
Abstract
Endocannabinoid-based therapies constitute an emerging tool for the potential treatment of neurodegenerative disorders, requiring characterization at the experimental level. The effects of URB597, an inhibitor of the fatty acid amide hydrolase (FAAH), were tested against the quinolinic acid (QUIN)-induced early toxic effects in rat cortical slices, and compared with those effects exerted by the endocannabinoid anandamide (AEA). URB597 prevented the QUIN-induced loss of mitochondrial function/cell viability and lipid peroxidation, while reduced necrosis, and to a lesser extent, apoptosis. The protective effects of URB597 were mediated by activation of cannabinoid receptor 1 (CB1r), as evidenced by their inhibition by the selective CB1r antagonist AM281. Similar effects were observed when testing AEA against QUIN toxicity. Our findings demonstrate the neuroprotective properties of URB597 during the early stages of excitotoxic damage to cortical tissue, suggesting that these properties are mediated by FAAH inhibition, and might be linked to the protective effects of AEA, or the combination of endocannabinoids.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
The endocannabinoid system (ECS) comprises endogenous cannabinoids, receptors, and enzymes for the synthesis and degradation of endocannabinoids. Altogether, components of this complex regulate neuromodulatory functions such as neurotransmission, immune control, and cell signaling. Many of these effects are mediated by the activation of G protein-coupled type 1 (CB1r) and type 2 (CB2r) receptors, of which CB1r are present in high levels in several brain regions, while CB2r are present in both glial cells and nerve endings (Aymerich et al. 2018). Neuronal signaling linked to CB1r and glial signaling linked to CB2r have been studied with special emphasis on their modulatory mechanisms in the ECS on physiological and pathophysiological processes, the later including neuroinflammatory and neurodegenerative diseases (Viscomi et al. 2010).
Endocannabinoids bind to both CB1r and CB2r and are recognized as regulators of the central and peripheral nervous systems. Endocannabinoids are released post-synaptically and act as retrograde signal modulators (Colín-González et al. 2016). Neurochemical and neuroanatomical studies have demonstrated a link between endogenous cannabinoids and the N-methyl-D-aspartate receptor (NMDAr), where CB1r activation produces glutamatergic hypofunction by a mechanism involving the dissociation and abduction of NR1 subunits (Sánchez-Blázquez et al. 2014). Modulation of the ECS can be explored through the use of several pharmacological tools, such as selective CB1r and CB2r agonists and antagonists. It has been suggested that pharmacological inhibition of endocannabinoid degradative enzymes may afford therapeutic efficacy during brain injury by modulating cannabinoid signaling and reducing excessive excitability (Cravatt et al. 1996; Carloni et al. 2012; Xu and Chen 2015; Fucich et al. 2020).
Fatty acid amide hydrolase (FAAH) is a membrane-bond serine hydrolase abundantly expressed in the CNS. FAAH hydrolyzes N-acylethanolamides such as anandamide (AEA) and palmitoylethanolamide (PEA), fatty acid primary amides such as oleamide (ODA), N-acyl taurines, and fatty acid esters such as 2-arachidonoylglycerol (2-AG) (Petrosino and Di Marzo 2010; Piomelli 2003). FAAH inhibition leads to increased levels of AEA and other endocannabinoids. In turn, these fatty acid ethanolamides play important physiological functions; so, their effects have been considered as an attractive therapeutic target for several disorders of the CNS (Mallet et al. 2016). Consequently, the FAAH pharmacological inactivation has been associated with analgesic, anti-inflammatory, and anti-depressive effects without producing the side effects of several CB1r agonists (Bambibo et al. 2009; Fowler et al. 2009; Naidu et al. 2010). These effects afford therapeutic potential in models of neurodegenerative disorders such as Alzheimer’s disease (AD), Huntington’s disease (HD), and neuroinflammation (Pascual et al. 2014; Maya-López et al. 2017; Kim et al. 2018; Nazıroğlu et al. 2019).
Frontal cortex is a brain region controlling major motor, sensorial, and cognitive functions, including working memory, language, etc.; therefore, the use of cortical slices in experiments mimicking the acute and chronic toxic conditions of neurodegenerative disorders provides a suitable model enriched in nerve connections, allowing dynamic functional studies linked to several brain disorders (Ting et al. 2018). In addition, it has been shown that the ECS modulates several functions in the prefrontal cortical area (Rea et al. 2019). Models involving complex cortical functions have been shown to be positively regulated by FAAH inhibition; for instance, a toxic model mimicking the degeneration of the cortico-striatal circuitry observed in HD that was produced in rats by the mitochondrial toxin 3-nitropropionic acid (3-NP) exhibited improvement in behavioral, morphological, and biochemical outcomes following FAAH inhibition (Maya-López et al. 2017).
Quinolinic acid (QUIN) is an endogenous glutamate agonist commonly used as model to induce both excitotoxicity and oxidative stress in in vivo and in vitro studies. QUIN interacts with NMDAr preferentially at NR2A and NR2B subunits. The overactivation of this receptor (excitotoxicity) produces an increase in intracellular Ca2+ levels and the subsequent activation of numerous enzymes (proteases, nitric oxide synthase, phospholipases, endonucleases, etc.) responsible for triggering a sequence of destructive events associated with neurodegenerative processes (Schwarcz et al. 2012). The short-term (minutes-hours) toxic effects elicited by QUIN in rat cortical slices have been described by our group (Colonnello et al. 2020; Maya-López et al. 2020; Reyes-Soto et al. 2020; Soltesova Prnova et al. 2020) and involve early excitotoxic events, oxidative damage, mitochondrial dysfunction, cell signaling deregulation, and cell damage.
