
Abstract
The transient gene expression of protoplasts in plant is a considerable tool for gene functional research that has been widely used in gene analysis and functional characterization. Therefore, the objectives of this study were to develop a protocol for the isolation and purification of sugarcane protoplasts (Saccharum spp. hybrids), conduct transient PEG-mediated protoplast transfection with D27, and localize the D27 protein in sugarcane protoplasts. Total yield and viability of protoplasts were optimized for enzyme combination, mannitol concentration, and duration and temperature of enzymatic hydrolysis. High production of intact protoplasts (10.94 × 106 protoplasts g−1 FW) and a survival rate of > 80.0% was achieved through enzymatic hydrolysis at constant temperature of 28 °C, 60–70 rpm min−1 for 8 h in a solution containing 2.0% cellulase R-10, 0.5% macerozyme R-10, 0.6% pectolyase Y-23, 20 mM 2-(N-morpholine) ethanesulfonic acid (MES), 20 mM KCl, and 400 mM mannitol (pH 5.7). Using GFP as the reporter gene, the protoplasts were transformed most efficiently with 25% PEG 4000 for 25 min and the ScD27 protein was localized in the chloroplasts. The localization of ScD27 protein in sugarcane protoplast demonstrated that the newly developed protocol was functionally effective. This optimized sugarcane protoplast isolation, purification, and transient expression protocol lays a foundation for future molecular biology research in sugarcane.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Sugarcane is a high yield sugar crop in the world that widely grows in tropical area. Due to its complex polyploid genome, there has been little genetic research on this crop. The yield formation of sugarcane is a complex process influenced by physiological, biochemical, and genetic factors as well as growth environment. Strigolactones (SLs) are a group of terpenoid lactone hormones produced in the plant roots that have diverse regulatory effects on plant growth and development, including inhibiting tillering or branching, shaping root morphology, promoting leaf senescence, and regulating secondary growth (Flematti et al. 2016; Gomez-Roldan et al. 2008; Umehara et al. 2008).
DWARF27 (D27) was previously proven to be involved in SL biosynthesis. The D27 protein is the first enzyme involved in the biosynthesis of strigolactone by converting all-trans- β-carotene into 9-cis-β-carotene in a reversible isomerization reaction (Vogel et al. 2010). The D27 gene encodes an iron-binding protein in rice, which catalyzes the isomerization of 9-cis/all-trans isomerization (Lin et al. 2009). In rice, expression of D27 may depend on auxin signal (Waters et al. 2012) and affect the regulation of the biosynthesis of SL (Waters et al. 2017). The expression of DgD27 in Chrysanthemum morifolium is inhibited by decapitation, but is induced by auxin. DgD27 is also sensitive to light, P deficiency, and low N (Wen et al. 2016). We reported earlier that the 1379 bp full-length sequence of β-carotene isomerase D27 (KP987221.1) from sugarcane (ScD27) contained a complete open reading frame of 867 bp encoding for 288 amino acids. ScD27 expression was tissue specific, higher in sugarcane shoot tips than axillary buds, and could be induced by drought, P deficiency, or salt stress (Wu et al. 2017a, b). We proposed that the ScD27 protein might locate in the chloroplast (Wu et al. 2017a, b).
Plant protoplasts are often used to study somatic hybridization, genome editing, transient gene expression, and subcellular protein localization, as they are totipotent and able to receive exogenous nucleic acid through transformation (Shen et al. 2014). Polyethylene glycol (PEG)-mediated protoplast transformation has been successful in a variety of plants to achieve high recombination frequency and homozygosity (Yu et al. 2017). Transient gene expression in protoplasts is a primary method to study the function of transgenes in genetic and physiological processes, such as cell wall regeneration, cell division, differentiation, cell genetic transformation, and somatic cell hybridization (Nanjareddy et al. 2016). Plant tissue, enzymatic hydrolysis, enzymolysis time, and temperature can affect the yield and viability of protoplasts (Wu et al. 2017a, b). Methods of protoplast isolation and transient gene expression have been established in many plants, such as Arabidopsis (Yoo et al. 2007), Zea mays (Chen et al. 2015), Manihot esculenta Crantz (Wu et al. 2017a, b), Brassica oleracae var. alboglabra Bailey (Wu et al. 2017a, b), and Cucumis sativus L (Sun et al. 2018). Sugarcane protoplasts have been isolated and cultured to obtain embryogenic calli (Sahab et al. 2019). However, until now, an efficient transient gene expression system has not been optimized for use on sugarcane protoplasts.
