
Abstract
The mechanism of [3 + 2] cycloaddition (32CA) reactions of phthalazinium-2-dicyanomethanide with electron-deficient and electron-rich ethylenes has been studied using density functional theory (DFT) methods at the M06-2X/cc-pVDZ level of theory. The energy results indicated that the regio- and stereoselectivity of the two 32CA reactions oppose one another, which is in agreement with the experimental report. Accordingly, in the 32CA reaction of phthalazinium-2-dicyanomethanide 1 with methyl vinyl ketone 2a, an electron poor dipolarophile, the meta/endo pathway was more stable than others, while the ortho/exo pathway was the most stable for the 32CA reaction of phthalazinium-2-dicyanomethanide 1 with 1-methoxy-4-vinyl benzene 2b, an electron-rich dipolarophile. Moreover, DFT reactivity indices were studied for the reagents to explain the nucleophilicity and electrophilicity of reagents and selectivity of the reactions. The electron rearrangements along the preferred pathways of these 32CA reactions were studied using the topological analysis of the electron localization function (ELF). The ELF analysis revealed that the reactions proceed through a two-stage one-step mechanism. The noncovalent interactions topological analysis of the possible pathways of the reactions revealed that in addition to strong attractive and repulsive interactions, the weak non-covalent interactions and the steric repulsive interactions have important roles in the determination of the preferred regio- and stereoselective pathway.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
1 Introduction
The [3 + 2] cycloaddition (32CA) reaction is one of the most important approaches for the synthesis of a wide range of heterocycles with high regio- and stereoselectivity [1]. Accordingly, the 32CA reactions of azinium ylide with alkenes and alkynes are well known [2,3,4,5,6,−7], and among them, there has been much focus on the phthalazinium ylide structures in recent years [8,9,−10]. Recently, molecular electron density theory (MEDT) studies of 32CA reactions involving representative three-atom components have allowed characterizing at least four different electronic structures, which experience a dissimilar chemical reactivity [11]. Then, the four simplest reactivity models are presented, providing a modern rationalization of 32CA reactions based on MEDT.
Phthalazinium-2-dicyanomethanide (1) is a Sustmann type II dipole, which can react via normal or inverse electron demand mechanisms with dipolarophiles, and the change in the mechanism is accompanied by a change in regiochemistry [9].
In this regard, the stereochemistry of the cycloaddition reaction of phthalazinium-2-dicyanomethanide (1) has been investigated using various symmetric and asymmetric alkenes [9]. In 2001, Butler et al. [8] indicated that in the reactions of phthalazinium-2-dicyanomethanide with the unsymmetrical electron poor monosubstituted alkenes and alkynes, the dicyanomethanide termins bonds to the unsubstituted carbon and give substituted pyrrole[2,1-α]phthalazines with preferred endo stereselectivity. On the other hand, in the treatment of 1 with the unsymmetrical electron-rich alkenes and aryl dipolarophiles the regio- and stereoselectivity is reversed and exo-2-substituted pyrole[2,1-a] phthalazines is produced as a major stereoisomer [9]
Moreover, kinetic [10,11,−13] studies and the effects of water on the cycloaddition reactions of phthalazinium-2- dicyanomethanide as a 1,3-dipole were examined extensively by experimental and theoretical methods.
Although several theoretical and experimental works have been devoted to establishing the effect of hydrophobic effects and special hydrogen bonding effects of water in the 32CA reactions [10,11,12], there are not any theoretical reports on the molecular mechanism and observed selectivities of these reactions in the literature. It is be noted that the understanding of the underlying principles of the polar 32CA reactions and the influence of the steric and electronic effects on the selectivity of these reactions have grown by theoretical methods.
In the present work, we perform a complete characterization of the reaction mechanism for the 32CA reactions of phthalazinium-2-dicyanomethanide (PDM, 1) with methyl vinyl ketone (MVK, 2a) as well as with 1-methoxy-4-vinyl-benzene (MVB, 2b), which were first reported by Butler et al. [9]. The mechanistic details were explored through computational analysis by the DFT method, and the origin of the observed experimental selectivity was investigated through analysis of the global and local reactivity indices. Also, the mechanism and selectivity of these 32CA reactions were studied using the electron localization function (ELF) [12,13,14] and noncovalent interactions (NCI) [15] topological analysis of TSs.
2 Computational methods
The M06-2X method has been utilized successfully for studying the mechanism and regioselectivity of 32CA reactions [14]. Therefore, all optimizations were carried out using the M06-2X functional along with the cc-pVDZ basis set. The stationary points were checked by frequency analysis in order to verify that the transition state structures (TSs) have only one imaginary frequency. The intrinsic reaction coordinate (IRC) paths were traced in order to obtain and check the energy profiles connecting each TS to the two associated minima of the proposed mechanism [15].
