
Abstract
A theoretical study of the regio- and stereoselectivities of the 1,3-dipolar cycloaddition reaction between methyl crotonate and pyrroline-1-oxide has been carried out using density functional theory (DFT) at the B3LYP/6-31G(d) level of theory. The reaction has been followed by performing transition state optimization, calculations of intrinsic reaction coordinate and activation energies; the molecular mechanism of the reactions is concerted and asynchronous. The regio- and exo/endo-selectivity have been explained in terms of frontier molecular orbital interactions, local and global electrophilicity and nucleophilicity indices and an analysis of the Wiberg bond indices in the transition state. The FMO analysis and DFT-based reactivity indices showed that the regioselectivity of this reaction is controlled by the HOMOdipole–LUMOdipolarophile interaction. The activation parameters indicated favoured endo approach along the meta-pathway in agreement with the experimental results.

The molecular mechanism of the 1,3-dipolar cycloaddition reaction between pyrroline-1-oxide and methyl crotonate is theoretically investigated by DFT method at the B3LYP/6-31G* level. The transition states corresponding to the possible stereoisomeric pathways along regioisomeric reaction channels were searched, localized and optimized. The cycloaddition is favored along the reaction channel with the meta-endo adduct as major diastereomer in agreement with experiment.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
1 Introduction
1,3-Dipolar cycloaddition (1,3-DC) is one of the simplest approaches for the construction of five-membered heterocyclic rings. [1] Reactions between nitrones and alkenes leading to isoxazolidines are well-known construction processes. [1, 2] Substituted isoxazolidines are interesting biological active compounds [3] and could be used as enzyme inhibitors. [4, 5] They are also frequently used as intermediates for the synthesis of a variety of compounds after cleavage of the N–O bond. [6]
In the context of the cycloadditions of nitrones with several dipolarophiles, we have reported a density functional theory (DFT) study of 1,3-dipolar cycloaddition reaction between simple nitrone with three-fluorinated dipolarophiles, analysis of the results on different reaction pathways shows that the reaction occurs through a concerted process and proceed more or less synchronously. [7] We have also studied the molecular mechanism for the 1,3-dipolar cycloaddition of nitrone with sulphonylethene chloride using ab initio and DFT methods at the HF, MP2 and B3LYP levels together with the 6-31G* basis set. Activation energies and asynchronicity are dependent on the computation level. Thus, while HF calculations gave large barriers, MP2 calculations tend to underestimate them. DFT calculations gave reasonable values. [8] Liu et al. [9] have performed DFT calculations at B3LYP/6-31G* level on the 1,3-dipolar cycloaddition reaction of the simplest nitrone to dipolarophiles containing electron-releasing substituents. Here, the endo approach is kinetically favoured because of stabilization of the secondary orbital interactions. In another study, Cossó et al. [10] have used B3LYP/6-31G* calculations to study the 1,3-dipolar cycloaddition reaction of unsubstituted nitrone with nitroethene. Asynchronicity in the bond formation process in the two regioisomeric approaches of the two reactants is found to be controlled by the electron-deficient dipolarophile. Domingo et al. have studied the 1,3-dipolar cycloaddition reaction of nitrones with several dipolarophiles using DFT methods at the B3LYP/6-31G* and B3LYP/6-31+G* levels. [11, 12] Their calculations predict an asynchronous concerted mechanism and both stereo and regioselectivity were found dependent on the computational model and computational level. Nacereddine et al. [13] have studied the regio- and stereoselectivities of the 1,3-dipolar cycloaddition of C-diethoxyphosphoryl-N-methylnitrone with substituted alkenes (allyl alcohol and methyl acrylate) using DFT method. An analysis of potential energy surfaces (PESs) shows that these 1,3-dipolar cycloaddition reactions favour the formation of the ortho-trans cycloadduct in agreement with experimental data. Stecko et al. have reported a DFT/B3LYP/6-31+G(d) study of the 1,3-dipolar cycloaddition of cyclic nitrones with electron-poor and electron-rich cyclic dipolarophiles: α, β-unsaturated lactones and vinyl ethers. Different reaction channels and reactants approaches, effective in regio- and stereochemical preferences are discussed; the results were compared to experimental data and found in good agreement. [14] Recently, Acharjee and Banerji [15] studied the 1,3-dipolar cycloaddition between C,N-diphenyl nitrone and an unsymmetrical disubstituted olefin at DFT/B3LYP/6-31G(d) level of theory. The analysis of FMO energies, reactivity indices and charge transfer in the transition states indicates a normal electron demand character for the reaction.
