
Abstract
Oxidative stress and neuroinflammation are the main physiopathological changes involved in the initiation and progression of various neurodegenerative disorders or brain injuries. Since the landmark finding reported in 2007 found that hydrogen reduced the levels of peroxynitrite anions and hydroxyl free radicals in ischemic stroke, molecular hydrogen’s antioxidative and anti-inflammatory effects have aroused widespread interest. Due to its excellent antioxidant and anti-inflammatory properties, hydrogen therapy via different routes of administration exhibits great therapeutic potential for a wide range of brain disorders, including Alzheimer’s disease, neonatal hypoxic-ischemic encephalopathy, depression, anxiety, traumatic brain injury, ischemic stroke, Parkinson’s disease, and multiple sclerosis. This paper reviews the routes for hydrogen administration, the effects of hydrogen on the previously mentioned brain disorders, and the primary mechanism underlying hydrogen’s neuroprotection. Finally, we discuss hydrogen therapy’s remaining issues and challenges in brain disorders. We conclude that understanding the exact molecular target, finding novel routes, and determining the optimal dosage for hydrogen administration is critical for future studies and applications.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Background
As the smallest gas molecule and the world’s most abundant element, molecular hydrogen comprises two electrons and two protons held together by a non-polar covalent molecule [1]. Hydrogen first showed its therapeutic effects in a mouse squamous cell carcinoma model after 2 weeks of hyperbaric hydrogen therapy [2]. In 2007, the antioxidative effect of hydrogen was demonstrated and aroused increasing concern by reducing cytotoxic oxygen radicals [3]. In the same study, the brain injury was significantly attenuated by inhaled hydrogen through buffering the cytotoxic oxidative stress-induced oxidative damage in an acute ischemia/reperfusion injury rat model [3]. Following this important finding, extensive work reported excellent anti-inflammatory effects and antioxidative stress of hydrogen in various brain disorders [1].
Oxidative stress is indispensable in aging and various neurological disorders [4]. In Alzheimer’s disease (AD), oxidative stress has been considered an early and essential pathogenic operator of AD [5]. In both transgenic and non-transgenic AD animal models, the reactive oxygen species (ROS) induce oxidative damage to proteins, lipids, and nucleic acids generating protein carbonyls, lipid peroxides, and DNA/RNA modifications [5,6,7]. ROS-induced oxidative damage contributes to neuronal degeneration in the cortex and hippocampus [5,6,7]. Similarly, oxidative stress contributes to the damage and degeneration of dopaminergic neurons in Parkinson’s disease (PD) [8]. The dysfunction of redox potential disrupts the normal function of essentially biological processes and finally leads to the loss of dopaminergic neurons [8, 9]. Additionally, substantial evidence suggests that oxidative stress is implicated in multiple brain injuries, including ischemic stroke, traumatic brain injury (TBI), and neonatal hypoxic-ischemic encephalopathy (HIE) [7, 10, 11]. For instance, excessive ROS contributes to secondary injury after TBI, and targeting the ROS generation attenuates secondary brain injury and inhibits epilepsy [12, 13]. Similar results are found in HIE and global cerebral ischemia [10, 14]. These findings suggest that oxidative stress constitutes essential pathophysiology in multiple brain diseases and is recognized as an important target for the prevention and treatment of brain disorders [7].
Neuroinflammation and oxidative stress are intimately related [15]. The excessive release of ROS and reactive nitrogen species (RNS) activates the signaling pathways that induce the activation of glial cells, including microglia and astrocytes [16]. The inflammatory cytokines released by excessively activated glial cells further increase oxidative stress and damage mitochondria. This process induces vicious cycles and maintains the increased or high secretion of proinflammatory cytokines [17, 18]. Like oxidative stress, neuroinflammation is a typical pathology that contributes to the initiation and progression of multiple neurodegenerative diseases and brain injury [10, 11, 19, 20]. According to previous studies, microglia mediate Aβ propagation at the early stage of AD and contribute to the accumulation of Aβ at the following stage [21,22,23]. Additionally, astrocyte, another primary glial cell type, mediates myelin phagocytosis and is implicated in the pathogenesis of ischemic neuronal death [24,25,26]. In the photothrombotic stroke model, the neuroinflammation-induced harsh microenvironment enhances stroke development and inhibits post-stroke recovery [11, 27]. Notably, anti-inflammatory therapy show promise in slowing down the disease progression in the animal model of multiple sclerosis (MS), a neurodegenerative disease characterized by neuroinflammation [28].