The solvent effects of water were calculated using full optimization of gas phase structures using the new solvation model density (SMD) solvent model [16]. The SMD model is based on the polarizable continuous quantum mechanical charge density of the solute, in the framework of the self-consistent reaction field (SCRF) [17]. Values of enthalpies, entropies and Gibbs free energies were calculated with standard statistical thermodynamics at 298 K and 1 atm. The global electron density transfer [18, 19] (GEDT) at the TSs was computed through a natural population analysis (NPA) [20]. The global electrophilicity index, ω, is given by the simple expression, ω = μ2/2η [21], in terms of the electronic chemical potential, μ, and the chemical hardness, η. These two quantities are evaluated in terms of the one-electron energies of the frontier molecular orbitals HOMO and LUMO, ɛH and ɛL, as μ = (ɛH + ɛL)/2 and η = εL˗ɛH [22, 23]. The global nucleophilicity index, N, [24, 25] based on the HOMO energies obtained within the Kohn Sham scheme [26] is defined as N = ɛH(NU)-ɛH(TCE), with tetracyanoethylene (TCE) used as a reference. In 2013, the Parr functions [27] were defined by Domingo as Pˉ(r) for electrophilic attack and P+(r) for nucleophilic attack. The local electrophilicity indices, ωk[27], and the local nucleophilicity indices, Nk [27], were calculated using ωk = ωPk+ and Nk = NPkˉ, respectively. All computations were carried out with the Gaussian 09 suite of programs [28].
The ELF topological analysis was carried out on the selected points of the IRC profile of TS1ma-en and TS2ob-ex using the TopMod program [29]. NCI analysis was performed for all possible TSs of the two reactions with the NCIPLOT program [30].
3 Results and discussion
The present study has been divided into three parts: firstly, the 32CA reactions of phthalazinium-2-dicyanomethanide (PDM, 1) with the electrophilic methyl vinyl ketone (MVK, 2a) as well as with the nucleophilic 1-methoxy-4-vinyl-benzene (MVB, 2b); secondly, an analysis of the DFT reactivity indices at the reactants is carried out; finally, ELF topological analysis was carried out focused on the formation of C–C σ-bonds along the most favorable pathways of 32CA reactions.
Due to the asymmetry of reactants, four reaction pathways are feasible for the 32CA reactions of 1 with 2a (Scheme 1) as well as with 5. The two stereoisomeric reaction pathways, endo (en) and exo (ex) , are related to the two stereoisomeric approach modes of the substituent, Y, of 2 and 5 relative to the sp2-hybridized N2 nitrogen of 1.

The possible reaction pathways for the 32CA reactions between phthalazinium-2-dicyanomethanide (PDM, 1), with methyl vinyl ketone (MVK, 2a) and 1-methoxy-4-vinyl-benzene (MVB, 2b)
The two possible regioisomeric reaction pathways, namely ortho (o) and meta (m), result from the forming of a bond between C4 or C5 of 2a or 2b and the C3 atom of 1 (with its two nitrile groups), respectively.
In order to analyze the regio- and stereoselectivity of these 32CA reactions, four reaction pathways were studied for each reaction. For each of the four competitive pathways, one TS and the corresponding cycloadduct was found. A schematic representation of the stationary points along all reaction pathways is presented in Scheme 1.
Firstly, the gas phase calculations for these 32CA reactions were not completely consistent with the experimental results. Then 32CA simulations were carried out in water as a solvent, which was expected have some influence on the relative energies and geometries of stationary points. The calculated energy results indicated that in water, the activation energies increased by between 3.60 and 5.74 kcal mol−1 and the exothermic character of the reaction decreased by between 3.62 and 8.03 kcal mol−1 as a consequence of greater solvation of the reagents than of the TSs. In contrast to the gas phase calculations, the results of the regio- and stereoselectivity of the studied 32CA reactions in water are completely in agreement with the experimental results in the solvent. Figures 1 and 2 present the geometries of TSs corresponding to those 32CA reactions in water. Therefore, energy discussions will be done using the relative energies in water and the relative energies of the stationary points in solvent (H2O) are reported in Tables 1 and 2, while for the gas phase are shown in Table S1.

The optimized geometries of the transition states for the 32CA reactions of phthalazinium-2-dicyanomethanide (PDM, 1) with methyl vinyl ketone (MVK, 2a) at the M06-2X/cc-pVDZ level of theory in the solvent phase. Bond distances are given in Å, wiberg bond indices are given in parentheses and the GEDT of TSs are also given

The geometry optimized transition states for the 32CA reactions of phthalazinium-2-dicyanomethanide (PDM, 1), with 1-methoxy-4-vinyl-benzene (MVB, 2b) at the M06-2X/cc-pVDZ level in the solvent phase. Bond distances are given in Å, wiberg bond indices are given in parentheses and the GEDT of TSs are also given
3.1 Energy analysis of the possible 32CA reaction pathways between phthalazinium-2-dicyanomethanide (PDM, 1), with methyl vinyl ketone (MVK 2a)
Firstly, the mechanism and selectivities of the 32CA reaction of 1 with 2a, which was experimentally performed by Butler et al. [9], were studied. As shown in Table 1, the relative energies of the TSs associated with the four possible cycloaddition modes are 5.03 (TS1ma-en), 6.62 (TS1ma-ex), 7.08 (TS2oa-en) and 7.32 (TS2oa-ex) kcalmol−1.
