
Abstract
Inhibition of enzymes responsible for endocannabinoid hydrolysis represents an invaluable emerging tool for the potential treatment of neurodegenerative disorders. Monoacylglycerol lipase (MAGL) is the enzyme responsible for degrading 2-arachydonoylglycerol (2-AG), the most abundant endocannabinoid in the central nervous system (CNS). Here, we tested the effects of the selective MAGL inhibitor JZL184 on the 3-nitropropinic acid (3-NP)-induced short-term loss of mitochondrial reductive capacity/viability and oxidative damage in rat brain synaptosomal/mitochondrial fractions and cortical slices. In synaptosomes, while 3-NP decreased mitochondrial function and increased lipid peroxidation, JZL184 attenuated both markers. The protective effects evoked by JZL184 on the 3-NP-induced mitochondrial dysfunction were primarily mediated by activation of cannabinoid receptor 2 (CB2R), as evidenced by their inhibition by the selective CB2R inverse agonist JTE907. The cannabinoid receptor 1 (CB1R) also participated in this effect in a lesser extent, as evidenced by the CB1R antagonist/inverse agonist AM281. In contrast, activation of CB1R, but not CB2R, was responsible for the protective effects of JZL184 on the 3-NP-iduced lipid peroxidation. Protective effects of JZL184 were confirmed in other toxic models involving excitotoxicity and oxidative damage as internal controls. In cortical slices, JZL184 ameliorated the 3-NP-induced loss of mitochondrial function, the increase in lipid peroxidation, and the inhibition of succinate dehydrogenase (mitochondrial complex II) activity, and these effects were independent on CB1R and CB2R, as evidenced by the lack of effects of AM281 and JTE907, respectively. Our novel results provide experimental evidence that the differential protective effects exerted by JZL184 on the early toxic effects induced by 3-NP in brain synaptosomes and cortical slices involve MAGL inhibition, and possibly the subsequent accumulation of 2-AG. These effects involve pro-energetic and redox modulatory mechanisms that may be either dependent or independent of cannabinoid receptors’ activation.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Mitochondrial dysfunction, oxidative damage, and excitotoxicity constitute confluent and interrelated toxic mechanisms triggering neurodegeneration. Combined, disrupted cell metabolism, reactive oxygen species (ROS) formation, and overactivation of the N-methyl-D-aspartate receptor (NMDAr) subtype of glutamate receptors lead to toxic cascades with ensuing excitotoxicity (Muddapu et al. 2020). 3-Nitropropionic acid (3-NP) is a mitochondrial toxin capable of resembling several pathological features of Huntington’s disease in cell and in mammal in vivo models (Túnez et al. 2010; Hariharan et al. 2014; Maya-López et al. 2017). Selective inhibition of succinate dehydrogenase (SDH, electron transport chain complex II) activity, ATP depletion, blockade of the Krebs cycle, increased ROS formation, and secondary excitotoxicity are among the toxic effects produced by 3-NP in the CNS (Brouillet et al. 2005; Chaturvedi and Beal 2013; Maya-López et al. 2017). The propensity of 3-NP to induce the aforementioned effects has been also extended to short-term in vitro studies (Reyes-Soto et al. 2022).
The endocannabinoid system (ECS, or endocannabinoidome) consists of endocannabinoids, receptors, and the enzymes for their synthesis and degradation, and regulates several modulatory processes, including neurotransmission, immune control, and cell signaling. The activation of cannabinoid type 1 (CB1R) and type 2 (CB2R) receptors by endocannabinoids has been suggested to be responsible for the abovementioned effects (Viscomi et al. 2010; Aymerich et al. 2018). The most abundant and physiologically relevant endocannabinoids 2-arachydonoylglycerol (2-AG) and anandamide (AEA) bind to CB1R and/or CB2R, thus acting as major regulators of the central and peripheral nervous systems (Colín-González et al. 2016; Aymerich et al. 2018). The enzymatic hydrolysis of 2-AG by monoacylglycerol lipase (MAGL), and of AEA by fatty acid amide hydrolase (FAAH), is a major physiological event coordinating the end of the endocannabinoid signaling (Cravatt et al. 1996; Dinh et al. 2002; Aymerich et al. 2016). The pharmacological inhibition of MAGL and FAAH affords neuroprotective efficacy by reducing excitability via modulation of cannabinoid signaling (Cravatt et al. 1996; Petrosino and Di Marzo 2010; Carloni et al. 2012; Xu and Chen 2015; Fucich et al. 2020).
Selective inhibition of MAGL activity by JZL184 has been shown to elicit anti-inflammatory, anti-hyperalgesic, and neuroprotective effects (Kerr et al. 2013; Khasabova et al. 2014; Zhang and Thayer 2018). These effects are likely produced by decreasing the formation of beta-amyloid in APdE9 mice and in glial cells (Pihlaja et al. 2015), releasing neuroprotective and anti-inflammatory molecules in a murine MPTP parkinsonian model (Fernández-Suárez et al. 2014). This may also occur in hippocampal neurons via activation of the Nrf2/ARE signaling pathway in a model of oxygen–glucose deprivation (Xu et al. 2021). In this regard, several groups have attributed the protective effects of JZL184 to selective activation of CB2R (Aymerich et al. 2016), which are mainly expressed in immune cells.
Despite neuroprotective effects elicited by the blockade of endocannabinoid hydrolysis have already been described, the protective properties of JZL184 under toxic conditions such as mitochondrial dysfunction, oxidative damage, and excitotoxicity have yet to be demonstrated at different experimental levels. Therefore, in this study, we investigated for the first time the effects of JZL184 on different markers of the short-term 3-NP-induced toxicity in two biological preparations: crude rat brain synaptosomal/mitochondrial fractions—nerve endings containing the whole biochemical machinery for neurotransmission and cannabinoid metabolism—and rat cortical slices—tissue slices comprising the whole cellular diversity of the brain cortex and its integral circuitry—and evaluated the role of CB1R and CB2R using specific antagonists/inverse agonists as a first pharmacological approach for the design of MAGL inhibition-based therapies. Both biological preparations employed in this study represent simple and representative fractions of the brain; they are functional and easy to obtain, thus constituting an optimal first experimental approach in toxicological and pharmacological studies. The 3-NP-induced loss of mitochondrial reductive capacity and oxidative damage were challenged with JZL184, which displayed protective effects.
Material and Methods
Reagents
AM281 (1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide) and JTE907 (N-(1,3-benzodioxol-5-ylmethyl)-1,2-dihydro-7- methoxy-2-oxo-8-(pentyloxy)-3-quinolinecarboxamide) were obtained from Tocris Bioscience (Bristol, UK). JZL184 (4-nitrophenyl 4-[di(2H-1,3-benzodioxol-5-yl)(hydroxy)methyl]piperidine-1-carboxylate), 3-NP (3-nitropropionic acid), thiobarbituric acid (TBA), MTT (3-(4,5-dimethylthizol-2-yl)-2,5-diphenyltetrazolium bromide), D-mannitol, sodium azide, and dichloroindophenol sodium salt were all purchased to Sigma Chemical Co. (Sigma-Aldrich, St. Louis Missouri, USA). Quinolinic acid (QUIN) was obtained from Spectrum Chemical (New Brunswick, NJ, USA). Other reagents were obtained from known commercial sources.
