
Abstract
The potential treatment of neurodegenerative disorders requires the development of novel pharmacological strategies at the experimental level, such as the endocannabinoid-based therapies. The effects of oleamide (OEA), a fatty acid primary amide with activity on cannabinoid receptors, was tested against mitochondrial toxicity induced by the electron transport chain complex II inhibitor, 3-nitropropionic acid (3-NP), in rat cortical slices. OEA prevented the 3-NP-induced loss of mitochondrial function/cell viability at a concentration range of 5 nM–25 µM, and this protective effect was observed only when the amide was administered as pretreatment, but not as post-treatment. The preservation of mitochondrial function/cell viability induced by OEA in the toxic model induced by 3-NP was lost when the slices were pre-incubated with the cannabinoid receptor 1 (CB1R) selective inhibitor, AM281, or the cannabinoid receptor 2 (CB2R) selective inhibitor, JTE-907. The 3-NP-induced inhibition of succinate dehydrogenase (mitochondrial Complex II) activity was recovered by 25 nM OEA. The amide also prevented the increased lipid peroxidation and the changes in reduced/oxidized glutathione (GSH/GSSG) ratio induced by 3-NP. The cell damage induced by 3-NP, assessed as incorporation of cellular propidium iodide, was mitigated by OEA. Our novel findings suggest that the neuroprotective properties displayed by OEA during the early stages of damage to cortical cells involve the converging activation of CB1R and CB2R and the increase in antioxidant activity, which combined may emerge from the preservation of the functional integrity of mitochondria.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Neuronal degeneration observed in several neurodegenerative disorders recruits major toxic events such as disrupted energy metabolism, oxidative stress, inflammation, excitotoxicity, and misfolded protein aggregation (Hensley et al. 2006; Hunter et al. 2007; Tilleux and Hermans 2007). As a triggering mechanism, transformed microglial cells and macrophages secrete and release several pro-inflammatory and neurotoxic metabolites during the onset and progression of inflammatory processes in the central nervous system (CNS), which are associated with the pathogenesis of neurodegenerative disorders (Muhl and Pfeilschifter 2003; Aguilera-Portillo et al. 2019). In this context, since it is widely known that the frontal cortex is a brain region regulating motor, sensorial, and cognitive functions, the use of cortical slices in experimental protocols resembling the acute and chronic toxic features of neurodegenerative disorders affords pertinent and complementary models, thus establishing dynamic functional protocols associated with several neurological disorders (Ting et al. 2018).
In turn, energy depletion and excitotoxicity also contribute to cell damage as triggering factors of neurodegeneration. While most of the cellular energy failure comes from mitochondrial dysfunction and ensuing disruption of cell metabolism, excitotoxicity is elicited by overactivation of the N-methyl-d-aspartate receptors (NMDAr) subtype of glutamate receptors with the consequent toxic cascade derived from a radical increase in intracellular Ca2+ levels (Sgambato-Faure and Cenci 2012; Schinder et al. 1996). Combined with inflammation, these processes trigger deleterious signaling cascades leading to brain cell death (Sgambato-Faure and Cenci 2012; Schinder et al. 1996).
The mitochondrial toxin 3-nitropropionic acid (3-NP) produces a neurotoxic model in mammals that mimics several major features of Huntington’s disease (HD), such as a pattern of cortico-striatal circuitry degeneration similar to the one observed in this disorder (Túnez et al. 2010; Hariharan et al. 2014; Maya-López et al. 2017). 3-NP inhibits succinate dehydrogenase (SDH, electron transport chain Complex II) activity by competing with succinate, thus depleting ATP levels, blocking the Krebs cycle, inducing secondary excitotoxicity and increasing the formation of reactive oxygen species (ROS), leading to cell death (Maya-López et al. 2017; Brouillet et al. 2005; Chaturvedi and Beal 2013; Burtscher et al. 2015).
Endogenous cannabinoids, membrane and organelle cannabinoid receptors (CB1R, CB2R and others), and enzymes for the synthesis and degradation of endocannabinoids compose the endocannabinoid system (ECS) (Bénard et al. 2012) or endocannabinoidome. Combined, these components coordinate neuromodulatory functions such as neurotransmission, immune responses, and cell signaling. The effects elicited by the regulation of the ECS are thought to be mostly related with the activation of G protein-coupled type 1 (CB1R) and type 2 (CB2R) receptors. While CB1R have been identified in high levels in several brain regions, CB2R are located in both glial cells and nerve endings (Aymerich et al. 2018). The signaling associated to CB1R and CB2R in neurons and glia has been intensely investigated with special emphasis on the modulatory effects of the ECS both under physiological and pathophysiological conditions resulting in neuroinflammatory and neurodegenerative diseases (Viscomi et al. 2010). ECS stimulation has been demonstrated to trigger neuroprotective responses through several mechanisms, such as a reduction in the release of excitatory neurotransmitters from the presynaptic compartment (Nazari et al. 2016), the coupling of CB1R to NR1 subunits of the NMDAr via histidine triad nucleotide-binding protein 1 (HINT-1) to reduce excitatory transmission at the postsynaptic level (Sánchez-Blázquez et al. 2013, 2014; Rodríguez-Muñoz et al. 2016), as well as the negative modulation of the G protein-coupled chemokine receptor CXCR4 via a physical heterodimeric association with CB2R and further inhibition of deleterious responses (Coke et al. 2016). Cannabinoids are also known to activate the peroxisome proliferator-activated receptors (PPARs), thus reducing inflammatory responses at the nuclear level (O’Sullivan 2007, 2016). Furthermore, it has been shown that the ECS modulates several functions in the prefrontal cortical area (Rea et al. 2019). Combined, these mechanisms support a modulatory/protective role of the ECS in the CNS and call for the design of novel pharmacological approaches to stimulate the ECS as a therapeutic target in neurotoxic models coursing with energetic disruption, inflammatory and pro-oxidant components. In this regard, a considerable number of emerging reports describe different cannabinoid-based therapies in neurological chronic disorders based on the broad neuroprotective profile of cannabinoids (Fernández-Ruiz et al. 2017; Lowe et al. 2021; Legare et al. 2022).