The energy results indicate that the 32CA reaction is regio- and stereoselective, where the most favorable TS, TS1ma-en, is about 2 and 1.6 kcal mol−1 more stable than TS2oa-en and TS1ma-ex, respectively. Therefore, the meta/endo pathway is kinetically favored, while the meta/exo pathway leads to the most stable product, 3ma-ex. All cycloaddition pathways are strongly exothermic in the range -26.98 to -29.28 kcal mol−1. Therefore, these cycloaddition reaction pathways can be considered irreversible. These results are in clear agreement with experimental finding, which reports 3ma-en as the major product [9].
A comparison between the activation enthalpies of the 32CA reaction of 1 with 2 in Table S2 indicates that the most favorable approach mode is the meta/endo pathway (TS1ma-en) with ΔH# = 4.59 kcal mol−1, which is in line with the calculated activation energies. An analysis of the activation Gibbs free energies showed that the meta/endo reaction pathway is a consequence of the negative activation entropy associated with it, ΔS# = -48.23 cal mol−1 K −1.
The geometries of all TSs in this reaction are shown in Fig. 1. The evaluation of the two forming bond lengths of the TSs indicated that all of the TSs is asynchronous. As shown in Fig. 1 for the meta pathways, the length of C1-C4 forming bonds is 2.028 Ǻ and 2.366 Ǻ for TS1ma-en and TS1ma-ex, and the distances between the C3 and C5 atoms in these TSs are 2.499 Ǻ and 2.126, respectively. However, the lengths of the C1-C5 and C3-C4 forming bonds in the TSs of the ortho pathways are 2.076 and 2.437 Ǻ at TS2oa-en and 2.471 and 2.027 Ǻ at TS2oa-ex, respectively. The bond order (BO) values of the forming bonds at TSs are also given in Fig. 1. The BO values validate the main conclusions, which were obtained from the analysis performed on the geometrical parameters. The analysis of the BO values for the forming bonds at TSs indicated that the bond formation at the C1 atom of 1 is more advanced than the C3 atom at the endo pathways, whereas it is reversed in the exo pathways.
Moreover, the electronic natures of the four possible reaction pathways are evaluated by computing the GEDT at the TSs in Fig. 1. The values of GEDT at the TSs are 0.214 at TS1ma-en, 0.142 at TS1ma-ex, 0.056 at TS2oa-en and 0.04 at TS2oa-ex. Hence, the most favorable meta/endo pathway (TS1ma-en) is the most polar one.
3.2 Energy analysis of the possible 32CA reaction pathways between phthalazinium-2-dicyanomethanide (PDM, 1) and 1-methoxy-4-vinyl-benzene (MVB, 2b)
In this part, the 32CA reaction between 1 and 2b, which was experimentally performed by Butler et.al. [9], is studied. A schematic representation of the stationary points along the four possible reaction pathways is shown in Scheme 1. Figure 2 shows the geometries of the TSs corresponding to the 32CA reactions of 1 with 2b, and Table 1 displays the relative energies for the TSs and cycloadducts.
Analysis of the relative energies for the TSs (Table 1) reveals that the ortho TSs, TS2ob-en and TS2ob-ex, are energetically more favored than the meta TSs (TS1mb-en and TS1mb-ex). Moreover, the activation energy for the ortho/exo approach via TS2ob-ex is lower than the endo one. Therefore, it can be concluded that the formation of cycloadduct 4ob-ex is kinetically favored, which is completely in agreement with the experimental results [9]. The formation of all cycloadducts is exothermic in the range -26.89 to -28.43 kcal mol−1, which makes these reaction pathways irreversible.
The optimized geometries of the four TSs corresponding to the reaction of 1 with 2b are shown in Fig. 2. In the case of the more favorable ortho-TSs, the formation of C3-C4 is less advanced than C1-C5, while for the meta transition structures, the length of C1-C4 is almost equal to the length of C3-C5. These values are confirmed with the analysis of the BO results, which indicate that in the case of ortho pathways, the BO values of the C3-C4 bond (0.456) are greater than for the C1-C5 bond (0.202–0.173). Moreover, for the meta pathways, the BO values of C1-C4 and C3-C5 are very close. These data show a different synchronicity in the bond formation process for the two regioisomeric pathways.
Finally, in order to evaluate the polar nature of the 32CA reaction of 1 with 2b, the GEDT was analyzed. In the 32CA reaction of 1 with 2b, the highest GEDT value is 0.194 e at the most favorable TS, TS2ob-ex, which indicates that this process has polar character, in agreement with the computed low activation energy of this pathway.