Animals
Adult male Wistar rats (250–300 g; N = 30 (total number of animals used); n = 6 experiments per group; one rat representing one whole experiment) were used for the preparation of synaptosomal/mitochondrial fractions (12 rats, 6 for mitochondrial function assays and 6 for lipid peroxidation assays) and for isolation of cortical slices (18 rats, 6 for mitochondrial function assays, 6 for lipid peroxidation assays, and 6 more for the estimation of the succinate dehydrogenase activity). Since experiments were not carried out in living organisms, but in biological preparations obtained from their dissected brains, each animal served on a given day to obtain the preparations (either synaptosomes or slices). All animals were obtained from the vivarium of the Universidad Autónoma Metropolitana-Iztapalapa, kindly donated by Dr. Socorro Retana-Márquez. Rats were housed at controlled conditions of room temperature (25 ± 3 °C), humidity (50%), and light–dark cycles (12:12 h), with food and water ad libitum. All experimental manipulations were carried out following the guidance of the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23) revised 1996, and the local Ethical Committees. Formal approval to conduct the experiments described was obtained from the animal subjects review board of the Instituto Nacional de Neurología y Neurocirugía (project numbers 17/20 and 73/20). All efforts were made to minimize animals’ suffering during the experiments.
Treatment of Crude Synaptosomal/Mitochondrial Fractions
Crude synaptosomal/mitochondrial P2 fractions were obtained from adult rat brains, according to a method reported previously by us (Rangel-López et al. 2015; Maya-López et al. 2020). Briefly, the collected brains (without the cerebellum) were homogenized in 10 volumes (g/ml) of sucrose (0.32 M) and centrifuged for 10 min at 1073 g (4 °C); supernatants were recovered and re-centrifuged for 15 min at 17,000 g (4 °C). The obtained pellets were resuspended in 40 volumes (ml) of HEPES buffer (0.1 M NaCl, 0.001 M NaH2PO4, 0.005 M NaHCO3, 0.001 M CaCl2, 0.006 M glucose, 0.01 M HEPES, pH 7.4). Aliquots of equal volumes were briefly stored at −70 °C until used throughout the experiments. Protein content in synaptosomal fractions was estimated by Bradford’s method (Bradford 1976). The collected fractions were incubated in the presence of 3-NP (0.1–1 mM), 100 μM QUIN, or 50 µM FeSO4 for 60 min. Additional fractions were preconditioned for 30 min with increasing concentrations JZL184 (0.1–10 nM), the selective CB1R antagonist/inverse agonist AM281 (10 nM, added 15 min before JZL184), and/or the selective CB2R inverse agonist JTE907 (0.13 nM, also added 15 min before JZL184). Although several reports in the literature have described the toxic concentrations of 3-NP (Reyes-Soto et al. 2022), we proceeded to determine our own concentration-dependent response curve to establish the optimal toxic concentration for our experiments. The toxic concentration of QUIN was chosen from previous reports (Rangel-López et al. 2015; Chavira-Ramos et al. 2021). The concentration range for JZL184 tested herein was inferred from a previous report in which it was demonstrated that 8 nM corresponded to the IC50 for MAGL inhibition in vitro, while 10 µM was optimal to inhibit FAAH (Long et al. 2009). The AM281 and JTE907 concentrations were also inferred from previous reports (Lan et al. 1999; Li et al. 2010; García-Morales et al. 2015; Sánchez-Rodríguez et al. 2018; Chavira-Ramos et al. 2021; Reyes-Soto et al. 2022). All samples were prepared for the experimental assays designed to estimate loss of mitochondrial function and oxidative damage to lipids following their incubation in the presence of the abovementioned treatments. The quantification of protein content in samples was carried out also according to Lowry’s method (Lowry et al. 1951).
Treatment of Cortical Slices
Cortical slices were obtained according to methods previously reported (Colín-González et al. 2014; Reyes-Soto et al. 2022). Cortical slices were collected and sectioned from the frontal cortex of rat brains using a chopper. The slices (~250-µm thickness) were incubated in Krebs–Ringer modified buffer (NaCl 135 mM, KCl 5 mM, MgSO4 1 mM, K2HPO4 0.4 mM, glucose 5.5 mM, HEPES 20 mM, and CaCl2 20 mM) for 30 min at 37 °C in the presence of 5% CO2. Samples were added with JZL184 (10 nM) as a pretreatment for 30 min, and then added with 3-NP (1 mM) and incubated for 60 min at 37 °C and 5% CO2. Some slices treated with JZL184 + 3-NP were also added with AM281 (10 nM) or JTE907 (0.13 nM) for 30 min prior to JZL184. Under the experimental conditions tested in this study, a higher concentration of AM281 (1 µM) decreased the mitochondrial reductive capacity (data not shown), denoting toxicity per se. Experimental groups were categorized as follows: (A) control (an experimental condition incubated only with vehicles for all compounds: H2O instead of AM281 or JTE907; DMSO 1% instead of JZL184; PBS instead of 3-NP); (B) 3-NP alone; (C) JZL184 alone; (D) JZL184 + 3-NP; (E) AM281 + J ZL184 + 3-NP; and (F) JTE907 + JZL184 + 3-NP. The concentrations of all compounds used in this biological preparation correspond to those described above for synaptosomal/mitochondrial fractions.
Functional Assessment of Mitochondrial Activity
The functional status of mitochondrial function was assessed by the MTT reduction assay, according to previous reports (Colín-González et al. 2014; Chavira-Ramos et al. 2021; Reyes-Soto et al. 2022). The treated synaptosomal/mitochondrial fractions and cortical slices were added to 300 µL Krebs’s buffer plus 15 µL MTT reagent (5 mg/ml) and incubated for 60 min at 37 °C. The content of formazan was estimated at 570 nm in a Cytation 3 Imaging Reader (BioTek Instruments, Winooski, VT, USA), and the results were expressed as percent of MTT reduction vs. the control.
Functional Assessment of Oxidative Damage to Lipids
Oxidative damage to lipids was assessed in cortical slices according to previous reports (Colín-González et al. 2014; Reyes-Soto et al. 2022). After treatment, the synaptosomal/mitochondrial fractions and the slices were homogenized in lysis buffer, and 100 µl-aliquots were added to 50 µl of thiobarbituric acid (TBA) reagent (0.75 g TBA + 15 g trichloroacetic acid + 2.53 ml HCl) and incubated in a boiled water-bath for 20 min. Samples were then centrifuged at 3000 g for 10 min at 4 °C. The absorbance was evaluated at 532 nm in a Cytation 3 Imagin Reader (BioTek) in the collected supernatants. Results, originally calculated as nmols of TBA-reactive substances (TBARS) formed per mg of protein, were finally expressed as percent of lipid peroxidation vs. the control.