The objectives of the present study, therefore, were to optimize the isolation, purification, and PEG-mediated transient transfection of sugarcane protoplasts and to determine the subcellular location of the ScD27 protein.
Materials and Methods
Plant Materials and Growth Conditions
Young leaves were obtained from the stem apex of ROC22, the most popular sugarcane cultivar in China that can be readily cultured in vitro. The leaves were disinfected, cut into 1-mm slices, and placed on MS culture media (pH 5.8.) containing 1 mg L−1 2,4-D, 3% sucrose, and 0.8% carrageenan. After 4 to 5 weeks, embryogenic calli were formed and seedlings were produced by transferring to differentiation and rooting media. Tissue culture was conducted in an artificial climate incubator at 25 ± 1 °C and photosynthetic active radiation (PAR) of 120 μ mol m−2 s−1 under a 16 h light/8 h dark cycle.
Protoplast Isolation
About 1 g of leaf tissue was collected from tissue culture seedlings, cut into 1-mm slices, and placed in a sterile 100 × 25 mm Petri dish containing 10 mL of enzyme mixture solution. A modified method of Yoo et al.(2007) and Huang et al. (2013) was used to prepare the enzyme mix: 1% or 2% (W/V) cellulase R-10 (Yakult, Japan), 0.3% or 0.5% macerozyme R-10 (Yakult, Japan), 0% or 0.6% pectolyase Y-23 (Yakult, Japan), 20 mM 2-(N-morpholine) ethanesulfonic acid (MES) (pH 5.7, preheating at 70 °C for 5 min before use), 20 mM potassium chloride (KCl), and 0.2, 0.4, 0.6, or 0.8 M mannitol, and ddH2O (Table 1). The enzyme solution was warmed to 55 °C for 10 min to improve enzyme solubility and allowed to cool to 25 °C, then 0.1% BSA, 10 mM CaCl2, 3–5 μl of β-mercaptoethanol were added and the pH was adjusted to 5.8 with 1 M KOH. The enzyme solution was filter sterilized through a 45 μM nylon membrane filter and was promptly used. The Petri dishes were incubated in darkness with gentle shaking at 50 rpm.
Enzymolysis was tested at 4, 6, 8, 12, and 16 h to determine the optimal time for protoplast isolation. Optimum culture temperature was tested at 25, 28, 30, and 35 °C. At each time and temperature point, a 10 μl sample of the enzyme-protoplast extract solution was taken for microscopic examination with an Olympus CX31 optical microscope (CX 31 Olympus, Japan).
Protoplast Purification
After enzymolysis, the enzyme-protoplast mixture was filtered through a 70 μM (200 mesh) nylon membrane to remove large tissue debris. The filtrate was collected in a sterile test tube and, centrifuged at 200 rpm at 4 °C for 5 min to collect protoplasts. The supernatant was eluted and an equal volume of pre-cooled sterile W5 buffer solution (154 mM NaCl, 125 mM CaCl2, 5 mM KCl, 2 mM MES, pH 5.7) was added with gentle shaking to wash the protoplasts. The collection and washing process was repeated once more and the protoplasts were stored in W5 solution in darkness at 22 °C. Number of protoplasts was determined using a double-chamber hemocytometer with an optical microscope (Olympus, Japan). Protoplast viability was measured using fluorescein diacetate (FDA) staining (Larkin 1976). The stain solution was prepared by dissolving 2 mg of FDA in 1 mL acetone and stored in a 4 °C refrigerator. Five mL of the FDA stain was added to 100 μL of protoplast suspension [final FDA concentration 0.01% (w/v)] and incubated for 10 min in darkness. Protoplast viability was assessed by examining an aliquot of stained protoplast suspension under a fluorescence microscope (Zeiss AxioImager A2m Carl Zeiss, Germany).
Protoplast yield was calculated as: protoplast g−1 fresh weight (FW) = the number of protoplasts in enzyme solution/weight of plant leaves used for enzymolysis. Protoplast viability was calculated as: Protoplast viability (%) = (the number of fluorescing protoplasts/total number of protoplasts) × 100.
Plasmid Construction and the ScD27 Protein Subcellular Localization
Competent cells of Escherichia coli strain DH5α were purchased from Beijing TransGen Biotech Co., Ltd. (Beijing, China). The sugarcane SL biosynthetic gene β-carotene isomerase, ScD27 was studied to evaluate the feasibility and practicability of this optimized transient expression system.