3.3 Analysis of the global reactivity indices of the reactants
Studies devoted to 32CA reactions have shown that the analysis of reactivity indices defined within conceptual DFT [31,32,−33] is a powerful tool to understand the reactivity in polar cycloadditions. The global indices, namely electronic chemical potential, µ, chemical hardness, η, global electrophilicity, ω, and nucleophilicity, N, for the involved reagents in these 32CA reactions are given in Table 2.
The electronic chemical potential of 1, µ = −4.08 eV, is lower than that of 2b, µ = − 3.19 eV, yet higher than that of 2a with µ = -4.24 eV. These values indicate that along the cycloaddition reactions, while the direction of electron flux is from 1 to the electron-deficient alkene, 2a, it will be from electron-rich 2b to 1.
A shown in Table 2, 1 presents both high electrophilicity, ω = 2.58 eV, and high nucleophilicity, N = 3.43 eV, being classified both as strong electrophile and strong nucleophile, based on the electrophilicity [34] and nucleophilicity [35] scales. This ambiphilic behavior can be a consequence of the presence of the cyanide and imine groups in 1. The nucleophilicity and electrophilicity indices of 2a are lower than 1, while it is classified as a strong electrophile.
In contrast, the inclusion of an electron-rich substituent in the ethylene framework (2b) notably increases the nucleophilicity, N = 3.47 eV, and consequently it is classified as a strong nucleophile. However, it is classified as a moderate electrophile, with an electrophilicity index ω = 1.03 eV.
These values together with the corresponding electronic chemical potentials, suggested that the corresponding 32CA reaction of 1 with 2a and 2b will have a polar character at the most stable pathways (meta/endo and ortho/exo), which is in clear agreement with the analysis of the GEDT computed at the most favorable TS.
In order to better understand the observed regioselectivity of these reactions, the DFT-based reactivity descriptors, such as Parr functions, local electrophilicity and nucleophilicity indices, have been used to explain the selectivity of the reactions [25]. For the polar reactions, the most favorable reaction pathways involve the initial interaction between the most electrophilic centers with the most nucleophilic center of the corresponding reactants [25].
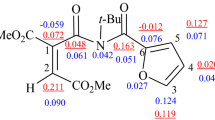
Therefore, a simple analysis of the Parr functions and ASD [27] allows us to characterize the most electrophilic and the most nucleophilic centers in the reactants, to study the regioselectivity of the studied reactions. Accordingly, the electrophilic, Pk+, and nucleophilic, Pk–, Parr functions of 1, based on the ASD map in the radical anion, and the Pk+ of 2a and Pk–of 2b, based on the ASD are analyzed (see Fig. 3 and Table 2). While the Pk– of 1 is mainly concentrated at C3 (Parr function of 0.61), C1 presents the most electrophilic center with an electrophilic Parr function of 0.41. Moreover, the electrophilic Parr function and analysis of the ASD of the radical anion of 2a indicates that the C4 atom is the most electrophilic center of this species, presenting the maximum value, PC4+ = 0.52, which will be the preferred position for a nucleophilic attack of the C3 atom of 1. Therefore, the most favorable electrophile–nucleophile interaction along the nucleophilic attack of the C3 atom of 1 to the electrophilic center of 2a (C4 atom) leading to the formation of the most stable regioisomer of 3ma-en.

Maps of ASD; a) the radical anion and b) the radical cation of 1, and the radical cation of 2a and of the radical anion of 2b
In the cycloaddition reaction of 1 with 2b, the most favorable interaction occurs between the C1 atom of 1 and C4 atom of 2b (PC4– = 0.37, NC4 = 1.46), which indicates that the C1-C4 bond formation is more advanced than the C3-C5 one. This behavior is similar to that found in analysis of the ASD map of the radical cations of 2b. Therefore, analysis of the Parr functions and ASD for the studied cycloaddition reactions can explain the source of the regioselectivities observed.
3.4 ELF analysis of the 32CA reaction of phthalazinium-2-dicyanomethanide (PDM, 1) with methyl vinyl ketone (MVK, 2a) and methyl vinyl benzene (MVB, 2b)
An appropriate tool to study the molecular mechanism of reactions is the ELF analysis along the reaction path [36,37,38]. The maximum probability of finding electron pairs, which are classified as core and valence basins, can result from the ELF analysis. Monosynaptic and disynaptic basins characterize valence basins which involve single and bonding electron pairs, respectively [39, 40]. The mechanism of the 32CA reaction of 1 with 2a or with 2b and C–C bond formations along these reactions has been studied using ELF analysis of the selected structures in the IRC curves of TS1ma-en and TS2ob-ex at the M06-2X/cc-pVDZ level of theory [41]. The related ELF valence attractors of the most important points together with their populations are shown in Figs. 4 and 5. The studied structures through ELF analysis were identified by Pi such as P1 and P2 where they were shown on the IRC map for better visualization.