Functional Assessment of Succinate Dehydrogenase Activity
Succinate dehydrogenase (SDH) activity was quantified in cortical slices according to a method recently reported (Reyes-Soto et al. 2022). Tissue samples were homogenized in buffer solution and centrifuged (3000 g); then, aliquots (400 µL) were incubated in an added solution of phosphate buffer assay reaction (650 µL; pH 7.0) containing 0.3 M d-mannitol + 5.0 mM magnesium soluble chloride + 125 µL of 0.004 M sodium azide + 125 µL of 0.50 mM dichloroindophenol + 125 µL of 0.2 M succinate. The reduction of dichlorophenolindophenol (DCPIP; assessed as the decay of color) was recorded over 40 min at 600 nm. An UV-1603 Shimadzu (Kyoto, Japan) spectrophotometer was used to detect optical density of the solutions. The content of protein in samples was measured according to previous protocols (Lowry et al. 1951), and results were expressed as units of reduction (SDH activity) per milligram of protein (U/mg protein), where one unit of SDH activity is defined as the amount of enzyme capable of reducing 1 µmol DCPIP per min at 25 °C (Popov et al. 2010).
Statistical Analysis
Results depict independent experiments (one experiment per animal) expressed as mean values ± standard error of n = 6 experiments per group. All experiments were performed in duplicate, and mean values were used for statistical calculations. Results were analyzed by one- or two-way analysis of variance (ANOVA) followed by post hoc Dunnett’s or Tukey’s tests (GraphPad, Scientific, San Diego, CA, USA). Values of p ≤ 0.05 were considered of statistical significance.
Results
JZL184 Ameliorated the 3-Nitropropionic Acid-Induced Loss of Mitochondrial Function in Synaptosomal/Mitochondrial Fractions in a CB2R-Related Manner
To establish and confirm the optimal concentrations of 3-NP and JZL184 to be tested, first we evaluated concentration–response effects of these compounds on the basal levels of mitochondrial function in synaptosomal/mitochondrial fractions.
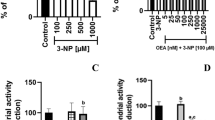
As shown in Fig. 1A, 3-NP produced a concentration-dependent decrease in mitochondrial function in a concentration range between 0.1 and 1 mM, with significant effects at 0.3 (p ≤ 0.05 vs. the control), 0.5, and 1 mM (p ≤ 0.01 vs. the control group). Since 1 mM exerted the strongest effect and this concentration is often used in other reports, we decided to use this concentration from this point on. As shown in Fig. 1B, JZL184 did not affect the basal (control; 0.6 OD units of MTT reduction) mitochondrial function at any of the concentrations tested. From this point on, we used the 10 nM concentration for subsequent experiments.

A, B Concentration-dependent effects of 3-nitropropionic acid (3-NP, in A) and JZL184 (JZL, in B) on basal mitochondrial function (mitochondrial reductive capacity) in rat brain synaptosomal/mitochondrial fractions. C, D Effect of JZL184 (JZL, 10 nM) on the 3-nitropropionic acid (3-NP, 1 mM, in A)- and quinolinic acid (QUIN, 100 µM, in B)-induced loss of mitochondrial reductive capacity in rat brain synaptosomal/mitochondrial fractions. AM281 (10 nM) and JTE907 (0.13 nM) were also used to investigate the role of cannabinoid receptors. Values represent the mean ± one SEM of n = 6 experiments per group. *p ≤ 0.05, **p ≤ 0.01, and ***p ≤ 0.001, different to the control group (CT); &&p ≤ 0.01, different to 3-NP; $$$p ≤ 0.001, different to QUIN; ##p ≤ 0.01, different to JZL+3-NP. For A and B: one-way ANOVA followed by Dunnett’s test. For C and D: two-way ANOVA followed by Tukey’s test
Next, we tested the effects of JZL184 on the 3-NP-induced loss of mitochondrial function in synaptosomes. As shown in Fig. 1C, 3-NP decreased mitochondrial function to ~55% vs. the control group (p ≤ 0.001). In turn, mitochondrial activity induced by 3-NP was restored to levels indistinguishable from controls (~90%; p ≤ 0.01 compared to the 3-NP group) upon treatment with JZL184. In synaptosomes preconditioned with JZL184 and exposed to 3-NP, the selective CB2R inverse agonist JTE907 returned mitochondrial activity to levels analogous to 3-NP treatment alone (p ≤ 0.01 compared to the JZL184 + 3-NP group). The CB1R antagonist/inverse agonist AM281 did not significantly alter the effect of JZL184 on 3-NP toxicity. In addition, no significant effect on basal mitochondrial function was noted upon JZL184 treatment.
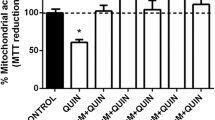
Figure 1D depicts the effect of JZL184 on QUIN-induced loss of mitochondrial function. The neurotoxic model induced by QUIN was performed for comparative purposes to confirm the protective effects of JZL184. As expected, QUIN was less effective in decreasing mitochondrial activity than 3-NP (~75% vs. the control group; p ≤ 0.001). JZL184 completely prevented the QUIN-induced loss of mitochondrial function, returning mitochondrial activity to basal levels (p ≤ 0.001 compared to the QUIN group); however, in contrast to the 3-NP toxic model, JTE907 did not modify the effect of JZL184 on QUIN toxicity. AM281 had no effect on the effects elicited by JZL184 in this toxic model. Furthermore, JZL184 alone did not modify the basal mitochondrial reductive capacity
JZL184 Prevented the Oxidative Damage to Lipids Induced by 3-Nitropropionic Acid in Synaptosomal/Mitochondrial Fractions in a CB1R-Related Manner
Next, we explored whether JZL184 could prevent 3-NP-induced lipid peroxidation (an index of oxidative cell damage induced by the noxious actions of reactive oxygen species) in synaptosomes as to determine whether redox modulatory effects are inherent to the protection afforded by MAGL inhibition.
As shown in Fig. 2A, 3-NP increased lipid peroxidation by ~20% vs. the control (p ≤ 0.01), while pretreatment with JZL184 decreased the effect of 3-NP to basal levels (p ≤ 0.001 compared to the 3-NP group). AM281, but not JTE907, abolished the protective effect of JZL184 on 3-NP-induced lipid peroxidation (p ≤ 0.001 compared to the JZL184 + 3-NP group). JZL184 did not produce oxidative damage to lipids compared to control values (17 nmol TBARS/mg protein).