In turn, the fatty acid primary amide oleamide (Cis-9,10-octadecenoamide; OEA) is an endocannabinoid-profiled compound abundantly found in the CNS (Fowler 2004) which is structurally similar to the endocannabinoid anandamide (Boger et al. 2000; Leggett et al. 2004), and was first described as an endogenous substance capable of inducing sleep (Cravatt et al. 1995). Later, OEA was characterized as an important physiological modulator in the CNS, evoking responses related with analgesia, memory (Murillo-Rodríguez et al. 2001; Akanmu et al. 2007) and locomotion (Huitrón-Resendiz et al. 2001). In addition, OEA has been shown to inhibit gap junction (connexin)-mediated cell–cell communication (Boger et al. 1998) while modulates serotonergic 5-HT1, 5HT2A/2C, and 5-HT7 receptors (Mueller and Driscoll 2009), and γ-amino butyric acid (GABAA) receptors (Verdon et al. 2000). OEA is also capable of inducing several neuroprotective effects in toxic models such as the reduction of amyloid-β (Aβ) peptide accumulation and the consequent reduction of the inflammatory response in a murine model of Alzheimer’s disease through calpain inhibition (Ano et al. 2015), as well as an antiepileptic and neuroprotective activity in the striatum of rats exposed to kainic acid (Nam et al. 2017). Furthermore, OEA attenuates the K( +) deprivation-induced apoptosis in cerebellar granule neurons (Yang et al. 2002), elicits CB1R-mediated antidepressant-like responses in the rat forced swimming test (Hill and Gorzalka 2005), suppresses inflammatory responses in murine microglia and human dendritic cells via activation of CB2R and P2Y receptors (Kita et al. 2019), and reduces the quinolinic acid-induced early excitotoxic damage in rat brain synaptosomes and cortical slices (Maya-López et al. 2020). Combined, this evidence supports a broad spectrum of neuroprotective actions evoked by OEA against several deleterious conditions affecting the CNS through different toxic mechanisms.
Here, we aimed to characterize additional neuroprotective properties and mechanistic actions of OEA. We investigated whether this amide is able to evoke protective effects in the toxic model of mitochondrial dysfunction produced by 3-NP in rat cortical slices, and if these effects are subordinated to cannabinoid receptors and/or other molecular mediators. Our results suggest that, in the toxic model studied here, OEA exerts its protective effects via the convergence of CB1R and CB2R activation and the increase in redox modulatory activity.
Material and Methods
Reagents
AM281 (1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide) and JTE-907 (N-(1,3-benzodioxol-5-ylmethyl)-1,2-dihydro-7-methoxy-2-oxo-8-(pentyloxy)-3-quinolinecarboxamide) were purchased from Tocris Bioscience (Bristol, UK). OEA, thiobarbituric acid (TBA), d-mannitol, sodium azide, dichloroindophenol sodium salt, and 3-(4,5-dimethylthizol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were purchased from Sigma-Aldrich (St. Louis Missouri, USA). Other reagents were obtained from other commercial sources.
Animals
Adult male Wistar rats (250–300 g; N = 20 (total number of animals used); n = 5 experiments per group; one rat representing one whole experiment) were used for the isolation of cortical slices. All animals were obtained as a donation from the vivarium of the Universidad Autónoma Metropolitana-Iztapalapa. Rats were housed under controlled conditions of temperature (25 ± 3 °C), humidity (50%), and light–dark cycles (12:12 h). Rats received food and water ad libitum. All experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23) revised 1996, and the local Ethical Committees. Formal approval to conduct the experimental procedures was obtained from the animal subjects review board of the Instituto Nacional de Neurología y Neurocirugía (Project number 126/17). All efforts were made to minimize animals pain suffering during the experiments.
Isolation and Treatment of Cortical Slices
Cortical slices were obtained according to a method previously described by us (Colín-González et al. 2014; Estrada-Valencia et al. 2022). The frontal cortices collected from rat brains were sectioned with a chopper to obtain the thin slices (~ 250 µm), which were immediately incubated in Krebs–Ringer modified buffer (NaCl 135 mM, KCl 5 mM, MgSO4 1 mM, K2HPO4 0.4 mM, glucose 5.5 mM, HEPES 20 mM, and CaCl2 20 mM) for 30 min at 37 °C in the presence of 5% CO2. The slices obtained from different animals were mixed for all experiments and incubated with OEA (5, 25, 50, 100, 250, 1000, or 25,000 nM) either as pre- or post-treatment for 30 min, and co-added with 3-NP (100–1000 µM; concentrations calculated according to previous reports (Colín-González et al. 2014) and incubated for 60 min at 37 °C and 5% CO2. Some slices treated with OEA + 3-NP also received the CB1R inverse agonist (AM281; 10 nM) or a selective CB2R inverse agonist (JTE-907; 0.13 nM) for 30 min prior to OEA incubation. Experimental groups were designed as follows: (A) Control (only vehicles for drugs: DMSO instead of AM281 or JTE-907; 10% EtOH instead of OEA; H2O instead of 3-NP); (B) OEA alone; (C) 3-NP alone; (D) OEA + 3-NP; (E) AM281 + OEA + 3-NP; (F) JTE-907 + OEA + 3-NP. The concentrations of cannabinoid receptor antagonists used herein were obtained or calculated on the basis of previous reports describing the biological activities of these agents (Gifford et al. 1997; Lan et al. 1999; Piomelli 2003; Gentili et al. 2019; Chavira-Ramos et al. 2021). All experiments were performed in a blind manner to avoid bias.
Reductive Capacity Assay as a Mitochondrial Functional Assessment
To assess the functional status of mitochondria, MTT reduction assay was performed according to previous reports (Colín-González et al. 2014; Chavira-Ramos et al. 2021). Slices were incubated with different treatments and then added to 300 µL Krebs’s buffer plus 15 µL MTT reagent (5 mg/mL) and incubated for 60 min at 37 °C. A Cytation 3 Imaging Reader (BioTek Instruments, Winooski, VT, USA) was used to estimate the content of formazan at 570 nm. The percentage levels of MTT reduction vs. the control values were calculated and expressed as final results.
Succinate Dehydrogenase Activity Assay
Succinate dehydrogenase (SDH) activity was quantified according to a method previously reported (Túnez et al. 2004; Strack et al. 2001). Briefly, aliquots (400 µL) of homogenized and centrifuged (3000 g) samples were incubated in the presence of a phosphate buffer assay reaction solution (650 µL; pH 7.0) containing 0.3 M d-mannitol + 5.0 mM magnesium soluble chloride + 125 µL of 4.0 mM sodium azide + 125 µL of 0.50 mM dichloroindophenol + 125 µL of 0.2 M succinate. The decay of color due to the reduction of dichloroindophenol was recorded over a 40-min period at 600 nm. The optical density was monitored in an UV-1603 Shimadzu (Kyoto, Japan) spectrophotometer. Protein content in samples was quantified according to previous protocols (Lowry et al. 1951). Results were expressed as units of reduction per milligram of protein (U/mg protein).
Assay of Oxidative Damage to Lipids
Lipid peroxidation was assessed as an index of the oxidative damage to lipids in the slices according to previous reports (Colín-González et al. 2014; Chavira-Ramos et al. 2021). Slices previously incubated with different treatments were homogenized in lysis buffer; then, 100 µL-aliquots were added to 50 µL of thiobarbituric acid (TBA) reagent (0.75 g TBA + 15 g trichloroacetic acid + 2.53 mL HCl) and incubated in a boiled water-bath for 20 min. All samples were centrifuged at 3000 g for 10 min at 4 °C. The optical density of the collected supernatants was registered at 532 nm in a Cytation 3 Imagin Reader (BioTek). The percents of lipid peroxidation calculated from the nmols of TBA-reactive substances (TBARS) formed per mg of protein, were expressed as final results.