Schematic representation of the ELF attractors of selected points on the IRC path of the most favored pathway of the 32CA reaction of PDM 1 and MVK 2a


Schematic representation of the ELF attractors of selected points on the IRC path of the most favored pathway of the 32CA reaction of PDM 1 and MVB 2b
ELF topological analysis of the first point of the IRC map of TS1ma-en shows two monosynaptic basins V(C3) and V´(C3) in the 1 fragment with the populations of 0.65 e and 0.51 e, and two disynaptic basins V(C4, C5) and V´(C4, C5) with total population of 3.30 e in the 2a fragment. However, at the C3-C4 distance of 2.39 Å (and C1-C5, 2.69 Å), two disynaptic basins V(C4, C5) and V´(C4, C5) and only a single monosynaptic basin V(C3) are observed with the populations 1.56 e, 1.69 e and 0.84 e, respectively. As shown in Fig. 3, before TS1ma-en and at a C3-C4 distance of 2.15 Å, the two disynaptic basins V(C4, C5) and V´(C4, C5) merge to a single disynaptic basin with a population of 3.16 e and the population of V(C3) has increased to 0.97 e. At this point, a monosynaptic basin has appeared on N2 with a population of 0.69 e. At TS1ma-en, the population associated with V(C4, C5) has decreased and a monosynaptic basin V(C4) appeared with the population of 0.04 e. After TS1ma-en and at a C3-C4 distance of 1.97 Å, another monosynaptic basin V(C1) with the population of 0.16 e has appeared and the population of V(C3) increased to 1.28 e. At this point, the population of disynaptic basin V(C4, C5) has decreased to 2.67 e and the asyn monosynaptic basin has disappeared. At C3-C4 = 1.90 Å (C1-C5 = 2.43 Å), a third monosynaptic basin V(C5) has appeared with a population of 0.32 e, and the population of V(C3) and V(C1) have increased to 1.37e and 0.2 e, respectively. The first new sigma bond appears between C3 and C4 at C3-C4 distance of 1.79 Å forming a disynaptic basin with a population of 1.52e. As shown in Fig. 4, it seems that the required electrons to form this sigma bond have been provided by C3. At this point, the populations of V(C1), V(C5) and V(N2) have also increased to 0.29 e, 0.42 e and 1.36 e, respectively. Then, two pseudoradical centers V(C1) and V(C5) merge to a disynaptic basin V(C1, C5) with a population of 1.30 e, so that the second single bond is formed. At this point, the population of V(C3, C4) and V(N2) increased to 1.75e and 1.83e, respectively. Finally, the population of V(C1, C5) and V(C3, C4) increase to 1.92 e and 1.85 e at 3ma-en. According to the ELF results, the reaction has a one-step two-stage mechanism [18, 42].
The mechanism and C–C bond formations along the 32CA reaction between 1 and 2b have also been studied using ELF analysis of selected structures in the IRC curve of TS2ob-ex at the M06-2X/cc-pVDZ level of theory [41]. The related ELF valence attractors of the most important points together with their populations are shown in Fig. 5.
Based on the ELF results, at the first point of the IRC map, two monosynaptic basins V(C3) and V´(C3) in the 1 fragment were found with populations of 0.67 e and 0.56 e, and two disynaptic basins V(C4, C5) and V´(C4, C5) with a total population of 3.37 e in the 2b fragment. However, at the C3–C5 distance of 2.67 Å (and C1–C4, 2.14 Å), the two disynaptic basins V(C4, C5) and V´(C4, C5) merge to generate a disynaptic basin with a population of 3.19 e, and two monosynaptic basins emerge on N2 and C1 with the population of 0.89 e and 0.28 e, respectively. At this point, the two monosynaptic basins V(C3) and V´(C3) are also observed with populations of 0.76 e and 0.32 e, respectively. Before TS2ob-ex and at C3-C5 distance of 2.64 Å, the population of V(C3) has increased to 0.78 e, and V´(C3) has disappeared. At this point, the populations of V(N2) and V(C1) have increased to 1.02 e and 0.36 e, respectively. At TS2ob-ex, another monosynaptic basin V(C4) with a population of 0.32 e has appeared, and the populations of V(C3) and V(C1) increased to 0.8 e and 0.46 e, respectively. At this point, the population associated with V(C4, C5) has decreased to 2.83 e, and the monosynaptic basin V(N2) has become a disynaptic basis V(N2, N) with a population of 1.57 e. After TS2ob-ex and at a C3–C5 distance of 2.49 Å (C1–C4 = 1.76 Å), the first new sigma bond has appeared between C1 and C4 by forming a disynaptic basin with a population of 1.42 e. At this point, the population of V(C3) has increased to 0.87e. Then, another monosynaptic basin, V(C5), has appeared at the C3–C5 distance of 2.28 Å (C1–C4 = 1.60 Å) with a population of 0.23 e. At this point, the population of V(C1, C4) and V(C3) increased to 1.70 e and 1.00 e, respectively. At the C3-C5, distance of 1.85 Å the second single bond has formed with a population of 1.64 e. Finally, the population of V(C3, C5) and V(C1, C4) increase to 1.86 e and 1.89 e at 4ob-ex. According to ELF results, this reaction also has a one-step two-stage mechanism [18].