In a nutshell, oxidative stress and neuroinflammation are prominent traits of various neurodegenerative diseases and brain injuries [29, 30]. The excellent anti-inflammatory effects and antioxidative stress of hydrogen suggest that hydrogen is a promising therapeutic medical gas for brain disorders [1]. Although hydrogen therapy’s effectiveness and underlying mechanism have witnessed tremendous progress since 2007, the current understanding of the exact underlying mechanism and the optimal administration of hydrogen in neurodegeneration and brain injuries is still limited. This review summarizes the existing routes for hydrogen administration, provides an extensive review of hydrogen in various brain disorders, and discusses the remaining issues and challenges in future studies and applications.
Routes for Hydrogen Administration
Hydrogen Gas Inhalation
Inhalation of hydrogen gas is one of the most commonly utilized hydrogen administration methods [31]. In previous studies, hydrogen in various concentrations has been employed, including 1% [3], 1.3% [32], 2% [3, 33], 2.1% [34], 4% [3], and 66.7% [35]. Inhaled hydrogen gas enters the lungs, diffuses into the alveoli, and then transfers throughout the body via the vascular system [36]. For safety concerns, the most widely studied hydrogen is maintained within 1–4% [36]. The inhaled hydrogen diffuses, transfers rapidly, and responds efficiently to defend against acute oxidative stress [36]. In terms of safety, eight healthy adult participants underwent 2.4% hydrogen for 72 h via a high-flow nasal cannula in a previous study [36]. Promisingly, hydrogen inhalation does not cause adverse effects, suggesting that hydrogen administration via gas inhalation is safe and well tolerated. However, a protocol with a higher concentration and longer time should be analyzed in the future [36].
Hydrogen-Rich Water Drinking
Although gas inhalation is one of the most straightforward approaches for hydrogen administration, it is not practical for continuous hydrogen therapy or preventive use [37]. Therefore, in previous studies, hydrogen-rich water has gained much attention [38]. Hydrogen administration via drinking hydrogen-rich water is more portable and safer than hydrogen gas inhalation for preventive use in daily life. The solubility of molecular hydrogen in water is up to 0.8 mM at room temperature under atmospheric pressure [37]. Because molecular hydrogen is a neutral molecule, its solubility is relatively low. Therefore, as reported previously, hydrogen with high pressure (0.4 MPa) was applied to increase the concentration of hydrogen to a supersaturated level and stored in an aluminum bag without dead volume [39]. Notably, the hydrogen-rich water should be stored in an aluminum container rather than a glass or plastic one because hydrogen penetrates the glass or plastic walls of the container rapidly and almost disappears around 8 h [37]. In addition, hydrogen-rich water can be made by electrolysis or by placing a magnesium metal or hydride into drinking water [40]. Although only 41% of hydrogen in hydrogen-rich water can be utilized within the body [41], water with a low concentration of hydrogen is also effective in improving cellular and metabolic processes [39].
Hydrogen-Dissolved Saline Injection
Although hydrogen administration via inhalation and drinking is portable, safe, and easy to use, the intake dose to a specific target area is limited [42]. Hydrogen-dissolved saline injection is an approach that can provide a substantial amount of hydrogen to the affected area. In previous animal studies, intraperitoneal injections of hydrogen-rich saline showed neuroprotective potential in multiple brain disorders [43, 44]. However, because the hydrogen-dissolved saline injection is invasive, it is challenging to be accepted as a way for preventive use or daily hydrogen treatment [42]. Furthermore, frequent injections of hydrogen-dissolved saline have the risk of cross-infection and would be very dangerous if hydrogen-dissolved saline were injected directly into the vein [42].
Primary Mechanism of the Neuroprotective Effect of Hydrogen
Anti-inflammatory Properties
The anti-inflammatory effect of hydrogen is one of the primary reasons for the application of molecular hydrogen in various brain diseases [45,46,47,48]. Usually, inflammation is considered a defense mechanism that protects the tissue from infection and damage and is involved in activating immune cells and releasing inflammatory cytokines [42]. In the brain, neuroinflammation is prevalent in neurodegenerative diseases and a common feature in brain injuries [7]. The overactivation of glial cells and excessive release of inflammatory factors contribute to the initiation of several neurodegenerative diseases (e.g., multiple sclerosis) [7]. They are also essential factors that affect the pathogenesis and progression of brain injury [7]. The anti-inflammatory effects of hydrogen have been widely studied [45,46,47,48]. For example, the increased proinflammatory cytokines, including IL-1β, IL-6, and TNF-α, and the reactive astrogliosis induced by spinal cord injury are attenuated by hydrogen-rich saline injection [49]. In addition, the overactivation of microglia is attenuated, and the microglia polarization is promoted toward the anti-inflammatory M2 phenotype in a middle cerebral artery occlusion model [50]. Similar phenotypes (A1 and A2 astrocytes) are also observed in astrocytes [51]. The M1/M2 microglial polarization affects the transformation of A1/A2 astrocytes [51], indicating that hydrogen has the potential to regulate A1/A2 transformation.
