main text for more details
Materials and methods
Bacterial strains and growth media
Strains and plasmids used in this study are listed in Table S1, Electronic Supplementary Materials (ESM). S. albus SAM2 (J1074 derivative with deletion of φC31 pseB4) was used in all experiments as the original strain (Bilyk and Luzhetskyy 2014). Antibiotic-sensitive test cultures Staphylococcus aureus 209P, Bacillus cereus ATCC19637, and Debaryomyces hansenii VKM Y-9 were grown at 30 °C during the bioassays. S. aureus and D. hansenii were grown on tryptic soy broth agar (TSA) medium (Merck KGaA, Darmstadt, Germany). B. cereus was grown on BCA (Bacillus cereus agar) medium (g/L: КH2PO4, 3; К2HPO4, 7; sodium citrate, 0.5; MgSO4, 0.1; (NH4)2SO4, 1; glucose, 2; aгap, 16; peptone, 0.2; distilled water to 1 L). E. coli strains were grown at 37 °C on LA medium supplemented with selective antibiotics (Kieser et al. 2000). For intergeneric matings, Streptomyces albus strains were grown on SFM or OM agar (Koshla et al. 2017) at 30 °C. To reveal endogenous antibacterial and antifungal activities, S. albus strains were grown on SG2, GYM, and R5 agar plates for 120 h; media recipes are described in Koshla et al. (2017).
Antibiotic activity assays
Conditions of S. albus antibiotic activity assays using agar plug and disk diffusion methods are described in Koshla et al. (2017). For HPLC–MS analyses, the S. albus strains were grown in R5 liquid medium for 120 h. Supernatant was extracted with ethyl acetate, and cells were extracted with methanol-acetone (1:1) as described earlier (Ahmed et al. 2017). Downstream analysis of the extracts was performed as described in Koshla et al. (2021). Culture broth samples and cell fractions taken from submerged fermentation were also analyzed with the help of well diffusion assay. For this assay, plates contained two layers of agar: The bottom layer was 20 mL of tryptic soy agar (TSA; 2% (w/v) agar), and the upper was 10 mL of TSA (0.7% (w/v) agar) where 200 μL of fresh D. hansenii cell suspension or B. cereus spore suspension (OD600 = 0.6) were re-suspended. Wells (Ø 7 mm) were made in the top layer, and 10 µL of the biological sample (either supernatant or cells suspended in water) was poured into the former. Bioassay plates were incubated at 30 °C and examined after 24 h of growth.
Antibiotic resistance assays
Spore suspensions of SAM2 and its derivatives were harvested from the lawns grown on SFM agar plates for 5 days. These suspensions were used to determine resistance spectra of the strains to common antibiotics using disk diffusion assay (Bauer et al. 1966). We also used resazurin assay in 96-well format, as described in Lee et al. (2020), to determine minimal inhibitory concentrations (MICs) of the lincomycin for the studied strains.
Genome sequencing and analysis
Total DNA of SAM2, KO-1295, and KO-1304 strains were purified from 24-h culture grown at 30 °C in tryptic soy broth according to salting out procedure No 4 (Kieser et al. 2000). DNA concentrations and quality were determined using Trinean Xpose (Gentbrugge, Belgium) and Agilent RNA Nano 6000 kit on Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA). The DNA samples that have passed all quality control checks (Trinean measurements, A260/A230 and A260/A280 values no less than 2.0, amount of DNA no less than 2 µg in 50 µL of water; Agilent 2100 measurements, DNA integrity number 8 ÷ 10) were used to prepare whole-genome sequencing library for Illumina (TruSeq DNA PCR-Free Kit). The Illumina library was sequenced on HiSeq platform. Raw data of S. albus strains, reference sequence of J1074, and Excel table with called variants and supplementary materials can be found at the in-house S. albus genomics database maintained by Lviv University research group: https://biotools.online/media/. At the quality control stage, the sequence reads were examined for overall quality and presence of Illumina adapters with FastQC (http://www.bioinformatics.babrahamac.uk/projects/fastqc). In order to omit poor quality data from further analysis, we trimmed low quality read ends and filtered low quality reads by using Trimmomatic version 0.36 (Bolger et al. 2014). Sequencing reads were aligned to reference J1074 genome (accession number CP004370) with Bowtie2 version 2.2.5 (Langmead and Salzberg 2012). SNP and DIP detection was performed with ReadXplorer (Hilker et al. 2014). Illumina coverage was 55–165 × for all strains (see the abovementioned in-house database for more data). We used primers described in (Tanaka et al. 2009) to check rsmG mutations.