Quantification of Reduced (GSH) and Oxidized (GSSG) Glutathione Levels
The levels of GSH and GSSG were quantified according to a method previously described (Galván-Arzate et al. 2005; Reyes-Soto et al. 2020). The slices previously exposed to the treatments were added to 500 μL of Krebs solution and stored at – 70 °C until analyzed. The unfrozen samples were added to 4 mL PBS for sonication. Aliquots of 500 µL were centrifuged at 12,000 rpm for 10 min, from which the supernatants were collected for measurement of GSH and GSSG contents. For GSH measurement, 100 μL of the samples were diluted in 1.8 mL PBS plus 100 μL o-phthaldialdehyde (OPA) and incubated for 15 min at RT. For GSSG measurement, 500 μL of the samples were diluted in 200 μL N-ethylmaleimide (NEM) plus 4.3 mL NaOH and incubated for 30 min at RT. Then, 100 μL of the first dilution were added to 1.8 mL PBS plus 100 μL OPA and incubated for 15 min at RT. The fluorescent signals of GSH and GSSG were detected in a Perkin-Elmer LS-50B Luminescence Spectrometer at excitation λ of 420 nm and emission λ of 350 nm. A standard curve with known concentrations of both molecules was constructed. GSH and GSSG contents in samples (relative fluorescence units) were recorded and expressed as the GSH/GSSG ratio.
Assessment of Cell Damage by Propidium Iodide (PI) Incorporation
A method based on fluorescence was employed to evaluate cell damage, according to a previous report (Maya-López et al. 2020). Slices exposed to the different treatments were added to a solution containing PI (100 μg/mL, Roche, Basel, Switzerland) for 10 min. Then, slices were washed with PBS thrice, fixed in p-formaldehyde (1%/PBS) for 60 min, washed with PBS thrice again, and mounted with a DAPI-resin for nuclei counterstaining. Images (10 ×, 20 ×, and 40 ×) were collected in a Cytation 3 Image Reader (BioTek), using the Gen5 v3.02.2 software. Simple and double staining (merge) for DAPI and PI were prepared, and the number of cells positive to PI per field were quantified and graphically depicted.
Statistical Analysis
Results represent independent experiments (one experiment per animal) expressed as mean values ± standard deviation of n = 3–6 independent experiments per group. All data were statistically analyzed either by one- or two-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc test (GraphPad, Scientific, San Diego, CA, USA). Values of P ≤ 0.05 were considered as statistically significant.
Results
OEA Prevents the Mitochondrial Dysfunction Induced by 3-NP in Cortical Slices
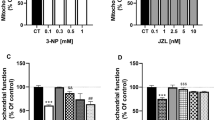
The MTT assay was used as a functional index of the status of mitochondrial activity/cell viability. First, a concentration–response effect was characterized with increased concentrations of 3-NP (100–1000 µM) to establish an optimum toxic concentration to use throughout the study. All tested concentrations led to a significant decrease (P ≤ 0.0001 in all cases) in mitochondrial reductive capacity compared to the control group (70%, 68%, 74%, and 78% below the control for 100, 250, 500, and 1000 µM, respectively; Fig. 1A).

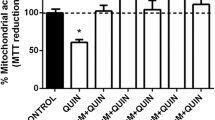
Effect of oleamide (OEA) on 3-nitropropionic acid (3-NP)-induced mitochondrial dysfunction in rat cortical slices. In panel A, the chart depicts the concentration–response effect (100–1000 µM) of 3-NP effect (60 min) on MTT reduction. In panel B, cortical slices were preconditioned for 30 min with increased concentrations of OEA (5–25,000 nM) and later exposed for 60 min to 3-NP (100 µM). In panel C, the chart shows the comparative effect of pre- vs. post-treatment of 3-NP-exposed slices with OEA (25 nM) on the levels of MTT reduction. In panel D, the chart depicts the effects of 10 nM AM281 (CB1R inverse agonist) and 0.13 nM JTE-907 (CB2R antagonist) on the OEA + 3-NP-induced changes in MTT reduction. Mean values ± S.D. of six independent experiments per group. aP ≤ 0.05, different of the control group; bP ≤ 0.05, different of 3-NP; cP ≤ 0.05, different of OEA + 3-NP; **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001, differences among groups. One- or two-way ANOVA followed by Bonferroni’s test
Next, the effect of increased concentrations of OEA (5, 25, 50, 100, 250, 1000, and 25,000 nM) were tested on the 3-NP-induced decrease in mitochondrial reductive capacity (Fig. 1B). While 3-NP alone (25 µM) decreased this endpoint by 51% compared to the control (P ≤ 0.05), all OEA concentrations tested completely prevented the toxic effect of 3-NP in a significant manner (P ≤ 0.05), returning mitochondrial activity to basal levels (all around 100%, except for 100 and 250 nM, which increased mitochondrial activity by 24% and 30%, compared to the control; P ≤ 0.05, different to 3-NP). None of these OEA concentrations affected mitochondrial function when tested alone (data not shown). From this point on, we choose 25 nM OEA as the appropriate concentration to be tested throughout the study.
The effect of OEA, either as pretreatment (30 min before the toxic insult, as assayed in Fig. 1B) or post-treatment (30 min after the toxic insult), was tested on the 3-NP-induced decrease in mitochondrial function (Fig. 1C). Again, 3-NP alone decreased the mitochondrial reductive capacity by 54% compared to the control (P ≤ 0.05). As pretreatment (preconditioning), OEA fully recovered the baseline of mitochondrial activity to levels statistically indistinguishable from controls when co-incubated with 3-NP (around 100%); in contrast, when administered as post-treatment, OEA did not modify the toxic effect of 3-NP (45% below the control).
We then explored the possible involvement of CB1R and CB2R on the effects of OEA in the toxic model produced by 3-NP. For this purpose, AM281 and JTE-907 (two agents capable of blocking CB1R and CB2R, respectively) were added to cortical slices before the addition of OEA and/or 3-NP (Fig. 1D). Once again, 3-NP alone decreased mitochondrial reductive capacity by 45% compared to the control (P ≤ 0.05, different to control), whereas OEA fully recovered this marker to levels statistically indistinguishable from controls (around 100% of activity, P ≤ 0.05 different to 3-NP). Both AM281 and JTE-907 inhibited the protective effect of OEA on 3-NP toxicity, returning the levels of mitochondrial activity close to 3-NP alone (35% and 40% below the control, respectively; P ≤ 0.05 different to the control and OEA + 3-NP).
The Loss of SDH Activity Induced by 3-NP Is Prevented by OEA
SDH activity was quantified to assess 3-NP toxicity at the mitochondrial level (Fig. 2). 3-NP produced a significant decrease in SDH activity in cortical slices compared to the control group (81% below; P ≤ 0.01). In contrast, co-incubation of slices with OEA and 3-NP prevented the loss of SDH activity produced by 3-NP in a significant manner (97% of activity compared to 3-NP; P ≤ 0.01, different to 3-NP). OEA alone did not change the baseline of SDH activity.