Cyclohexylcarbamic acid 3′-carbamoylbiphenyl-3-yl ester (URB597) is an irreversible inhibitor of FAAH with therapeutic potential. URB597 has shown protective effects in animals exposed to injections of mitochondrial and parkinsonian neurotoxins 3-NP, 6-hydroxydopamine (6-OHDA), and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) (Escamilla-Ramírez et al. 2017; Maya-López et al. 2017), as well as in in vivo QUIN toxicity (Aguilera-Portillo et al. 2019). In these models, URB597 prevented oxidative damage to lipids, attenuated motor disturbances, and ameliorated neurochemical alterations induced by these toxins, and most of these effects may be attributed to its inhibitory action on FAAH activity and, consequently, to the increase in the intracellular levels of AEA and other endocannabinoids, then producing activation of CB1r and/or other protective signals. However, both the protective effect of URB597 and the active role of CB1r as a mediator of neuroprotection have yet to be demonstrated at different experimental levels. In this study, we investigated the protective properties of URB597 on early markers of QUIN-induced toxicity in rat cortical slices—a simple biological preparation comprising the whole cellular complexity of the brain—and evaluated the possible involvement of CB1r activation in this protective pattern using specific pharmacological approaches. The effects of URB597 were explored on the following toxic outcomes: mitochondrial function/dysfunction as an index of cell viability, oxidative damage to lipids as an index of oxidative stress, and the levels of apoptosis and necrosis as indexes of short-term programmed cell and abrupt cell membrane rupture, respectively.
Material and Methods
Reagents
URB597, AM281 (1-(2,4-dichlorophenyl)-5-(4-iodophenyl)-4-methyl-N-4-morpholinyl-1H-pyrazole-3-carboxamide), and JTE-907 (N-(1,3-benzodioxol-5-ylmethyl)-1,2-dihydro-7-methoxy-2-oxo-8-(pentyloxy)-3-quinolinecarboxamide) were purchased to Tocris Bioscience (Bristol, UK). AEA, thiobarbituric acid (TBA), and 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were obtained from Sigma Chemical Co. (Sigma-Aldrich, St. Louis Missouri, USA). QUIN was purchased from Spectrum Chemical (New Brunswick, NJ, USA). Other reagents were obtained from other commercial sources.
Animals
Adult male Wistar rats (250–300 g; N = 12 (total number of animals used); n = 3 experiments per group; one rat representing one whole experiment) were used for the isolation of cortical slices. All animals were obtained from the vivarium of the Instituto Nacional de Neurología y Neurocirugía. Rats were housed at controlled room temperature (25 ± 3 °C), humidity (50%), and light-dark cycles (12:12 h). Food and water were available ad libitum. The experimental manipulation was carried out in accordance with the National Institute of Health Guide for the Care and Use of Laboratory Animals (NIH Publications No. 80-23) revised 1996 and the local Ethical Committees. Formal approval to conduct the experiments described has been obtained from the animal subjects review board of the Instituto Nacional de Neurología y Neurocirugía (Project number 126/17) and could be provided upon request. During the experiments, all efforts were made to minimize animal suffering.
Isolation of Cortical Slices and Treatments
Cortical slices were obtained according to a method previously described by us (Colín-González et al. 2014). Animals were decapitated and the frontal cortices were collected and sectioned. Tissue slices (~ 250 µm) were obtained with a chopper and incubated in Krebs-Ringer modified buffer (NaCl 135 mM, KCl 5 mM, MgSO4 1 mM, K2HPO4 0.4 mM, glucose 5.5 mM, HEPES 20 mM, and CaCl2 20 mM) for 30 min at 37 °C in the presence of 5% CO2. Slices were collected from different animals and mixed for all experiments. Tissue samples were incubated with URB597 (1–25 nM) or AEA (500 nM) as pretreatments for 30 min, and then co-added with QUIN (100 µM) and incubated for 60 min at 37 °C and 5% CO2. Some slices treated with URB597 + QUIN or AEA + QUIN also received a CB1r reverse agonist (AM281; 10 nM) or a selective CB2r agonist (JTE-907; 0.13 nM) for 30 min prior to URB597 or AEA incubation. Experimental groups are designed as follows: (a) control (an experimental condition with no drugs but only vehicles for drugs: H2O instead of AM281 or JTE-907; DMSO 1% instead of URB597 or AEA; PBS instead of QUIN); (b) URB597 alone; (c) AEA alone; (d) QUIN alone; (e) URB597 + QUIN; (f) AEA + QUIN; (g) AM281 + URB597 + QUIN; (h) AM281 + AEA + QUIN; (i) JTE-907 + URB597 + QUIN; (j) JTE-907 + AEA + QUIN. The concentrations of cannabinoid agonists and antagonists used herein were obtained or calculated on the basis of previous reports describing biological activities of these agents (Gifford et al. 1997; Lan et al. 1999; Piomelli et al. 2006; Landucci et al. 2011; Gentili et al. 2019; Zubeyir et al. 2020). All experiments were performed in a blind manner to avoid bias.
Mitochondrial Functional Assessment by MTT Reduction Assay
MTT reduction assay was performed to assess the functional status of mitochondrial function, according to a previous report (Colín-González et al. 2014). After being incubated with different treatments, slices were added to 300-µL Krebs’s buffer plus 15-µL MTT reagent (5 mg/mL) and incubated for 60 min at 37 °C. The content of formazan at 570 nm was estimated with a Cytation 3 Imaging Reader (BioTek Instruments, Winooski, VT, USA). Results were expressed as the percentage of MTT reduction vs control values.