Plasmid construction
All vectors and plasmids are listed in Table S1, ESM. The XNR_2146 and XNR_2147 genes were amplified from the SAM2 genome with primer pairs Xnr2146_xbaI_up (AAATCTAGATCCACCAGAACGCAGGTAC), Xnr2146_ecoRI_rp (AAAGAATTC GTCGGTCAGCCCAGGTGGAC) and Xnr2147_xbaI_up (AAATCTAGA GTTTGGAAGCGATGTCCGACG), and Xnr2147_ecoRI_rp (AAAGAATTCGAA ACGGGGAGGAAAGGGTG), respectively. Sites for restriction endonucleases within the primers are underlined. Except for rsmG primers mentioned above, the rest of the oligonucleotides were designed in this work using S. albus J1074 genome as a reference with the help of Geneious Prime software version 2017 R11.0 (Biomatters Ltd, New Zealand). PCR products were digested with XbaI and EcoRI (Thermo Fisher Scientific, USA) and ligated into pTES vector pre-digested with the same enzymes. The recombinant plasmids pVMC and pTET carrying XNR_2146 and XNR_2147 genes, respectively, were thus generated. The intergenic region containing XNR_2146 promoter was amplified from the SAM2 genome using primers xnr2146prom_up (AAATCTAGACTTCGTCCACCGCCACATCC) and xnr2146prom_rp1 (AAAGGTACCGTGGACATAGGGGTACCTGC). PCR products and pGUS vector were digested with XbaI and KpnI restriction enzymes. The recombinant plasmid pPROM was obtained by ligation of the aforementioned DNA fragments. Identity of all plasmids was confirmed via restriction mapping and DNA sequencing.
Protein structure modelling and bioinformatics analysis of Xnr_2147
Structure inference pipeline AlphaFold2 version 2.2.3 + 49 (Jumper et al., 2021) used to model Xnr_2147 was accessed via https://console.latch.bio/workflows. The N-terminal DNA-binding domain was identified by alignment with the corresponding domain from the E. coli TetR family transcriptional regulator (GHL31221.1). Multiple sequence alignment was performed by MUSCLE with default parameters. Superposition of proteins was performed on RCSB PDB 3D Viewer resource (https://www.rcsb.org/3d-view).
Phylogenetic reconstruction
Sequences of Xnr_2146 orthologs, paralogs, and known lincomycin efflux proteins were collected from GenBank as a result of extensive literature search; the information on these proteins is summarized in ESM, Table S2. Complete amino acid sequences of the proteins were used for analysis. The optimal evolutionary model for the resulting dataset was estimated according to Akaike index AIC on IQTree server (http://iqtree.cibiv.univie.ac.at/). The tree was built with the help of phylogeny.fr v.2 server (Dereeper et al. 2006) using the evolutionary model parameters estimated by IQTree server. Reliability of tree topology was estimated with the help of approximate likelihood ratio test (aLRT), as described earlier (Anisimova and Gascuel 2006).
Results
Generation and initial characterization of S. albus KO-1295 and KO-1304 strains
Based on our knowledge of the benefits of different mutations and combinations thereof, we first plated S. albus R94G (~ 4 × 108 CFU) on GYM agar plates supplemented with 5 µg/mL of streptomycin. Out of ten streptomycin-resistant (Strr) colonies, we picked one, referred to as KO-1295, and confirmed, through targeted sequencing, that it carried missense mutations within 16S rRNA methyltransferase gene rsmG which at the amino acid level lead to substitutions R15S and G40E. RsmG is a known target for mutations leading to low-level Str resistance (Nishimura et al. 2007; Lopatniuk et al. 2019), and we determined that for KO-1295 streptomycin MIC was 10 µg/mL. Next, we plated KO-1295 spore suspensions on GYM agar supplemented with 100 µg/mL of lincomycin (Lin). Three stable lincomycin-resistant (Linr) clones were isolated, of which one, referred to as KO-1304, was taken for further analysis. KO-1304 retained the ability to grow on agar plates supplemented with 300 µg/mL Lin and showed superior antibiotic activities when grown on SG2, R5, and GYM media and tested against the fungal test culture D. hansenii and the bacterial test culture B. cereus, as compared to its parental strains (Table 1 and Fig. 2). In the disc diffusion assay, SAM2 and KO-1304 did not differ in their resistance to common antibiotics, such as beta-lactams, aminoglycosides, and macrolides. Yet, KO-1304 did show increased resistance to rifampicin after 72 h of growth, as compared to SAM2 (Table S3, ESM).