Effect of oleamide (OEA; 25 nM) on 3-nitropropionic acid-induced succinate dehydrogenase (SDH) decreased activity in cortical slices. Values are expressed as mean ± S.D. of six independent experiments per group. aP ≤ 0.01, different of the control group; bP ≤ 0.01, different of 3-NP. Two-way ANOVA followed by Bonferroni’s test
OEA Ameliorates the Oxidative Stress Induced by 3-NP
Next, we explored whether the pretreatment with OEA can prevent the 3-NP-induced lipid peroxidation (an index of oxidative damage to lipids due to enhanced production of reactive oxygen species) and the reduced/oxidized glutathione (GSH/GSSG) ratio (an index of the redox status of the cortical tissue) (Fig. 3).

Effect of oleamide (OEA; 25 nM) on 3-nitropropionic acid (3-NP; 100 µM)-induced oxidative damage to lipids (TBA-reactive substances formation; panel A) and changes in the reduced/oxidized glutathione (GSH/GSSG) ratio (panel B) in cortical slices. Values are expressed as mean ± S.D. of six independent experiments per group. aP ≤ 0.05, different of the control group; bP ≤ 0.05, different of 3-NP. Two-way ANOVA followed by Bonferroni’s test
As shown in Fig. 3A, 3-NP alone increased lipid peroxidation by 25% compared to the control (P ≤ 0.05), whereas the pretreatment with OEA reduced the effect of 3-NP by 36% (P ≤ 0.05, different to 3-NP). OEA alone decreased the baseline of lipid peroxidation by 25% (P ≤ 0.05, different to the control).
In Fig. 3B, 3-NP alone decreased the GSH/GSSG ratio by 40% compared to the control (P ≤ 0.05), whereas OEA prevented the effect of 3-NP by 24% (P ≤ 0.05, different to 3-NP). OEA alone did not modify the baseline GSH/GSSG.
OEA Reduces the 3-NP-Induced Cell Damage
The effect of OEA on the 3-NP-induced cell damage is shown in the fluorescence micrographs in Fig. 4A, with its corresponding quantitative analysis in Fig. 4B. Bright fields of all treatments are shown in the first column, where it can be observed that the general appearance of the 3-NP condition differs from all others, showing diffuse tissue. In the first line, the control condition depicts intense DAPI staining contrasting with almost absent PI staining, thus supporting the concept that cell permeability remains intact, which is demonstrated in the merged image (Fig. 4A). In contrast, slices exposed to 3-NP (second line) show intense PI staining merging with DAPI labeling (PI/DAPI ratio = 344% above the control; P ≤ 0.001, different from the control group; Fig. 4B). Similar to the control group, OEA alone (third line) produced only moderate PI staining (Fig. 4A). In turn, the OEA + 3-NP treatment (fourth line) depicts slight increase in PI staining compared with the control (PI/DAPI ratio = 55% above the control; Fig. 4B), suggesting cell protection compared to the 3-NP group (P ≤ 0.001, different to 3-NP).

Effect of oleamide (OEA) on 3-nitropropionic acid (3-NP)-induced cell damage in rat cortical slices. In the first column, bright field images of all experimental conditions are shown, whereas 4′,6-diamidino-2-phenylindole (DAPI) and propidium iodide (PI) stains are shown in the second and third columns. Merged images are depicted in the fourth column. Fluorescence micrographs show cell nuclei (DAPI in blue and PI in red) in slices exposed to vehicles (control), 3-NP (100 μM), OEA (25 nM), and 3-NP + OEA. Bar lines correspond to 100 μm scale. All fields correspond to 20 × magnifications, except for the fifth column which depicts a 40 × magnification (panel A). The graphic representation of the densitometric analysis of the images from the upper panels is shown below (panel B). Mean values ± S.D. of three independent experiments per group. ***P ≤ 0.001, different of the control group; ▲▲▲P ≤ 0.001, different of 3-NP. Two-way ANOVA followed by Bonferroni’s test
Discussion
Pharmacological approaches aimed at reducing the deleterious events occurring in neurodegenerative disorders via the modulation of the ECS have gained attention in biomedical research (Robson 2014), since neuroprotective effects can be evoked by the activation of CB1R, CB2R, and other receptors linked to the ECS. For instance, the CB1R-mediated modulation of excitatory responses via the reduction of NMDAr activity (Sánchez-Blázquez et al. 2014) results in protective effects in different neurotoxic protocols with excitotoxic components (Aguilera-Portillo et al. 2019; Maya-López et al. 2020; Rangel-López et al. 2015; Kotlar et al. 2019). In addition, the CB2R activation by selective or non-selective cannabinoid receptor agonists also participates in several neuroprotective responses mostly via reduction of inflammatory responses (Du et al. 2022; El-Atawneh and Goldblum 2022; Young and Denovan-Wright 2022).
Here, we describe for the first time the neuroprotective effects of nM–µM concentrations of OEA in a model of Complex 2 inhibition and mitochondrial energy depletion induced by the well-known mycotoxin 3-NP in a biological preparation that resembles the whole complexity of the brain by encompassing not only neurons but all other cell types: cortical slices. OEA showed efficacy in protecting this preparation against the 3-NP-induced loss of mitochondrial reductive capacity concomitantly decreasing oxidative stress and cell damage, thus confirming its broad neuroprotective potential, which was recently demonstrated also in an excitotoxic model produced in cortical slices by the endogenous metabolite quinolinic acid (QUIN) (Maya-López et al. 2020). However, it is noteworthy that, in contrast to the previous report, these protective effects were also achieved by concentrations ranging from 5 nM to 25 µM. The evidence from our previous report combined with the present study is relevant as, in both studies, OEA has been able to exert neuroprotection in a partial or complete cannabinoid receptors-dependent manner. In fact, in these two different paradigms (excitotoxicity vs. direct mitochondrial dysfunction), the protective effects of OEA were dependent on both CB1R and CB2R activation (as evidenced by the effects of their corresponding antagonists/inverse agonists AM281 and JTE-907, respectively). Whether the protective effects exerted by CB1R and CB2R observed in this report are either complementary or excluding remains to be investigated in future studies. Therefore, OEA is a promising candidate for the design of therapeutic approaches against neurotoxic insults as it exerts protection in a wide range of concentrations (nM–µM) and under different toxic conditions. In this regard, following our own recommendation to test lower concentrations of this amide in other neurotoxic paradigms (Maya-López et al. 2020), here we have demonstrated that OEA concentrations as low as 5–25 nM can be neuroprotective.