The experimental investigations of Asrof et al. [16] implied that the cycloaddition between pyrroline-1-oxide 1 to dipolarophile 3b (methyl crotonate) gives a mixture of substituted isoxazolidines 4b and 5b in a 93:7 ratio, respectively (scheme 1). In what follows, we present a DFT study on the cycloaddition reaction involving cyclic 5-membered nitrone with methyl crotonate. Our results are presented and discussed on the basis of the generated trends in terms of detailed conceptual DFT-based reactivity indices and the analysis of stationary points on the potential energy surfaces. This analysis allows to elucidate the regio- and stereoselectivities of the 1,3-dipolar cycloaddition and to explain the experimental observations.

1,3-Dipolar cycloaddition of pyrroline-1-oxide with methylcrotonate.
2 Computational details
All calculations reported in this paper were performed using Gaussian 03 suite of programs [17] along with the graphical interface GaussView3.08. The full geometrical optimization of all structures and transition states structures (TSs) were carried out with DFT by applying the Becke’s [18] three-parameter hybrid functional and Lee-Yang-Parr’s [19] correlation functional. The basis set 6-31G(d) [20] has been employed for the prediction of activation energies of cycloaddition reactions and to provide geometries and electronic properties. The stationary points were characterized by frequency calculations in order to check that the TSs had one and only one imaginary frequency with the corresponding eigenvector involving the formation of the newly created C–C and C–O bonds. Furthermore, the intrinsic reaction coordinate (IRC) [21, 22] path was mapped to authenticate the connection of a TS to the two associated minima of the proposed mechanism. Electronic structures of critical points were analysed by the natural bond orbital (NBO) method. [23] Global reactivity indices were estimated according to the equations recommended by Parr and Yang. [24] In particular, the electronic chemical potentials (μ) and chemical hardness (η) of the reactants studied were evaluated in terms of the one-electron energies of the frontier molecular orbital HOMO and LUMO, using the following equations:
The values of μ and η were then used for the calculation of global electrophilicity (ω) according to the formula:
The global nucleophilicity (N) [25] is referred to tetracyanoethylene (TCE) because it presents the lowest HOMO energy and a very large electrophilicity (ω = 5. 95 eV) in a large series of molecules already investigated in the context of polar cycloadditions. The global nucleophilicity can then be expressed as:
The local electrophilicity (ω k ) [26] condensed to atom k was calculated by projecting the indice ω onto any reaction centre k of the molecule by using Fukui function \(f_{\mathrm {k}}^+ \). [27]
The local nucleophilicity (N k) [28] condensed to atom k was calculated using global nucleophilicity N and Fukui function \(f_{\mathrm {k}}^- \) according to the formula:
For an atom k in a molecule, depending upon the type of electron transfer, we have three different types of condensed Fukui function defined as follows. [29]
where ρ k (N+1), ρ k (N) and ρ k (N-1) are defined as the gross electronic populations of the site k in the anionic, neutral and cationic species, respectively.
3 Results and discussions
For this 1,3-DC reaction between cyclic nitrone: pyrroline-1-oxide and methyl crotonate, we first evaluated the geometrical parameters and energies of all the stationary points (reactants, transition structures and cycloadducts) at DFT/B3LYP/6-31G(d). Population analysis at the transition structures in terms of bond orders and natural charges was performed, together with an analysis based on the global and local reactivity indices of the reactants involved during the cycloadditions.