Assay of Lipid Peroxidation
Lipid peroxidation was assessed in cortical slices according to a previous report (Colín-González et al. 2014). After being incubated with different treatments, slices were homogenized in lysis buffer, and 100-µL aliquots were added to 50 µL of TBA reagent (0.75 g TBA + 15 g trichloroacetic acid + 2.53 mL HCl) and incubated in a boiled water bath for 20 min. Then, samples were centrifuged at 3000g for 10 min at 4 °C. The pink chromophore produced was indicative of the amount of peroxidized lipid. The supernatants were collected and the absorbance was registered at 532 nm in a Cytation 3 Imagin Reader (BioTek). Results were expressed as nmols of TBA-reactive substances (TBARS) formed per mg of protein.
Flow Cytometry (Annexin/Propidium Iodide) Assay
The apoptotic/necrotic cell death detection assay was carried out by surface labeling with phosphatidylserine-binding protein annexin V and propidium iodide (Pierozan et al. 2018). After incubation, slices were mechanically disaggregated using fine needle aspirations in Krebs-Ringer modified buffer (NaCl 135 mM, KCl 5 mM, MgSO4 1 mM, K2HPO4 0.4 mM, glucose 5.5 mM, HEPES 20 mM, and CaCl2 20 mM), assisted with microscopy. Cells were collected using a disposable pipette and transferred into a processing tube. Samples were washed once with phosphate-buffered saline (PBS). Cells were then labeled by incubation with annexin/propidium iodide (Annexin V-FLUOS Staining kit) for 15 min at room temperature in the dark, following manufacturer’s instructions. Stained cells were acquired (10,000 events) on a FACS Calibur flow cytometer. Analysis was performed in the FlowJo Software. Results are presented as % cell positive to annexin and/or propidium iodide.
Statistical Analysis
Results represent independent experiments (one experiment per animal) expressed as mean values ± standard deviation of n = 4 experiments per group. Assays were performed minimally in quadruplicate and the mean was used for statistical calculation. Results of mitochondrial function and lipid peroxidation were analyzed by one-way analysis of variance (ANOVA) followed by post hoc Duncan’s test. Data of the annexin/propidium iodide assay were statistically analyzed by two-way ANOVA followed by Bonferroni’s post hoc test (GraphPad, Scientific, San Diego, CA, USA). Values of P ≤ 0.05 were considered as statistically significant.
Results
URB597 Prevented the QUIN-Induced Mitochondrial Dysfunction and Oxidative Damage to Lipids
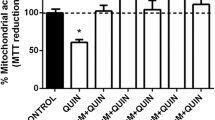
Mitochondrial function/cell viability was estimated by the MTT assay to address whether QUIN-induced mitochondrial dysfunction could be attenuated by URB597 (Fig. 1a). First, cortical slices were incubated in the presence of increased concentrations of URB597 alone (1–25 nM). MTT reduction remained unchanged at 1–20 nM URB597, compared with the control (0 nM); however, at 25 nM, URB597 decreased this marker by 32% compared with the control (p ≤ 0.001). In the presence of 100 µM QUIN alone (concentration obtained from Rangel-López et al. 2015), mitochondrial function decreased 22% below the control (p ≤ 0.001, different from control). Then, cortical slices were incubated with increased concentrations of URB597 (1, 5, 10, and 20 nM) as a pretreatment for 30 min prior to co-incubation with QUIN. While 1 and 20 nM URB597 did not prevent the effect of QUIN, incubation of slices with 5 and 10 nM resulted in a significant prevention of QUIN toxicity (22% and 13% above QUIN, respectively; p ≤ 0.001 and 0.05, different from QUIN, respectively).

Effect of URB597 on the quinolinic acid (QUIN)-induced mitochondrial dysfunction and lipid peroxidation in rat cortical slices. a Bars in the graph depict the percent of MTT reduction in cortical slices preconditioned for 30 min with URB597 (1–20 nM) and later exposed for 30 min to QUIN (100 µM). b Bars depict the levels of TBA-reactive substances in slices pretreated with URB597 (5 nM) for 30 min before QUIN (100 µM). Values are expressed as mean ± SD of n = 4 experiments per group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, different from the control; &p ≤ 0.05, &&&p ≤ 0.001, different from QUIN (for MTT reduction). One-way ANOVA followed by Duncan’s test.
Next, we explored whether the QUIN-induced oxidative damage to lipids—a major source of cell damage involving the toxic actions of reactive oxygen species (ROS)—can be prevented by URB597 (Fig. 1b). QUIN alone increased lipid peroxidation by 40% compared with the control (p ≤ 0.05). Pretreatment for 30 min with URB597 (5 nM, the concentration exhibiting protective effects on mitochondrial function) reduced the QUIN-induced lipid peroxidation by 53% (p ≤ 0.01, different from QUIN alone). URB597 per se did not produce oxidative damage to lipids.
URB597 Decreased the QUIN-Induced Necrotic and Apoptotic Cell Death
We also evaluated the role of URB597 on cell death patterns triggered by QUIN in the slices taking advantage of the annexin-V/PI assay (Fig. 2). In Fig. 2a, flow cytometric dot-plots show the distribution of cells toward the quadrants, where Q4 represents living cells, Q3 and Q2 cells in early and late apoptosis, respectively, and Q1 cells in necrosis. Flow cytometric dot-plots also contain the percent of cells in each quadrant per treatment, which served to construct the graph shown in Fig. 2b. QUIN decreased the number of living cells compared with the control (15% below; p ≤ 0.01, different from the control), and increased the number of cells in apoptosis (56% above; p ≤ 0.05, different from the control) and necrosis (376% above; p ≤ 0.001, different from the control). The QUIN-induced increase in necrosis was significantly prevented by pre-incubation with 5 nM URB597 (81% below QUIN; p ≤ 0.001), and apoptosis was also decreased, but did not attain statistical significance. It is noteworthy that slices incubated with URB597 alone showed an increase in necrotic cells (12% below QUIN; p ≤ 0.01, different from the control).