Effect of JZL184 (JZL, 10 nM) on 3-nitropropionic acid (3-NP, 1 mM, in A)- and iron sulfate (FeSO4, 50 µM, in B)-induced lipid peroxidation (TBARS formation) in rat brain synaptosomal/mitochondrial fractions. AM281 (10 nM) and JTE907 (0.13 nM) were also used to investigate the role of cannabinoid receptors. Values represent the mean ± one SEM of n = 6 experiments per group. **p ≤ 0.01 and ***p ≤ 0.001, different to the control (CT); &&&p ≤ 0.001, different to 3-NP; %%%p ≤ 0.001, different to JZL + 3-NP; ap ≤ 0.05, different to FeSO4. Two-way ANOVA followed by Tukey’s test

Effect of JZL184 (JZL, 10 nM) on 3-nitropropionic acid (3-NP, 1 mM)-induced loss of mitochondrial reductive capacity (in A) and lipid peroxidation (TBARS formation; in B) in rat brain cortical slices. AM281 (10 nM) and JTE907 (0.13 nM) were also used to investigate the role of cannabinoid receptors. Values represent the mean ± one SEM of n = 6 experiments per group. ***p ≤ 0.001, different to the control (CT); &&p ≤ 0.01 and &&&p ≤ 0.001, different to 3-NP. Two-way ANOVA followed by Tukey’s test
Figure 2B shows the effect of JZL184 on the FeSO4-induced lipid peroxidation in synaptosomal/mitochondrial fractions. The pro-oxidant effect of FeSO4 was tested as a positive control of lipid peroxidation to evaluate whether the protective profile of JZL184 involved direct antioxidant properties. As expected, FeSO4 increased lipid peroxidation by ~100% compared to the control group (p ≤ 0.001), whereas JZL184 not only was unable to reduce the effect of FeSO4, but also increased oxidative damage in synaptosomes (p ≤ 0.05 compared to the FeSO4 group).
JZL184 Ameliorated the 3-NP-Induced Loss of Mitochondrial Function and Lipid Peroxidation in a CB1R- and CB2R-Independent Manner
To confirm the protective effect of JZL184 in a more complex biological preparation containing multiple types of cells with an integral circuitry and well-preserved cytoarchitecture, the effects of JZL184 on 3-NP-induced mitochondrial dysfunction/loss of cell viability and lipid peroxidation were tested in fresh cortical slices.
As shown in Fig. 3A, 3-NP decreased the basal mitochondrial reductive capacity by ~50% compared to the control group (p ≤ 0.001), while JZL184 attenuated this effect by ~80% (p ≤ 0.01 compared to the 3-NP group alone). Neither AM281, nor JTE907, modified the effect of JZL184 on the 3-NP-induced loss of mitochondrial function/cell viability. JZL184 did not modify the basal levels of mitochondrial function (control; 0.73 OD units of MTT reduction).
As shown in Fig. 3B, 3-NP increased lipid peroxidation by ~95% compared to the control group (p ≤ 0.001), while JZL184 attenuated this effect to basal levels (p ≤ 0.001 compared to the 3-NP group). Again, neither AM281, nor JTE907, modified the effect of JZL184 on the 3-NP-induced oxidative damage to lipids. JZL184 did not modify the basal levels of lipid peroxidation (control; 1.15 nmol TBARS/mg protein).
JZL184 Recovered the 3-NP-Induced Inhibition of SDH Activity
We also explored the effect of JZL184 on 3-NP-induced inhibition of SDH activity (Fig. 4). 3-NP alone decreased the levels of SDH activity by 65% compared to the control (p ≤ 0.0001), whereas JZL184 attenuated this effect by 95% compared to the 3-NP group (p ≤ 0.001, different to 3-NP). JZL184 alone did not change the baseline SDH activity (control; 17.5 units of SDH activity).

Effect of JZL184 (JZL; 10 nM) on 3-nitropropionic acid (3-NP, 1 mM)-induced succinate dehydrogenase (SDH) decreased activity in cortical slices. Values represent the mean ± one SEM of n = 6 experiments per group. ****p ≤ 0.0001, different to the control (CT); &&&p ≤ 0.001, different to 3-NP. Two-way ANOVA followed by Tukey’s test
Discussion
2-AG is present in several brain regions, including the cerebral cortex, hippocampus, hypothalamus, diencephalon, striatum, mesencephalon, and cerebellum (Sugiura et al. 2006). It is also found in the spinal cord and the hypophysis. This endocannabinoid is synthesized on demand by the action of diacylglycerol lipase at the cytoplasmatic and nuclear membrane levels (García del Caño et al. 2015) and has been shown to evoke neuroprotective effects through the activation of both CB1R and CB2R, being CB1R the main regulator of neurotransmitters release as it is abundantly found in presynaptic terminals of the substantia nigra, striatum, hippocampus, cerebellum, and cerebral cortex (Kawamura et al. 2006; Tsuboi et al. 2018). The main enzyme responsible for 2-AG hydrolysis is MAGL (Scalvini et al. 2016), which has recently emerged as a possible therapeutic tool, as it can control not only the levels of 2-AG, but also of other monoacylglycerides, and indirectly, free fatty acids, including lipids with proinflammatory effects (Gil-Ordóñez et al. 2018). MAGL is localized in cytoplasmatic membranes as well as in the membrane of the endoplasmic reticulum and is equally distributed among the cytosolic and membrane fractions (Grabner et al. 2017). Notably, the genetic and pharmacological inhibition of MAGL has been shown to increase 2-AG levels, leading to desensitization of CB1R (Chanda et al. 2010; Schlosburg et al. 2010; Navia-Paldanius et al. 2015). In a chronic pharmacological model, mice treated with JZL184 (40 mg/kg/day i.p.) for six consecutive days showed a significant decrease in the number and function of CB1R (Schlosburg et al. 2010). In addition, in a MAGL-KO model, mice exhibited a compensatory response in CB1R signaling in brain areas associated with anxiety (Imperatore et al. 2015). Altogether this evidence suggests that 2-AG is responsible for the negative regulation of CB1R, it should be considered that these studies were carried out under in vivo and chronic conditions. In contrast, our findings suggest that under in vitro and short-term conditions, at least in synaptosomal fractions, the inhibition of MAGL recruits the activation of CB2R for protection against the loss of mitochondrial function, and CB1R for protection against oxidative damage, likely due to a direct action of 2-AG on these receptors, and this conclusion was drawn upon the use of the selective CB1R antagonist/inverse agonist AM281 and/or the selective CB2R inverse agonist JTE907, two pharmacological tools capable of revealing the involvement of specific cannabinoid receptors in paradigms involving cannabinoid signaling (Pertwee 2010), like the one explored in this report.
Short-term studies are relevant to identify primary targets for compounds in biological preparations containing receptors and signaling molecules that regulate cytoprotective responses. In this novel report, we demonstrate for the first time that selective inhibition of MAGL by JZL184 affords protection against mitochondrial dysfunction and oxidative damage to lipids induced by 3-NP in two biological preparations, both containing the biochemical machinery to metabolically process endocannabinoids and their receptors. These protective effects were confirmed also in the excitotoxic model produced by the NMDA agonist, QUIN. This pharmacological strategy was effective in preventing the loss of mitochondrial reductive capacity induced by 3-NP and QUIN, as well as the 3-NP- and FeSO4-induced lipid peroxidation in synaptosomal/mitochondrial fractions, both in a cannabinoid receptor-dependent manner. Notably, JZL184 also restored mitochondrial reductive capacity, decreased the oxidative damage to lipids, and prevented the loss of SDH activity induced by 3-NP in cortical slices in a cannabinoid receptor-independent manner. This broad spectrum of protective effects, as well as the differential involvement of cannabinoid receptors, suggests wide efficacy for JZL184 as a therapeutic tool, while recognizing that its mechanisms of action need more detailed characterization. In previous reports, we assessed the efficacy of URB597, a selective inhibitor of FAAH, in protecting cellular fractions and murine models against the toxic effects induced by 3-NP and QUIN by CB1R- or CB2R-dependent mechanisms (Maya-López et al. 2017; Aguilera-Portillo et al. 2019; Chavira-Ramos et al. 2021); however, MAGL inhibition and further 2-AG accumulation offers a more promising strategy as 2-AG is the most abundant endocannabinoid in the CNS, and is a full agonist for cannabinoid receptors, also holding several other targets for a broader regulation of physiological responses (Sugiura et al. 2006; Aymerich et al. 2016; Zhu et al. 2023). One weakness of our study is the lack of analysis of 2-AG levels in our preparations, as it may provide unequivocal evidence for the endocannabinoid’s role in the protective effects described in this report. Nonetheless, there is a considerable amount of evidence on the induction of 2-AG levels by MAGL inhibition, but this approach should be considered in further studies.