The evidence collected here highlights the role of both CB1R and CB2R as mediators of the protective responses of OEA against the mitochondrial dysfunction elicited by 3-NP. Traditionally, the effects of OEA have been attributed to its role as an endogenous agonist of CB1R (Leggett et al. 2004). Whether the protective effects evoked by the amide on the 3-NP-induced SDH inhibition described in this study might also involve the regulation of mitochondrial CB1R (mtCB1R)—a population of CB1R located in the mitochondrial membrane which are responsible of regulating neuronal energy metabolism (Bénard et al. 2012; Hebert-Chatelain et al. 2014)—remains to be investigated in future studies. In the interim, activation of CB2R receptors by OEA have been reported to induce protective effects probably by reducing the noxious signals derived from a deregulation of intracellular Ca2+ levels and mitochondrial energy disruption, and this concept is based on the reported ability of CB2R to form heterodimers with the G protein-coupled chemokine receptor (CXCR4), a protein which is in charge of intracellular Ca2+ mobilization (Coke et al. 2016) and is widely expressed in the brain (van der Meer et al. 2000). In addition, since CB2R are mostly located in glial cells (more prominently in microglia) and in nerve endings in a functional manner (Pascual et al. 2014), they are responsible of modulating inflammatory responses by reducing the expression of proinflammatory cytokines via inhibition of microglia overactivation (Yang et al. 2022), which in turn constitutes an additional potential mechanism accounting for OEA-induced protection in a biological preparation expressing several cell types, including neurons and all glial cells. Therefore, albeit occurring at different cellular levels, a coordinated action of mechanisms regulated by both CB1R and CB2R might contribute to the patterns of protection elicited by OEA in the brain.
To our knowledge, there is only one report in the literature linking OEA with 3-NP. This group reported decreased levels of OEA as a result of an intoxication with 3-NP in a Norwegian human case (Bendiksen Skogvold et al. 2022). Decreased OEA was interpreted as part of the toxic changes observed in the cerebrospinal fluid, plasma and urine of this patient, but no further implications of its meaning were offered. In addition, besides this report, there are only a few more articles available exploring the neuroprotective properties of OEA. Among them, antiepileptic and neuroprotective effects were demonstrated in the striatum of rats administered with the excitotoxin kainic acid (Nam et al. 2017). In the above referenced report, calpain inhibition partially dependent of cannabinoid receptors activation was described as part of the protective mechanisms of OEA. Blockade of gap junctions by OEA has been shown to reduce the propagation of apoptosis under toxic conditions (Mueller and Driscoll 2009). Moreover, OEA may be responsible of the coordinated regulation of signaling pathways responsible of anti-inflammatory responses evoked in a model of inflammation induced by lipopolysaccharide and NF-κB activation (Oh et al. 2010), though this effect might also be eventually linked to its role as a ligand of peroxisome proliferator-activated receptor gamma (PPARγ) (Dionisi et al. 2012). OEA also suppresses inflammatory events in microglial and human dendritic cells via CB2R activation (Kita et al. 2019). Finally, we recently demonstrated that OEA mitigates the toxic effects of QUIN in an excitotoxic model in synaptosomes and cortical slices in a CB1R- and CB2R-dependent manner, as already discussed above (Maya-López et al. 2020). Together, this evidence and the findings of this study strengthen the concept that OEA protects the brain against toxic insults via cannabinoid receptors activation and other mechanisms through several signaling pathways.
In regard to the protective effect of OEA on the 3-NP-induced oxidative damage to lipids and the disruption of GSH/GSSG, these might be linked to the prevention of the loss of mitochondrial membrane potential and the subsequent leakage of free radicals. Indeed, an active role of endocannabinoids and lipid analogues as modulators of oxidative stress has been recently suggested (Gallelli et al. 2018); thus, a direct effect of OEA as reactive oxygen species (ROS) scavenger should be considered and addressed in future studies. Combined, these results suggest an indirect redox modulatory activity by OEA secondary to the preservation of the mitochondrial functional integrity seems to be the most plausible explanation. Nonetheless, the antioxidant effects evoked by this amide contributes to its protective pattern in maintaining cell homeostasis and preservation of cellular integrity.
Based on the present results and previous reports in the scientific literature, we hypothesize that the pharmacological manipulation of the ECS by endocannabinoids such as OEA represents an important and novel strategy to intervene neurodegenerative events in neurological disorders with energy depletion/excitotoxic components.
Concluding
The findings gathered in this work support the concept that the protective effects elicited by OEA in cortical slices exposed to the mitochondrial insult produced by 3-NP are the result of a combined action of cannabinoid receptors (CB1R and CB2R) activation and the stimulation of antioxidant responses directed to ameliorate ROS generation secondary to dysfunctional mitochondrial activity and the associated toxic events derived from this process, such as oxidative damage, altered redox status, and excitotoxicity. Given that several other diverse effects have been ascribed to OEA in the literature, such as the regulation of thigh junction proteins, additional mechanisms cannot be excluded and might contribute to the protective pattern elicited by this amide in the tested toxic paradigm. Such additional mechanisms deserve detailed characterization in further studies. In the interim, the experimental evidence collected herein, combined with previous reports in the literature, lead us to posit that the design of therapies against neurodegenerative disorders with energetic dysfunction, pro-oxidant and excitotoxic components should also consider the use of cannabinoid-profiled amides such as OEA.
Data Availability
The data that support the findings of this study are available from the corresponding author, AS, upon reasonable request.
References