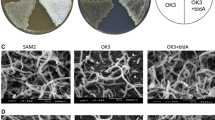
Strain KO-1304 exhibits increased antibiotic activity under conditions of submerged fermentation as compared to other strains used in this work. S. albus strains were grown in GYM for 120 h, and then supernatant and cell fractions were added to the wells of the B. cereus lawn. Antibiotic activity was analyzed by measuring zones of growth inhibition after 24 h of incubation. The two zones from the broth samples are marked by a dashed circle and an arrow to emphasize the difference between KO-1304 and its parental strain KO-1295. A similar situation was observed for the cell samples. The photograph represents typical result of four biological repeats
Uncovering the genetic basis for lincomycin resistant phenotype of KO-1304
Whereas the nature of Strr-conferring mutations is well studied (Ochi 2017), much less is known about the genetic rearrangements that lead to Linr phenotypes (Wang et al. 2017). We therefore sequenced and compared the genomes of SAM2, KO-1295, and KO-1304 (see “Materials and methods” section for more details and raw sequencing data access). The KO-1295 genome sequencing revealed no new mutations (besides altered rsmG, see above) which would contribute to its Strr phenotype or be associated with the specialized metabolism. The only difference between KO-1295 and KO-1304 genetic makeups lied within gene XNR_2147 for TetR-like protein. In KO-1304 mutant, this gene carries three closely situated single-nucleotide deletions. They alter C-terminus of the KO-1304 protein and make it one amino acid shorter than wild type Xnr_2147 (Fig. 3A). From the available and very detailed structural information on TetR protein (Orth et al. 1998), one may suggest that mutations within XNR_2147 affect its ligand-binding/dimerization domain (LBDD). The domains boundaries within Xnr_2147 were proposed on the basis of the archetypal E. coli TetR protein (Fig. 3B) and supported by multiple sequence alignment of Xnr_2147 with a few structurally characterized the TetR/AcrR family proteins shown in Fig. 3C. The latter included putative TetR/AcrR family transcriptional repressor of Salmonella typhimurium Lt2 (accession number: 1T33_A), TetR of E. coli (GHL31221.1), TetR homolog of unknown function from Mycobacterium tuberculosis H37Rv (CCP43032.1), E. coli AcrR (CAD6020473.1), and Klebsiella pneumoniae AcrR (QUQ60756.1). Alignment of these proteins and Xnr_2147 revealed amino acid residue conservation within the N-terminal DNA-binding domain, while C-terminal domain was poorly conserved around the site where mutations occurred (see Fig. 3C). It was not surprising if one takes into account a diversity of TetR/AcrR ligands. We modeled Xnr_2147 as well as its mutated version using AlphaFold pipeline (see Methods). Superposition of Xnr_2147 and SCO0520 (the most similar AcrR-like regulator from S. coelicolor A3(2) with a known crystal structure (Filippova et al. 2011)) demonstrated their overall similarity (Fig. 3D). The mutations affected residues 205–210 of the Xnr_2147 protein, and this region did not correspond to any of the SCO0520 helices due to the shorter length of the latter protein (194 aa) versus Xnr_2147 (215 aa). We also compared Xnr_2147 models with the other well-studied TetR proteins, yet these attempts have not yielded any conclusive information (data not shown). Hence, available structural information about TetR proteins does not provide insight into the functional significance of the mutated region of Xnr_2147, underscoring the need for experimental interrogation. At this point, we suppose that amino acid substitutions within the LBDD alter the Xnr_2147 dimerization properties. Ultimately, this would preclude mutated Xnr_2147 from binding to its operator sequences, as the dimerization of TetR monomers is essential for TetR protein to interact with DNA.