The Takara MiniBEST Universal RNA Extraction Kit (Takara, Japan) was used to extract RNA and PrimeScript® 1st Strand cDNA Synthesis Kit (Takara, Japan) was used to synthesize cDNA following the manufacturer’s instructions. Based on the entire coding sequence of ScD27 (Wu et al. 2017a, b), two primers, each containing NcoI or BglII enzyme restriction site at its 5′end, were designed and synthesized by Shenggong Biotech Co., Ltd (Shanghai, China). The nucleotide sequence (5′ to 3) of Primer 1302D27-forward was CATGCCATGGATGCTCCTTGCTCACGCT and the nucleotide sequence (5′ to 3′) of Primer 1302D27-reverse was GAAGATCTTCAAATAGAGCAATTCACTTGACG. The ScD27 gene was amplified by PCR using pMD18-T-ScD27 plasmid as the template. The reaction mixture contained 5 μl 1 pmol of each primer, 40 μM dNTP, 0.1U Taq DNA polymerase (Takara, Japan), 1.5 mM MgCl2, and 1 × buffer supplied with the enzyme. The thermal cycling program was 94 °C, 3 min; 30 cycles of (94 °C,30 s; 60 °C, 30 s; 72 °C, 1 min); and 72 °C, 10 min. The PCR product 927 bp and Nco I and Bgl II-double digested pCMBIA1302 vector (1302 bp, a kind gift of Shu-Zhen Zhang, the Institute of Tropical Bioscience and Biotechnology Chinese Academy of Tropical Agricultural Sciences, Haikou, Hainan, China) were gel purified using T4 DNA ligase. The ligation mixture was used to transform E. coli DH5α cells. Recombinant colonies were cultured to amplify the recombinant pCMBIA1302-ScD27 plasmids, which were verified by restriction enzyme digestion and DNA sequencing by Sango Biotech Co., Ltd (Shanghai, China). Sequencing results showed that the ScD27 fragment was inserted between the 35S promoter and the GFP gene. The subcellular localization of the ScD27 protein was predicted using the website WoLF PSORT (http://www.genscript.com/wolf.psort.html) and determined using laser scanning confocal microscopy.
Protoplast Transformation and PEG-Mediated Gene Transient Expression
The methods of Yoo et al. (2007) and Sun et al. (2018) were used in PEG-mediated transient expression experiments. About 1–5 × 104 protoplasts/mL 100 μl solution in MMG buffer (0.4 M mannitol, 15 mM MgCl2, and 4.0 mM MES, pH 5.7) were mixed with 110 μl PEG solution [40% PEG 4000 (W/V), 2.0 M mannitol, and 0.1 M CaCl2,] and transformed with recombinant pCAMBIA1302-35S-GFP plasmids.
Protoplast transformation was conducted at room temperature in darkness by mixing 10 μl of pCAMBIA1302-35S-GFP (1 μg/μl) with 100 μl of protoplast solution and 110 μl of PEG 4000 solution. The PEG concentrations tested were 10%, 15%, 20%, 25%, 30%, and 35% (w/v). Transformation times were tested at 10, 15, 20, 25, 30, and 35 min. Transformation was terminated by adding 800 μl W5 solution. After termination, transformed solutions were centrifuged at 500 rpm for 2 min. The supernatant was discarded. The protoplast pellet was resuspended in 1 mL W5 solution, which was then transferred to a sterile BSA-coated Petri dish and incubated in the dark at 25 ± 1 °C for 16–24 h.
The expression of the GFP fusion protein was observed using fluorescence microscope. Transformation efficiency was calculated as: transformation efficiency (%) = (number of fluorescent protoplasts/total protoplasts) × 100.
Data Analysis
Experiments were repeated three times, and analysis of variance was conducted using SPSS software Version 18.0 (SPSS Inc. Chicago, IL, USA). The least significant difference (LSD) was P ≤ 0.05.