3.5 NCI topological analysis of the TSs
NCI is a good tool for qualitatively identify non-covalent interactions. To evaluate the favored and unfavored noncovalent interactions, the NCI and the RDG plots for the TSs of the possible pathways of the two 32CA reactions of 1 with 2a, and 1 with 2b were studied [43] (Figs. 6 and 7). At the NCI and RDG maps, strong attractive interactions are represented by blue surfaces and strong repulsive interactions are shown with red surfaces, while the green areas correspond to weak favorable or unfavorable van der Waals interactions. The single blue surfaces in Fig. 6 of the NCI gradient isosurfaces for the TSs are related to the strong and attractive interactions between the carbon atoms of 1 and 2a, which are necessary for the formation of the sigma bonds. NCI analysis of the TSs has also shown the presence of the weak non-covalent interactions (green surfaces) which is more extended for the most stable TS, TS1ma-en. Therefore, the weak non-covalent interactions have a crucial effect on the regio- and stereoselectivity of this reaction.

NCI gradient isosurfaces, represented at an isovalue of 0.5 a.u. and plots of the RDG versus the electron-density multiplied by the sign of the second Hessian eigenvalue for TS1ma-en, TS1ma-ex, TS2oa-en and TS2oa-ex at the M06-2X/cc-pVDZ level of theory

NCI gradient isosurfaces, represented at an isovalue of 0.5 a.u. and plots of the RDG versus the electron-density multiplied by the sign of the second Hessian eigenvalue for TS1mb-en, TS1mb-ex, TS2ob-en and TS2ob-ex at the M06-2X/cc-pVDZ level of theory
The NCI and the RDG plots for the TSs of the possible pathways of the 32CA reaction of 1 with 2b are shown in Fig. 7. Similarly, the single blue surfaces of the NCI gradient isosurfaces for TS2ob-en and TS2ob-ex are related to the strong and attractive interactions between the carbon atoms of 1 and 2b, which are necessary for the formation of the second sigma bonds. It is noticeable that the blue surfaces of TS1mb-en and TS1mb-ex, which are related to the unfavored regioisomeric channel, do not have a steep slope in their RDG maps. NCI analysis of the TSs also shows the presence of weak non-covalent interactions (green steep slope in the RDG maps) which are more extended for the endo TSs, TS1mb-en and TS2ob-en. Therefore, TS1mb-ex is the most unstable TS, because of the absence of strong and weak attractive interactions. Finally, based on the NCI results and RDG maps, there is a weaker repulsive interaction, resulting from steric effects, between 1 and 2b in the most stable TS, TS2ob-ex.
4 Conclusion
The regio-and stereoselectivity of the 32CA reactions of phthalazinium-2-dicyano methanide 1 with methyl vinyl ketone 2a and 1-methoxy-4-vinyl-benzene 2b has been studied through the M06-2X/cc-pVDZ level of theory. Based on the calculated kinetic and thermodynamic parameters, for the 32CA reaction of 1 with 2a, meta/endo is most favorable pathway, while for the reaction of 1 with 2b the most preferred pathway is the ortho/exo. The regioselectivity of these 32CA reactions has also been investigated through global and local reactivity indices, and the results are in agreement with the experimental observations. The ELF analysis shows that both reactions proceed through a two-stage one-step mechanism and the first sigma bond for both reactions is formed between the most nucleophilic and electrophilic centers of the corresponding reactants, based on the analysis of the global indices and Parr functions. NCI analysis of the TSs has shown the critical role of the strong attractive and repulsive interactions, as well as weak non-covalent interactions in the regio- and stereoselectivity of these reactions.