Antioxidant Properties
Since the discovery of hydrogen’s cytoprotective and antioxidant effects in a focal stroke model [3], molecular hydrogen has been widely studied in neuroscience due to its ability to scavenge powerful oxidants in brain diseases [52]. The disequilibrium between the production of free radicals and the endogenous antioxidant defense system is considered one of the essential pathological processes in most brain diseases [7, 10]. The hydrogen worked as an electron donor for RNS and ROS to scavenge ONOO- and ·OH [3]. In most cases, hydrogen only selectively scavenges the excessive hydroxyl radical by reaction of H2 + •OH = H2O + •H. The produced •H is removed by another reaction of •H + O2− = HO2− [53]. It is also possible that •H reacts with •OH to form H2O [54]. Other necessary ROS and RNS for normal signaling regulation are preserved [53]. In addition, the regulation of the KEAP1/NRF2/ARE signaling pathway contributes to the antioxidative effects of hydrogen [55]. Under normal physiological conditions, the KEAP1 binds to NRF2 and modulates NRF2 levels by ubiquitination and proteasomal degradation within the cytoplasm [56]. In response to oxidative stress, the cysteine modification-induced conformational changes in KEAP1 allow NRF2 to evade degradation and escape from KEAP1 trapping [5]. The phosphorylated NRF2 moves from the cytoplasm to the nucleus and binds to ARE, promoting the transcription of detoxification and cytoprotective genes [5]. Hydrogen-rich water, hydrogen gas, and the injection of hydrogen-rich saline activate the KEAP1/NRF2/ARE pathway and promote the antioxidant capacity in various brain diseases, including TBI [57], depression [58], and anxiety [58].
Endogenous Hydrogen Produced by Gut Microbiota
Gut bacteria are one of the primary sources of endogenous hydrogen [59]. Members of the Enterobacteriaceae family, strains of the genus Clostridium, and anaerobic cocci contribute to the most percentage of hydrogen production released by gut bacteria [59]. As mentioned previously, hydrogen gas can reduce the expression of proinflammatory cytokines, neutralize hydroxyl radicals, and exerts cytoprotective effects [3, 51]. The role of endogenous hydrogen in brain disorders has been investigated in neurodegenerative disorders. For example, a previous study found that PD patients lacked Prevotella and Clostridium (hydrogen-releasing bacteria) and compromised gut microbiota was always accompanied by a worse motor ability [59, 60]. This finding indicates that endogenous hydrogen released by gut microbiota is involved in the progression of PD. Similarly, endobacteria-produced hydrogen is fundamental for proper neuronal function, and the changes in endobacteria-produced hydrogen also contribute to AD pathogenesis [61]. Although there are relatively few studies regarding the role of endogenous hydrogen produced by gut microbiota in brain disorders, the findings mentioned previously suggest that exogenous hydrogen may represent a potential agent for neurodegenerative diseases and brain injury.
The solubility of hydrogen depends on temperature and pressure. As mentioned previously, the solubility of molecular hydrogen in water is up to 0.8 mM at room temperature under atmospheric pressure [37]. The hydrogen solubility in the plasma and the blood is around 6.44 μmol/L/kPa at 37 °C [62]. The hydrogen solubility and the half-life of hydrogen in the body fluids are significant for the endogenous/exogenous hydrogen conferring its neuroprotective and practical application. However, the hydrogen solubility in cerebrospinal fluid and the half-life of hydrogen in various body fluids remain unclear and deserve further investigation.
Molecular Hydrogen Therapy in Alzheimer’s Disease
As one of the most common forms of dementia in the elderly, AD affects more than 6 million individuals in the USA and more than 50 million worldwide [6, 63]. It is characterized by progressive cognitive decline, extracellular β-amyloid plaques, intracellular neurofibrillary tangles, neuroinflammation, mitochondrial dysfunction, and oxidative stress [6]. In AD, the Aβ accumulation and tau pathology contribute to mitochondrial function, leading to the excessive production of reactive oxidative species and ATP depletion, which promote Aβ deposition and tau hyperphosphorylation [6]. Evidence indicates that maintaining the cellular redox balance is essential for AD prevention and therapy [6, 64].