3.1 Regiochemistry of 1,3-dipolar cycloaddition reaction based on FMOs and reactivity indices analysis
3.1a FMOs analysis:
In this section, the frontier molecular orbital theory [30–39] was applied to explain the regioselectivity and reactivity in 1,3-dipolar cycloaddition of methyl crotonate and the nitrone.
Analysis of the frontier molecular orbitals of the reactants show that in the nitrone, both HOMO and LUMO are π molecular orbitals (MOs). However, in the methyl crotonate, the HOMO is a nonbonding MO localized essentially on the oxygen of the carbonyl group and, in consequence, it is expected that it will not be directly involved in the 1,3-DC process. The HOMO-1 of dipolarophile is a bonding π molecular orbital with a large contribution of the atoms present at the active sites, thus the relative reactivity can be explained with analysis of this MO.
For a better visualization of the FMO approach, we have presented in figure 1 the two possible interactions: HOMOdipole–LUMOdipolarophile(ΔE 1 = 4. 649 eV) and (HOMO-1)dipolarophile–LUMOdipole(ΔE 2 = 7. 112eV). The FMO analysis for this cycloaddition shows that the main interactions occur between the HOMO dipole and the LUMO dipolarophile, thereby revealing normal electronic demand character of the cycloaddition reaction. This agrees with Sustmann’s type I reactions. [40, 41] According to Houk’s rule [42] in general, regioselectivity of these cycloadditions can be rationalized in terms of more favourable FMO interactions between the largest coefficient centres of the dipole and the dipolarophile. Table 1 shows that the larger orbital coefficients for the binding atoms are that of C4 of the dipolarophile and O1 of the dipole, indicating that the important orbital overlap should be between these two atoms. The oxygen of dipole favours an interaction with C4 of dipolarophile and carbon of dipole interacts with C5 of dipolarophile to give the meta-regioisomer, which is the major product, obtained experimentally. [16] This result agrees with the 1,3-DC of the cyclic nitrone to methyl propiolate studied by Marco and Domingo [11] who predicted the experimentally observed meta-regioselectivity.

Frontier molecular orbital (B3LYP/6-31G*)) interaction in the 1,3-DC between pyrroline-1-oxide and methyl crotonate.
3.1b Analysis in terms of global and local reactivity of the reactants:
The energies of frontier molecular orbitals, electronic chemical potential (μ), chemical hardness (η), global electrophilicity (ω) and global nucleophilicity (N) for the two reactants have been calculated using equations (1)–(4) and are reported in table 2.
In section 3.1a, we saw that for the dipolarophile, the HOMO cannot be used for interpreting the 1,3-DC, in this case, the chemical potential, hardness, nucleophilicity and electrophilicity have been evaluated using this HOMO-1 and the LUMO energy values.
Electronic chemical potential of pyrroline-1-oxide (−0.106685 a.u.) is higher than that of dipolarophile methyl crotonate (−0.15195 a.u.), indicating thereby that in this 1,3-dipolar cycloaddition, the charge transfer will take place from the dipole to the dipolarophile in agreement with the FMO energy predictions. Values of global electrophilicity of reactants are 1.358 and 0.770 eV, respectively, for methyl crotonate and nitrone. Thus, nitrone 1 will act as a nucleophile whereas alkene 3b will act as an electrophile, and therefore indicate that the electronic flux is from the dipole to the dipolarophile. The global electrophilicity difference Δω (0.588 eV) is characteristic of pericyclic reactions, [43, 44] which indicates a lower polar character for this cycloaddition. This is also revealed by the low charge transfer observed along this cycloaddition reaction (see section 3.2d). Several studies related to cycloaddition reactions have shown that analysis of the local electrophilicity indice, ω K at the electrophilic reagent and the nucleophilic indice N K at the nucleophilic compound explain the observed regioselectivity. [45] Values of the Fukui indices f k, local and global electrophilicity indices ω K are reported in table 3 and for better visualization, we have depicted the most favourable two-centre interaction in figure 2. Dipolarophile methyl crotonate has the largest electrophilic activation at the C4carbon atom, ω K = 0. 340eV, whereas the dipole pyrroline-1-oxide has the largest nucleophilic activation at the O1oxygen atom N K = 1. 428 eV. Therefore, C4 atom of the dipolarophile will be the preferred position for a nucleophilic attack by O1 of the dipole, leading to the formation of the meta-regioisomer which is in good agreement with the experimental data. [16] This fact agrees with the asynchronicity observed at the transition states (see section 3.2b). The O1-C4 or C3-C4 bonds formed at the meta-TSs or ortho-TSs are shorter and more advanced than the C3-C5 or O1-C5bonds. This is in agreement with other DFT cycloaddition studies [45] hsuggesting that the most electrophilic reagents control the asynchronicity of the process by a larger bond-formation process at the most electrophilic site of the molecule.