In turn, direct evidence on the neuroprotective effects of JZL184 has been reported under different experimental conditions. JZL184 was shown to reduce amyloid beta-induced neurodegeneration and apoptosis in hippocampal neurons in culture by increasing the levels of 2-AG, which acted via CB1R-dependent suppression of extracellular signal-regulated kinases 1 and 2 (ERK1/2) and nuclear factor-κB (NF-κB) phosphorylation and cyclooxygenase-2 (COX-2) expression (Chen et al. 2011). The exogenous application of 2-AG and/or the increase in endogenous 2-AG levels by JZL184 abolished the interleukin-1β- and lipopolysaccharide-induced reduction of PPARγ expression in mouse hippocampal neurons in culture, suggesting that these receptors may be involved in the protective properties of 2-AG (Du et al. 2011). In addition, cerebral MAGL activity, via an alternative pathway, has been shown to promote neuroinflammatory prostaglandin production through the hydrolysis of 2-AG, thus highlighting the importance of MAGL inhibition as an experimental strategy to reduce neuroinflammation and neurodegeneration (Nomura et al. 2011). More recently, JZL184 was shown to improve cell survival in SH-SY5Y neuroblastoma dopaminergic neurons in a CB2R-dependent mechanism (Aymerich et al. 2016). Notably, these protective effects were produced at a concentration much higher (100 nM) that the one we used herein (10 nM), which is closer to the IC50 reported for JZL184 in mice (8 nM); this condition was justified by the fact that SH-SY5Y are human neuroblastoma cells, therefore being less sensitive than murine cells. Moreover, although these authors suggested that neuroprotection induced by JZL184 might be selective to dopaminergic cells, as it failed to protect cells with striatal phenotype, here we have demonstrated for the first time that this compound affords neuroprotection in toxic models typically linked to Huntington’s disease by mechanisms dependent and independent of cannabinoid receptors. In further support to our findings that protective effects of JZL184 do not necessarily involve CB2R activation, recent evidence by Zhang and Chen (2018) suggested that the expression of APP and β-secretase, as well as production of total Aβ and Aβ42, was significantly reduced in APP transgenic mice lacking CB2R (TG-CB2-KO) treated with JZL184, thus suggesting the activation of alternative protective mechanisms by this compound. Furthermore, the protective effects in vivo of JZL184 have been extended to the attenuation of infarct volume and hemispheric swelling in a model of experimental ischemic stroke in rats via inhibition of inflammatory responses and eicosanoid production rather than activation of cannabinoid receptors (Choi et al. 2018), thus supporting the concept of alternative mechanisms for JZL184-induced neuroprotection. In line with this evidence, in hippocampal neurons in culture exposed to oxygen/glucose deprivation/reoxygenation representing an in vitro model of cerebral ischemia/reperfusion, JZL184 was shown to improve cell viability, inhibit ROS formation, increase antioxidant defense, and decrease proinflammatory cytokines (Xu et al. 2021). These authors suggested that direct activation of the antioxidant Nrf2/ARE signaling pathway was responsible for the neuroprotective responses observed. Indeed, this specific mechanism may help to explain the differential protective mechanisms observed in our study, as activation of the Nrf2/ARE pathway may occur in cortical slices conformed by integral cell bodies (Reyes-Soto et al. 2022), whereas activation of cannabinoid receptors is more likely to occur in synaptosomal fractions, a biological preparation lacking most of the components of the Nrf2/ARE pathway. Thus, additional studies are warranted to investigate whether JZL184 selectively activates one or more of the aforementioned mechanisms to induce these protective responses.
Emphasis should also be directed to the fact that CB1R and CB2R are not the only receptors activated by 2-AG, as it has been demonstrated that this endocannabinoid can also act on nuclear peroxisome proliferator-activated receptor-gamma (PPARγ) (Pertwee et al. 2010; Du et al. 2011), which is an important mediator in signaling against neuroinflammation (Xu and Chen 2015), thus representing a potential alternative mechanism to explain the effects observed in cortical slices. In addition, 2-AG binds and activates the member 1 of subfamily V of transient receptor potential vanilloid 1 (TRPV1) cationic channel both in vitro and in vivo (Di Marzo 2018; Cristino et al. 2020), as well as GABA receptors and G protein-coupled receptor 55 (GPR55), thereby regulating the activity of excitatory hippocampal neurons (Sylantyev et al. 2013). The lack of effect of the CB1R and CB2R antagonists/inverse agonists found in cortical slices suggests that in contrast to what was observed in synaptosomal/mitochondrial fractions, MAGL inhibition in slices might induce preferential activation of non-classical cannabinoid receptors, such as PPARγ, the most plausible candidates; therefore, our study should be extended to explore the role of both PPARγ and TRPV1 receptors using the same toxic paradigm. Moreover, short-term 2-AG accumulation noted in our study should retain the endocannabinoid preferentially at the intracellular level, where it can reach these organelle receptors. In addition, at this point, we cannot discard the possibility that some of the effects reported herein might also derive from the transformation of 2-AG in prostaglandin-glyceryl esters by COX-2 (Hu et al. 2008), which adds a possible key mechanism to cannabinoid receptor-independent effects. Of further consideration, decreased 2-AG degradation by MAGL inhibition also means lower generation of arachidonic acid from 2-AG, which may also affect the transformation of this fatty acid into different eicosanoids (Yui et al. 2015), thus displaying several other pharmacological effects needing further investigation. These alternative mechanisms are particularly relevant for the toxic model induced by QUIN, where neither CB1, nor CB2 blockade modified the protective effect of JZL184. Probably, in this excitotoxic model, JZL184 exerts its protective effects preferentially through cannabinoid receptor-independent mechanisms, this finding deserving further investigation.
Based on these considerations, JZL184-induced pro-energetic, anti-inflammatory, and antioxidant effects elicited by MAGL inhibition, and the subsequent 2-AG accumulation might contribute to the protective responses against the toxic effects evoked by 3-NP and QUIN. Finally, it is clear that, either dependently or independently of cannabinoid receptors, the preservation of mitochondrial reductive capacity and the reduction of oxidative damage are both early protective processes that should be linked to the preservation of SDH activity, which in turn display sufficient efficacy to protect the biological preparations assessed in this study.