A growing number of studies reveal that hydrogen possesses antioxidant and anti-inflammatory effects, indicating its therapeutic potential in AD [1, 65]. In a rat model of Aβ intracerebroventricular injection-induced AD, hydrogen-rich saline exhibited its beneficial effect by significantly improving spatial learning and memory [66]. Moreover, long-term potentiation, an essential process in the context of synaptic plasticity supporting learning and memory functions, was impaired in the Aβ-treated group. However, interestingly, hydrogen-rich saline significantly enhanced long-term potentiation and improved the synaptic information storage processes [66]. Hydrogen’s beneficial role in AD is inseparable from its antioxidant and anti-inflammatory effects. For instance, Aβ intracerebroventricular injection induces pronounced lipid peroxidation, oxidative DNA damage, and neuroinflammation, which were alleviated by hydrogen-rich saline treatment [66, 67]. Furthermore, previous studies have indicated that inhibiting the c-Jun N-terminal kinases signaling pathway, an imperative in stress signaling pathways implicated in neuronal plasticity, neuronal degeneration regeneration, and cellular apoptosis, is a potential approach for AD treatment [68]. Intriguingly, the hydrogen inhibits the JNKs pathway, indicating that the therapeutic effects of hydrogen in AD may be mediated by the inhibition JNK pathway [68].
Moreover, the gender-dependent neuroprotective effect is found in a transgenic AD mouse model. As reported by a previous study, hydrogen-rich water administration only improves cognitive function in the female APP/PS1 mice without affecting male transgenic AD mice [69]. Consistent with this finding, the hydrogen-rich water attenuated oxidative stress and neuroinflammation. However, these effects are more profound in female AD mice than males [69]. Further analysis suggested that the sex-specific effects of hydrogen may rely on the changes in brain estrogen levels, estrogen receptor beta (ERβ), and the brain-derived neurotrophic factor (BDNF) [69]. The estrogen levels, Erβ, and BDNF levels in the female AD mice decreased, but no apparent changes are detected in male AD mice [69]. Although the change of BDNF in the male AD mice is inconsistent with other studies and needs further analysis in this mouse model [70, 71], the hydrogen reverses the changes in estrogen levels, Erβ, and BDNF levels. These findings suggest that the gender-dependent neuroprotective effect of hydrogen may partly depend on the activation of ERβ-BDNF signaling in AD pathogenesis [69].
Furthermore, an in vitro study using cultured human neuronal cells confirms the neuroprotective role of hydrogen [72]. For example, H2O2 exposure induces excessive hydroxyl radicals in human neuroblastoma, which is ameliorated prominently by hydrogen [72]. Notably, in the in vitro model of AD, the Aβ-induced cellular apoptosis is significantly attenuated in the hydrogen-rich cell culture medium [72]. The mechanistic study found that the activation of the AMPK/SIRT1/FOXO3a signaling pathway contributes to the protective effect of hydrogen [72]. Worked as a critical molecular sensor and modulator, AMPK is involved in the anti-aging signaling network. Similar to AMPK, the Sirt1-FoxO3a axis is a well-studied pathway that responds to oxidative stress and favors cell survival [72, 73].
Molecular Hydrogen Therapy in Neonatal Hypoxic-Ischemic Encephalopathy
Neonatal hypoxic-ischemic encephalopathy (HIE) is one of the most common but severe brain injuries that result in a high morbidity and mortality rate [10]. The HIE occurs when the neonatal brain is deprived of the blood and oxygen [10]. The impaired cerebral blood flow and oxygen deprivation lead to mitochondrial dysfunction, ATP depletion, oxidative stress, cellular damage, and neuronal apoptosis [74]. The progression of HI injury can be divided into primary and secondary energy failure [75]. The primary energy failure occurs when the hypoxic-ischemic insults initiate. When the ATP is deprived rapidly due to the decreased oxidative phosphorylation, the neurons change to anaerobic metabolism, resulting in lactic acid and hypoxanthine accumulation [75]. The rapid depletion of ATP causes the failure of numerous essential process that maintains cellular integrity, particularly the failure of sodium/potassium pumps. Next, the failure of Na/K pumps and the accumulation of metabolites induce depolarization of neurons, followed by excessive release of excitatory amino acids on the extracellular side and an additional influx of sodium and calcium [75, 76]. These changes finally induced cellular edema and early cell apoptosis.
Moreover, the primary energy failure-induced changes contribute to the secondary energy failure phase several hours to days after ischemia and hypoxia after the initial injury. During this process, mitochondrial dysfunction, excessive production of free radicals, and increased inflammation are involved in secondary energy failure and contribute to late cell death [75, 76]. Mitochondrial dysfunction and oxidative stress are crucial in brain damage following hypoxia and ischemia [77]. Although the low concentrations of reactive oxygen species ROS and RNS worked as signaling molecules under physiological conditions, excessive free radicals induced oxidative damage to DNA, protein, and lipids during hypoxic-ischemic injury [77]. However, the mitochondria changes and oxidative damage contribute to the secondary phase of injury after ischemia and hypoxia, which induces persistent inflammation and mitochondrial dysfunction, exacerbating the neuronal injury [78].