Aguilera-Portillo G, Rangel-López E, Villeda-Hernández J, Chavarría A, Castellanos P, Elmazoglu Z, Karasu Ç, Túnez I, Pedraza G, Königsberg M, Santamaría A (2019) The pharmacological inhibition of fatty acid amide hydrolase prevents excitotoxic damage in the rat striatum: possible involvement of CB1 receptors regulation. Mol Neurobiol 56(2):844–856. https://doi.org/10.1007/s12035-018-1129-2
Akanmu MA, Adeosun SO, Ilesanmi OR (2007) Neuropharmacological effects of oleamide in male and female mice. Behav Brain Res 182(1):88–94. https://doi.org/10.1016/j.bbr.2007.05.006
Ano Y, Ozawa M, Kutsukake T, Sugiyama S, Uchida K, Yoshida A, Nakayama H (2015) Preventive effects of a fermented dairy product against Alzheimer’s disease and identification of a novel oleamide with enhanced microglial phagocytosis and anti-inflammatory activity. PLoS ONE 10(3):e0118512. https://doi.org/10.1371/journal.pone.0118512
Aymerich MS, Aso E, Abellanas MA, Tolon RM, Ramos JA, Ferrer I, Romero J, Fernández-Ruiz J (2018) Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem Pharmacol 157:67–84. https://doi.org/10.1016/j.bcp.2018.08.016
Bénard G, Massa F, Puente N, Lourenço J, Bellocchio L, Soria-Gómez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutiérrez S, Martín-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, López-Rodríguez ML, Grandes P, Rossignol R, Marsicano G (2012) Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat Neurosci 15(4):558–564. https://doi.org/10.1038/nn.3053
Bendiksen Skogvold H, Yazdani M, Sandås EM, Vassli AØ, Kristensen E, Haarr D, Rootwelt H, Prestø Elgstøen KB (2022) A pioneer study on human 3-nitropropionic acid intoxication: contributions from metabolomics. J Appl Toxicol 42(5):818–829. https://doi.org/10.1002/jat.4259
Boger DL, Fecik RA, Patterson JE, Miyauchi H, Patricelli MP, Cravatt BF (2000) Fatty acid amide hydrolase substrate specificity. Bioorg Med Chem Lett 10(23):2613–2616. https://doi.org/10.1016/s0960-894x(00)00528-x
Boger DL, Patterson JE, Guan X, Cravatt BF, Lerner RA, Gilula NB (1998) Chemical requirements for inhibition of gap junction communication by the biologically active lipid oleamide. Proc Natl Acad Sci USA 95(9):4810–4815. https://doi.org/10.1073/pnas.95.9.4810
Brouillet E, Jacquard C, Bizat N, Blum D (2005) 3-Nitropropionic acid: a mitochondrial toxin to uncover physiopathological mechanisms underlying striatal degeneration in Huntington’s disease. J Neurochem 95(6):1521–1540. https://doi.org/10.1111/j.1471-4159.2005.03515.x
Burtscher J, Zangrandi L, Schwarzer C, Gnaiger E (2015) Differences in mitochondrial function in homogenated samples from healthy and epileptic specific brain tissues revealed by high-resolution respirometry. Mitochondrion 25:104–112. https://doi.org/10.1016/j.mito.2015.10.007
Chaturvedi RK, Beal MF (2013) Mitochondrial disease of the brain. Free Radic Biol Med 63:1–29. https://doi.org/10.1016/j.freeradbiomed.2013.03.018
Chavira-Ramos K, Orozco-Morales M, Karasu C, Tinkov AA, Aschner M, Santamaría A, Colín-González AL (2021) URB597 prevents the short-term excitotoxic cell damage in rat cortical slices: role of cannabinoid 1 receptors. Neurotox Res 39(2):146–155. https://doi.org/10.1007/s12640-020-00301-1
Coke CJ, Scarlett KA, Chetram MA, Jones KJ, Sandifer BJ, Davis AS, Marcus AI, Hinton CV (2016) Simultaneous activation of induced heterodimerization between CXCR4 chemokine receptor and cannabinoid receptor 2 (CB2) reveals a mechanism for regulation of tumor progression. J Biol Chem 291(19):9991–10005. https://doi.org/10.1074/jbc.M115.712661
Colín-González AL, Maya-López M, Pedraza-Chaverrí J, Ali SF, Chavarría A, Santamaría A (2014) The Janus faces of 3-hydroxykynurenine: dual redox modulatory activity and lack of neurotoxicity in the rat striatum. Brain Res 1589:1–14. https://doi.org/10.1016/j.brainres.2014.09.034
Cravatt BF, Prospero-Garcia O, Siuzdak G, Gilula NB, Henriksen SJ, Boger DL, Lerner RA (1995) Chemical characterization of a family of brain lipids that induce sleep. Science 268(5216):1506–1509. https://doi.org/10.1126/science.7770779
Dionisi M, Alexander SP, Bennett AJ (2012) Oleamide activates peroxisome proliferator-activated receptor gamma (PPARγ) in vitro. Lipids Health Dis 11:51. https://doi.org/10.1186/1476-511X-11-51
Du W, Zhang T, Yang F, Gul A, Tang Z, Zhang H, Jiang S, Wang S, Dong J (2022) Endocannabinoid signalling/cannabinoid receptor 2 is involved in icariin-mediated protective effects against bleomycin-induced pulmonary fibrosis. Phytomedicine 103:154187. https://doi.org/10.1016/j.phymed.2022.154187
El-Atawneh S, Goldblum A (2022) Candidate therapeutics by screening for multitargeting ligands: combining the CB2 receptor with CB1, PPARγ and 5-HT4 receptors. Front Pharmacol 13:812745. https://doi.org/10.3389/fphar.2022.812745
Estrada-Valencia R, Hurtado-Díaz ME, Rangel-López E, Retana-Márquez S, Túnez I, Tinkov A, Karasu C, Ferrer B, Pedraza-Chaverri J, Aschner J, Santamaría A (2022) Alpha-mangostin alleviates the short-term 6-hydroxydopamine-induced neurotoxicity and oxidative damage in rat cortical slices and in Caenorhabditis elegans. Neurotox Res 40(2):573–584. https://doi.org/10.1007/s12640-022-00493-8
Fernández-Ruiz J, Gómez-Ruiz M, García C, Hernández M, Ramos JA (2017) Modeling neurodegenerative disorders for developing cannabinoid-based neuroprotective therapies. Methods Enzymol 593:175–198. https://doi.org/10.1016/bs.mie.2017.06.021
Fowler CJ (2004) Oleamide: a member of the endocannabinoid family? Br J Pharmacol 141(2):195–196. https://doi.org/10.1038/sj.bjp.0705608
Gallelli CA, Calcagnini S, Romano A, Koczwara JB, de Ceglia M, Dante D, Villani R, Giudetti AM, Cassano T, Gaetani S (2018) Modulation of the oxidative stress and lipid peroxidation by endocannabinoids and their lipid analogues. Antioxidants 7(7):93. https://doi.org/10.3390/antiox7070093
Galván-Arzate S, Pedraza-Chaverrí J, Medina-Campos ON, Maldonado PD, Vázquez-Román B, Ríos C, Santamaría A (2005) Delayed effects of thallium in the rat brain: regional changes in lipid peroxidation and behavioral markers, but moderate alterations in antioxidants, after a single administration. Food Chem Toxicol 43(7):1037–1045. https://doi.org/10.1016/j.fct.2005.02.006
Gentili M, Ronchetti S, Ricci E, Di Paola R, Gugliandolo E, Cuzzocrea S, Bereshchenko O, Migliorati G, Riccardi C (2019) Selective CB2 inverse agonist JTE907 drives T cell differentiation towards a Treg cell phenotype and ameliorates inflammation in a mouse model of inflammatory bowel disease. Pharmacol Res 141:21–31. https://doi.org/10.1016/j.phrs.2018.12.005