Prediction of favoured interactions between dipole and dipolarophile using DFT-based indices.
3.2 Mechanistic study of the cycloaddition reaction based on activation energy along the different paths of the reaction
3.2a Energies of the transition structures:
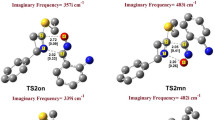
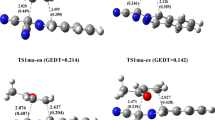
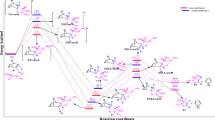
In the present study, the structures of transition states are located through vibrational frequency analysis. Each transition state is characterized by a single imaginary frequency. We have considered four reactive channels corresponding to the endo and exo-approaches of the dipolarophile towards the dipole via two regioisomeric pathways – ortho and meta. A convenient naming system has been employed for the structures of the transition states and the cycloadducts (scheme 2). So, TS-oen and TS-oex are the transition structures for the endo and exo approaches of the dipolarophile on the dipole, respectively, along the ortho-channels leading to products, P-oen and P-oex. Similarly, TS-men and TS-mex are the TSs for the endo and exo approaches of the dipolarophile on the nitrone, respectively, along the meta-channels leading to products, P-men and P-mex. The ortho-pathways correspond to the O1/dipole–C5/dipolarophile and C3/dipole–C4/dipolarophile bond-formation process (5-substituted-CO2Me-isoxazolidine), whereas the meta-channels correspond to the formation of the 4-substituted-CO2Me-isoxazolidine by the formation of the O1/dipole–C4/dipolarophile and C3/dipole–C5/dipolarophile bonds. A schematic representation of the four optimized transition state structures is presented with the atom numbering in figure 3. Total energies (in a.u.) for the all species and relative energies (in kcal/mol) for the TSs and cycloadducts are summarized in table 4. The PESs, corresponding to all the reaction channels, are illustrated in figure 4.

The exo and endo approaches of pyrroline-1-oxide to alkene 3b.

Transition structures corresponding to the regioisomeric path of the 1,3-DC reaction between pyrroline-1-oxide and methyl crotonate. Distances directly involved in the forming-bond process are given in angstroms.

Relative energy profiles (to reactants, in kcal/mol) of stationary points in gas and solvent phases.