Evidence suggests antioxidant treatments may alleviate neuronal damage following hypoxia–ischemia [78]. As mentioned previously, molecular hydrogen is one such antioxidant therapy for HIE [79]. In a neonatal HIE rat model, hydrogen therapy with 2% hydrogen is employed immediately following the hypoxic-ischemic insult. Intriguingly, hydrogen inhalation significantly reduced the infarct brain area and alleviated neuronal apoptosis time dependently in the cortex and hippocampus [80]. The neuroprotective effects of the hydrogen can be detected even with 30-min hydrogen inhalation [80]. As reported in the same study, 30-min hydrogen therapy significantly inhibits the activity of caspase-3 and caspase-12. Notably, caspase-12 is a vital regulator of ER stress-induced cellular apoptosis. It is proteolytically activated under ER stress-induced cellular apoptosis [81]. Therefore, the inhibitive effect of hydrogen on caspase-12 suggests the potential role of hydrogen in alleviating ER stress-induced apoptosis [80]. Another work using 3% hydrogen supports the neuroprotective effects of hydrogen in neonatal rats following HI insults [82]. The motor deficits and impaired learning and memory function were significantly attenuated by 3% hydrogen [82]. Moreover, similar to hydrogen therapy with 2% hydrogen, hydrogen therapy with 3% also exhibited a time-dependent neuroprotective effect, showing the most significant protective result with 90-min hydrogen inhalation, compared to 30-min and 60-min therapy immediately after HI insults [82]. Notably, hydrogen therapy may have a neuroprotective time window in HIE treatment [82]. For example, the protective effect of 90-min hydrogen therapy decreased if the treatment is initiated 12 h after HI insults and nearly disappeared 24 h following injury [82]. In vitro study further confirmed the neuroprotective role of hydrogen in the oxygen–glucose deprivation/reperfusion (OGD/R) model using cultured PC12 cells [82]. The OGD/R-induced PC12 apoptosis was prominently attenuated by 90 min H2 therapy, and the protective role of hydrogen is abolished by heme oxygenase-1 (HO-1) knockdown. HO-1 is a critical enzyme in protecting against oxidative stress [83]. A further mechanistic study supports the MAPK/HO-1/PGC-1α pathway as one of the possible molecular mechanisms underlying hydrogen therapy’s anti-inflammatory, anti-apoptotic, and antioxidative effects [82].
Therapeutic hypothermia is a standard treatment for HIE. However, hypothermia has limited efficacy, and nearly 50% of the neonates receiving the treatment still suffer from severe disability and death [10]. However, hydrogen can enhance the therapeutic benefits of hypothermia therapy against HI insults [84]. In a neonatal HI piglet model, hydrogen ventilation combined with mild hypothermia is administered 24 h after HI insults [84]. Interestingly, the combination therapy with hypothermia and hydrogen significantly alleviates the neurological deficits, and animals with hypothermia therapy alone only displayed a tendency for improvement [84]. Although hypothermia therapy alone cannot alleviate cellular apoptosis in the dorsal cortex, combination therapy with hypothermia and hydrogen ventilation significantly reduced the cellular apoptosis induced by HI insults [84].
However, hydrogen does not always show its neuroprotective effect in HIE. In addition to the treatment duration and therapeutic time window, the results of hydrogen therapy may also depend on the disease severity. For example, a previous study finds that post-hydrogen treatment with 2.9% hydrogen inhalation is ineffective in protecting neonates against moderate and severe neonatal HIE [85]. No significant changes are detected in the infarct volume and the lipid peroxidation marker following hydrogen therapy in HIE [85]. Unlike other preconditioning or pretreatment regimens, hydrogen preconditioning exacerbates the HI insult-induced brain damage, suggesting hydrogen therapy may only work as a treatment rather than a pretreatment approach to improve resistance to hypoxic-ischemic insults [85].
Molecular Hydrogen Therapy in Depression and Anxiety
Growing evidence suggests that oxidative stress and neuroinflammation are pivotal in the pathogenesis of depression and anxiety [86]. Patients or animals with depression or anxiety display increased neuroinflammation and oxidative stress in the brain and the periphery systems [87,88,89]. Consistent with this, studies found that pharmacological agents that induce oxidative stress in rats cause anxiety-like behavior and depression [90,91,92], suggesting that antioxidants may be a potential treatment approach for depression and anxiety [91, 93].