Gifford AN, Tang Y, Gatley SJ, Volkow ND, Lan R, Makriyannis A (1997) Effect of the cannabinoid receptor SPECT agent, AM 281, on hippocampal acetylcholine release from rat brain slices. Neurosci Lett 238(1–2):84–86. https://doi.org/10.1016/s0304-3940(97)00851-3
Hariharan A, Shetty S, Shirole T, Jagtap AG (2014) Potential of protease inhibitor in 3-nitropropionic acid induced Huntington’s disease like symptoms: mitochondrial dysfunction and neurodegeneration. Neurotoxicology 45:139–148. https://doi.org/10.1016/j.neuro.2014.10.004
Hebert-Chatelain E, Reguero L, Puente N, Lutz B, Chaouloff F, Rossignol R, Piazza P-V, Benard G, Grandes P, Marsicano G (2014) Cannabinoid control of brain bioenergetics: exploring the subcellular localization of the CB1 receptor. Mol Metab 3(4):495–504. https://doi.org/10.1016/j.molmet.2014.03.007
Hensley K, Mhatre M, Mou S, Pye QN, Stewart C, West M, Williamson KS (2006) On the relation of oxidative stress to neuroinflammation: lessons learned from the G93A-SOD1 mouse model of amyotrophic lateral sclerosis. Antioxid Redox Signal 8(11–12):2075–2087. https://doi.org/10.1089/ars.2006.8.2075
Hill MN, Gorzalka BB (2005) Pharmacological enhancement of cannabinoid CB1 receptor activity elicits an antidepressant-like response in the rat forced swim test. Eur Neuropsychopharmacol 15(6):593–599. https://doi.org/10.1016/j.euroneuro.2005.03.003
Huitrón-Resendiz S, Gombart L, Cravatt BF, Henriksen SJ (2001) Effect of oleamide on sleep and its relationship to blood pressure, body temperature, and locomotor activity in rats. Exp Neurol 172(1):235–243. https://doi.org/10.1006/exnr.2001.7792
Hunter RL, Dragicevic N, Seifert K, Choi DY, Liu M, Kim HC, Cass WA, Sullivan PG, Bing G (2007) Inflammation induces mitochondrial dysfunction and dopaminergic neurodegeneration in the nigrostriatal system. J Neurochem 100(5):1375–1386. https://doi.org/10.1111/j.1471-4159.2006.04327.x
Kita M, Ano Y, Inoue A, Aoki J (2019) Identification of P2Y receptors involved in oleamide-suppressing inflammatory responses in murine microglia and human dendritic cells. Sci Rep 9(1):3135. https://doi.org/10.1038/s41598-019-40008-8
Kotlar I, Rangel-López E, Colonnello A, Aguilera-Portillo G, Serratos IN, Galván-Arzate S, Pedraza-Chaverri J, Túnez I, Wajner M, Santamaría A (2019) Anandamide reduces the toxic synergism exerted by quinolinic acid and glutaric acid in rat brain neuronal cells. Neuroscience 401:84–95. https://doi.org/10.1016/j.neuroscience.2019.01.014
Lan R, Gatley J, Lu Q, Fan P, Fernando SR, Volkow ND, Pertwee R, Makriyannis A (1999) Design and synthesis of the CB1 selective cannabinoid antagonist AM281: a potential human SPECT ligand. AAPS PharmSci 1(2):E4. https://doi.org/10.1208/ps010204
Legare CA, Raup-Konsavage WM, Vrana KE (2022) Therapeutic potential of cannabis, cannabidiol, and cannabinoid-based pharmaceuticals. Pharmacology 107(3–4):131–149. https://doi.org/10.1159/000521683
Leggett JD, Aspley S, Beckett SR, D’Antona AM, Kendall DA, Kendall DA (2004) Oleamide is a selective endogenous agonist of rat and human CB1 cannabinoid receptors. Br J Pharmacol 141(2):253–262. https://doi.org/10.1038/sj.bjp.0705607
Lowe H, Toyang N, Steele B, Bryant J, Ngwa W (2021) The endocannabinoid system: a potential target for the treatment of various diseases. Int J Mol Sci 22(17):9472. https://doi.org/10.3390/ijms22179472
Lowry OH, Rosebrough NJ, Farr AL, Randall RJ (1951) Protein measurement with the Folin phenol reagent. J Biol Chem 193(1):265–275. PMID: 14907713
Maya-López M, Colín-González AL, Aguilera G, de Lima ME, Colpo-Ceolin A, Rangel-López E, Villeda-Hernández J, Rembao-Bojórquez D, Túnez I, Luna-López A, Lazzarini-Lechuga R, González-Puertos VY, Posadas-Rodríguez P, Silva-Palacios A, Königsberg M, Santamaría A (2017) Neuroprotective effect of WIN55,212–2 against 3-nitropropionic acid-induced toxicity in the rat brain: involvement of CB1 and NMDA receptors. Am J Transl Res 9(2):261–274. PMID: 28337258
Maya-López M, Rubio-López LC, Rodríguez-Alvarez IV, Orduño-Piceno J, Flores-Valdivia Y, Colonnello A, Rangel-López E, Túnez I, Prospéro-García O, Santamaría A (2020) A cannabinoid receptor-mediated mechanism participates in the neuroprotective effects of oleamide against excitotoxic damage in rat brain synaptosomes and cortical slices. Neurotox Res 37(1):126–135. https://doi.org/10.1007/s12640-019-00083-1
Mueller GP, Driscoll WJ (2009) Biosynthesis of oleamide. Vitam Horm 81:55–78. https://doi.org/10.1016/S0083-6729(09)81003-0
Muhl H, Pfeilschifter J (2003) Endothelial nitric oxide synthase: a determinant of TNF-alpha production by human monocytes/macrophages. Biochem Biophys Res Commun 310(3):677–680. https://doi.org/10.1016/j.bbrc.2003.09.039
Murillo-Rodríguez E, Giordano M, Cabeza R, Henriksen SJ, Méndez Díaz M, Navarro L, Prospéro-García O (2001) Oleamide modulates memory in rats. Neurosci Lett 313(1–2):61–64. https://doi.org/10.1016/s0304-3940(01)02256-x
Nam HY, Na EJ, Lee E, Kwon Y, Kim HJ (2017) Antiepileptic and neuroprotective effects of oleamide in rat striatum on kainite induced behavioral seizure and excitotoxic damage via calpain inhibition. Front Pharmacol 8:817. https://doi.org/10.3389/fphar.2017.00817
Nazari M, Komaki A, Karamian R, Shahidi S, Sarihi A, Asadbegi M (2016) The interactive role of CB1 and GABA B receptors in hippocampal synaptic plasticity in rats. Brain Res Bull 120:123–130. https://doi.org/10.1016/j.brainresbull.2015.11.013
O’Sullivan S (2007) Cannabinoids go nuclear: evidence for activation of peroxisome proliferator-activated receptors. Br J Pharmacol 152(5):576–582. https://doi.org/10.1038/sj.bjp.0707423
O’Sullivan S (2016) An update on PPAR activation by cannabinoids. Br J Pharmacol 173(12):1899–1910. https://doi.org/10.1111/bph.13497
Oh YT, Lee JY, Lee J, Lee JH, Kim J-E, Ha J, Kang I (2010) Oleamide suppresses lipopolysaccharide-induced expression of iNOS and COX-2 through inhibition of NF-κB activation in BV2 murine microglial cells. Neurosci Lett 474(3):148–153. https://doi.org/10.1016/j.neulet.2010.03.026
Pascual AC, Gaveglio VL, Giusto NM, Pasquaré SJ (2014) Cannabinoid receptor-dependent metabolism of 2-arachidonoylglycerol during aging. Exp Gerontol 55:134–142. https://doi.org/10.1016/j.exger.2014.04.008
Piomelli D (2003) The molecular logic of endocannabinoid signaling. Nat Rev Neurosci 4(11):873–884. https://doi.org/10.1038/nrn1247