Effect of URB597 on the quinolinic acid (QUIN)-induced apoptotic and necrotic cell death. Cell loss was estimated by the annexin V-FICT and PI assay. a Cytometrograms of control (CT), URB597 (URB), quinolinic acid (QUIN), and URB + QUIN are shown. b Their corresponding densitometric analyses are depicted. Cortical slices were pretreated for 30 min with URB597 (5 nM) and further exposed for 30 min to QUIN (100 µM). Values are expressed as mean ± SD of n = 4 experiments per group. *p ≤ 0.05, **p ≤ 0.01, ***p ≤ 0.001, different from the control; ###p ≤ 0.001, different from QUIN. Two-way ANOVA followed by Bonferroni’s test.
URB597 and AEA Ameliorated the QUIN-Induced Loss of Mitochondrial Function in a CB1r-Dependent Manner
To further investigate whether the protective effect of URB597 was mediated by CB1r and/or CB2r, we incubated slices with URB597 plus QUIN in the presence of a CB1r antagonist/inverse agonist (AM281; 10 nM) or a selective CB2r inverse agonist (JTE-907; 0.1 nM) for 30 min prior to URB597 (Fig. 3a). QUIN alone decreased the mitochondrial function/cell viability by 24% below of the control (p ≤ 0.001). Preconditioning of URB597 + QUIN-treated slices with AM281 partially recovered the QUIN-induced loss of cell viability by decreasing mitochondrial function compared with the URB597 + QUIN treatment (18% below; p ≤ 0.05, different from the URB597 + QUIN treatment). In contrast, preconditioning of slices exposed to URB597 + QUIN with JTE-907 did not induce changes in the levels of cell viability observed with URB597 + QUIN. AM281 and JTE alone did not modify the basal mitochondrial function. Then, we conducted the next experiment to determine to what extent another substrate of FAAH, AEA, can prevent the QUIN-induced mitochondrial dysfunction.

Effects of URB597, anandamide (AEA), AM281 (AM), and JTE-907 (JTE) on the quinolinic acid (QUIN)-induced mitochondrial dysfunction in rat cortical slices. Bars in the graphs depict the percent of MTT reduction in slices pretreated for 30 min with URB597 (5 nM) (a) or AEA (500 nM; b), and/or the CB1r and CB2r antagonists (10 nM AM281 and 0.1 nM JTE-907, respectively; a, b) added 30 min before URB or AEA, and later exposed for 30 min to QUIN (100 µM). Values are expressed as mean ± SD of n = 4 experiments per group. ***p ≤ 0.001, different from the control; &p ≤ 0.05, &&&p ≤ 0.001, different from QUIN; #p ≤ 0.05, different from URB + QUIN or AEA + QUIN. Two-way ANOVA followed by Bonferroni’s test.
At the concentration tested in this study, AEA (500 nM) partially prevented the QUIN-induced toxicity (10% above QUIN; p < 0.05, different from QUIN treatment), whereas preconditioning the slices exposed to AEA + QUIN-treated partially recovered the effect of QUIN alone by decreasing mitochondrial function in 8% compared with the AEA + QUIN treatment (p ≤ 0.05, different from the AEA + QUIN treatment) (Fig. 3b). JTE-907 improved the effect of AEA by increasing the mitochondrial function in 10% above the AEA + QUIN treatment (p ≤ 0.01, different from the AEA + QUIN treatment).
Discussion
This report supports the concept that activation of CB1r is involved in the neuroprotective actions of URB597. Herein, we explored the potential involvement of CB1r and CB2r on the protective pattern evoked by URB597 against the early QUIN-induced toxicity. For these purposes, we incubated cortical slices in the presence of QUIN to produce excitotoxicity. URB597, at 5 nM, was highly efficacious in attenuating the toxicity, and moderate protection was noted at 10 nM, corroborating a previous report demonstrating neuroprotective effects of this compound on the neurotoxic effects elicited in the same toxic paradigm under in vivo conditions (Aguilera-Portillo et al. 2019). Indeed, the present study confirms the neuroprotective potential of FAAH inhibitors to counteract the deleterious actions of excitotoxic events and provides evidence that this protection can be attained at the early stages of toxicity via CB1r activation. In turn, the loss of basal cell viability/mitochondrial function produced by 25 nM URB597 suggests that this compound has reached a toxic concentration in this biological preparation by saturation. Therefore, this concentration was not used in further experiments.