Results
Isolation of Sugarcane Protoplasts
Enzyme Composition
Yield of isolated protoplasts ranged from 0.05 to 10.94 × 106 protoplasts/g FW depending on enzyme composition (Fig. 1). Enzyme composition 8 (2.0% cellulase R-10, 0.5% macerozyme R-10, and 0.6% pectolyase Y-23) yielded significantly more protoplasts (10.94 × 106 g−1 FW) than the other enzyme compositions (Table 1; Fig. 1). However, there was no predictable change in viability of protoplasts due to enzyme composition, which ranged from 46.96 to 85.6% (Fig. 1). Enzyme compositions 5 to 8, all contained 0.6% pectolyase Y-23, and they yielded significantly more sugarcane protoplasts than compositions 1 to 4 that contained no pectolyase (Fig. 1).

Effect of enzymatic composition on yield (bars) and viability (line) of ROC 22 protoplasts. Different letters represent a statistically significant difference at P ≤ 0.05, and bars represent standard errors (SEs)
Mannitol Concentration and Enzymolysis Time
To determine the optimal enzymatic osmotic pressures, mannitol at different concentration was set. Mannitol at 0.4 M resulted in significantly higher protoplast yields (6.30 × 106 protoplasts/g FW, P ≤ 0.05) with 86% viability (Fig. 2a). Compared to that of 0.4 mM mannitol, however, yield and viability of protoplasts were all significantly lower at 0.2 M, 0.6 M, or 0.8 M mannitol concentration, with 3.76 × 106, 1.47 × 106, or 1.37 × 106 protoplasts/g FW and 58%, 50%, or 32% viability, respectively. Thus, 0.4 M mannitol was the optimal concentration for enzymolytic protoplast production.
Protoplast viability was significantly higher at 4, 6, or 8 h enzymolysis (69%, 76%, or 67%) compared to 12 or 16 h enzymolysis (56% or 35%) (Fig. 2b). Significantly, higher protoplast yield of 4.52 × 106 protoplasts/g FW was obtained at 12 h enzymolysis (Fig. 3). Similarly, 12 h enzymolysis significantly increased protoplast yield (Fig. 2b) with less debris (data not shown).

Effects of mannitol concentration and enzymolysis time on the total protoplast yield (106 protoplasts/g FW) and protoplast viability (%) from sugarcane. Different letters represent a statistically significant difference at P ≤ 0.05, and bars represent standard errors (SEs)

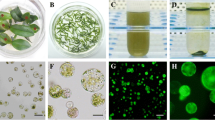
Photomicrographs of isolated sugarcane protoplasts. a, b, c, d show protoplasts isolated after 4, 6, 8, and 12 h, respectively
Influence of Temperature
Enzymolysis temperature (25, 28, 30, or 35 °C) affected protoplast yield and viability (Fig. 4). The highest mean yield of protoplasts (4.86 × 106 protoplasts/g FW) was obtained when enzymolysis was conducted at 28 °C (Fig. 4). Viability did not differ significantly among temperature treatments, although that at 28 °C (75.23%) tended to be greater than at 25, 30, or 35 °C. The FDA dye test further demonstrated the protoplast yield and viability were sufficiently optimized to attempt transformation studies (Fig. 5). We achieved these results by using 0.4 M mannitol concentration, 12 h enzymolysis at 28 °C, and enzyme composition 8 (2% cellulase R-10, 0.5% macerozyme R-10, and 0.6% pectolyase Y-23).

Effect of enzymolysis temperature on the yield and viability of ROC22 protoplasts. Different letters represent a statistically significant difference at P ≤ 0.05, and bars represent standard errors (SEs)

The viability of protoplasts isolated from sugarcane mesophyll cells using bright-field (a) and ultraviolet light (b). Scale bar indicates 50 μm
Transformation Efficiency of Sugarcane Protoplasts
The transformation efficiency of sugarcane protoplasts was significantly greater at 25% PEG4000 concentration compared to other higher (30%, 35%) or lower concentrations (10%, 15%, or 20%) (Fig. 6). In terms of duration of transformation, a significantly greater efficiency was achieved at 25 min (41.0%) in comparison to other duration, i.e., 10(23.6%), 15(30.1%), 20(36.1%), 30(30.4%), or 35(19.1%) min (Fig. 7).

Effect of PEG4000 concentration on protoplast transformation efficiency. Different letters represent a statistically significant difference at P ≤ 0.05, and bars represent standard errors (SEs). Transformation efficiency was also significantly greater at 25 min compared to the other times, range 8.0 to 39.4%

Effect of transformation time on protoplast transformation efficiency. Different letters represent a statistically significant difference at P ≤ 0.05, and bars represent standard errors (SEs)
Subcellular Localization of GFP-Fused ScD27 Protein in Sugarcane Protoplasts
Using our optimized protocol, the pCAMBIA1302-35S-GFP recombinant plasmids were able to express transiently in transformed sugarcane protoplasts (Fig. 8). Furthermore, the ScD27-GFP also detected the co-expression of specific GFP fusion protein in the sugarcane protoplasts. In contrast, the expression of reporter gene GFP alone was detected in all organelles, indicating that sugarcane protoplasts had been transfected. This success will greatly facilitate molecular biology studies in sugarcane.