Rangel-López E, Colín-González AL, Paz-Loyola AL, Pinzón E, Torres I, Serratos IN, Castellanos P, Wajner M, Souza DO, Santamaría A (2015) Cannabinoid receptor agonists reduce the short-term mitochondrial dysfunction and oxidative stress linked to excitotoxicity in the rat brain. Neuroscience 285:97–106. https://doi.org/10.1016/j.neuroscience.2014.11.016
Rea K, McGowan F, Corcoran L, Roche M, Finn DP (2019) The prefrontal cortical endocannabinoid system modulates fear-pain interactions in a subregion-specific manner. Br J Pharmacol 176(10):1492–1505. https://doi.org/10.1111/bph.14376
Reyes-Soto CY, Rangel-López E, Galván-Arzate S, Colín-González AL, Silva-Palacios A, Zazueta C, Pedraza-Chaverri J, Ramírez J, Chavarria A, Túnez I, Ke T, Aschner M, Santamaría A (2020) S-Allylcysteine protects against excitotoxic damage in rat cortical slices via reduction of oxidative damage, activation of Nrf2/ARE binding, and BDNF preservation. Neurotox Res 38(4):929–940. https://doi.org/10.1007/s12640-020-00260-7
Robson PJ (2014) Therapeutic potential of cannabinoid medicines. Drug Test Analysis 6(1–2):24–30. https://doi.org/10.1002/dta.1529
Rodríguez-Muñoz M, Sánchez-Blázquez P, Merlos M, Garzón Niño J (2016) Endocannabinoid control of glutamate NMDA receptors: the therapeutic potential and consequences of dysfunction. Oncotarget 7(34):55840–55862. https://doi.org/10.18632/oncotarget.10095
Sánchez-Blázquez P, Rodríguez-Muñoz M, Garzón J (2014) The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: implications in psychosis and schizophrenia. Front Pharmacol 4:169. https://doi.org/10.3389/fphar.2013.00169
Sánchez-Blázquez P, Rodríguez-Muñoz M, Vicente-Sánchez A, Garzón J (2013) Cannabinoid receptors couple to NMDA receptors to reduce the production of NO and the mobilization of zinc induced by glutamate. Antioxid Redox Signal 19(15):1766–1782. https://doi.org/10.1089/ars.2012.5100
Schinder AF, Olson EC, Spitzer NC, Montal M (1996) Mitochondrial dysfunction is a primary event in glutamate neurotoxicity. J Neurosci 16(19):6125–6133. https://doi.org/10.1523/JNEUROSCI.16-19-06125.1996
Sgambato-Faure V, Cenci MA (2012) Glutamatergic mechanisms in the dyskinesias induced by pharmacological dopamine replacement and deep brain stimulation for the treatment of Parkinson’s disease. Prog Neurobiol 96(1):69–86. https://doi.org/10.1016/j.pneurobio.2011.10.005
Strack A, Duffy CF, Malvey M, Arriaga EA (2001) Individual mitochondrion characterization: a comparison of classical assays to capillary electrophoresis with laser-induced fluorescence detection. Anal Biochem 294(2):141–147. https://doi.org/10.1006/abio.2001.5148
Tilleux S, Hermans E (2007) Neuroinflammation and regulation of glial glutamate uptake in neurological disorders. J Neurosci Res 85(10):2059–2070. https://doi.org/10.1002/jnr.21325
Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, Lein E (2018) Preparation of acute brain slices using an optimized N-methyl-D-glucamine protective recovery method. J vis Exp 132:e53825. https://doi.org/10.3791/53825
Túnez I, Montilla P, Muñoz MC, Drucker-Colín R (2004) Effect of nicotine on 3-nitropropionic acid-induced oxidative stress in synaptosomes. Eur J Pharmacol 504(3):169–175. https://doi.org/10.1016/j.ejphar.2004.09.061
Túnez I, Tasset I, Pérez-De La Cruz V, Santamaria A (2010) 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: past, present and future. Molecules 15(2):878–916. https://doi.org/10.3390/molecules15020878
van der Meer P, Ulrich AM, González-Scarano F, Lavi E (2000) Immunohistochemical analysis of CCR2, CCR3, CCR5, and CXCR4 in the human brain: potential mechanisms for HIV dementia. Exp Mol Pathol 69(3):192–201. https://doi.org/10.1006/exmp.2000.2336
Verdon B, Zheng J, Nicholson RA, Ganellin CR, Lees G (2000) Stereoselective modulatory actions of oleamide on GABAA receptors and voltage-gated Na+ channels in vitro: a putative endogenous ligand for depressant drug sites in CNS. Br J Pharmacol 129(2):283–290. https://doi.org/10.1038/sj.bjp.0703051
Viscomi MT, Oddi S, Latini L, Bisicchia E, Maccarrone M, Molinari M (2010) The endocannabinoid system: a new entry in remote cell death mechanisms. Exp Neurol 224(1):56–65. https://doi.org/10.1016/j.expneurol.2010.03.023
Yang JY, Abe K, Xu NJ, Matsuki N, Wu CF (2002) Oleamide attenuates apoptotic death in cultured rat cerebellar granule neurons. Neurosci Lett 328(2):165–169. https://doi.org/10.1016/s0304-3940(02)00460-3
Yang L, Li Z, Xu Z, Zhang B, Liu A, He Q, Zheng F, Zhan J (2022) Protective effects of cannabinoid type 2 receptor activation against microglia overactivation and neuronal pyroptosis in sepsis-associated encephalopathy. Neuroscience 493:99–108. https://doi.org/10.1016/j.neuroscience.2022.04.011
Young AP, Denovan-Wright EM (2022) Synthetic cannabinoids reduce the inflammatory activity of microglia and subsequently improve neuronal survival in vitro. Brain Behav Immun 105:29–43. https://doi.org/10.1016/j.bbi.2022.06.011
Funding
This work was supported by the CONACYT-TUBITAK collaborative agreement (grant 265991 given to AS) and the National Institute of Environmental Health Sciences (grants R01ES03771 and R01ES10563 given to MA). None of the sponsors were involved in design, collection, analysis or interpretation of data, neither in writing of the report or decision to submit the article for publication.
Author information
Authors and Affiliations
Contributions
M.A. and A.S. designed the whole study. C.Y.R.-S., M.V.-F., E.A.O.-N., J.N.-O., S.G.-A., E.R.-L., M.M.-L., and T.K. performed all experiments and prepared Figs. 1, 2, 3, and 4. S.R.-M., I.T., and A.A.T. provided critical comments to design, interpretation of results and discussion. S.R.-M., I.T., and A.A.T. also provided reagents. M.A. and A.S. wrote the main manuscript. All authors reviewed the manuscript.
Corresponding author
Ethics declarations
Ethics Approval
All experiments were carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80–23) revised 1996, and the local Ethical Committees. Formal approval to conduct the experimental procedures was obtained from the animal subjects review board of the Instituto Nacional de Neurología y Neurocirugía (Project number 126/17). All efforts were made to minimize animals pain suffering during the experiments.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Reyes-Soto, C.Y., Villaseca-Flores, M., Ovalle-Noguez, E.A. et al. Oleamide Reduces Mitochondrial Dysfunction and Toxicity in Rat Cortical Slices Through the Combined Action of Cannabinoid Receptors Activation and Induction of Antioxidant Activity. Neurotox Res 40, 2167–2178 (2022). https://doi.org/10.1007/s12640-022-00575-7
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-022-00575-7