
Abstract
The 4th edition of the World Health Organization’s Classification of Head and Neck Tumours was published in January of 2017. This article provides a summary of the changes to Chapter 4 Tumours of the oral cavity and mobile tongue and Chapter 8 Odontogenic and maxillofacial bone tumours. Odontogenic cysts which were eliminated from the 3rd 2005 edition were included in the 4th edition as well as other unique allied conditons of the jaws. Many new tumors published since 2005 have been included in the 2017 classification.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
The next edition of the WHO’s classification of odontogenic cysts, tumors and maxillofacial bone tumors was published in early 2017 (Table 1). The new edition, like those earlier, has a profound impact on the practice of head and neck surgical pathology throughout the world. The goal of this paper is to summarize the changes in the new classification since the 2005 edition. Like all classifications, the final product was a consensus of invited experts from around the world with extensive experience with odontogenic cysts and tumors as well as bone pathology. The final classification represents a consensus only of those selected to participate at a fixed point in time. Debate at times was lively, and we acknowledge the inclusion of some subjectivity depending on the experience and training of those selected. WHO invited participants in the Consensus and Editorial Panel included Prof Takashi Takata, Japan, Chair; Prof Daniel Baumhoer, Switzerland; Prof Samir El-Mofty, USA; Prof Edward Odell, United Kingdom; Prof Paul Speight, United Kingdom; Prof John Wright, USA, Prof Rosnah Zain, Malaysia. The panel was guided by the principles of simplicity, clinical relevance, scientific validity and utility for nonspecialist pathologists. Numerous changes were considered and incorporated to provide a contemporary, consensus classification that should provide the practicing worldwide community of head and neck pathologists with a working framework for the diagnosis of odontogenic cysts, tumors and other allied bone tumors at this point in time.
Odontogenic cysts, conceptually inseparable from odontogenic tumors which can be cystic, were omitted from the 2005 classification. Odontogenic cysts have been reincorporated into the 2017 classification and updated significantly from the 1992 classification. The overall classification of odontogenic tumors focuses on those that are biologically benign and those that are malignant. The 2005 classification divided the benign tumors into “Odontogenic epithelium with mature, fibrous stroma without odontogenic ectomesenchyme, Odontogenic epithelium with odontogenic ectomesenchyme, with or without hard tissue formation, and Mesenchyme and/or odontogenic ectomesenchyme with or without odontogenic epithelium.” While accurate, this seemed overly complex and the 2017 version will recognize only epithelial, mesenchymal (ectomesenchymal), and mixed odontogenic tumors.
Malignant Odontogenic Tumors
The 2005 classification divides ameloblastic carcinoma into three types; primary and secondary intraosseous tumors and secondary peripheral tumors. Primary peripheral lesions which do exist were not included. There seemed little justification to divide such a rare tumor which in 2017 continues simply as a single entity; ameloblastic carcinoma. In 2005, primary intraosseous squamous cell carcinoma was divided into entities based on their histogenesis. In 2017, this group of lesions will be represented by a single entity; primary intraosseous carcinoma. There is no clinical relevance for the histogenesis of these carcinomas at this time and while many of the intraosseous carcinomas show squamous differentiation, not all do.
Sclerosing odontogenic carcinoma has been added to the 2017 classification. First reported in 2008 [1, 2] approximately ten cases have been published to date. Sclerosing odontogenic carcinoma is a cytologically bland epithelial tumor with significant stromal sclerosis (Figs. 1, 2) and characterized by aggressive infiltrative growth into muscle and nerve. The epithelial component tends to be small single-file cords and strands, which are variably conspicuous on routine sections and best demonstrated with cytokeratin IHC (Fig. 3). To date, no cases have metastasized and all patients are alive with only one recurrence after initial curettage.

Sclerosing odontogenic carcinoma with slender cords of epithelium in a desmoplastic stroma (H&E Original mag × 33)

Sclerosing odontogenic carcinoma where the epithelial component shows minimal cytologicatypia (H&E Original mag × 66)

Sclerosing odontogenic carcinoma (IHC AE1/3)
Clear cell odontogenic carcinoma has been continued with updated IHC and genetic profiles. More than 80% of cases demonstrate EWSR1 rearrangement, most often with ATF1 [3, 4].
While sensitive, the translocation is not specific and has been documented in hyalinizing clear cell carcinoma of salivary origin as well as other clear cell malignancies.
In 2005, odontogenic carcinosarcoma was eliminated from the classification because many if not most of the reported cases predated IHC confirmation and many of the reported cases likely represented epithelial–mesenchymal transition into spindle cell carcinoma. Odontogenic carcinosarcoma was reinstituted in the 2017 classification as improved documentation reconfirmed the existence of the lesion [5–7].