Subcellular localization of ScD27-GFP fusion. The empty pCAMBIA1302 vector with GFP and the pCAMBIA1302 vector with ScD27–GFP fusion were transiently expressed in sugarcane mesophyll protoplasts. The upper part is the intracellular location of ScD27–GFP fusion, and the lower part is the intracellular location of GFP. Individual and merged images of GFP (green), and bright field in transformed protoplasts shown. The length of the scale bars is indicated. The scale bar is 5 µm (color figure online)
Discussion
Transient gene expression is an efficient technique for assessing gene function in eukaryotes, such as calcium signaling, protein–protein interactions, cellular interaction analyses (Zhang et al. 2011a), signal transduction and regulation, ion channel regulation through light, stress, and hormone responses (Im and Yoo 2014; Rose 2019; Xiong et al. 2019), and even further, in high throughput CRISPR/Cas9 screening application (Andersson et al. 2017).
Protocols for protoplasts isolation and transient gene expression have been established in various monocots, such as Oryza sativa (Kim et al. 2015), Zea mays (Chen et al. 2015). However, the efficiency of protoplast isolation and transformation has been low in sugarcane (Chen et al. 1987, 1988; Smith et al. 1992) and few report is available on gene transient expression in sugarcane protoplasts in the recent 10 years (Wang et al. 2019). Disadvantages such as abnormal protein expression in a heterogeneous system or false positive localization often exist in sugarcane gene functional studies that use tobacco (Nicotiana tabacum L.) or Arabidopsis thaliana’s protoplast transient expression system (Figueiredo et al. 2011). There is a need to develop an optimized protocol for the isolation and purification of sugarcane protoplasts, and to use these protoplasts for gene localization was essential.
A technical protocol for highly efficient isolation and purification of sugarcane protoplast has been established in this study, which was also used to study the transient expression and the subcellular localization of sugarcane D27 gene.
Protoplasts of high quality are very critical for transient expression studies. Several factors can impact the generation process of plant protoplasts, while the universal transient gene expression system used in model plants do not work well directly on sugarcane protoplasts, therefore, the universal transient gene expression system was optimized in this study and large numbers of viable sugarcane protoplasts were isolated.
Compared to the well-established protoplast isolation techniques of Arabidopsis (Yoo et al. 2007), relatively higher concentrations of cellulase R-10 (2.0%) and macerozyme R-10 (0.5%) with an additional digestion enzyme pectolyase Y-23(0.6%) were required for obtaining the highest yield of sugarcane protoplasts (Table 1). Different organs and tissues can be used as the starting materials for protoplast isolation in plants (He et al. 2016), including leaves (Shu et al. 2017; Shen et al. 2017; Zhang et al. 2011b), roots (Lin 1980), callus (Titouh et al. 2017), and shoots (Ortiz-Ramirez et al. 2018). However, in sugarcane, leaves or shoots are usually used as a reliable source for the isolation of high yield and relatively uniform protoplasts. The quality of protoplasts from the leaf mesophyll cells was better than that from the shoots (data are not shown).
Plant protoplasts remain intact only at a proper osmotic pressure. Different concentrations of mannitol have been used to maintain optimal osmotic pressure and high protoplast viability for the study of gene transient expression (Sun et al. 2018; Sahab et al. 2019; Wu et al. 2017a, b). The study found that when mannitol concentration was 0.4 mol L−1, the sugarcane protoplasts did not absorb water and there were few cell fragments. If the mannitol concentration was less than 0.4 mol L−1, the viability of the sugarcane protoplasts decreased.
The study demonstrated that the ScD27 gene protein was localized in the chloroplast. PEG-mediated protoplast transformation method is a mature, widely applied technology for studying transient expression in plant cells (Sahab et al. 2019). It has been established that PEG concentration, plasmid DNA concentration, and mass and viability of protoplasts affect the efficiency of protoplast transformation (Cao et al. 2016). Another key factor is the stability of the cell osmotic environment because the cytoplasm is fragile and uneven in concentration and the protoplasts are easy to break. In this study, the transformation efficiency and the number of transformed protoplasts increased gradually up to 25% PEG400 concentration. Other experimental factors might be investigated to further optimize the gene transient expression system in sugarcane.