Hydrogen is a potential therapeutic gas for depression and anxiety due to its antioxidative, anti-inflammatory, and anti-apoptotic effects [42]. In a previous study, water or hydrogen-rich water is supplied to the animal at the commencement of the chronic unpredictable mild stress (CUMS) procedure [94]. Rats receiving a 4-week CUMS procedure exhibited apparent depressive-like behavior, including decreased sucrose preference and extended immobility [94]. Interestingly, hydrogen-rich water significantly prevents the CUMS-induced depressive-like behavior [94]. Further studies found significantly decreased IL-1β levels, caspase-1 activity, and ROS production in the hippocampus and prefrontal cortex [94]. The caspase-1 is a cysteine protease that promotes the secretion of proinflammatory, including IL-1β [95]. The inhibited activation of caspase-1 in the hydrogen-treated group alleviates neuroinflammation and contributes to the attenuation of oxidative stress [94].
Repeated inhaling of high concentrations of hydrogen can also increase stress resilience [58]. For example, the mice receiving 1-h or 3-h daily hydrogen treatment with high concentrations (67%) for 14 days exhibit increased resilience to acute and chronic stress-induced anxious-depressive-like behavior [58], as evidenced by decreased immobility in the tail suspension and forced swimming test, improved novelty-suppressed feeding, and increased time spent in the central zone assed by the open field test [58]. Consistent with the previously mentioned time-dependent neuroprotective effect in HIE, mice receiving 3-h daily hydrogen are more resilient to acute stress [58]. The hypothalamic–pituitary–adrenal (HPA) axis is an indispensable adaptive neuroendocrine system in stress resilience and vulnerability [48]. Repeated inhalation of hydrogen significantly inhibited the chronic stress-induced changes of corticosterone and adrenocorticotropic hormone [48], the major hormones contributing to the HPA axis’s response to stress [96, 97]. In line with the previous study, chronic stress-induced neuroinflammation is alleviated by hydrogen inhalation [48]. More interestingly, hydrogen inhalation-induced resilience to acute stress in adolescence can be detected in early adulthood, suggesting the benefits of hydrogen have long-lasting effects [48].
Molecular Hydrogen Therapy in Multiple Sclerosis
MS is a chronic and progressive inflammatory disease characterized by mistaken attacks of reactive inflammatory cells against the brain tissue, resulting in pronounced demyelination, axonal degeneration, and extensive inflammation [98]. Indeed, multiple inflammatory cells play a pathogenic role in MS, including CD4 and CD8 T lymphocytes, macrophages, and microglia [99]. Currently, the most widely accepted concept of MS lesion formation is that acute demyelination is induced by phagocytes that internalize and degrade myelin sheaths with infiltrating T cells. The recruitment of inflammatory cells is an early or initial event in MS progression. Other studies proposed that extensive apoptosis of oligodendrocytes and excessive activation of glial cells are the major pathological changes in the newly formed MS lesion [100, 101]. Therefore, inflammation-related changes play a central role in the initiation and progression of MS [100]. The inflammatory changes in MS also induce the exaggerated expression and release of reactive oxygen species [100]. For example, the activation of microglia and infiltration of macrophages can contribute to the release of large amounts of oxidative stress-related molecules, including superoxides, nitric oxide, hydrogen peroxide, and hydroxyl radicals [100]. In addition, numerous studies revealed the association between oxidative stress and MS lesion formation [100]. For example, prominent immunoreactivity for oxidized DNA and lipids was found in the area of initial demyelination, suggesting the essential role of oxidative stress in the early stages of MS [102]. Notably, in an animal model of MS, experimental autoimmune encephalomyelitis (EAE) rats treated with ROS scavengers significantly alleviated the autoimmune inflammatory lesions, indicating that therapeutic manipulation of oxidative stress may be a potential approach for the treatment of MS [103]. Many currently used MS medications are expensive and often have side effects [104], which prompted efforts to identify novel therapies for MS patients.
Molecular hydrogen’s anti-inflammatory and antioxidant properties have attracted increased research attention and interest in MS prevention and treatment [104]. In the experimental autoimmune encephalomyelitis model, one of the most widely used animal models for MS, hydrogen-rich water orally twice a day significantly delayed the initiation and attenuated the severity EAE and attenuated the severity of EAE, as evidenced by prominently improved clinical scores in the hydrogen-treated group [104]. Further analysis confirmed the prophylactic and therapeutic effects of hydrogen on demyelination following hydrogen administration [104]. Notably, the effects of hydrogen on the disease severity are exhibited in a dose-dependent manner within a specific range [104]. Consistent with the previous study, another study using the EAE animal model also found the effects of hydrogen on alleviating the severity of EAE and demyelination [46]. Intriguingly, the expression of CNPase, a myelin-associated enzyme, is preserved by hydrogen treatment, indicating that hydrogen administration inhibited myelin-associated changes in EAE [46]. In addition, hydrogen-rich saline inhibits glial activation and reduces the levels of inflammatory cytokines (i.e., TNF-α, IL-1β, IL-6, and HMGB1), suggesting that the anti-inflammatory effects were involved in alleviating disease severity [46]. Furthermore, the antioxidant capacity also contributed to the beneficial effects of the hydrogen in MS [46]. Hydrogen-rich saline improved the activity of antioxidant enzymes and reduced the generation of oxidative stress-induced lipid peroxidation and oxidative DNA damage, confirming the antioxidant effect of hydrogen in MS [46]. Further analysis found that the activation of the NRF2-ARE signaling pathway was responsible for the upregulated antioxidant capacity, and the NRF2 inhibitor abolished the improved antioxidant capacity [46].