References
Butler RN, Coyne AG, Moloney EM (2007) Organic synthesis in water: 1,3-dipolar cycloaddition reactions at ambient temperature with aqueous suspensions of solid reactants. Tetrahedron Lett 48:3501–3503. https://doi.org/10.1016/j.tetlet.2007.03.119
Druta II, Andrei MA, Ganj CI, Aburel PS (1999) Synthesis of indolizine derivatives by the reaction of 2-(2′-pyridyl)-pyridinium ylides with ethylenic dipolarophiles. Tetrahedron 55:13063–13070. https://doi.org/10.1016/S0040-4020(99)00798-X
Elender K, Nöth H, Riebel P et al (2000) 1,3-Dipolar cycloaddition reactions of stable bicyclic and monocyclic azomethine ylides: preparative aspects. Tetrahedron 56:5443–5463. https://doi.org/10.1016/S0040-4020(00)00453-1
Tsuge O, Kanemasa S, Sakamoto K, Takenaka S (1988) Cycloaddition reactions of highly stabilized isoquinolinium methylides to nonactivated olefins and electron-rich olefins. Bull Chem Soc Jpn 61:2513–2524. https://doi.org/10.1246/bcsj.61.2513
Miki Y, Hachiken H, Takemura S, Ikeda M (1984) Novel synthesis and 1,3-dipolar cycloaddition reaction of pyridinium N-methylide. Heterocycles 22:701. https://doi.org/10.3987/R-1984-04-0701
Ikemi YM (1983) 1, 3-Dipolar cycloaddition of pyridinium and diazinium dicyanomethylids with Bis (trimethylsilyl) acetylene: synthesis of tri-methylsilyl substituted indolizines and bis (trimethylsilyl) acetylene as an equivalent in 1, 3-dipolar cycloaddition. Heterocycleseterocycles 1983(20):1009
Huisgen R, Hauck H, Grashey R, Seidl H (1969) 1.3-Dipolare Cycloadditionen, XLIV. Nitrone und α.β-ungesättigte Dicarbonylverbindungen; Stereospezifität der Cycloadditionen. Chem Ber 102:736–745. https://doi.org/10.1002/cber.19691020305
Butler RN, Coyne AG, McArdle P et al (2001) Regioselectivity and endo/exo selectivity in the cycloadditions of the phthalazinium dicyanomethanide 1,3-dipole with unsymmetrical alkene and alkyne dipolarophiles. Unexpected reversals of regiochemistry: a combined experimental and DFT theoretical study. J Chem Soc Perkin Trans 1:1391–1397. https://doi.org/10.1039/b101150m
Butler RN, Coyne AG, Cunningham WJ, Burke LA (2002) Kinetic and synthetic influences of water and solvent-free conditions on 1,3-dipolar cycloaddition reactions: the phthalazinium and pyridazinium dicyanomethanide 1,3-dipoles: surprisingly successful synthetic methodsElectronic supplementary information. J Chem Soc Perkin Trans 2(2):1807–1815. https://doi.org/10.1039/b206028k
Antoci V, Moldoveanu C, Danac R et al (2020) Huisgen [3 + 2] dipolar cycloadditions of phthalazinium ylides to activated symmetric and non-symmetric alkynes. Molecules 25:4416. https://doi.org/10.3390/molecules25194416
Ríos-Gutiérrez M, Domingo LR (2019) Unravelling the Mysteries of the [3+2] Cycloaddition Reactions. Eur J Org Chem 2019:267–282. https://doi.org/10.1002/ejoc.201800916
Butler RN, Coyne AG, Cunningham WJ, Burke LA (2002) Kinetic and synthetic influences of water and solvent-free conditions on 1,3-dipolar cycloaddition reactions: the phthalazinium and pyridazinium dicyanomethanide 1,3-dipoles: surprisingly successful synthetic methodsElectronic supplementary information. J Chem Soc Perkin Trans 2:1807–1815. https://doi.org/10.1039/b206028k
Butler RN, Cunningham WJ, Coyne AG, Burke LA (2004) The influence of water on the rates of 1,3-dipolar cycloaddition reactions: Trigger points for exponential rate increases in water-organic solvent mixtures. Water-super versus water-normal dipolarophiles. J Am Chem Soc 126:11923–11929. https://doi.org/10.1021/ja040119y
Lan Y, Zou L, Cao Y, Houk KN (2011) Computational methods to calculate accurate activation and reaction energies of 1,3-dipolar cycloadditions of 24 1,3-dipoles. J Phys Chem A 115:13906–13920. https://doi.org/10.1021/jp207563h
Gonzalez C, Schlegel HB (1990) Reaction path following in mass-weighted internal coordinates. J Phys Chem 94:5523–5527. https://doi.org/10.1021/j100377a021
Marenich AV, Cramer CJ, Truhlar DG (2009) Universal solvation model based on solute electron density and on a continuum model of the solvent defined by the bulk dielectric constant and atomic surface tensions. J Phys Chem B 113:6378–6396. https://doi.org/10.1021/jp810292n
Cancès E, Mennucci B, Tomasi J (1997) A new integral equation formalism for the polarizable continuum model: theoretical background and applications to Isotropic and anisotropic dielectrics. J Chem Phys 107:3032–3041. https://doi.org/10.1063/1.474659
Domingo LR (2014) A new C-C bond formation model based on the quantum chemical topology of electron density. RSC Adv 4:32415. https://doi.org/10.1039/C4RA04280H
Domingo LR, Ríos-Gutiérrez M, Pérez P (2016) A new model for C-C bond formation processes derived from the Molecular Electron Density Theory in the study of the mechanism of [3+2] cycloaddition reactions of carbenoid nitrile ylides with electron-deficient ethylenes. Tetrahedron 72:1524–1532. https://doi.org/10.1016/j.tet.2016.01.061