The group of odontogenic sarcomas have been continued in 2017 as “odontogenic sarcomas” because of the lack of evidence showing clinical relevance to subclassification. Lastly, it is important to note that odontogenic carcinoma with dentinoid was introduced as a new entity in 2014, [8] but it was not included in the 2017 classification in order to allow more time for additional cases to be published with peer review. Odontogenic carcinoma with dentinoid is discussed with clear cell odontogenic carcinoma.
Benign Odontogenic Tumours: Epithelial
Ameloblastoma
Ameloblastoma has undergone modifications in terminology and classification with introduction of prospective views based on updates on current genetic studies. The debate of benign vs malignant ameloblastoma was debated. Acknowledging its local aggressiveness and propensity to recur, ameloblastoma remained benign, despite the incredibly rare variant known as malignant ameloblastoma.
In the 2005 WHO, Ameloblastomas were classified as solid/multicystic, extraosseous/peripheral, desmoplastic and unicystic types. Currently, the classification has been simplified and narrowed to ameloblastoma, unicystic ameloblastoma and extraosseous/peripheral types. The adjective “solid/multicystic” for the conventional ameloblastoma was dropped because it has no biologic significance and can lead to confusion with unicystic ameloblastoma. Desmoplastic ameloblastoma will be reclassified as a histologic subtype and not a clinicopathologic entity. Despite its unique clinical and sometimes radiographic features, it behaves like any conventional ameloblastoma. Odontoameloblastoma was included in the classification in 2005. The association of ameloblastoma with odontoma is well established and accepted, but the consensus group did not think it justified being separated as an entity. There is no evidence that these tumors begin as odontoameloblastomas or recur as odontoameloblastomas; they recur as ameloblastomas. We believe that these ameloblastomas arise in an odontoma from primitive ectoderm just as they arise from primitive ectoderm involved in odontogenesis. Accordingly, the association is discussed under ameloblastoma and is more accurately described as an ameloblastoma arising in an odontoma, not odontoameloblastoma.
The year 2014 constituted a turning point in our understanding the etiopathogenesis of ameloblastoma as important studies on the genetics of this tumor were published [9–11]. These studies reported the identification of highly recurrent somatic, activating mutations in the signaling pathways of the mitogen-activated protein kinase (MAPK) and Hedgehog in ameloblastoma. The interest in these two pathways was stirred as they are known to be active during tooth development [12–14] and more specifically, mutations in MAPK components (i.e., BRAF, KRAS and FGFR2) have been identified in both benign [15] and malignant tumors [16–18] In 2015, an additional investigation regarding BRAF and Hedgehog related-SMO mutations was published, which examined not only ameloblastomas (ameloblastoma and unicystic ameloblastoma), but also a series of odontogenic carcinomas [19].
Within the MAPK pathway, BRAF has the role of a serine-threonine kinase. The V600E mutation is the most frequent and it has been identified in many cancer types, such as melanoma, hairy cell leukemia, papillary thyroid carcinoma and colorectal cancer [16]. The mutated BRAF constitutively activates downstream signaling that finally results in increased cell proliferation, survival and neoplastic transformation.
According to the cytogenetic studies on ameloblastoma performed so far, using real-time PCR often enhanced by Sanger sequencing, the incidence of the BRAFV600E mutation ranged from 43% [10] to 82% [19] with a combined frequency from all studies of 59% [9–11, 19]. Other MAPK-related mutations comprised mainly of RAS and FGFR-2 showed a combined incidence of only 28% [10, 11] Collectively, the incidence of BRAF, RAS and FGFR-2 mutations in the studied cases of ameloblastoma was approximately 79%. Furthermore, these mutations were mutually exclusive, but for one case that showed simultaneous BRAFV600E and FGFR-2 mutations [10]. It is suggested that mutations in the MAPK pathways are an early and critical event in the etiopathogenesis of ameloblastoma [20].
On clinical grounds, patients with BRAFV600E ameloblastoma have a mean age at diagnosis of 34.5 years compared to 53.6 years in BRAF wild-type cases [11].
The SMO mutation in ameloblastoma was either absent [19] or ranged between 17% [11] and 39%, [10] with a combined incidence of 22%. More interestingly, all BRAFV600E mutations and SMO mutations showed mutual exclusivity except two cases, [10, 11] suggesting that these genetic alterations may define two independent genetic etiologies for ameloblastoma [10]. Ameloblastoma with SMO mutations are predominantly found in the maxilla (57%) while those with BRAF mutations are mainly located in the mandible (75%) [10, 11, 19]. The emerging genetic dichotomy between the mandibular and maxillary tumours in regard to BRAF and SMO is not yet elucidated. The microenvironment of the different anatomic sites could participate in the divergent mutations although from the published cases, it cannot be determined if SMO and BRAF mutations correspond to histologic subtypes. Correlations between the mutational status of the ameloblastoma and clinical outcomes await further studies with large, multicenter series. Since the mutational status of BRAFV600E usually conforms to the immunohistochemical staining of its protein product, including fixed tissues that have undergone decalcification, [9, 11]. it is expected that comprehensive data on clinico-pathological correlations will be available in the near future.