Bioinformatic analysis of the Xnr_2147 protein. A BLASTP-assisted pairwise alignment of C-terminal amino acid sequences of the wild type Xnr_2147 protein (Xnr_2147_wt) and its mutant form found in KO-1304 (Xnr_2147_ko1304). Altered sequences of Ko-1304 version of Xnr_2147 is boxed. B Domain boundaries of Xnr_2147 based on the E. coli TetR protein data and multiple sequence alignment (see below part C). DBD, DNA-binding domain; LBDD, ligand-binding and dimerization domain; aa, amino acid(s). C Identification of DNA-binding domain by multiple sequence alignment of TetR/AcrR proteins (see text for details). The C-terminus shows moderate conservation across some but not all aligned sequences; mutated region of Xnr_2147 is marked by an arrow. D Structural comparison of SCO0520 (turquoise), wild-type Xnr_2147 (blue), and mutated Xnr_2147 (purple). The mutated region and the corresponding region in wild type Xnr_2147 are indicated by arrows and green color. E A model of Xnr_2147-based regulation. The wild-type Xnr_2147 repressor binds the XNR_2146 promoter and may also repress its own transcription. The mutated Xnr_2147 repressor is deficient in dimerization, which is obligatory for TetR proteins to bind DNA. Hence, transcription of XNR_2146 is relieved of Xnr_2147 repressing action, which leads to increased resistance of KO-1304 to lincomycin
The XNR_2147 is flanked by genes XNR_2148 for putative metalloprotease and XNR_2146 for transmembrane efflux protein of major facilitator superfamily. Orthologs of the aforementioned three genes are ubiquitously found in streptomycete genomes, forming a highly syntenous region (ESM Fig. S1). We hypothesized that XNR_2147 and XNR_2146 constitute a two-gene module that functions by analogy to paradigmatic tetR-tetA pair (Cuthbertson and Nodwell 2013), where loss of or decreased activity of repressor protein Xnr_2147 would lead to increased expression of XNR_2146, and, consequently, to Lin resistance (Fig. 3E). This idea is circumstantially supported by available RNA-seq data showing that higher transcription of XNR_2147 is associated with lower or zero transcription of efflux gene XNR_2146 (Zaburannyi et al. 2014) (see Fig. S2, ESM). Also, the involvement of efflux protein in lincomycin resistance is known for pathogenic and producer bacteria. Nevertheless, Xnr_2146 protein is clearly different from known lincomycin pumps (denoted as Lmr and Lsa), as could be judged from phylogenetic reconstruction (Fig. 4). In the tree Xnr_2146 grouped primarily with its orthologs found in different Streptomyces genomes (see Fig. S1, ESM) and not with Lmr proteins (see Fig. 4).

Unrooted maximum-likelihood tree of amino acids sequences of efflux protein Xnr_2146 (marked with arrow), its orthologs, paralogs, and known lincomycin efflux proteins. Confidence indices aLRT are shown only on the selected nodes of interest for the sake of clarity of the figure. On the other nodes aLRT values were at least 0.5. The scale bar represents 2.0 aa substitution per aa position. Source data are given in Supplementary Table S2 (ESM)
To verify our model of XNR_2147 action in S. albus, we cloned XNR_2146 promoter (xnr2146p) into reporter plasmid pGUS (Myronovskyi et al. 2011) so that xnr2146p would drive the expression of glucuronidase (GusA) reporter gene gusA. The resultant plasmid pPROM was introduced into KO-1304 and its parental strain KO-1295. S. albus KO-1295 pPROM+ strain was expected to show low to zero expression of the reporter gene due to the presence of the functional repressor Xnr_2147. Since we suppose that Xnr_2147 has lost its repressing function in KO-1304, xnr2146p should drive the transcription of reporter gene gusA in this mutant. In KO-1295, xnr2146p exhibited low albeit detectable GusA activity after 72 h of growth and no activity after 120 h, as could be judged from plate assay of GusA activity. In contrast, KO-1304 pPROM+ showed GusA activity after 72 h of growth (higher than that of KO-1295 at the same time point) which significantly increased at 120-h time point (Fig. 5).

Mutations within XNR_2147 gene prompt active transcription from XNR_2146 promoter in late stages of growth. Top row, GusA-mediated assay of the transcription from XNR_2146 promoter (plasmid pPROM) in KO-1304 mutant and its parental strain KO-1295. Strain SAM2 carrying plasmid pSETGUS (tipAp-gusA) was used as a positive control. Also, 6-µL drops of chromogenic solution (X-Gluc, 60 µg/µL) were applied to the lawns of the aforementioned strains after 72 h and 120 h of growth, and the plates (top and bottom faces) were photographed on 123rd h. Note strong dark-blue coloring of pROM+ KO-1304 lawn upon X-Gluc application after 120 h of growth, while the parental pROM+ strain remains colorless at this time point. Bottom row, negative controls; note the absence of chromogenic signal upon application of X-Gluc to the lawns of strains carrying empty vector pGUS. Photographs represent typical result of four experiments
Evidence for involvement of XNR_2146 in lincomycin resistance and enhancement of specialized metabolism of S. albus
We cloned genes XNR_2146 and XNR_2147 individually into integrative expression vector pTES (Herrmann et al. 2012) to yield plasmids pVMC and pTET, respectively (see Table S1). In these plasmids, expression of the target gene is under control of strong constitutive promoter ermEp. Also, expression of transporter gene XNR_2146 would be decoupled from regulatory influences of XNR_2147, as only coding sequence of XNR_2146, along with its RBS, has been cloned into the vector. The plasmids were introduced into SAM2 and KO-1304 strains, and their Lin minimal inhibitory concentrations (liquid culture in 96-well format, see the Methods) were compared to the control (carrying empty vector pTES) strains. The data are summarized in Table 2. The introduction of the extra copy of transporter gene XNR_2146 clearly increased Lin resistance of S. albus, both in wild type (SAM2) and KO-1304 genetic backgrounds. This observation supports the hypothesis about implication of aforementioned gene in Linr phenotype of KO-1304. Interestingly, the introduction of wild type copy of XNR_2147 gene into KO-1304 did not restore the Lin resistance of KO-1304 to the level of the wild type. This observation will be treated in more details in the “Discussion” section.