Conclusion
An optimized protocol for sugarcane mesophyll protoplast isolation, purification, and transient gene expression was developed in this study using 2% cellulase R-10, 0.5% macerozyme R-10, and 0.6% pectolyase Y-23, enzymolysis for 12 h at 28 °C in the dark with constant oscillation at 60–70 rpm/min. Protoplast yield was as high as 10.94 × 106 protoplasts/g FW, and protoplast viability was 85.12%. Sugarcane protoplasts were transformed overnight at 28 °C with 40% PEG-4000 and 5 μg of plasmid DNA. The ScD27 protein was localized in the chloroplasts. This simple method using protoplasts will be very useful for future research to elucidate sugarcane gene function.
References
Andersson, M., H. Turesson, A. Nicolia, A.S. Falt, M. Samuelsson, and P. Hofvander. 2017. Efficient targeted multiallelic mutagenesis in tetraploid potato (Solanum tuberosum) by transient CRISPR-Cas9 expression in protoplasts. Plant Cell Reports 36(1): 117–128.
Cao, Y., H. Li, A.Q. Pham, and G. Stacey. 2016. An improved transient expression system using arabidopsis protoplasts. Current Protocols in Plant Biology 1(2): 285–291.
Chen, W.H., K.M. Gartland, M.R. Davey, R. Sotak, J.S. Gartland, B.J. Mulligan, J.B. Power, and E.C. Cocking. 1987. Transformation of sugarcane protoplasts by direct uptake of a selectable chimaeric gene. Plant Cell Reports 6(4): 297–301.
Chen, W.H., M.R. Davey, J.B. Power, and E.C. Cocking. 1988. Sugarcane protoplasts: Factors affecting division and plant regeneration. Plant Cell Reports 7(5): 344–347.
Chen, J., Q. Yi, Q. Song, Y. Gu, J. Zhang, Y. Hu, H. Liu, Y. Liu, G. Yu, and Y. Huang. 2015. A highly efficient maize nucellus protoplast system for transient gene expression and studying programmed cell death-related processes. Plant Cell Reports 34(7): 1239–1251.
Figueiredo, J.F., P. Romer, T. Lahaye, J.H. Graham, F.F. White, and J.B. Jones. 2011. Agrobacterium-mediated transient expression in citrus leaves: a rapid tool for gene expression and functional gene assay. Plant Cell Reports 30(7): 1339–1345.
Flematti, G.R., A. Scaffidi, M.T. Waters, and S.M. Smith. 2016. Stereospecificity in strigolactone biosynthesis and perception. Planta 243(6): 1361–1373.
Gomez-Roldan, V., S. Fermas, P.B. Brewer, V. Puech-Pages, E.A. Dun, J.P. Pillot, F. Letisse, R. Matusova, S. Danoun, J.C. Portais, H. Bouwmeester, G. Becard, C.A. Beveridge, C. Rameau, and S.F. Rochange. 2008. Strigolactone inhibition of shoot branching. Nature 455(7210): 189–194.
He, F., S. Chen, Y. Ning, and G.L. Wang. 2016. Rice (Oryza sativa) protoplast isolation and its application for transient expression analysis. Current Protocols in Plant Biology 1(2): 373–383.
Huang, H., Z. Wang, J. Cheng, W. Zhao, X. Li, H. Wang, Z. Zhang, and X. Sui. 2013. An efficient cucumber (Cucumis sativus L.) protoplast isolation and transient expression system. Scientia Horticulturae 150: 206–212.
Im, J.H., and S.D. Yoo. 2014. Transient expression in Arabidopsis leaf mesophyll protoplast system for cell-based functional analysis of MAPK cascades signaling. Methods in Molecular Biology 1171: 3–12.
Kim, N., S.J. Moon, M.K. Min, E.H. Choi, J.A. Kim, E.Y. Koh, I. Yoon, M.O. Byun, S.D. Yoo, and B.G. Kim. 2015. Functional characterization and reconstitution of ABA signaling components using transient gene expression in rice protoplasts. Frontiers in Plant Science 6: 614.
Larkin, P.J. 1976. Purification and viability determinations of plant protoplasts. Planta 128(3): 213–216.