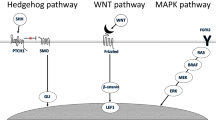
Elucidation of the activated molecular pathways opens exciting opportunities for targeted therapy, either adjunctively or perhaps exclusively. It has been shown that in the AM-1 ameloblastoma cell line that carries the BRAFV600E mutation, the abnormally activated MAPK pathway has been abolished by pharmacological inhibition of BRAF [10, 11]. Similarly, human ameloblastoma are potentially responsive to molecularly targeted therapies, like those that have already been in clinical use against BRAFV600E mutation [21]. BRAF and SMO pathways are illustrated in Fig. 4.

Schematic illustration of BRAF a and Sonic Hedgehog b pathways that are involved in the pathogenesis of ameloblastoma
The unicystic ameloblastoma (UAM) was recognized as a distinct subtype of ameloblastoma about 4 decades ago, based on clinical and radiological features as well as distinct histopathological findings [22]. At that time the separation seemed warranted as UAM responded satisfactorily to conservative approaches in contrast to its conventional counterpart that demanded extensive surgery. UAM was divided into three subtypes according to the pattern of proliferation of the ameloblastomatous epithelium: luminal, intraluminal and mural. Since then, there has been a general agreement that while the first two subtypes “deserve” to be treated conservatively, the latter (i.e., the mural type) should be treated as ameloblastoma [23, 24]. (Fig. 5) In 2000, mural involvement in UAM was shown to have a recurrence rate after conservative surgical removal approaching that of conventional ameloblastoma and the luminal variants showed recurrence under 10% [25]. As a consequence, in the 2017 WHO, it is recommended that mural infiltration in UAM be recognized as having the same biological aggressiveness as conventional ameloblastoma. Whether this should be classified as a variant of UAM or as conventional ameloblastoma will require further documentation.

Unicystic ameloblastoma with mural infiltration (H&E stain Original mag × 33)
The mutation status of BRAF has been examined in a small number of unicystic ameloblastomas (n = 15) [11, 15, 19] and that of SMO in even a smaller number (n = 7) [19]. In total, 73% of the examined unicystic ameloblastomas (all located in the mandible) were found to bear the BRAFV600E mutation, however none of them showed mutations in SMO. The BRAF mutation was found in all histopathological subtypes of unicystic ameloblastoma, implying that a direct relationship to the biological behavior of the tumor is not always present. It can be suggested that additional factors, such as those related to tumor microenvironment [26], may contribute to the aggressiveness of these tumors.
Squamous odontogenic tumor, calcifying epithelial odontogenic tumor, and adenomatoid odontogenic tumor have been updated but without significant modifications.
Keratocystic Odontogenic Tumor
The most controversial decision in the 2017 classification was to move keratocystic odontogenic tumor back into the cyst category as odontogenic keratocyst (OKC). The consensus panel acknowledged that there was some evidence to reclassify the cyst in 2005, but we did not believe the evidence was sufficient at the time to justify the reclassification as a neoplasm. The evidence for reclassification was based on “aggressive growth”, recurrence after treatment, the rare occurrence of a “solid” variant of OKC, and most importantly, mutations in the PTCH gene. PTCH gene mutations have been documented in up to 85% of syndromic (Nevoid basal cell carcinoma syndrome, NBCCS) and around 30% of nonsyndromic OKCs [27–33]. Because the NBCCS is caused by PTCH mutation, every nucleated cell in the body would contain the mutation in vertically transmitted syndromic patients. Accordingly, finding PTCH mutations in OKCs of syndromic patients is not only not surprising, it’s predictable. The roughly 15–20% of syndromic patients in which the mutation cannot be demonstrated can be explained by acquiring the phenotype through somatic mutation. That leaves the justification for neoplastic pathogenesis based on the mutation in nonsyndromic patients in which the majority of patients do not show the mutation. Additionally, because odontogenic cysts were not included in the 2005 classification, it was unclear if the authors intended to classify the mutated cysts as neoplasms and the nonmutated ones as cysts. The mutations in OKCs are not limited to PTCH, as mutations in CDKN2A, TP53, MCC, CADMI and FHIT have also been reported [34–36]. While there has been a sea change in our understanding of the molecular pathogenesis, which underlies neoplasms, currently neoplasia continues to be defined by clinical phenotype in all medical dictionaries and current medical pathology texts. Almost every definition includes the concept of autonomy with the neoplasm continuing to grow after the stimulus which produced it is removed. Neoplasms should not regress spontaneously and yet, OKCs are well documented to completely regress following decompression [37] and the lining of many decompressed cysts appears more like oral mucosa than OKC histologically. While the material and methods were not “gold standard,” loss of heterozygosity has been demonstrated in other odontogenic cysts [38]. Cutaneous cysts identical histologically to OKCs have been reported in both syndromic and nonsyndromic patients and yet reclassification as neoplasms in the medical or dermatopathology communities has not be recommended. It is important to note that the consensus panel is not necessarily saying OKCs are not neoplastic but we believe the evidence currently is lacking to justify the continuation of keratocystic odontogenic tumor as a tumor.