Introduction of additional copy of XNR_2146 gene into parental (SAM2) strain also enhanced its specialized metabolism, as judged from agar plug assays (Table 3). To gain more insight into this observation, we subjected the extracts from SAM2 pVMC+ and SAM2 carrying empty vector pTES (biomass and spent medium separately) grown in liquid medium R5 for 120 h to LC–MS analysis (see Methods). Our findings are summarized in Fig. 6. Overall, both analyzed strains did not differ in the pattern of specialized metabolites. Increased (approximately threefold) accumulation of butenolide (cation [M + H]+ m/z, calculated, 225.1491 Da; observed, 225.1487 Da; mass error tolerance 5 ppm) in the supernatant of SAM2 pVMC+ as compared to SAM2 strain was one prominent exception. We revealed the presence of unknown 255 Da peak in the spent medium of SAM2 pVMC+. The extracts from the cells of SAM2 pVMC+ also revealed the presence of several unknown mass peaks which are significantly more abundant as compared to SAM2.

Total ion chromatograms of the extracts prepared from either supernatant (A) or biomass (B) of S. albus SAM2 and SAM2 pVMC+ (VMC) strains. The x axis, retention time; y axis, peak intensity, in arbitrary units. Interrogation signs mark unknown mass peaks. The chromatograms represent typical pattern of peaks observed in four independent experiments, biomass dry weight differed by no more than 10% of the mean value
Discussion
Identification of novel genes and mutations that enhance specialized metabolism of streptomycetes is of great interest as they are valuable tools to increase the production of useful compounds and potentially may lead to activation of cryptic biosynthetic gene clusters (Ochi 2017). Such a research direction is especially relevant for recognized Streptomyces expression chassis, which S. albus J1074 arguably belongs to. Here, we show that sequential introduction of streptomycin and lincomycin resistance mutations into rpsL-engineered strain R94G improves specialized metabolism of the latter. We identified that Linr phenotype of KO-1304 is caused by mutation within gene XNR_2147 for TetR type repressor. Our data support the scenario where this mutation relieves the transcription of nearby exporter gene XNR_2146, which in its turn increases resistance to Lin via its active efflux. Our data also suggest that XNR_2146 promoter is rather weak, as we observed significant accumulation of GusA activity only after prolonged (120 h) cultivation.
Introduction of native XNR_2147 into KO-1304 did not lead to decrease in Lin resistance of the mutant. In this regard, we note that the Lin concentrations being used in our work far exceed the inducing concentrations. So, the amount of drug could be enough to disable all active Xnr_2147 dimers, thus cancelling out its effects on XNR_2146 expression. Part of active Xnr_2147 monomers could also be sequestered into nonfunctional dimers by mutant Xnr_2147 proteins. The TetR-based regulatory circuits are known for exquisite sensitivity towards effector molecules and often show paradoxical effects on resistance levels because of combination of promoter strength, differential affinity towards operators, and other environmental factors (Batchelor et al. 2004; Bertram and Hillen 2008). Further in-depth analysis of XNR_2147 and XNR2146 regulatory relationship is needed to fully understand the mechanism of their interaction.
It is likely that the export function of Xnr_2146 is also the reason for enhanced specialized metabolism of S. albus KO-1304. Perhaps, being the member of multidrug transporters, Xnr_2146 facilitates the extrusion of other endogenous specialized metabolites (such as butenolide) accumulated by S. albus, which might trigger the production of the other bioactive compounds (Ahmed et al. 2017). Whatever the real scenario is, we find it remarkable that efflux-associated genes showed up for the first time as a cause of spontaneous antibiotic resistance mutation that influence specialized metabolism. We believe that specialized metabolite transport processes deserve more attention as a tool to manipulate natural product biosynthesis. Indeed, several recent reports support the idea that better knowledge of transporters and their manipulation is vital to attain high production of valuable metabolites (Peng et al. 2018; Chen et al. 2019; Chu et al. 2021).
Data availability
All data described in the manuscript are available either in Electronic Supplementary Materials associated with this manuscript or in genomic database described in the Methods section.
Code availability
Not applicable.