Molecular Hydrogen Therapy in Parkinson’s Disease
As the second most common neurodegenerative disease, Parkinson’s disease (PD) is characterized by motor dysfunction and non-motor symptoms, including sleep disturbances, depression, and constipation [105]. The pathological hallmarks of PD include the loss of dopaminergic neurons in the substantia nigra pars compacta (SNc) and the abnormal intracellular accumulation of misfolded and aggregated α-synuclein-induced Lewy bodies [106]. Like other neurodegenerative diseases, oxidative stress and inflammation play an indispensable role in PD’s generation and progression [7]. Several lines of evidence suggest that pathological changes in dopamine metabolism, inflammation, and mitochondrial dysfunction contribute to oxidative stress and damage in PD [107].
According to previous studies, dopamine metabolism disruption is one of the sources of oxidative stress in PD [108]. Under normal conditions, dopamine is generated from tyrosine by aromatic amino acid decarboxylase and tyrosine hydroxylase and then stored in synaptic vesicles. However, under pathological changes, the cytosolic dopamine in damaged neurons is metabolized by auto-oxidation and monoamine oxidase, generating excessive ROS [108, 109]. Mitochondria dysfunction is another primary source of oxidative stress in PD. For example, complex I deficiency has been detected in the substantia nigra pars compacta of PD patients and results in unfavorable neuronal apoptosis [110]. As one of the most widely used neurotoxicant inducers of PD, 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) is a complex I inhibitor [107]. MPTP can be transferred into MPP+ by monoamine oxidase-B and accumulated in the dopaminergic neurons of SNc, which result in ROS release by the mitochondria, including hydroxyl radicals, hydrogen peroxide, nitric oxide, and superoxide anion [108]. Moreover, mitochondria-associated gene mutations affect mitochondrial function and structure [111]. For example, parkin mutations impair mitochondrial complex I activity [112]. However, parkin overexpression attenuates dopamine neuronal loss induced by MPTP through mitochondrial protection and alleviates α-synuclein aggregation [113]. These findings indicate that mitochondria-related changes and excessive oxidative stress are essential in neuronal dopamine loss and PD progression [108].
Due to the prominent role of excessive oxidative stress in the pathological changes of PD, increasing studies detected the potential therapeutic effects of antioxidants in PD treatment [114]. As mentioned previously, hydrogen is one of the most potent natural antioxidants. It has recently garnered heightened attention due to its antioxidant and anti-inflammatory properties [115, 116]. In an MPTP-induced PD model, hydrogen-rich water alleviated acute MPTP administration-induced neurotoxicity, as evidenced by a significant attenuation in the loss of dopaminergic neurons in substantia nigra pars [115]. Moreover, hydrogen-rich water significantly improved chronic MPTP administration-induced behavioral deficits [115]. Notably, the mechanism underlying the beneficial effects of hydrogen-rich water is involved in alleviating oxidative stress. For example, the ROS-derived oxidative products, including 8-oxoguanine (a marker of DNA damage) and 4-hydroxynonenal (a marker of lipid peroxidation), were inhibited by hydrogen-rich water, confirming the antioxidant effects of hydrogen in the PD [115]. Similarly, the effect of hydrogen-rich water in the PD animal model was also dose dependent, with a better effect at a lower concentration than the saturated concentration [115]. The dose-dependent effects of hydrogen are also confirmed in clinical trials. For example, PD patients who received 1000 mL of hydrogen water per day for 48 weeks significantly improved Parkinson’s features [117]. However, a more extended hydrogen water treatment did not show any effects in patients with PD [118]. The negative results of hydrogen therapy are also found in one of the clinical trials using inhaled hydrogen [119], indicating that the duration, concentration, and routes for hydrogen administration are essential in hydrogen therapy. Interestingly, a Si-based agent that can generate hydrogen continually further enriched the routes of hydrogen therapy [120]. In a 6-hydroxydopamine (6-OHDA)-induced PD mouse model, Si-based agent treatment alleviated dopaminergic neurodegeneration and ameliorated 6-OHDA-induced behavioral impairment, suggesting Si-based agent that continues generating molecule hydrogen may be a potential approach to treat PD [120].