Mixed Odontogenic Tumors
There is considerable evidence that some ameloblastic fibromas (AF) are neoplastic and do not produce dental hard tissues while other histologically identical lesions begin to mature and ultimately produce dental hard tissues, maturing into odontomas with time. BRAFV600E mutations and low frequency of fractional allelic loss of tumor suppressor gene loci have been reported in AF [39, 40]. Maturation is characterized by morphologic as well as functional changes and when dental hard tissue are produced, the tumors have been referred to as ameloblastic fibro-dentinoma (AFD) or ameloblastic fibro-odontoma (AFO). While it was agreed that a small number of AFDs and AFOs could conceptually be neoplastic because of their exceptionally large size, the consensus was that once dental hard tissues are produced, these tumors are likely maturing into odontomas. Accordingly, it was concluded that there was little evidence to justify classifying AFD and AFO as independent entities and the decision was made to group them under odontomas as developing odontomas as others have suggested [41–43].
Odontomas, both compound and complex have been updated but remained relatively unchanged with the exception of recognizing AFD and AFO within the histologic spectrum of developing odontomas.
The ghost cell lesions presented a similar challenge because the 2005 classification moved the calcifying odontogenic cyst into the tumor classification and renamed it calcifying cystic odontogenic tumor. The narrative which accompanied calcifying cystic odontogenic tumor offered little to no justification for including the cystic lesions as neoplasms. While not as contentious as the OKC debate, the consensus panel were unanimous in returning the calcifying cystic odontogenic tumor to the cyst classification. An international collaborative study in 2008 reviewed the WHO classification of ghost cell lesions and suggested further work needed to be done to define their biologic behavior [44]. This same international group showed that just under 90% of all ghost cell lesions are either entirely cystic or associated with odontomas, lesions for which there is no justification for classifying as neoplastic. The 2017 classification clarifies the relationship of COC to odontomas and other odontogenic tumors which was not addressed in 2005.
The solid tumor containing ghost cells was updated and continued as dentinogenic ghost cell tumor.
A new odontogenic tumor described as primordial odontogenic tumor in 2014 will be included in the 2017 classification [45, 46]. Only seven cases have been reported and most have been in children and affected the mandible. All cases have been well circumscribed pericoronal radiolucencies. Histologically, most of the tumor consists of an immature loose fibrous connective tissue resembling dental papilla or cellular odontogenic ectomesenchyme. The entire surface of the tumor is rimmed by cuboidal to columnar epithelium resembling the inner enamel epithelium of the enamel organ (Figs. 6, 7). Tumors have been removed conservatively with no recurrences reported.

Primordial odontogenic tumor with primitive ectomesenchymal stroma lined by odontogenic epithelium (H&E stain Original mag × 6.6)

Primordial odontogenic tumor showing cellular detail of the stromal and epithelial components (H&E Original mag × 33)
Mesenchymal Odontogenic Tumors
In 2005 the odontogenic fibroma was defined as “a rare neoplasm characterized by varying amounts of inactive-looking odontogenic epithelium embedded in a mature, fibrous stroma” and divided into an epithelium poor type, referred to as the simple type, and an epithelial rich type, often called the WHO type. After considerable debate, the consensus group concluded that the epithelial poor or simple type was poorly defined and documented, and decided to drop the subclassification. In 2017, the odontogenic fibroma is defined as “a rare neoplasm of mature fibrous connective tissue, with variable amounts of inactive-looking odontogenic epithelium with or without evidence of calcification.”
Odontogenic myxoma and cementoblastoma have been updated and continued in the 2017 classification.
In 2005, ossifying fibroma was not included in the classification of odontogenic tumors. It was however discussed under “bone related lesions.” There is no debate that an ossifying fibroma occurs in the jaws, perhaps exclusively in the jaws, which is neoplastic and histologically distinct from juvenile trabecular or juvenile psammomatoid ossifying fibromas. The tumor is arguably of periodontal ligament origin and therefore odontogenic. Although the only definition of cementum is based on its anatomic association with tooth roots and the matrix produced by the tumor has no clinical or biological significance, the cemento-ossifying fibroma will be included in the 2017 classification under odontogenic tumors to distinguish it from the juvenile categories of ossifying fibroma, but for practical reasons, it will be discussed with the other ossifying fibromas under fibro-osseous lesions in Chap. 8.