As shown in table 4, the activation barriers associated with the cycloaddition reactions are: 10.4 kcal/mol (TS-men), 12.54 kcal/mol (TS-mex), 12.77 kcal/mol (TS-oen) and 14.84 kcal/mol (TS-oex). Accordingly, it can be predicted that the meta/endo-regioisomer will be formed preferentially. Formation of the four cycloadducts P-men, P-mex, P-oen and P-oex are exothermic by −18.79, −12.54, −10. 08 and −8.83 kcal/mol, respectively. These values reveal that meta-approaches are favoured in regard to ortho ones along the cycloaddition process, in agreement with the FMO analysis. In this case, the kinetic and thermodynamic products are coincident and then only the formation of P-men is noticeable, in good agreement with the experimental observations. [16] The favoured formation of the endo-cycloadduct can be attributed to the stabilizing secondary orbital interactions of the ester carbonyl group in the LUMO of the dipolarophile with the HOMO lobe on the nitrogen atom of the cyclic nitrone. This fact is also in agreement with the study of Houk et al. [9] who have performed an ab initio study on the 1,3-DC of a simple nitrone to dipolarophiles containing electron-releasing substituent. Once again, the endo-cycloadduct was kinetically favoured owing to stabilizing secondary orbital interactions. To take into account the solvent effects, single-point calculations at the B3LYP/6-31G* gas phase optimized geometries have been performed. A self-consistent reaction field (SCRF) [30, 46] model based on the polarizable continuum model (PCM) of Tomasi’s group [47] have been applied. The solvent used in the experimental study is dichloromethane, so we have used the dielectric constant, at 298 K, ε = 8. 93. In dichloromethane solvent, the reactants are more stabilized than TSs and cycloadducts. As a consequence, the activation barriers associated with the four TSs: TS-men, TS-mex, TS-oen and TS-oex increase to 14.14, 16.22, 18.28 and 19.49 kcal/mol, respectively. Solvent effects decreased the exothermicity of the process because of the greater solvation of the polar nitrone than that of the TSs and cycloadducts. [10, 48] We can conclude that the solvent effect produces minor changes of regio and stereoselectivity relative to the gas-phase since the trends of the relatives energies are the same.
3.2b Geometries of the transition structures:
The optimized geometries of four TSs corresponding to the reaction of pyrroline-1-oxide and methyl crotonate are depicted in figure 3. The corresponding selected geometric parameters, illustrated in table 5, reveal that all transition structures are asynchronous. For both transition structures TS-men and TS-mex, the Lengths of the C–C forming bonds (2.29 and 2.27 Å) are markedly longer than that of the C–O forming bonds (1.89 and 1.93 Å) showing a significantly dissymmetry for the two newly formed bonds in these TSs. However, in the ortho-pathways, C–O forming bond in TS-oen and TS-oex (2.18 and 2.22 Å) is longer than C–C forming bonds (2.04 and 2.03 Å). This shows a change of the asynchronicity on the bond formation process for the two regioisomeric pathways. The degree of asynchronicity, Δd, can be determined by considering the difference between the lengths of the two forming bonds such that Δd = | d(C–O) – d(C–C) | as shown in table 5. It appears clearly that the meta-TSs are more asynchronous (Δd ≈ 0.34–0.40 Å) than the ortho ones (Δd ≈ 0.14–0.18 Å). The transition state associated with the more favourable stereoisomeric channel: TS-men is much more asynchronous than those associated with the other channels. This fact supports the empirical rule that holds for a variety of Diels–Alder cycloadditions that ‘for dissymetrically substituted dienophiles, the more asynchronous transition state has the lower energy’. [45–51]
3.2c Transition vectors and frequencies analysis:
Transition vectors (TVs) analyses allow us to understand the chemical process associated with each transitions structure involved in this 1,3-DC. We have reported in table 6, the imaginary frequencies, the main TVs components and their corresponding geometric parameters for the four transition states. For the two meta-TSs (TS-men and TS-mex), the dominant TV components are associated with the C3–C5 ( ≈ 0.38) and O1–C4 ( ≈ 0.43) bond distances, which correspond to the two newly σ-bonds formed in these 1,3-DC processes. For the ortho-TSs, the values of the C3–C4 components ( ≈ 0.46–0.51) are larger than for the O1–C5 ones ( ≈ 0.29). It can appear, from these values, the asynchronicity in the bond formation process along this 1,3-DC because the TVs components associated with the C3–C5 and O1–C5 bonds are different. Several dihedral angles also participate in the transition vectors. The Ha4–C4–C5–Ha5dihedral angle is associated with the hybridization change that is developing in the C4 and C5 centres from sp 2 to sp 3. The Ha3–C3–N2–O1 dihedral angle shows the sp 2 to sp 3 re-hybridization taking place at the N2 nitrogen centre along the 1,3-DC reactions. The imaginary frequency values from TS-men, TS-mex, TS-oen and TS-oex are 406.9i, 406.6i, 412.4i and 424.1i cm−1, respectively. These values are lower than those for the Diels–Alder cycloadditions (500 cm−1) and indicate that these processes are associated with heavy atom motions and are also related to the earlier TSs.