References
Ahmed Y, Rebets Y, Tokovenko B, Brötz E, Luzhetskyy A (2017) Identification of butenolide regulatory system controlling secondary metabolism in Streptomyces albus J1074. Sci Rep 7:9784. https://doi.org/10.1038/s41598-017-10316-y
Anisimova M, Gascuel O (2006) Approximate likelihood-ratio test for branches: a fast, accurate, and powerful alternative. Syst Biol 55:539–552. https://doi.org/10.1080/10635150600755453
Batchelor E, Silhavy TJ, Goulian M (2004) Continuous control in bacterial regulatory circuits. J Bacteriol 22:7618–7625. https://doi.org/10.1128/jb.186.22.7618-7625.2004
Bauer A, Kirby W, Sherris J, Turck M (1966) Antibiotic susceptibility testing by a standardized single disk method. Am J Clin Pathol 36:493–496
Bertram R, Hillen W (2008) The application of Tet repressor in prokaryotic gene regulation and expression. Microb Biotechnol 1:2–16. https://doi.org/10.1111/j.1751-7915.2007.00001.x
Bilyk B, Luzhetskyy A (2014) Unusual site-specific DNA integration into the highly active pseudo-attB of the Streptomyces albus J1074 genome. Appl Microbiol Biotechnol 98:5095–5104. https://doi.org/10.1007/s00253-014-5605-y
Bolger A, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Chater KF, Wilde LC (1980) Streptomyces albus G mutants defective in the SalGI restriction-modification system. J Gen Microbiol 116:323–334. https://doi.org/10.1099/00221287-116-2-323
Chen H, Wang J, Cui J, Wang C, Liang S, Liu H, Wen J (2019) Negative regulation of bleomycins biosynthesis by ArsR/SmtB family repressor BlmR in Streptomyces verticillus. Appl Microbiol Biotechnol 103:6629–6644. https://doi.org/10.1007/s00253-019-09923-8
Chu L, Li S, Dong Z, Zhang Y, Jin P, Ye L, Wang X, Xiang W (2021) Mining and engineering exporters for titer improvement of macrolide biopesticides in Streptomyces. Microb Biotechnol. https://doi.org/10.1111/1751-7915.13883
Cuthbertson L, Nodwell JR (2013) The TetR family of regulators. Microbiol Mol Biol Rev 77:440–475. https://doi.org/10.1128/mmbr.00018-13
Dereeper A, Guignon V, Blanc G, Audic S, Buffet S, Chevenet F, Dufayard JF, Guindon S, Lefort V, Lescot M, Claverie JM, Gascuel O (2006) Phylogeny.fr: robust phylogenetic analysis for the non-specialist. Nucleic Acids Res 36(Web Server issue):W465–W469. https://doi.org/10.1093/nar/gkn180
Filippova E, Chruszcz M, Cymborowski M, Gu J, Savchenko A, Edwards A, Minor W (2011) Crystal structure of a putative transcriptional regulator SCO0520 from Streptomyces coelicolor A3(2) reveals an unusual dimer among TetR family proteins. J Struct Funct Genomics 12(3):149–157. https://doi.org/10.1007/s10969-011-9112-4
Gromyko O, Rebets Y, Ostash B, Luzhetskyy A, Fukuhara M, Bechthold A, Nakamura T, Fedorenko V (2004) Generation of Streptomyces globisporus SMY622 strain with increased landomycin E production and it’s initial characterization. J Antibiot 57:383–389. https://doi.org/10.7164/antibiotics.57.383
Herrmann S, Siegl T, Luzhetska M, Petzke L, Jilg C, Welle E, Erb A, Leadlay PF, Bechthold A, Luzhetskyy A (2012) Site-specific recombination strategies for engineering actinomycete genomes. Appl Environ Microbiol 78:1804–1812. https://doi.org/10.1128/AEM.06054-11
Hilker R, Stadermann KB, Doppmeier D, Kalinowski J, Stoye J, Straube J, Winnebald J, Goesmann A (2014) ReadXplorer—visualization and analysis of mapped sequences. Bioinformatics 30:2247–2254. https://doi.org/10.1093/bioinformatics/btu205
Horbal L, Fedorenko V, Luzhetskyy A (2014) Novel and tightly regulated resorcinol and cumate-inducible expression systems for Streptomyces and other actinobacteria. Appl Microbiol Biotechnol 98:8641–8655. https://doi.org/10.1007/s00253-014-5918-x
Iqbal HA, Low-Beinart L, Obiajulu JU, Brady SF (2016) Natural product discovery through improved functional metagenomics in Streptomyces. J Am Chem Soc 138:9341–9344. https://doi.org/10.1021/jacs.6b02921
Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, Bridgland A, Meyer C, Kohl SAA, Ballard AJ, Cowie A, Romera-Paredes B, Nikolov S, Jain R, Adler J, Back T, Petersen S, Reiman D, Clancy E, Zielinski M, Steinegger M, Pacholska M, Berghammer T, Bodenstein S, Silver D, Vinyals O, Senior AW, Kavukcuoglu K, Kohli P, Hassabis D (2021) Highly accurate protein structure prediction with AlphaFold. Nature 596(7873):583–589. https://doi.org/10.1038/s41586-021-03819-2
Kallifidas D, Jiang G, Ding Y, Luesch H (2018) Rational engineering of Streptomyces albus J1074 for the overexpression of secondary metabolite gene clusters. Microb Cell Fact 17:25. https://doi.org/10.1186/s12934-018-0874-2
Kieser T, Bibb MJ, Buttner MJ, Chater KF, Hopwood DA (2000) Practical Streptomyces genetics. The John Innes Foundation, Norwich
Koshla O, Lopatniuk M, Rokytskyy I, Yushchuk O, Dacyuk Y, Fedorenko V, Luzhetskyy A, Ostash B (2017) Properties of Streptomyces albus J1074 mutant deficient in tRNALeuUAA gene bldA. Arch Microbiol 199:1175–1183. https://doi.org/10.1007/s00203-017-1389-7
Koshla O, Lopatniuk M, Borys O, Misaki Y, Kravets V, Ostash I, Shemediuk A, Ochi K, Luzhetskyy A, Fedorenko V, Ostash B (2021) Genetically engineered rpsL merodiploidy impacts secondary metabolism and antibiotic resistance in Streptomyces. World J Microbiol Biotechnol 37:62. https://doi.org/10.1007/s11274-021-03030-5
Langmead B, Salzberg S (2012) Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. https://doi.org/10.1038/nmeth.1923
Lee JH, Yoo JS, Kim Y, Kim JS, Lee EJ, Roe JH (2020) The WblC/WhiB7 transcription factor controls intrinsic resistance to translation-targeting antibiotics by altering ribosome composition. mBio 11:e00625–20. https://doi.org/10.1128/mbio.00625-20
Lopatniuk M, Myronovskyi M, Nottebrock A, Busche T, Kalinowski J, Ostash B, Fedorenko V, Luzhetskyy A (2019) Effect of “ribosome engineering” on the transcription level and production of S. albus indigenous secondary metabolites. Appl Microbiol Biotechnol 103:7097–7110. https://doi.org/10.1007/s00253-019-10005-y
Myronovskyi M, Welle E, Fedorenko V, Luzhetskyy A (2011) Beta-glucuronidase as a sensitive and versatile reporter in actinomycetes. Appl Environ Microbiol 77(15):5370–5383. https://doi.org/10.1128/AEM.00434-11
Myronovskyi M, Rosenkränzer B, Nadmid S, Pujic P, Normand P, Luzhetskyy A (2018) Generation of a cluster-free Streptomyces albus chassis strains for improved heterologous expression of secondary metabolite clusters. Metab Eng 49:316–324. https://doi.org/10.1016/j.ymben.2018.09.004
Myronovskyi M, Luzhetskyy A (2019) Heterologous production of small molecules in the optimized Streptomyces hosts. Nat Prod Rep 36:1281–1294. https://doi.org/10.1039/c9np00023b
Nishimura K, Hosaka T, Tokuyama S, Okamoto S, Ochi K (2007) Mutations in rsmG, encoding a 16S rRNA methyltransferase, result in low-level streptomycin resistance and antibiotic overproduction in Streptomyces coelicolor A3(2). J Bacteriol 189:3876–3883. https://doi.org/10.1128/jb.01776-06
Ochi K (2017) Insights into microbial cryptic gene activation and strain improvement: principle, application and technical aspects. J Antibiot 70:25–40. https://doi.org/10.1038/ja.2016.82
Orth P, Cordes F, Schnappinger D, Hillen W, Saenger W, Hinrichs W (1998) Conformational changes of the Tet repressor induced by tetracycline trapping. J Mol Biol 279:439–447. https://doi.org/10.1006/jmbi.1998.1775
Peng Q, Gao G, Lü J, Long Q, Chen X, Zhang F, Xu M, Liu K, Wang Y, Deng Z, Li Z, Tao M (2018) Engineered Streptomyces lividans strains for optimal identification and expression of cryptic biosynthetic gene clusters. Front Microbiol 9:3042. https://doi.org/10.3389/fmicb.2018.03042
Shashkov A, Streshinskaya G, Tul’skaya E, Senchenkova S, Baryshnikova L, Dmitrenok A, Ostash B, Fedorenko V (2016) Cell wall glycopolymers of Streptomyces albus, Streptomyces albidoflavus and Streptomyces pathocidini. Antonie Van Leeuwenhoek 109:923–936. https://doi.org/10.1007/s10482-016-0691-8
Siegl T, Tokovenko B, Myronovskyi M, Luzhetskyy A (2013) Design, construction and characterisation of a synthetic promoter library for fine-tuned gene expression in actinomycetes. Metab Eng 19:98–106. https://doi.org/10.1016/j.ymben.2013.07.006