Odontogenic Cysts
The most significant change affecting odontogenic cysts was the reincorporation of odontogenic keratocyst and calcifying odontogenic cyst in the cyst classification when they had been classified in 2005 as neoplasms. There were additional significant changes since other cysts had not appeared in the WHO classification since 1992. Under inflammatory cysts, inflammatory collateral cysts are included which defines and subdivides these cysts into paradental cysts and buccal bifurcation cysts. Paradental cysts are typically found distal to mandibular third molars and buccal bifurcation cysts are typically found buccal to erupting mandibular first or second molars in children (Fig. 8). Primordial cysts have been dropped and are no longer used synonymously for odontogenic keratocysts. Orthokeratinized odontogenic cysts are now recognized as an odontogenic cyst distinct from OKC. The cystic lining consists of a mature stratified squamous epithelium without rete ridge development which exhibits orthokeratosis and a prominent granular cell layer. The basal cells tend to be flattened to cuboidal but not palisaded and hyperchromatic. In contradistinction to OKCs, orthokeratinized odontogenic cysts are not particularly aggressive biologically, do not have a significant recurrence rate after removal and are typically not associated with the nevoid basal cell carcinoma syndrome. Most of the other odontogenic cysts have been updated but not significantly altered. New diagnostic criteria for glandular odontogenic cysts (GOC) are presented, and the histologic overlap between GOC and cystic mucoepidermoid carcinomas acknowledged, with caution expressed in providing a definitive diagnosis based on a small incisional biopsy. Initial studies have not shown MAML2 gene rearrangements in GOCs that are seen in many mucoepidermoid carcinomas [47]. The only non-odontogenic cyst included in the 2017 classification is incisive canal cyst.

Buccal bifurcation cyst which is typically buccal to an erupting first or second mandibularmolar
Non-odontogenic Maxillofacial Bone Tumors
Most bone pathology can be seen in the gnathic and craniofacial bones. There were no new entities introduced and most lesions were updated, particularly as the genetic landscape of all neoplasms has evolved since 2005. A summary of the genetic alterations reported in odontogenic and maxillofacial bone tumors in presented in Table 2.
Chondrosarcoma grading is discussed and its relationship to prognosis. IDH1/2 mutations have been demonstrated in approximately half of the cases of chondrosarcoma [48]. Mesenchymal chondrosarcomas show HEY-NCOA2 fusion rather than mutations of IDH1/2 [49]. Osteosarcoma and its histologic variants have been updated. MDM2 amplification and the utility of MDM2 and CDK4 IHC is discussed. This appears most useful and relevant to aid in distinguishing low grade well differentiated osteosarcomas from benign fibro-osseous lesions [50]. Gnathic osteosarcomas are less aggressive and metastasize considerably less frequently than those of the extragnathic skeleton. The role of neoadjuvant therapy for gnathic tumors is discussed and questioned.
Chondroma, osteoma, and melanotic neuroectodermal tumor of infancy were updated without significant change. Chondroblastoma shows specific H3F3B driver point mutations [51]. Chondromyxoid fibroma, osteoid osteoma, and osteoblastoma are updated. Desmoplastic fibroma shows driver mutations of CTNNB1 or APC [52, 53].
The ossifying fibromas, including cemento-ossifying fibroma, juvenile trabecular ossifying fibroma and juvenile psammomatoid ossifying fibroma, were discussed as a group because of their similar and overlapping fibro-osseous appearance histologically. Fibrous dysplasia (FD) was updated and linked to postzygotic GNAS1 gain of function mutations [54].Most patients who develop malignancy associated with FD have been previously radiated but spontaneous transformation has been reported [55]. The cemento-osseous dysplasias were updated, as was osteochondroma. Mutations of EXT1 or EXT2 occur in osteochondroma [56].
The giant cell lesions of the jaws were updated and peripheral and central giant cell granulomas discussed. The H3F3A mutation in giant cell tumors of bone are not found in gnathic giant cell granulomas [57]. Cherubism, which is autosomal dominant, shows a strong association with SH3BP2 mutations [58]. A strong association with CDH11 and/or USP6 mutations is/are seen in primary, but not secondary, aneurysmal bone cysts [59]. Simple bone cysts and solitary plasmacytomas of bone were included and updated.
References
Koutlas IG, Allen CM, Warnock GR, Manivel JC. Sclerosing odontogenic carcinoma: a previously unreported variant of a locally aggressive odontogenic neoplasm without apparent metastatic potential. Am J Surg Pathol. 2008;39:1613–19.
Irie T, Ogawa I, Takata T, Toyosawa S, Saito N, Akiba M, et al. Sclerosing odontogenic carcinoma with benign fibro-osseous lesion of the mandible: an extremely rare case report. Pathol Int. 2010;60:694–700.