It has been suggested that URB597 might act through cannabinoid receptor-independent mechanisms, such as antioxidant and free radical superoxide radical scavenger (Pelição et al. 2016). However, we can discard that the neuroprotective and antioxidant effects of this compound might be due to intrinsic properties as a direct antioxidant or free radical scavenger since there is evidence in literature demonstrating that URB597 is unable to trap different radicals under in vitro conditions per se (Elmazoglu et al. 2020); therefore, its neuroprotective role is likely to be related with the accumulation of AEA. In this regard, our results demonstrate that URB597, at the concentration tested, is capable of reducing the QUIN-induced oxidative damage to lipids, and these effects might correspond to the reported antioxidant properties of AEA, which have been shown to modulate the activity of antioxidant enzymes in a CB1r- and CB2r-dependent mechanism (Gallielli et al. 2018). In addition, it has been demonstrated that FAAH inhibition by URB597, and the concomitant accumulation of endogenous AEA, as well as the administration of exogenous AEA, may both lead to the expression of endogenous antioxidants likely related with transcription of Nrf2 and the expression of heme oxygenase-1 (Li et al. 2013), though these effects were described in breast cancer cells. Therefore, whether URB597 protection is associated with changes in redox status and antioxidant signaling has yet to be demonstrated in neurons. It is also noteworthy that, though FAAH is the principal AEA hydrolase in nervous tissue, the physiological actions of this and other N-acylethanolamides can also be terminated by alternative lipid amidases such as N-acylethanolamine acid amidase (NAAA) (Piomelli et al. 2020); therefore, further studies should include pharmacological strategies oriented to characterize the precise role of this enzyme in excitotoxic paradigms.
Inhibition of FAAH leading to accumulation of AEA and the subsequent activation of CB1r constitutes an axis that has been shown to prevent cell damage and apoptotic/necrotic death in different experimental models, including traumatic brain injury (Katz et al. 2015), retinal damage after high intraocular pressure-induced ischemia (Nucci et al. 2007), an in vitro seizure model (Nazıroğlu et al. 2019), and striatal excitotoxic damage (Aguilera-Portillo et al. 2019). Altogether, this evidence supports the concept that this axis is responsible for triggering cascades of protective signals aimed to reduce necrosis and apoptosis. Accordingly, in our study, in quantitative terms (percent of cells), the predominant type of cell death detected, at the basal level, was apoptosis, followed by necrosis, and this was due to the fact that early and late apoptotic events were added each other and considered together for graphical purposes. On the other hand, it is noteworthy that, under the experimental conditions used in this report, QUIN exerted a more prominent effect on necrosis than apoptosis, and this makes sense when considering that our experiments attended the short-term phase of toxic events in the excitotoxic paradigm. Under this scenario, early necrotic cell death—a sudden and disorganized process—is expected to predominate over programmed apoptotic cell death, though both may take place in excitotoxic insults (Ferrer et al. 1995). In order to better appreciate the course of apoptosis in this paradigm, studies at longer times of exposure to QUIN should be considered (for instance, in organotypic cultures). In the interim, since our interest in this study was focused on exploring the early toxic events underlying the exposure of slices to an excitotoxic insult, this first approach describes initial necrotic cell damage over apoptosis. It is therefore not surprising at all that, despite URB597 slightly attenuated apoptosis, its more prominent effect was directed to ameliorate necrosis as the predominant early event in course, thus emphasizing its neuroprotective potential. Based on our findings, we suggest that the lack of protective effect of URB597 on the QUIN-induced decrease in living cells might correspond to the lack of effect of this compound on the early toxic induction of apoptosis, since QUIN-induced necrosis was completely abolished by URB597. Moreover, since necrosis was more prominently induced by QUIN and prevented by URB597, it is likely that the inflammatory component of necrosis, but not apoptosis, might also be a primary target for this compound, since it has been suggested that FAAH inhibition can reduce inflammatory outcomes in some models of brain cell damage (Elmazoglu et al. 2020) via inhibition of the pro-inflammatory NF-κB pathway (Su et al. 2017).
We also evaluated whether CB1r and CB2r are involved in the actions of URB597. Based on our findings, we conclude that, under the experimental conditions applied herein, the effects of this compound involve the activation of CB1r. In a similar study, URB597 completely prevented the toxic effect of QUIN in cultured neuronal cells (Kotlar et al. 2019). In addition, the effect of the CB1r antagonist AM281 supports the concept that the protective effects of URB597 could be explained by accumulation of AEA in combination with other bioactive amides after FAAH inhibition, and further activation of CB1r by endocannabinoids, as FAAH is the primary catabolic regulator of AEA and ODA (De Petrocellis et al. 2004). In further support of this concept, it has been demonstrated that the QUIN-induced excitotoxic damage is prevented by activation of CB1r specifically sitting in glutamatergic terminals (Chiarlone et al. 2014). In this context, we suggest that CB1r activation might induce protection by five already reported mechanisms: (1) reduction of the NMDA receptor-mediated excitability through a mechanism linked to NR1 subunit abduction by the CB1r-coupled protein HINT1 (Sánchez-Blázquez et al. 2014), (2) inhibition of glutamate release, (3) decreased intracellular Ca2+ levels, (4) regulation of ion channels, and (5) activation of MAPK and phosphatidylinositol-3-kinase pathways (van der Stelt and Di Marzo 2005). Together, these mechanisms may exert protection against QUIN-induced toxicity by reducing NMDAr overactivation, regulation of glutamate release, and mitochondrial energy metabolism, while decreasing the ensuing oxidative stress (Pintor et al. 2006; Bénard et al. 2012).
AEA partially prevented the loss of cell viability induced by QUIN, and we also investigated if this effect was mediated by CB1r and/or CB2r. According to our results, the protective effect of AEA against QUIN toxicity is CB1r-dependent, as previously demonstrated (Rangel-López et al. 2015; Kotlar et al. 2019). Nonetheless, a participation of the CB2r in the toxic paradigm elicited by QUIN should not be discarded. Finally, our results also show that JTE-907 had no effect on the changes in mitochondrial function induced by the URB597 + QUIN and AEA + QUIN conditions, suggesting that, at least under these experimental conditions, CB2r does not mediate the protective actions of URB597 and AEA.