Concluding Remarks
Our novel findings support and confirm the concept that the modulation of the ECS focused on the accumulation of endocannabinoids constitutes a valuable strategy to prevent the toxic effects of neurotoxic compounds triggering degenerative events. Specifically, JZL184 represents an experimental tool useful to ameliorate the early toxic effects of the mitochondrial toxin 3-NP and the excitotoxin QUIN, in a mechanism involving the inhibition of MAGL and the subsequent accumulation of 2-AG. While both CB1R and CB2R participate in a differential manner in the protective mechanisms exerted by JZL184 in synaptosomal/mitochondrial fractions, none of these receptors seem to be responsible for its protective effects in cortical slices, suggesting that other mechanisms might be involved in protecting these biological preparations. The collected evidence herein presses for more detailed investigations on the protective mechanisms evoked by JZL184 and suggests the design of therapies against neurodegenerative disorders based on agents aimed to reduce endocannabinoids’ hydrolysis.
Availability of Data and Materials
The data that support the findings of this study are available from the corresponding authors, ALCG and AS, upon reasonable request.
References
Aguilera-Portillo G, Rangel-López E, Villeda-Hernández J, Chavarría A, Castellanos P, Elmazoglu Z, Karasu Ç, Túnez I, Pedraza G, Königsberg M, Santamaría A (2019) The pharmacological inhibition of fatty acid amide hydrolase prevents excitotoxic damage in the rat striatum: possible involvement of CB1 receptors regulation. Mol Neurobiol 56:844–856. https://doi.org/10.1007/s12035-018-1129-2
Aymerich MS, Aso E, Abellanas MA, Tolon RM, Ramos JA, Ferrer I, Romero J, Fernández-Ruiz J (2018) Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem Pharmacol 157:67–84. https://doi.org/10.1016/j.bcp.2018.08.016
Aymerich MS, Rojo-Bustamante E, Molina C, Celorrio M, Sánchez-Arias JA, Franco R (2016) Neuroprotective effect of JZL184 in MPP(+)-treated SH-SY5Y cells through CB2 receptors. Mol Neurobiol 53:2312–2319. https://doi.org/10.1007/s12035-015-9213-3
Bradford MM (1976) A rapid and sensitive method for the quantitation of microgram quantities of protein utilizing the principle of protein-dye binding. Anal Biochem 72:248–254. https://doi.org/10.1006/abio.1976.9999
Brouillet E, Jacquard C, Bizat N, Blum D (2005) 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem 95:1521–1540. https://doi.org/10.1111/j.1471-4159.2005.03515.x
Carloni S, Alonso-Alconada D, Girelli S, Duranti A, Tontini A, Piomelli D, Hilario E, Alvarez A, Balduini W (2012) Pretreatment with the monoacylglycerol lipase inhibitor URB602 protects from the long-term consequences of neonatal hypoxic-ischemic brain injury in rats. Pediatr Res 72:400–406. https://doi.org/10.1038/pr.2012.91
Chanda PK, Gao Y, Mark L, Btesh J, Strassle BW, Lu P, Piesla MJ, Zhang M-Y, Bingham B, Uveges A, Kowal D, Garbe D, Kouranova EV, Ring RH, Bates B, Pangalos MN, Kennedy JD, Whiteside GT, Samad TA (2010) Monoacylglycerol lipase activity is a critical modulator of the tone and integrity of the endocannabinoid system. Mol Pharmacol 78(6):996–1003. https://doi.org/10.1124/mol.110.068304
Chaturvedi RK, Beal MF (2013) Mitochondrial disease of the brain. Free Radic Biol Med 63:1–29. https://doi.org/10.1016/j.freeradbiomed.2013.03.018
Chavira-Ramos K, Orozco-Morales M, Karasu C, Tinkov AA, Aschner M, Santamaría A, Colín-González AL (2021) URB597 prevents the short-term excitotoxic cell damage in rat cortical slices: role of cannabinoid 1 receptors. Neurotox Res 39:146–155. https://doi.org/10.1007/s12640-020-00301-1
Chen X, Zhang J, Chen C (2011) Endocannabinoid 2-arachidonoylglycerol protects neurons against β-amyloid insults. Neuroscience 178:159–168. https://doi.org/10.1016/j.neuroscience.2011.01.024
Choi S-H, Arai AL, Mou Y, Kang B, Yen CC-C, Hallenbeck J, Silva AC (2018) Neuroprotective effects of MAGL (monoacylglycerol lipase) inhibitors in experimental ischemic stroke. Stroke 49(3):718–726. https://doi.org/10.1161/STROKEAHA.117.019664
Colín-González AL, Aguilera G, Santamaría A (2016) Cannabinoids: glutamatergic transmission and kynurenines. Adv Neurobiol 12:173–198. https://doi.org/10.1007/978-3-319-28383-8_10
Colín-González AL, Maya-López M, Pedraza-Chaverrí J, Ali SF, Chavarría A, Santamaría A (2014) The Janus faces of 3-hydroxykynurenine: dual redox modulatory activity and lack of neurotoxicity in the rat striatum. Brain Res 1589:1–14. https://doi.org/10.1016/j.brainres.2014.09.034
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87. https://doi.org/10.1038/384083a0
Cristino L, Bisogno T, Di Marzo V (2020) Cannabinoids and the expanded endocannabinoid system in neurological disorders. Nature Rev Neurology 16(1):9–29. https://doi.org/10.1038/s41582-019-0284-z
Di Marzo V (2018) New approaches and challenges to targeting the endocannabinoid system. Nature Rev Drug Discovery 17(9):623–639. https://doi.org/10.1038/nrd.2018.115
Dinh TP, Carpenter D, Leslie FM, Freund TF, Katona I, Sensi SL, Kathuria S, Piomelli D (2002) Brain monoglyceride lipase participating in endocannabinoid inactivation. Proc Natl Acad Sci USA 99:10819–10824. https://doi.org/10.1073/pnas.152334899
Du H, Chen X, Zhang J, Chen C (2011) Inhibition of COX-2 expression by endocannabinoid 2-arachidonoylglycerol is mediated via PPAR-γ. Br J Pharmacol 163(7):1533–1549. https://doi.org/10.1111/j.1476-5381.2011.01444.x
Fernández-Suárez D, Celorrio M, Riezu-Boj JI, Ugarte A, Pacheco R, González H, Oyarzabal J, Hillard CJ, Franco R, Aymerich MS (2014) Monoacylglycerol lipase inhibitor JZL184 is neuroprotective and alters glial cell phenotype in the chronic MPTP mouse model. Neurobiol Aging 35:2603–2616. https://doi.org/10.1016/j.neurobiolaging.2014.05.021
Fucich EA, Stielper ZF, Cancienne HL, Edwards S, Gilpin NW, Molina PE, Middleton JW (2020) Endocannabinoid degradation inhibitors ameliorate neuronal and synaptic alterations following traumatic brain injury. 123:707–717. https://doi.org/10.1152/jn.00570.2019