Bilodeau EA, Weinreb I, Antonescu CR, Zhang L, Dacic S, Muller S, et al. Clear cell odontogenic carcinomas show EWSR1 rearrangements: a novel finding and a biological link to salivary clear cell carcinoma. Am J Surg Pathol. 2013;37:1001–5.
Yancoskie AE, Sreekantaiah C, Jacob J, Rosenberg A, Edelman M, Antonescu CR, et al. EWSR1 and ATF1 rearrangements in clear cell odontogenic carcinoma: presentation of a case. Oral Surg Oral Med Oral Pathol Oral Radiol. 2014;118:e115–e8.
Kunkel M, Ghalibafian M, Radner H, Reichert TE, Fischer B, Wanger W. Ameloblastic fibrosarcoma or odontogenic carcinosarcoma: a matter of classification. Oral Oncol. 2004;40:444–9.
Slater LJ. Odontogenic sarcoma and carcinosarcoma. Semin Diagn Pathol. 1999;16:325–32.
DeLair D, Bejarano PA, Peleg M, El-Mofty SK. Ameloblastic carcinosarcoma of the mandible arising in ameloblastic fibroma: a case report and review of the literature. Oral Surg Oral Med Oral Pathol Oral Radiol Endod. 2007;103:516–20.
Mosqueda-Taylor A, Neville BW, Tatemoto Y, Ogawa I, Takata T. Odontogenic carcinoma with dentinoid: a new odontogenic carcinoma. Head Neck Pathol. 2014;8:421–31.
Kurppa KF, Caton J, Morgan PR, Ristimaki A, Ruhin B, Kellokoski J, Elenius K, Heikinheimo K. High frequency of BRAF V600E mutations in ameloblastoma. J Pathol. 2014;232:492–8.
Sweeney RT, McClary AC, Myers BR, Biscocho J, Neahring L, Kwei KA, Qu K, Gong X, Ng T, Jones CD, Varma S, Odegaard JI, Sugiyama T, Koyota S, Rubin BP, Troxell ML, Pelham RJ, Zehnder JL, Beachy PA, Pollack JR, West RB. Identification of recurrent SMO and BRAF mutations in ameloblastomas. Nat Genet. 2014;46:722–5.
Brown NA, Rolland D, McHugh JB, Weigelin HC, Zhao L, Lim MS, Elenitoba-Johnson KS, Betz BL. Activating FGFR2-RAS-BRAF mutations in ameloblastoma. Clin Cancer Res. 2014;20:5517–26.
Kumamoto H, Ohki K, Ooya K. Expression of Sonic hedgehog (SHH) signaling molecules in ameloblastomas. J Oral Pathol Med. 2004;33:185–90.
Dassule HR, Lewis P, Bei M, Maas R, McMahon AP. Sonic hedgehog regulates growth and morphogenesis of the tooth. Development. 2000;127:4775–85.
Heikinheimo K, Jee KJ, Niini T, Aalto Y, Happonen RP, Leivo I, Knuutila S. Gene expression profiling of ameloblastoma and human tooth germ by means of a cDNA microarray. J Dent Res. 2002;81:525–30.
Pereira NB, Pereira KM, Coura BP, Diniz MG, de Castro WH, Gomes CC, Gomez RS. BRAFV600E mutation in the diagnosis of unicystic ameloblastoma. J Oral Pathol Med. 2016. doi:10.1111/jop.12443.
Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, Teague J, Woffendin H, Garnett MJ, Bottomley W, Davis N, Dicks E, Ewing R, Floyd Y, Gray K, Hall S, Hawes R, Hughes J, Kosmidou V, Menzies A, Mould C, Parker A, Stevens C, Watt S, Hooper S, Wilson R, Jayatilake H, Gusterson BA, Cooper C, Shipley J, Hargrave D, Pritchard-Jones K, Maitland N, Chenevix-Trench G, Riggins GJ, Bigner DD, Palmieri G, Cossu A, Flanagan A, Nicholson A, Ho JW, Leung SY, Yuen ST, Weber BL, Seigler HF, Darrow TL, Paterson H, Marais R, Marshall CJ, Wooster R, Stratton MR, Futreal PA. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–54.
Parada LF, Land H, Weinberg RA, Wolf D, Rotter V. Cooperation between gene encoding p53 tumour antigen and ras in cellular transformation. Nature. 1984;312:649–51.
Li Y, Mangasarian K, Mansukhani A, Basilico C. Activation of FGF receptors by mutations in the transmembrane domain. Oncogene. 1997;14:1397–406.
Diniz MG, Gomes CC, Guimarães BV, Castro WH, Lacerda JC, Cardoso SV, de Faria PR, Dias FL, Eisenberg AL, Loyola AM, Gomez RS. Assessment of BRAFV600E and SMOF412E mutations in epithelial odontogenic tumours. Tumour Biol. 2015;36:5649–53.