Concluding Remarks
In summary, the novel findings collected in this work demonstrate that (1) the modulation of the ECS constitutes a potential tool to prevent the noxious effects of excitotoxic agents; (2) the use of URB597 represents a useful experimental strategy capable of ameliorating the early excitotoxic effects of QUIN; (3) CB1r activation participates in the mechanism of protection induced by URB597 in the experimental conditions tested; and (4) the neuroprotective effect of URB597 might involve the protective properties of AEA and other accumulated endocannabinoids. We therefore suggest that the design of therapies against neurodegenerative disorders with excitotoxic components should consider the use of FAAH inhibitors.
References
Aguilera-Portillo G, Rangel-López E, Villeda-Hernández J, Chavarría A, Castellanos P, Elmazoglu Z, Karasu Ç, Túnez I, Pedraza G, Königsberg M, Santamaría A (2019) The pharmacological inhibition of fatty acid amide hydrolase prevents excitotoxic damage in the rat striatum: possible involvement of CB1 receptors regulation. Mol Neurobiol 56:844–856
Aymerich MS, Aso E, Abellanas MA, Tolon RM, Ramos JA, Ferrer I, Romero J, Fernández-Ruiz J (2018) Cannabinoid pharmacology/therapeutics in chronic degenerative disorders affecting the central nervous system. Biochem Pharmacol 157:67–84
Bambico FR, Duranti A, Tontini A, Tarzia G, Gobbi G (2009) Endocannabinoids in the treatment of mood disorders: evidence from animal models. Curr Pharm Des 15:1623–1646
Bénard G, Massa F, Puente N, Lourenço J, Bellocchio L, Soria-Gómez E, Matias I, Delamarre A, Metna-Laurent M, Cannich A, Hebert-Chatelain E, Mulle C, Ortega-Gutiérrez S, Martín-Fontecha M, Klugmann M, Guggenhuber S, Lutz B, Gertsch J, Chaouloff F, López-Rodríguez ML, Grandes P, Rossignol R, Marsicano G (2012) Mitochondrial CB1 receptors regulate neuronal energy metabolism. Nat Neurosci 15:558–564
Carloni S, Alonso-Alconada D, Girelli S, Duranti A, Tontini A, Piomelli D, Hilario E, Alvarez A, Balduini W (2012) Pretreatment with the monoacylglycerol lipase inhibitor URB602 protects from the long-term consequences of neonatal hypoxic–ischemic brain injury in rats. Pediatr Res 72:400–406
Chiarlone A, Bellocchio L, Blázquez C, Resel E, Soria-Gómez E, Cannich A, Ferrero JJ, Sagredo O, Benito C, Romero J, Sánchez-Prieto J, Lutz B, Fernández-Ruiz J, Galve-Roperh I, Guzmán M (2014) A restricted population of CB1 cannabinoid receptors with neuroprotective activity. Proc Natl Acad Sci USA 111:8257–8262
Colín-González AL, Maya-López M, Pedraza-Chaverrí J, Ali SF, Chavarría A, Santamaría A (2014) The Janus faces of 3-hydroxykynurenine: dual redox modulatory activity and lack of neurotoxicity in the rat striatum. Brain Res 1589:1–14
Colín-González AL, Aguilera G, Santamaría A (2016) Cannabinoids: glutamatergic transmission and kynurenines. Adv Neurobiol 12:173–198
Colonnello A, Aguilera-Portillo G, Rubio-López LC, Robles-Bañuelos B, Rangel-López E, Cortez-Núñez S, Evaristo-Priego Y, Silva-Palacios A, Galván-Arzate S, García-Contreras R, Túnez I, Chen P, Aschner M, Santamaría A (2020) Comparing the neuroprotective effects of caffeic acid in rat cortical slices and Caenorhabditis elegans: involvement of Nrf2 and SKN-1 signaling pathways. Neurotox Res 37:326–337
Cravatt BF, Giang DK, Mayfield SP, Boger DL, Lerner RA, Gilula NB (1996) Molecular characterization of an enzyme that degrades neuromodulatory fatty-acid amides. Nature 384:83–87
De Petrocellis L, Cascio MG, Di Marzo V (2004) The endocannabinoid system: a general view and latest additions. Br J Pharmacol 141:765–774
Elmazoglu Z, Rangel-López E, Medina-Campos ON, Pedraza-Chaverri J, Túnez I, Aschner M, Santamaría A, Karasu Ç (2020) Cannabinoid-profiled agents improve cell survival via reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Aβ1-42 peptide in rat hippocampal neurons. Neurochem Int. https://doi.org/10.1016/j.neuint.2020.104817
Escamilla-Ramírez A, García E, Palencia-Hernández G, Colín-González AL, Galván-Arzate S, Túnez I, Sotelo J, Santamaría A (2017) URB597 and the cannabinoid WIN55,212–2 reduce behavioral and neurochemical deficits induced by MPTP in mice: possible role of redox modulation and NMDA receptors. Neurotox Res 31:532–544
Ferrer I, Martin F, Serrano T, Reiriz J, Pérez-Navarro E, Alberch J, Macaya A, Planas AM (1995) Both apoptosis and necrosis occur following intrastriatal administration of excitotoxins. Acta Neuropathol 90:504–510
Fowler CJ, Naidu PS, Lichtman A, Onnis V (2009) The case for the development of novel analgesic agents targeting both fatty acid amide hydrolase and either cyclooxygenase or TRPV1. Br J Pharmacol 156:412–419