Lin, W. 1980. Corn root protoplasts: Isolation and general characterization of ion transPORT. Plant Physiology 66(4): 550–554.
Lin, H., R. Wang, Q. Qian, M. Yan, X. Meng, Z. Fu, C. Yan, B. Jiang, Z. Su, J. Li, and Y. Wang. 2009. DWARF27, an iron-containing protein required for the biosynthesis of strigolactones, regulates rice tiller bud outgrowth. Plant Cell 21(5): 1512–1525.
Nanjareddy, K., M. Arthikala, L. Blanco, E.S. Arellano, and M. Lara. 2016. Protoplast isolation, transient transformation of leaf mesophyll protoplasts and improved Agrobacterium-mediated leaf disc infiltration of Phaseolus vulgaris: Tools for rapid gene expression analysis. BMC Biotechnology 16(1): 53.
Ortiz-Ramirez, C., E.D. Arevalo, X. Xu, D.P. Jackson, and K.D. Birnbaum. 2018. An efficient cell sorting protocol for maize protoplasts. Current Protocols in Plant Biology 3(3): e20072.
Rose, R.J. 2019. Somatic embryogenesis in the medicago truncatula model: Cellular and molecular mechanisms. Frontiers in Plant Science 10: 267.
Sahab, S., M.J. Hayden, J. Mason, and G. Spangenberg. 2019. Mesophyll protoplasts and peg-mediated transfections: Transient assays and generation of stable transgenic canola plants. Methods in Molecular Biology 1864: 131–152.
Shen, J., J. Fu, J. Ma, X. Wang, C. Gao, C. Zhuang, J. Wan, and L. Jiang. 2014. Isolation, culture, and transient transformation of plant protoplasts. Current Protocols in Plant Biology 63: 2–8.
Shen, Y., D. Meng, K. McGrouther, J. Zhang, and L. Cheng. 2017. Efficient isolation of Magnolia protoplasts and the application to subcellular localization of MdeHSF1. Plant Methods 13: 44.
Shu, Y., L. Huang, M. Chen, Y. Tao, Z. Wang, and H. Ma. 2017. Establishment and optimization of systems for protoplasts isolation of soybean and chickpea that used in subcellular location. Sheng Wu Gong Cheng Xue Bao 33(6): 976–985.
Smith, G.R., R. Ford, M.J. Frenkel, D.D. Shukla, and J.L. Dale. 1992. Transient expression of the coat protein of sugarcane mosaic virus in sugarcane protoplasts and expression in Escherichia coli. Archives of Virology 125(1–4): 15–23.
Sun, B., F. Zhang, N. Xiao, M. Jiang, Q. Yuan, S. Xue, H. Miao, Q. Chen, M. Li, X. Wang, Q. Wang, and H. Tang. 2018. An efficient mesophyll protoplast isolation, purification and PEG-mediated transient gene expression for subcellular localization in Chinese kale. Scientia Horticulturae 241: 187–193.
Titouh, K., N. Boufis, and L. Khelifi. 2017. Microcalli induction in protoplasts isolated from embryogenic callus of date palm. Methods in Molecular Biology 1637: 227–237.
Umehara, M., A. Hanada, S. Yoshida, K. Akiyama, T. Arite, N. Takeda-Kamiya, H. Magome, Y. Kamiya, K. Shirasu, K. Yoneyama, J. Kyozuka, and S. Yamaguchi. 2008. Inhibition of shoot branching by new terpenoid plant hormones. Nature 455(7210): 195–200.
Vogel, J.T., M.H. Walter, P. Giavalisco, A. Lytovchenko, W. Kohlen, T. Charnikhova, A.J. Simkin, C. Goulet, D. Strack, H.J. Bouwmeester, A.R. Fernie, and H.J. Klee. 2010. SlCCD7 controls strigolactone biosynthesis, shoot branching and mycorrhiza-induced apocarotenoid formation in tomato. The Plant Journal 61(2): 300–311.
Wang, J., T. Zhao, B. Yang, W. Wang, C. Feng, X. Feng, L. Shen, and S. Zhang. 2019. Characteristics, expression pattern and intracellular localisation of sugarcane cytoplasmic hexokinase gene ShHXK8. Sugar Tech 21(6): 909–916.