Brown NA, Betz BL. Ameloblastoma: a review of recent molecular pathogenetic discoveries. Biomark. Cancer. 2015;7(Suppl 2):19–24.
Kaye FJ, Ivey AM, Drane WE, Mendenhall WM, Allan RW. Clinical and radiographic response with combined BRAF-targeted therapy in stage 4 ameloblastoma. J Natl Cancer Inst. 2014;107:378.
Robinson L, Martinez MG. Unicystic ameloblastoma: a prognostically distinct entity. Cancer. 1977;40:2278–85.
Ackermann GL, Altini M, Shear M. The unicystic ameloblastoma: a clinicopathological study of 57 cases. J Oral Pathol. 1988;17:541–6.
Philipsen HP, Reichart PA. Unicystic ameloblastoma. A review of 193 cases from the literature. Oral Oncol. 1998;34:317–25.
Li T-J, Wu Y-T, Yu S-F, Yu G-Y. Unicystic ameloblastoma. A clinicopathologic study of 33 Chinese patients. Am J Surg Pathol. 2000;10:1385–92.
Vered M, Shohat I, Buchner A, Dayan D. Myofibroblasts in stroma of odontogenic cysts and tumors can contribute to variations in the biological behavior of lesions. Oral Oncol. 2005;41:1028–33.
Gu XM, Zhao HS, Sun LS, Li TJ. PTCH mutations in sporadic and Gorlin–syndrome-related odontogenic keratocysts. J Dent Res. 2006;85:859–63.
Li TJ, Yuan JW, Gu XM, Zhao HS. PTCH germline mutations in Chinese nevoid basal cell carcinoma syndrome patients. Oral Dis. 2008;25:174–9.
Sun LS, Li XF, Li TJ. PTCH1 and SMO gene alterations in keratocystic odontogenic tumors. J Dent Res. 2008;87:575–9.
Pan S, Li TJ. Mechanisms of inactivation of PTCH1 gene in keratocystic odontogenic tumors: modifivation of the two-hit hypothesis. Clin Cancer Res. 2010;16:442–50.
Barreto DC, Gomez RS, Bale AE, Boson WL, De Marco L. PTCH gene mutations in odontogenic keratocysts. J Dent Res. 2000;79:1418–22.
Ohki K, Kumamoto H, Ichinohasama R, Sato T, et al. PTC gene mutations and expression of SHH, PTC, SMO, and GLI-1 in odontogenic keratocysts. Int J Oral Maxillofac Surg. 2009;33:584–92.
Song YL, Zhang WF, Peng B, Wang CN, et al. Germline mutations of the pTCH ggene in families with odontogenic keratocysts and nevoid basal cell carcinoma syndrome. Tumor Biol. 2006;27:175–80.
Agaram NP, Collins B, Barnes L, Lomago D, et al. Molecular analysis to demonstrate that odontogenic keratocysts are neoplastic. Arch Pathol Lab Med. 2004;128:313–7.
Henley J, Summerlin D-J, Tomich C, Zhang S, Cheng L. Molecular evidence supporting the neoplastic nature of odontogenic keratocyst: a laser capture microdissection study of 15 cases. Histopathology. 2005;47:582–6.
Malcic´ A, Jukic´ S, Anic´ I, Pavelic´ B, et al. Alterations of FHIT and p53 genes in keratocystic odontogenic tumours, dentigerous cyst and radicular cyst. J Oral Pathol Med. 2008;37:294–301.
Pogrel MA, Jordan RC. Marsupialization as a definitive treatment for the odontogenic keratocyst. J Oral Maxillofac Surg. 2004;62:651–5.
Diniz MG, Galvao CF, Macedo PS, Gomes CC, Gomez RS. Evidence of loss of heterozygosity of the PTCH gene in orthokeratinized odontogenic cyst. J Oral Pathol Med. 2011;40:277–80.
Brown NA, Rolland D, McHugh JB, Weigelin HC, Zhao L, Lim MS, et al. Activating FGFR2-RAS-BRAF mutations in ameloblastoma. Clin Cancer Res. 2014;20:5517–26.
Galvão CF, Gomes CC, Diniz MG, Vargas PA, de Paula AM, Mosqueda-Taylor A, et al. Loss of heterozygosity (LOH) in tumour suppressor genes in benign and malignant mixed odontogenic tumours. J Oral Pathol Med. 2012;41:389–93.
Slootweg PJ. An analysis of the interrelationship of the mixed odontogenic tumors-ameloblastic fibroma, ameloblastic fibroodontoma, and the odontomas. Oral Surg Oral Med Oral Pathol. 1981;51:266–77.
Philipsen HP, Reichart PA, Praetorius F. Mixed odontogenic tumours and odontomas. Considerations on interrelationship. Review of the literature and presentation of 134 new cases of odontomas. Oral Oncol. 1997;33:86–99.