3.2d Bond order and charge analysis:
In connection with the structure of the transition states, the bond order (BO) values are used to analyse of the evolution of bond formation or bond breaking along the reaction pathway. To also understand the molecular mechanism in this study, the Wiberg bond indices [52] have been computed using the NBO population analysis, the results are reported in table 7. General analysis of the bond order values for all the transition structures showed that the cycloaddition process is particularly asynchronous. For the meta-TSs, the values of percentage of forming O1–C4 bonds, in the ranges of 51.68–49.01, are greater than those for the forming C3–C5 ones (34.24-33.74). For the ortho-TSs, however, the percentage of forming bond O1–C5(35.19–32.03) has lesser values than that of C3–C4 bond (49.57–48.10).
A comparative study of the values of activation energies, bond orders and frequencies corroborates the fact that the more asynchronous TSs presents a lower activation barrier and a lower imaginary frequency. [11, 51] The charge transfer evaluated by the natural population analysis in terms of the residual charge on the pyrroline-1-oxide fragment, for all the optimized TSs, are listed in table 7. Positive values are indicative of an electron flow from the HOMO/dipole to the LUMO/dipolarophile, in agreement with the electronic chemical potential values, but their magnitudes reveal an almost neutral reaction.
4 Conclusion
In summary, we have checked in the present theoretical study, the regioselectivity and stereoselectivity of the 1,3-dipolar cycloaddition reaction of the cyclic nitrone: pyrroline-1-oxide and methyl crotonate using both frontier molecular orbitals analysis and complete exploration of the potential energy surface at DFT/B3LYP level using the 6-31G* basis set. For this cycloaddition, four reactive pathways have been characterized relative to the endo and exo approaches of the dipolarophile to the dipole along the ortho and meta-regioisomeric pathways. Analysis of FMO energies, electronic chemical potentials and charge transfer at the transition states indicates a normal electron demand character for the reaction. Interaction energies for the global–global and local–global interactions have been investigated revealing a clear preference for the meta-regioselectivity of the cycloaddition process. Geometrical parameter and Wiberg bond indices indicate that the cycloaddition reaction follows a concerted mechanism with asynchronous transition states. General analysis of the bond order values for all the TSs structures showed that the cycloaddition process is asynchronous. This theoretical study shows a clear preference for the meta-regioselectivity of the cycloaddition process in conformity with the experimental findings. This study also demonstrates that B3LYP/6-31G(d) calculations can be used for description of the cycloaddition reaction between the 5-membered cyclic nitrone and an unsymmetrically disubstituted olefin.