Tamehiro N, Hosaka T, Xu J, Hu H, Otake N, Ochi K (2003) Innovative approach for improvement of an antibiotic-overproducing industrial strain of Streptomyces albus. Appl Environ Microbiol 69:6412–6417. https://doi.org/10.1128/aem.69.11.6412-6417.2003
Tan GY, Deng K, Liu X, Tao H, Chang Y, Chen J, Chen K, Sheng Z, Deng Z, Liu T (2017) Heterologous biosynthesis of spinosad: an omics-guided large polyketide synthase gene cluster reconstitution in Streptomyces. ACS Synth Biol 6:995–1005. https://doi.org/10.1021/acssynbio.6b00330
Tanaka Y, Komatsu M, Okamoto S, Tokuyama S, Kaji A, Ikeda H, Ochi K (2009) Antibiotic overproduction by rpsL and rsmG mutants of various actinomycetes. Appl Environ Microbiol 75:4919–4922. https://doi.org/10.1128/AEM.00681-09
Tanaka Y, Kasahara K, Hirose Y, Murakami K, Kugimiya R, Ochi K (2013) Activation and products of the cryptic secondary metabolite biosynthetic gene clusters by rifampin resistance (rpoB) mutations in actinomycetes. J Bacteriol 195:2959–2970. https://doi.org/10.1128/jb.00147-13
van Bergeijk DA, Terlouw BR, Medema MH, van Wezel GP (2020) Ecology and genomics of Actinobacteria: new concepts for natural product discovery. Nat Rev Microbiol 18:546–558. https://doi.org/10.1038/s41579-020-0379-y
Wang G, Hosaka T, Ochi K (2008) Dramatic activation of antibiotic production in Streptomyces coelicolor by cumulative drug resistance mutations. Appl Environ Microbiol 74:2834–2840. https://doi.org/10.1128/aem.02800-07
Wang G, Izawa M, Yang X, Xu D, Tanaka Y, Ochi K (2017) Identification of a novel lincomycin resistance mutation associated with activation of antibiotic production in Streptomyces coelicolor A3(2). Antimicrob Agents Chemother 61:e02247-e2316. https://doi.org/10.1128/aac.02247-16
Wright GD (2017) Opportunities for natural products in 21st century antibiotic discovery. Nat Prod Rep 34:694–701. https://doi.org/10.1039/c7np00019g
Yuzawa S, Mirsiaghi M, Jocic R, Fujii T, Masson F, Benites VT, Baidoo EEK, Sundstrom E, Tanjore D, Pray TR, George A, Davis RW, Gladden JM, Simmons BA, Katz L, Keasling JD (2018) Short-chain ketone production by engineered polyketide synthases in Streptomyces albus. Nature Communications 9(1):4569. https://doi.org/10.1038/s41467-018-07040-0
Zaburannyi N, Rabyk M, Ostash B, Fedorenko V, Luzhetskyy A (2014) Insights into naturally minimised Streptomyces albus J1074 genome. BMC Genomics 15:97. https://doi.org/10.1186/1471-2164-15-97
Acknowledgements
We thank Prof. Colin P. Smith (University of Manchester, UK) for providing us with E. coli ET12567 (pUZ8002) strain.
Funding
This work was supported by grants F60/2 + F60/52 from State Fund for Fundamental Research, Bg-80F and Bg-21F from the Ministry of Education, and Science of Ukraine (MESU), DAAD fellowship 57313677 (to B.O.). V.F. acknowledges MESU for grant Bg-09F.
Author information
Authors and Affiliations
Contributions
BO and KO conceived and designed research, with conceptual input from VF and AL. V.-MT, BD, IO, ML, and TB performed research and analyzed data. JK provided genome sequencing assistance and critical reagents. BO, VF, and V.-MT wrote the paper.
Corresponding author
Ethics declarations
Ethics approval
This work did not involve the studies of human or animal subjects.
Consent to participate
Not applicable.
Consent for publication
All authors agreed on the publication.
Conflict of interest
The authors declare no competing interests.
Additional information
Communicated by Agnieszka Szalewska-Palasz
Publisher's note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Tseduliak, VM., Dolia, B., Ostash, I. et al. Mutations within gene XNR_2147 for TetR-like protein enhance lincomycin resistance and endogenous specialized metabolism of Streptomyces albus J1074. J Appl Genetics 64, 185–195 (2023). https://doi.org/10.1007/s13353-022-00738-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13353-022-00738-4