Buchner A, Vered M. Ameloblastic fibroma: a stage in the development of a hamartomatous odontoma or a true neoplasm? Critical analysis of 162 previously reported cases plus 10 new cases. Oral Surg Oral Med Oral Pathol Oral Radiol. 2013;116:598–606.
Ledesma-Montes C, Gorlin RJ, Shear M, Praetorius F, et al. International collaborative study on ghost cell odontogenic tumours: calcifying cystic odontogenic tumour, dentinogenic ghost cell tumour and ghost cell odontogenic carcinoma. J Oral Pathol Med. 2008;37:302–8.
Mosqueda-Taylor A, Pires FR, Aguirre- Urízar JM, Carlos-Bregni R, de la Piedra- Garza JM, Martínez-Conde R, et al. Primordial odontogenic tumour: clinicopathological analysis of six cases of a previously undescribed entity. Histopathology. 2014;65:606–12.
Slater LJ, Eftimie LF, Herford AS. Primordial odontogenic tumor: report of a case. J Oral Maxillofac Surg. 2016;74:547–51.
Bishop JA, Yonescu R, Batista D, Warnock GR, Westra WH. Glandular odontogenic cysts (GOCs) lack MAML2 rearrangements: a finding to discredit the putative nature of GOC as a precursor to central mucoepidermoid carcinoma. Head Neck Pathol. 2014;8:287–90.
Amary MF, Ye H, Forbes G, Damato S, Halai D, Berisha F, et al. Isocitrate dehydrogenase 1 mutations (IDH1) and p16/CDKN2A copy number change in conventional chondrosarcomas. Virchows Arch. 2015;466:217–22.
Wang L, Motoi T, Khanin R, Olshen A, Mertens F, Bridge J, et al. Identification of a novel, recurrent HEY1-NCOA2 fusion in mesenchymal chondrosarcoma based on a genome-wide screen of exon-level expression data. Genes Chromosomes Cancer. 2012;51:127–39.
Yoshida A, Ushiku T, Motoi T, Shibata T, Beppu Y, Fukayama M, et al. Immunohistochemical analysis of MDM2 and CDK4 distinguishes low-grade osteosarcoma from benign mimics. Mod Pathol. 2010;23:1279–88.
Behjati S, Tarpey PS, Presneau N, Scheipl S, Pillay N, Van Loo P, et al. Distinct H3F3A and H3F3B driver mutations define chondroblastoma and giant cell tumor of bone. Nat Genet. 2013;45:1479–82.
Flucke U, Tops BB, van Diest PJ, Slootweg PJ. Desmoid-type fibromatosis of the head and neck region in the paediatric population: a clinicopathologic and genetic study of seven cases. Hstopathology. 2014;64:769–76.
E Horvai A, C Jordan R. Fibro-osseous lesions of the craniofacial bones; β –catenin immunohistochemical analysis and CT-NNB1 and APC mutation analysis. Head Neck. 2014;8:291–97.
Marie PJ. Cellular and molecular basis of fibrous dysplasia. Histol Histopathol. 2001;16:981–8.
Ruggieri P, Sim FH, Bond JR, Unni KK. Malignancies in fibrous dysplasia. Cancer. 1994;73:1411–24.
Romeo S, Hogendoorn PC, Dei Tos AP. Benign cartilaginous tumors of bone: from morphology to somatic and germline genetics. Adv Anat Pathol. 2009;16:307–15.
Presneau N, Baumhoer D, Behjati S, Pillay N, Tarpey P, Campbell P, et al. Diagnostic value of H3F3A mutations in giant cell tumor of bone compared to osteoclast-rich mimics. J Pathol. 2015;1:113–23.
Mangion J, Rahman N, Edkins S, Barfoot R, Nguyen T, Sigurdsson A, et al. The gene for cherubism maps to chromosome 4p16.3. Am J Hum Genet. 1999;65:151–7.
Oliveira AM, Perez-Atayde AR, Inwards CY, Medeiros F, Derr V, Hsi BL, et al. USP6 and CDH11 oncogenes identify the neoplastic cell in primary aneurysmal bone cysts and are absent in so-called secondary aneurysmal bone cysts. Am J Pathol. 2004;165:1773–80.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
Neither Drs. Wright nor Vered have anything to disclose regarding potential conflicts of interest.
Ethical Approval
This article does not contain any studies with human participants or animals performed by any of the authors.
Informed Consent
This article does not require Informed consent.
Additional information
Special Issue: World Health Organization Classification Update
Rights and permissions
About this article

Cite this article
Wright, J.M., Vered, M. Update from the 4th Edition of the World Health Organization Classification of Head and Neck Tumours: Odontogenic and Maxillofacial Bone Tumors. Head and Neck Pathol 11, 68–77 (2017). https://doi.org/10.1007/s12105-017-0794-1
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12105-017-0794-1