Waters, M.T., P.B. Brewer, J.D. Bussell, S.M. Smith, and C.A. Beveridge. 2012. The Arabidopsis ortholog of rice DWARF27 acts upstream of MAX1 in the control of plant development by strigolactones. Plant Physiology 159(3): 1073–1085.
Waters, M.T., C. Gutjahr, T. Bennett, and D.C. Nelson. 2017. Strigolactone signaling and evolution. Annual Review of Plant Biology 68: 291–322.
Wen, C., Q. Zhao, J. Nie, G. Liu, L. Shen, C. Cheng, L. Xi, N. Ma, and L. Zhao. 2016. Physiological controls of chrysanthemum DgD27 gene expression in regulation of shoot branching. Plant Cell Reports 35(5): 1053–1070.
Wu, Z.D., X.L. Liu, J.Y. Liu, F.G. Zan, X.J. Li, H.B. Liu, X.Q. Lin, X.K. Chen, H.S. Su, P.F. Zhao, and C.W. Wu. 2017a. Cloning and expression analysis of key gene ScD27 in strigolactones biosynthesis pathway. Acta Agronomica Sinica 48: 1554–1559.
Wu, J.Z., Q. Liu, X.S. Geng, K.M. Li, L.J. Luo, and J.P. Liu. 2017b. Highly efficient mesophyll protoplast isolation and PEG-mediated transient gene expression for rapid and large-scale gene characterization in cassava (Manihot esculenta Crantz). BMC Biotechnology 17(1): 29.
Xiong, L., C. Li, H. Li, X. Lyu, T. Zhao, J. Liu, Z. Zuo, and B. Liu. 2019. A transient expression system in soybean mesophyll protoplasts reveals the formation of cytoplasmic GmCRY1 photobody-like structures. Science China Life Sciences 62: 1070–1077.
Yoo, S.D., Y.H. Cho, and J. Sheen. 2007. Arabidopsis mesophyll protoplasts: A versatile cell system for transient gene expression analysis. Nature Protocols 2(7): 1565–1572.
Yu, G., Q. Cheng, Z. Xie, B. Xu, B. Huang, and B. Zhao. 2017. An efficient protocol for perennial ryegrass mesophyll protoplast isolation and transformation, and its application on interaction study between LpNOL and LpNYC1. Plant Methods 13: 46.
Zhang, Y., J. Su, S. Duan, Y. Ao, J. Dai, J. Liu, P. Wang, Y. Li, B. Liu, D. Feng, J. Wang, and H. Wang. 2011. A highly efficient rice green tissue protoplast system for transient gene expression and studying light/chloroplast-related processes. Plant Methods 7(1): 30.
Acknowledgments
This study was supported by Fund for the National Natural Science Foundation of China (31860405); Earmarked Fund for China Agriculture Research System (CARS-170101); Provincial Innovation Team of Sugarcane Germplasm Innovation and New Variety Breeding of Yunnan Academy of Agricultural Sciences (2019HC013); Central Public-interest Scientific Institution Basal Research Fund for Chinese Academy of Tropical Agricultural Sciences (No. 1630052017020-4); The Applied Basic Research Projects in Yunnan Province (2016FB071); Applied Basic Research Projects of Yunnan Academy of Agricultural Sciences (YJM201705); Overseas Top Talents Project “Sugarcane genetic improvement and extension”; Yunnan Provincial Science and Technology Cooperation Program, China (Yunnan) -Sri Lanka Sugarcane International Joint Research Center (2018IA076); The Key Research and Development Program of Yunnan Province, the Joint Research and Development Center of Sugarcane Variety Improvement in South and Southeast Asia (2019IB008).
Author information
Authors and Affiliations
Contributions
CWW and FGZ, ZDW, XKC conceived and designed the experiments. ZDW, FGZ, XH, and ZYL performed the experiments. FGZ, ZDW, and XKC result analysis, and manuscript drafting. YBP, XKC, and DMB revised the manuscript. All authors have read and approved the final manuscript. XKC, JYL, LPZ, LY, YZ, XLL, HMX, KY, JZ, PFZ, and WQ participated in experimental work.
Corresponding authors
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
About this article

Cite this article
Wu, Zd., Hu, X., Zan, Fg. et al. Subcellular Localization of the D27 Protein in Sugarcane (Saccharum spp. Hybrids) Using an Optimized Protoplast Isolation, Purification, and Transient Gene Expression Protocol. Sugar Tech 23, 316–325 (2021). https://doi.org/10.1007/s12355-020-00879-y
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12355-020-00879-y