References
Padw A 1984 1,3-Dipolar cycloaddition chemistry, vols 1-2 (New York: Wiley Interscience)
(a) Padwa A, Tomioka Y and Venkatramanan MK 1987 Tetrahedron Lett. 28 755; (b) Breuer E, Aurich H G and Nielsen 1989 Nitrones, nitronates and nitroxides (New York: Wiley); (c) Padwa A 1991 Comprehensive Organic Synthesis 4 1069; (d) Torssell K B G 1998 Nitrile oxides, nitrones and nitronates in organic synthesis; (New York: VCH); (e) Gothelf K V and Jørgensen K A 1998 Chem. Rev. 98 863; (f) Bortolini O, Mulani I, De Nin A, Maiuolo L, Nard M, Russo B and Avnet S 2011 Tetrahedron 67 563; (g) Majumder S and Bhuyan P J 2012 Tetrahedron Lett. 53 762
(a) Minter A R, Brennan B B and Mapp A K 2004 J. Am. Chem. Soc. 126 10504; (b) Palmer G C, Ordy M J, Simmons R D, Strand J C, Radov L A, Mullen G, Kinsolving C R, Mitchell J T and Alle S D 1989 Antimicrob. Agents Chemother. 33 895
Ding P, Miller M, Chen Y, Helquist P, Oliver A J and Wiest O 2004 Org. Lett. 6 1805
Wess G, Kramer W, Schuber G, Enhsen A, Baringhaus K H, Globmik H, Müller S, Bock K, Klein H, John M, Neckermann G and Hoffmann A 1993 Tetrahedron Lett. 34 819
Padwa A and Pearson W H 2002 Synthetic applications of 1,3-dipolar cycloaddition chemistry toward heterocycles and natural products (New York: Wiley & Sons)
Marakchi K, Kabbaj O K and Komiha N 2002 J. Fluorine Chem. 114 81
Marakchi K, Kabbaj O K, Komiha N, Jalal R and Esseffar M 2003 J. Mol. Struct. (Theochem) 620 271
Liu J, Niwayama S, You Y and Houk K N 1998 J. Org. Chem. 63 1064
Cossó F P, Marao I, Jiao H and Schleyer P V R 1999 J. Am. Chem. Soc. 121 6737
Cadra V, Portoleś R, Murga J, Uriel S, Marco J A, Domingo L R and Zaragoza R J 2000 J. Org. Chem. 65 7000
Domingo L R 2000 Eur. J. Org. Chem. 2265
Nacereddine A K, Yahia W, Bouacha S and Djerourou A 2010 Tetrahedron Lett. 51 2617
Stecko S, Michel C, Milet A, Pérez S and Chmielewski M 2008 Tetrahedron: Asymmetry 19 2140
Acharjee N and Banerji A 2011 Comput. Theor. Chem. 967 50
Asrof Ali Sk, Khan J H, Wazeer M I M and Perzanowski H P 1989 Tetrahedron 45 5979
Frisch M J et al. Gaussian 03, Revision B.04 Gaussian: Pittsburgh PA 2003
Becke A D J 1988 Chem. Phys. 38 3098
Lee C Yang and W Parr R G 1988 Phys. Rev. B37 785
Hehre W J, Radom L, Schleyer P R and Pople J A 1986 Ab initio molecular orbital theory (New York: Wiley)
Gonzalez C and Schlegel H B 1989 J. Chem. Phys. 90 2154
Gonzalez C and Schlegel H B 1990 J. Phys. Chem. 94 5523
(a) Reed A E, Curtiss L A and Weinhold F 1988 Chem. Rev. 88 899; (b) Reed A E Weinstock R B and Weinhold F 1985 J. Chem. Phys. 83 735
Parr R G and Yang W 1989 Density functional theory of atoms and molecules (New York: Oxford University)
(a) Domingo L R, Chamorro E and Pérez P J 2088 J. Org. Chem. 73 4615; (b) Domingo L R and Picher M T 2004 Tetrahedron 60 5053
(a) Domingo L R, Aurell M J, Pérez P and Contreras R 2002 J. Phys. Chem. A106 6871
Geerlings P, De Prof F and Langenaeker W 2003 Chem. Rev. 103 1793
Pérez P, Domingo L R, Duque-Norna M and Chamorro E 2009 J. Mol. Struct. (Theochem) 895 86
Yang W and Mortier W J 1986 J. Am. Chem. Soc. 108 5708