García del Caño G, Aretxabala X, González-Burguera I, Montaña M, López de Jesús M, Barrondo S, Barrio RJ, Sampedro C, Goicolea MA, Sallés J (2015) Nuclear diacylglycerol lipase-α in rat brain cortical neurons: evidence of 2-arachidonoylglycerol production in concert with phospholipase C-β activity. J Neurochem 132(5):489–503. https://doi.org/10.1111/jnc.12963
García-Morales V, Montero F, Moreno-López B (2015) Cannabinoid agonists rearrange synaptic vesicles at excitatory synapses and depress motoneuron activity in vivo. Neuropharmacology 92:69–79. https://doi.org/10.1016/j.neuropharm.2014.12.036
Gil-Ordóñez A, Martín-Fontecha M, Ortega-Gutiérrez S, López-Rodríguez ML (2018) Monoacylglycerol lipase (MAGL) as a promising therapeutic target. Biochem Pharmacol 157:18–32. https://doi.org/10.1016/j.bcp.2018.07.036
Grabner GF, Zimmermann R, Schicho R, Taschler U (2017) Monoglyceride lipase as a drug target: at the crossroads of arachidonic acid metabolism and endocannabinoid signaling. Pharmacol Ther 175:35–46. https://doi.org/10.1016/j.pharmthera.2017.02.033
Hariharan A, Shetty S, Shirole T, Jagtap AG (2014) Potential of protease inhibitor in 3-nitropropionic acid induced Huntington’s disease like symptoms: mitochondrial dysfunction and neurodegeneration. Neurotoxicology 45:139–148. https://doi.org/10.1016/j.neuro.2014.10.004
Hu SS-J, Bradshaw HB, Chen JS-C, Tan B, Walker JM (2008) Prostaglandin E2 glycerol ester, an endogenous COX-2 metabolite of 2-arachidonoylglycerol, induces hyperalgesia and modulates NFκB activity. Br J Pharmacol 153(7):1538–1549. https://doi.org/10.1038/bjp.2008.33
Imperatore R, Morello G, Luongo L, Taschler U, Romano R, Gregorio D, De Belardo C, Maione S, Marzo V, Di Cristino L (2015) Genetic deletion of monoacylglycerol lipase leads to impaired cannabinoid receptor CB1R signaling and anxiety-like behavior J Neurochem 135(4):799–813 https://doi.org/10.1111/jnc.13267
Kawamura Y, Fukaya M, Maejima T, Yoshida T, Miura E, Watanabe M, Ohno-Shosaku T, Kano M (2006) The CB1 cannabinoid receptor is the major cannabinoid receptor at excitatory presynaptic sites in the hippocampus and cerebellum. J Neurosci 26(11):2991–3001. https://doi.org/10.1523/JNEUROSCI.4872-05.2006
Kerr DM, Harhen B, Okine BN, Egan LJ, Finn DP, Roche M (2013) The monoacylglycerol lipase inhibitor JZL184 attenuates LPS-induced increases in cytokine expression in the rat frontal cortex and plasma: differential mechanisms of action. Br J Pharmacol 169:808–819. https://doi.org/10.1111/j.1476-5381.2012.02237.x
Khasabova IA, Yao X, Paz J, Lewandowski CT, Lindberg AE, Coicou L, Burlakova N, Simone DA, Seybold VS (2014) JZL184 is antihyperalgesic in a murine model of cisplatin-induced peripheral neuropathy. Pharmacol Res 90:67–75. https://doi.org/10.1016/j.phrs.2014.09.008
Lan R, Gatley J, Lu Q, Fan P, Fernando SR, Volkow ND, Pertwee R, Makriyannis A (1999) Design and synthesis of the CB1 selective cannabinoid antagonist AM281: a potential human SPECT ligand. AAPS PharmSci 1:E4. https://doi.org/10.1208/ps010204
Li Q, Yan H, Wilson WA, Swartzwelder HS (2010) Modulation of NMDA and AMPA-mediated synaptic transmission by CB1 receptors in frontal cortical pyramidal cells. Brain Res 1342:127–137. https://doi.org/10.1016/j.brainres.2010.04.029
Long JZ, Li W, Booker L, Burston JJ, Kinsey SG, Schlosburg JE, Pavón FJ, Serrano AM, Selley DE, Parsons LH, Lichtman AH, Cravatt BF (2009) Selective blockade of 2-arachydonoylglycerol hydrolysis produces cannabinoid behavioral effects. Nat Chem Biol 5(1):37–44. https://doi.org/10.1038/nchembio.129
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193:265–275. PMID:14907713
Maya-López M, Rubio-López LC, Rodríguez-Alvarez IV, Orduño-Piceno J, Flores-Valdivia Y, Colonnello A, Rangel-López E, Túnez I, Prospéro-García O, Santamaría A (2020) A cannabinoid receptor-mediated mechanism participates in the neuroprotective effects of oleamide against excitotoxic damage in rat brain synaptosomes and cortical slices. Neurotox Res 37:126–135. https://doi.org/10.1007/s12640-019-00083-1
Maya-López M, Ruiz-Contreras HA, Negrete-Ruíz MJ, Martínez-Sánchez JE, Benítez-Valenzuela J, Colín-González AL, Villeda-Hernández J, Sánchez-Chapul L, Parra-Cid C, Rangel-López E, Santamaría A (2017) URB597 reduces biochemical, behavioral and morphological alterations in two neurotoxic models in rats. Biomed Pharmacother 88:745–753. https://doi.org/10.1016/j.biopha.2017.01.116
Muddapu VR, Dharshini SAP, Chakravarthy VS, Gromiha MM (2020) Neurodegenerative diseases—is metabolic deficiency the root cause? Front Neurosci 14:213. https://doi.org/10.3389/fnins.2020.00213
Navia-Paldanius D, Aaltonen N, Lehtonen M, Savinainen JR, Taschler U, Radner FPW, Zimmermann R, Laitinen JT (2015) Increased tonic cannabinoid CB1R activity and brain region-specific desensitization of CB1R Gi/o signaling axis in mice with global genetic knockout of monoacylglycerol lipase. Eur J Pharmaceutical Sci 77:180–188. https://doi.org/10.1016/j.ejps.2015.06.005
Nomura DK, Morrison BE, Blankman JL, Long JZ, Kinsey SG, Marcondes MCG, Ward AM, Hahn YK, Lichtman AH, Conti B, Cravatt BF (2011) Endocannabinoid hydrolysis generates brain prostaglandins that promote neuroinflammation. Science 334(6057):809–813. https://doi.org/10.1126/science.1209200
Pertwee RG (2010) Receptors and channels targeted by synthetic cannabinoid receptor agonists and antagonists. Curr Med Chem 17(14):1360–1381. https://doi.org/10.2174/092986710790980050
Pertwee RG, Howlett AC, Abood ME, Alexander SPH, Di Marzo V, Elphick MR, Greasley PJ, Hansen HS, Kunos G, Mackie K, Mechoulam R, Ross RA (2010) International union of basic and clinical pharmacology. LXXIX. Cannabinoid receptors and their ligands: beyond CB1 and CB2. Pharmacol Rev 62(4):588–631. https://doi.org/10.1124/pr.110.003004
Petrosino S, Di Marzo V (2010) FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs 11:51–62. PMID:20047159
Pihlaja R, Takkinen J, Eskola O, Vasara J, López-Picón FR, Haaparanta-Solin M, Rinne JO (2015) Monoacylglycerol lipase inhibitor JZL184 reduces neuroinflammatory response in APdE9 mice and in adult mouse glial cells. J Neuroinflammation 12:81. https://doi.org/10.1186/s12974-015-0305-9