Fucich EA, Stielper ZF, Cancienne HL, Edwards S, Gilpin NW, Molina PE, Middleton JW (2020) Endocannabinoid degradation inhibitors ameliorate neuronal and synaptic alterations following traumatic brain injury 123:707–717
Gallelli CA, Calcagnini S, Romano A, Koczwara JB, de Ceglia M, Dante D, Villani R, Giudetti AM, Cassano T, Gaetani S (2018) Modulation of the oxidative stress and lipid peroxidation by endocannabinoids and their lipid analogues. Antioxidants 7:93
Gentili M, Ronchetti S, Ricci E, Di Paola R, Gugliandolo E, Cuzzocrea S, Bereshchenko O, Migliorati G, Riccardi C (2019) Selective CB2 inverse agonist JTE907 drives T cell differentiation towards a Treg cell phenotype and ameliorates inflammation in a mouse model of inflammatory bowel disease. Pharmacol Res 141:21–31
Gifford AN, Tang Y, Gatley SJ, Volkow ND, Lan R, Makriyannis A (1997) Effect of the cannabinoid receptor SPECT agent, AM 281, on hippocampal acetylcholine release from rat brain slices. Neurosci Lett 238:84–86
Katz PS, Sulzer JK, Impastato RA, Teng SX, Rogers EK, Molina PE (2015) Endocannabinoid degradation inhibition improves neurobehavioral function, blood-brain barrier integrity, and neuroinflammation following mild traumatic brain injury. J Neurotrauma 32:297–306
Kim MJ, Tanioka M, Um SW, Hong S-K, Lee BH (2018) Analgesic effects of FAAH inhibitor in the insular cortex of nerve-injured rats. Mol Pain 14:1744806918814345
Kotlar I, Rangel-López E, Colonnello A, Aguilera-Portillo G, Serratos IN, Galván-Arzate S, Pedraza-Chaverri J, Túnez I, Wajner M, Santamaría A (2019) Anandamide reduces the toxic synergism exerted by quinolinic acid and glutaric acid in rat brain neuronal cells. Neuroscience 401:84–95
Lan R, Gatley J, Lu Q, Fan P, Fernando SR, Volkow ND, Pertwee R, Makriyannis A (1999) Design and synthesis of the CB1 selective cannabinoid antagonist AM281: a potential human SPECT ligand. AAPS PharmSci 1:E4
Landucci E, Scartabelli T, Gerace E, Moroni F, Pellegrini-Giampietro DE (2011) CB1 receptors and post-ischemic brain damage: studies on the toxic and neuroprotective effects of cannabinoids in rat organotypic hippocampal slices. Neuropharmacology 60:674–682
Li H, Wood JT, Whitten KM, Vadivel SK, Seng S, Makriyannis A, Avraham HK (2013) Inhibition of fatty acid amide hydrolase activates Nrf2 signalling and induces heme oxygenase 1 transcription in breast cancer cells. Br J Pharmacol 170:489–505
Mallet C, Dubray C, Dualé C (2016) FAAH inhibitors in the limelight, but regrettably. Int J Clin Pharmacol Ther 54:498–501
Maya-López M, Ruiz-Contreras HA, Negrete-Ruíz MJ, Martínez-Sánchez JE, Benítez-Valenzuela J, Colín-González AL, Villeda-Hernández J, Sánchez-Chapul L, Parra-Cid C, Rangel-López E, Santamaría A (2017) URB597 reduces biochemical, behavioral and morphological alterations in two neurotoxic models in rats. Biomed Pharmacother 88:745–753
Maya-López M, Rubio-López LC, Rodríguez-Alvarez IV, Orduño-Piceno J, Flores-Valdivia Y, Colonnello A, Rangel-López E, Túnez I, Prospéro-García O, Santamaría A (2020) A cannabinoid receptor-mediated mechanism participates in the neuroprotective effects of oleamide against excitotoxic damage in rat brain synaptosomes and cortical slices. Neurotox Res 37:126–135
Naidu PS, Kinsey SG, Guo TL, Cravatt BF, Lichtman AH (2010) Regulation of inflammatory pain by inhibition of fatty acid amide hydrolase. J Pharmacol Exp Ther 334:182–190
Nazıroğlu M, Taner AN, Balbay E, Çiğ B (2019) Inhibitions of anandamide transport and FAAH synthesis decrease apoptosis and oxidative stress through inhibition of TRPV1 channel in an in vitro seizure model. Mol Cell Biochem 453:143–155
Nucci C, Gasperi V, Tartaglione R, Cerulli A, Terrinoni A, Bari M, De Simone C, Agrò AF, Morrone LA, Corasaniti MT, Bagetta G, Maccarrone M (2007) Involvement of the endocannabinoid system in retinal damage after high intraocular pressure-induced ischemia in rats. Invest Ophthalmol Vis Sci 48:2997–3004
Pascual AC, Martín-Moreno AM, Giusto NM, de Ceballos ML, Pasquaré SJ (2014) Normal aging in rats and pathological aging in human Alzheimer’s disease decrease FAAH activity: modulation by cannabinoid agonists. Exp Gerontol 60:92–69
Pelição R, Santos MC, Freitas-Lima LC, Meyrelles SS, Vasquez EC, Nakamura-Palacios EM, Rodrigues LCM (2016) URB597 inhibits oxidative stress induced by alcohol binging in the prefrontal cortex of adolescent rats. Neurosci Lett 624:17–22
Petrosino S, Di Marzo V (2010) FAAH and MAGL inhibitors: therapeutic opportunities from regulating endocannabinoid levels. Curr Opin Investig Drugs 11:51–62
Pierozan P, Colín-González AL, Biasibetti H, Camacho da Silva J, Wyse A, Wajner M, Santamaria A (2018) Toxic synergism between quinolinic acid and glutaric acid in neuronal cells is mediated by oxidative stress: insights to a new toxic model. Mol Neurobiol 55:5362–5376