Tomasi J and Persico M 1994 Chem. Rev. 94 2027
Kumar Das T, Salampuria S and Banerjee M 2010 J. Mol. Struct. (Theochem) 959 22
Ohgaki E, Motoyoshiya J, Narita S, Kakurai T, Hayashi S and Hirakawa K 1990 J. Chem. Soc. Perkin Trans. 1 3109
Aso M, Ojida A, Yang G, Cha O, Osawa E and Kanematsu K 1993 J. Org. Chem. 58 3960
Steck S, Michel C, Milet A, Perez S and Chmielewski M 2008 Tetrahedron: Asymmetry 19 1660
Wei D, Zhu Y, Zhang C, Sun D, Zhang W and Tang M 2011 J. Mol. Catal. A: Chem. 334 108
Marakchi K, Kabbaj O K, Komiha N and Chraibi M 2001 J. Fluorine Chem. 109 163
Marakchi K, Kabbaj O K, Komiha N and Abou El Makarim H 2010 Phys. Chem. News 52 128
Naji N, Marakchi K, Kabbaj O K, Komiha N, Chraibi M, Joffre J and Soufiaoui M 1999 J. Fluorine Chem. 94 127
Sheng Y H, Fang D C, Wuc Y D, Fub X Y and Jianga Y 1999 J. Mol. Struct. (Theochem) 467 31
Sustman R and Sicking W 1987 Chem. Ber. 120 1653
Sustman R and Sicking W 1987 Chem. Ber. 120 1471
Houk K N 1975 Acc. Chem. Res. 8 361
Merino P, Revuelta J, Tejero T, Chiacchio U, Rescifina A and Romeo G 2003 Tetrahedron 59 3581
Pérez P, Domingo L R, Aurell M J and Conteras R 2003 Tetrahedron 59 3117
(a) Domingo L R, Aurell M J, Pérez P and Contreras R 2002 Tetrahedron 58 4417; (b) Domingo L R, Asensio A and Arroyo P 2002 J. Phys. Org. Chem. 15 660; (c) Domingo L R, Arno M, Contreras R and Pérez P 2002 J. Phys. Chem. A 106 952; (d) Domingo LR 2002 Tetrahedron 58 3765; (e) Domingo L R, Aurell M J, Pérez P and Contreras R 2003 J. Org. Chem. 68 3884; (f) Domingo L R and Andres J 2003 J. Org. Chem. 68 8662; (g) Jasiński R, Koifman O I and Barański A 2011 Mendeleev Commun. 21 262; (h) Domingo L R, Pérez P and Sáez J A 2004 Tetrahedron 60 11503
Simkin B Y and Sheikhet I I 1995 Quantum chemical and statistical theory of solutions. A computational approach (London: Ellis Horwood Ltd.)
(a) Cances M T, Mennunci V and Tomasi J 1997 J. Chem. Phys. 107 3032; (b) Cossi M, Barone V, Cammi R and Tomasi J 1996 J. Chem. Phys. Lett. 255 327; (c) Barone V, Cossi M and Tomasi J 1998 J. Comput. Chem. 19 404
Mendez F, Tamariz J and Geerlings P 1998 J. Phys. Chem. A102 6292
Froese R D J, Organ M G, Goddard J D, Stack T D P and Trost B M 1995 J. Am. Chem. Soc. 117 10931
Garcia J I, Martinez-Merino V, Mayoral J A and Salvatella L 1998 J. Am. Chem. Soc. 120 2415
Domingo L R 1999 J. Org. Chem. 64 3922
Wiberg K B 1968 Tetrahedron 24 1083
Acknowledgement
This work was supported by the Ministry of High Education of Morocco (SCH09/09) project ‘Plan d’Urgence’ and European PF7 Marie Curie PIRSES-GA-2012-317544 project CAPZEO.
Author information
Authors and Affiliations
Corresponding author
Rights and permissions
About this article
Cite this article
MARAKCHI, K., GHAILANE, R., KABBAJ, O.K. et al. DFT study of the mechanism and stereoselectivity of the 1,3-dipolar cycloaddition between pyrroline-1-oxide and methyl crotonate. J Chem Sci 126, 283–292 (2014). https://doi.org/10.1007/s12039-013-0563-y
Received:
Revised:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12039-013-0563-y