Popov VN, Eprintsev AT, Fedorin DN, Igamberdiev AU (2010) Succinate dehydrogenase in Arabidopsis thaliana is regulated by light via phytochrome A. FEBS Lett 584(1):199–202. https://doi.org/10.1016/j.febslet.2009.11.057
Rangel-López E, Colín-González AL, Paz-Loyola AL, Pinzón E, Torres I, Serratos IN, Castellanos P, Wajner M, Souza DO, Santamaría A (2015) Cannabinoid receptor agonists reduce the short-term mitochondrial dysfunction and oxidative stress linked to excitotoxicity in the rat brain. Neuroscience 285:97–106. https://doi.org/10.1016/j.neuroscience.2014.11.016
Reyes-Soto CY, Villaseca-Flores M, Ovalle-Noguez EA, Nava-Osorio J, Galván-Arzate S, Rangel-López E, Maya-López M, Retana-Márquez S, Túnez I, Tinkov AA, Ke T, Aschner M, Santamaría A (2022) Oleamide reduces mitochondrial dysfunction and toxicity in rat cortical slices through the combined action of cannabinoid receptors activation and induction of antioxidant activity. Neurotox Res 40:2167–2178. https://doi.org/10.1007/s12640-022-00575-7
Sánchez-Rodríguez MA, Gómez O, Esteban PF, García-Ovejero D, Molina-Holgado E (2018) The endocannabinoid 2-arachidonoylglycerol regulates oligodendrocyte progenitor cell migration. Biochem Pharmacol 157:180–188. https://doi.org/10.1016/j.bcp.2018.09.006
Scalvini L, Piomelli D, Mor M (2016) Monoglyceride lipase: structure and inhibitors. Chem Phys Lipids 197:13–24. https://doi.org/10.1016/j.chemphyslip.2015.07.011
Schlosburg JE, Blankman JL, Long JZ, Nomura DK, Pan B, Kinsey SG, Nguyen PT, Ramesh D, Booker L, Burston JJ, Thomas EA, Selley DE, Sim-Selley LJ, Liu Q, Lichtman AH, Cravatt BF (2010) Chronic monoacylglycerol lipase blockade causes functional antagonism of the endocannabinoid system. Nature Neurosci 13(9):1113–1119. https://doi.org/10.1038/nn.2616
Sugiura T, Kishimoto S, Oka S, Gokoh M (2006) Biochemistry, pharmacology and physiology of 2-arachidonoylglycerol, an endogenous cannabinoid receptor ligand. Prog Lipid Res 45(5):405–446. https://doi.org/10.1016/j.plipres.2006.03.003
Sylantyev S, Jensen TP, Ross RA, Rusakov DA (2013) Cannabinoid- and lysophosphatidylinositol-sensitive receptor GPR55 boosts neurotransmitter release at central synapses. Proc Natl Acad Sci USA 110(13):5193–5198. https://doi.org/10.1073/pnas.1211204110
Tsuboi K, Uyama T, Okamoto Y, Ueda N (2018) Endocannabinoids and related N-acylethanolamines: biological activities and metabolism. Inflamm Regen 38:28. https://doi.org/10.1186/s41232-018-0086-5
Túnez I, Tasset I, Pérez-De La Cruz V, Santamaria A (2010) 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: past, present and future. Molecules 15(2):878–916. https://doi.org/10.3390/molecules15020878
Viscomi MT, Oddi S, Latini L, Bisicchia E, Maccarrone M, Molinari M (2010) The endocannabinoid system: a new entry in remote cell death mechanisms. Exp Neurol 224:56–65. https://doi.org/10.1016/j.expneurol.2010.03.023
Xu J, Guo Q, Huo K, Song Y, Li N, Du J (2021) JZL184 protects hippocampal neurons from oxygen-glucose deprivation-induced injury via activating Nrf2/ARE signaling pathway. Human Exp Toxicol 40:1084–1094. https://doi.org/10.1177/0960327120984220
Xu JY, Chen C (2015) Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 21:152–168. https://doi.org/10.1177/1073858414524632
Yui K, Imataka G, Nakamura H, Ohara N, Naito Y (2015) Eicosanoids derived from arachidonic acid and their family prostaglandins and cyclooxygenase in psychiatric disorders. Curr Neuropharmacol 13(6):776–785. https://doi.org/10.2174/1570159X13666151102103305
Zhang J, Chen C (2018) Alleviation of neuropathology by inhibition of monoacylglycerol lipase in APP transgenic mice lacking CB2 receptors. Mol Neurobiol 55(6):4802–4810. https://doi.org/10.1007/s12035-017-0689-x
Zhang X, Thayer SA (2018) Monoacylglycerol lipase inhibitor JZL184 prevents HIV-1 gp120-induced synapse loss by altering endocannabinoid signaling. Neuropharmacology 128:269–281. https://doi.org/10.1016/j.neuropharm.2017.10.023
Zhu D, Zhang J, Hashem J, Gao F, Chen C (2023) Inhibition of 2-arachidonoylglycerol degradation enhances glial immunity by single-cell transcriptomic analysis. J Neuroinflammation 20(1):17. https://doi.org/10.1186/s12974-023-02701-4
Acknowledgements
We appreciate the excellent technical assistance of Jade Nava-Osorio, Enid Ovalle-Noguez and Marisol Maya-López.
Funding
This work was supported by the CONACYT-TUBITAK collaborative agreement (grant 265991 given to AS) and the National Institute of Environmental Health Sciences (grants R01ES03771 and R01ES10563 given to MA). None of the sponsors were involved in design, collection, analysis, or interpretation of data, neither in writing of the report or decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
A.L.C.-G. and A.S. designed the whole study. K.J.P.-R., K.C.-R., and S.G.-A. performed all experiments and prepared the figures. S.G.-A., E.R.-L., C.K., I.T., A.V.S., M.A., and M.O.-M. provided critical comments to design, interpretation of results and discussion, as well as reagents. M.A. and A.S. wrote the main manuscript. All authors reviewed the manuscript.
Corresponding authors
Ethics declarations
Ethics Approval
All experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 80–23) revised 1996, and the local Ethical Committees. Formal approval to conduct the experimental procedures was obtained from the animal subjects review board of the Instituto Nacional de Neurología y Neurocirugía (Project numbers 17/20 and 73/20). All efforts were made to minimize animals pain suffering during the experiments.
Conflict of Interest
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
This manuscript was submitted to the journal for consideration for the first time prior to the change of affiliation of the corresponding author.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Paredes-Ruiz, K.J., Chavira-Ramos, K., Galvan-Arzate, S. et al. Monoacylglycerol Lipase Inhibition Prevents Short-Term Mitochondrial Dysfunction and Oxidative Damage in Rat Brain Synaptosomal/Mitochondrial Fractions and Cortical Slices: Role of Cannabinoid Receptors. Neurotox Res 41, 514–525 (2023). https://doi.org/10.1007/s12640-023-00661-4
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-023-00661-4