Reed AE, Weinstock RB, Weinhold F (1985) Natural population analysis. J Chem Phys 83:735–746. https://doi.org/10.1063/1.449486
Parr RG, Szentpály LV, Liu S (1999) Electrophilicity index. J Am Chem Soc 121:1922–1924. https://doi.org/10.1021/ja983494x
Blanchard P, Brüning E (2015) Density functional theory of atoms and molecules. Prog Math Phys 69:563–573
Parr RG, Pearson RG (1983) Absolute hardness: companion parameter to absolute electronegativity. J Am Chem Soc 105:7512–7516. https://doi.org/10.1021/ja00364a005
Domingo LR, Chamorro E, Pérez P (2008) Understanding the reactivity of captodative ethylenes in polar cycloaddition reactions. A theoretical study. J Org Chem 73:4615–4624. https://doi.org/10.1021/jo800572a
Domingo LR, Pérez P (2011) The nucleophilicity N index in organic chemistry. Org Biomol Chem 9:7168–7175. https://doi.org/10.1039/c1ob05856h
Kohn W, Sham LJ (1965) Self-consistent equations including exchange and correlation effects. Phys Rev. https://doi.org/10.1103/PhysRev.140.A1133
Domingo LR, Pérez P, Sáez JA (2013) Understanding the local reactivity in polar organic reactions through electrophilic and nucleophilic Parr functions. RSC Adv 3:1486–1494. https://doi.org/10.1039/c2ra22886f
Frisch M, Trucks G, Schlegel H et al (2009) Gaussian 09, Revision A Gaussian, Inc, Wallingford, CT
Noury S, Krokidis X, Fuster F, Silvi B (1999) Computational tools for the electron localization function topological analysis. Comput Chem 23:597–604. https://doi.org/10.1016/S0097-8485(99)00039-X
Contreras-García J, Johnson ER, Keinan S et al (2011) NCIPLOT: a program for plotting noncovalent interaction regions. J Chem Theory Comput 7:625–632. https://doi.org/10.1021/ct100641a
Parr RG, Yang W (1995) Density-functional theory of the electronic structure of molecules. Annu Rev Phys Chem 46:701–728. https://doi.org/10.1146/annurev.pc.46.100195.003413
Chermette H (1999) Chemical reactivity indexes in density functional theory. J Comput Chem 20:129–154. https://doi.org/10.1002/(SICI)1096-987X(19990115)20:1%3c129::AID-JCC13%3e3.0.CO;2-A
Ess DH, Jones GO, Houk KN (2006) Conceptual, qualitative, and quantitative theories of 1,3-dipolar and Diels-Alder cycloadditions used in synthesis. Adv Synth Catal 348:2337–2361
Domingo LR, Aurell MJ, Pérez P, Contreras R (2002) Quantitative characterization of the global electrophilicity power of common diene/dienophile pairs in Diels-Alder reactions. Tetrahedron 58:4417–4423. https://doi.org/10.1016/S0040-4020(02)00410-6
Jaramillo P, Domingo LR, Chamorro E, Pérez P (2008) A further exploration of a nucleophilicity index based on the gas-phase ionization potentials. J Mol Struct THEOCHEM 865:68–72. https://doi.org/10.1016/j.theochem.2008.06.022
Andrés J, González-Navarrete P, Safont VS (2014) Unraveling reaction mechanisms by means of Quantum Chemical Topology Analysis. Int J Quantum Chem 114:1239–1252
Andres J, Berski S, Domingo LR et al (2011) Describing the molecular mechanism of organic reactions by using topological analysis of electronic localization function. Curr Org Chem 15:3566–3575. https://doi.org/10.2174/138527211797636156
Polo V, Andres J, Berski S et al (2008) Understanding reaction mechanisms in organic chemistry from catastrophe theory applied to the electron localization function topology. J Phys Chem A 112:7128–7136. https://doi.org/10.1021/jp801429m
Silvi B, Savin A (1994) Classification of chemical bonds based on topological analysis of electron localization functions. Nature 371:683–686. https://doi.org/10.1038/371683a0
Silvi B (2002) The synaptic order: a key concept to understand multicenter bonding. J Mol Struct 614:3–10
Becke AD, Edgecombe KE (1990) A simple measure of electron localization in atomic and molecular systems. J Chem Phys 92:5397–5403. https://doi.org/10.1063/1.458517
Domingo L (2016) Molecular electron density theory: a modern view of reactivity in organic chemistry. Molecules 21:1319. https://doi.org/10.3390/molecules21101319
Johnson ER, Keinan S, Mori-Sánchez P et al (2010) Revealing noncovalent interactions. J Am Chem Soc 132:6498–6506. https://doi.org/10.1021/ja100936w
Acknowledgements
The authors wish to acknowledge Dr Louise S. Price, University College London, UK, for reading the manuscript and providing valuable suggestions.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Conflict of interest
The authors declare that they have no conflict of interest.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Lotfi, M., Hamzehloueian, M. & Haghdadi, M. A DFT study on the mechanism and selectivity of [3 + 2] cycloaddition reactions leading to pyrole[2,1-a] phthalazine compounds. Theor Chem Acc 140, 57 (2021). https://doi.org/10.1007/s00214-021-02756-7
Received:
Accepted:
Published:
DOI: https://doi.org/10.1007/s00214-021-02756-7