Pintor A, Tebano MT, Martire A, Grieco R, Galluzo M, Scattoni L, Pèzzola A, Coccurello R, Felici F, Cuomo V, Piomelli D, Calamandrei G, Popoli P (2006) The cannabinoid receptor agonist WIN 55,212–2 attenuates the effects induced by quinolinic acid in the rat striatum. Neuropharmacology 51:1004–1012
Piomelli D (2003) The molecular logic of endocannabinoid signaling. Nat Rev Neurosci 4:873–884
Piomelli D, Tarzia G, Duranti A, Tontini A, Mor M, Compton TR, Dasse O, Monaghan EP, Parrott JA, Putman D (2006) Pharmacological profile of theselective FAAH inhibitor KDS-4. CNS Drug Rev 12:21-38
Piomelli D, Scalvini L, Fotio Y, Lodola A, Spadoni G, Tarzia G, Mor M (2020) N-acylethanolamine acid amidase (NAAA): structure, function, and inhibition. J Med Chem. https://doi.org/10.1021/acs.jmedchem.0c00191
Rangel-López E, Colín-González AL, Paz-Loyola AL, Pinzón E, Torres I, Serratos IN, Castellanos P, Wajner M, Souza DO, Santamaría A (2015) Cannabinoid receptor agonists reduce the short-term mitochondrial dysfunction and oxidative stress linked to excitotoxicity in the rat brain. Neuroscience 285:97–106
Rea K, McGowan F, Corcoran L, Roche M, Finn DP (2019) The prefrontal cortical endocannabinoid system modulates fear-pain interactions in a subregion-specific manner. Br J Pharmacol 176:1492–1505
Reyes-Soto CY, Rangel-López E, Galván-Arzate S, Colín-González AL, Silva-Palacios A, Zazueta C, Pedraza-Chaverri J, Ramírez J, Chavarria A, Túnez I, Ke T, Aschner M, Santamaría A (2020) S-Allylcysteine protects against excitotoxic damage in rat cortical slices via reduction of oxidative damage, activation of Nrf2/ARE binding, and BDNF preservation. Neurotox Res. https://doi.org/10.1007/s12640-020-00260-7
Sánchez-Blázquez P, Rodríguez-Muñoz M, Garzón J (2014) The cannabinoid receptor 1 associates with NMDA receptors to produce glutamatergic hypofunction: implications in psychosis and schizophrenia. Front Pharmacol 4:169
Schwarcz R, Bruno JP, Muchowski PJ, Wu HQ (2012) Kynurenines in the mammalian brain: when physiology meets pathology. Nat Rev Neurosci 13:465–477
Soltesova Prnova M, Medina-Campos ON, Pedraza-Chaverri J, Colín-González AL, Piedra-García F, Rangel-López E, Kovacikova L, Ceylan A, Karasu C, Santamaria A, Stefek M (2020) Antioxidant mechanisms in the neuroprotective action of cemtirestat: studies in chemical models, liposomes and rat brain cortical slices. Neuroscience 443:206–217
Su S-H, Wu Y-F, Lin Q, Hai J (2017) Cannabinoid receptor agonist WIN55,212–2 and fatty acid amide hydrolase inhibitor URB597 ameliorate neuroinflammatory responses in chronic cerebral hypoperfusion model by blocking NF-κB pathways. Naunyn Schmiedebergs Arch Pharmacol 390:1189–1200
Ting JT, Lee BR, Chong P, Soler-Llavina G, Cobbs C, Koch C, Zeng H, Lein E (2018) Preparation of acute brain slices using an optimized N-methyl-D-glucamine protective recovery method. J Vis Exp 132:e53825
van der Stelt M, Di Marzo V (2005) Cannabinoid receptors and their role in neuroprotection. Neuromolecular Med 7:37–50
Viscomi MT, Oddi S, Latini L, Bisicchia E, Maccarrone M, Molinari M (2010) The endocannabinoid system: a new entry in remote cell death mechanisms. Exp Neurol 224:56–65
Xu JY, Chen C (2015) Endocannabinoids in synaptic plasticity and neuroprotection. Neuroscientist 21:152–168
Zhou J, Burkovskiy I, Yang H, Sardinha J, Lehmann C (2016) CB2 and GPR55 receptors as therapeutic targets for systemic immune dysregulation. Front Pharmacol 7:264
Zubeyir E, Rangel-López E, Medina-Campos ON, Pedraza-Chaverri J, Túnez I, Aschner M, Santamaría A, Karasu C (2020) Cannabinoid-profiled agents improve cell survival via reduction of oxidative stress and inflammation, and Nrf2 activation in a toxic model combining hyperglycemia+Aβ1-42 peptide in rat hippocampal neurons. Neurochem Int. https://doi.org/10.1016/j.neuint.2020.104817
Funding
This work was supported by the CONACYT-TUBITAK collaborative agreement (grant 265991 given to AS) and the National Institute of Environmental Health Sciences (grants R01ES03771, R01ES10563, and R01ES020852 given to MA). None of the sponsors were involved in design, collection, analysis, or interpretation of data, neither in writing of the report or decision to submit the article for publication.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of Interest
The authors declare that they have no conflict of interest.
Additional information
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Chavira-Ramos, K., Orozco-Morales, M., Karasu, Ç. et al. URB597 Prevents the Short-Term Excitotoxic Cell Damage in Rat Cortical Slices: Role of Cannabinoid 1 Receptors. Neurotox Res 39, 146–155 (2021). https://doi.org/10.1007/s12640-020-00301-1
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12640-020-00301-1