Molecular Hydrogen Therapy in Ischemic Stroke
Stroke is one of the most common cerebrovascular diseases with high morbidity and disability rate [121]. It can be divided into ischemic and hemorrhagic strokes [121]. Ischemic stroke accounts for 85% of total strokes and occurs when blood vessels are blocked by a blood clot or other particles [122]. When an ischemic stroke occurs, initial inflammation is triggered by cellular debris and dying cells in the ischemic area [123]. Increasing evidence indicates that postischemic inflammation exacerbates brain injury and contributes to the secondary damage of neurons [124].
Oxidative damage is another specific change induced by ischemic insults [125]. Rapid production of ROS following acute ischemic stroke overwhelms the antioxidant capacity, causing further brain damage [125]. Excessive oxidative stress damaged cellular macromolecules and led to autophagy, cellular apoptosis, and necrosis [125]. Furthermore, the rapid and prolonged reperfusion leads to the second burst of ROS generation and contributes to reperfusion-induced secondary neuronal damage [7, 125]. Neuroinflammation and oxidative stress in ischemic stroke are interactive and play an essential role in ischemic/reperfusion-induced injury [126]. Substantial evidence suggests that approaches targeting neuroinflammation and oxidative stress are potential treatment options for ischemic stroke [126].
The antioxidative and anti-inflammatory effects of molecular hydrogen have attracted increasing attention in ischemic stroke over the past few years [127]. In a previous study, the primary culture of neocortical cells underwent oxygen–glucose deprivation (OGD) followed by reperfusion with a cell medium containing normal O2 and glucose was employed to mimic ischemia–reperfusion injury [3]. The cell culture model with ischemia–reperfusion induced prominently increased hydroxyl radicals (.OH), the most reactive oxygen species [3]. Intriguingly, the cell culture medium with dissolved molecular hydrogen notably alleviated this increase and promoted neuronal vitality and survival, indicating that molecular hydrogen protects neurons against oxidative stress-induced neuronal death [3]. Furthermore, ROS was generated in a rat model of focal ischemic stroke and caused apparent brain injury. Notably, hydrogen inhalation exhibited concentration-dependent protection on this brain injury, as evidenced by the most substantial reduction in infarct volume with hydrogen inhalation at 2–4% [3]. Of particular interest, hydrogen only exhibited its neuroprotective effect when the treatment is employed during reperfusion and had no significant effect on infarct volume during ischemia, indicating that hydrogen therapy has its “therapeutic window” [3]. In addition, the protective effects of hydrogen on the brain injury were not only limited to the initial stage (ischemia and perfusion). After the ischemic-perfusion insults, hydrogen also alleviated its progressive damage, as evidenced by significant improvement in the functional behavioral assessment and suppression of oxidative stress and inflammation [3]. Similar findings are found in the global cerebral ischemia and reperfusion mouse model [32]. Mice subjected to global cerebral ischemia and reperfusion exhibited neurological deficits and severe neuronal injury in the hippocampal CA1 region, which are significantly reduced in the hydrogen treatment group [32]. This neuroprotection of hydrogen involvs the alleviation of oxidative DNA damage, lipid peroxidation, and postischemic autophagy in the CA1 region [32].
Another study clarified another possible mechanism underlying hydrogen neuroprotection using OGD/reperfusion damaged hippocampal neurons [128]. Similarly, the study found that hydrogen significantly alleviated ROS levels and cellular apoptosis following OGD/R insults. However, the hydrogen was provided by a cell culture incubator consisting of 60% hydrogen, suggesting neuroprotection of hydrogen can be achieved through different hydrogen administration routes [128]. Notably, the decreased mitochondrial membrane potential induced by OGD/R insults was attenuated by hydrogen therapy, indicating that hydrogen protects against mitochondrial dysfunction [128]. However, the neuroprotective effect of hydrogen is lost entirely when the mitophagy inhibitor is added, suggesting that mitophagy may mediate the improved mitochondrial function and the neuroprotective effects of hydrogen [128]. Further analysis detects the activation of the PINK1/Parkin pathway, a well-understood mitophagy-associated pathway, which confirmed the essential role of mitophagy in hydrogen therapy [128]. Although further studies are still needed on how hydrogen affects the expressions of mitophagy-related proteins, these findings provided one of the possible mechanisms underlying the neuroprotection of hydrogen.
Molecular Hydrogen Therapy in Traumatic Brain Injury
Traumatic brain injury (TBI) is one of the leading causes of disability and mortality among people of all age groups [129]. More than 50 million people suffer from TBI yearly, and almost 50% of the world’s population will experience mild TBI more than once [130]. TBI occurs when an external force causes damage to the head, including closed head injury induced by a blow, jolt, or bump to the head and penetrating injury induced by objects penetrating the skull [131]. TBI-induced injury includes primary and secondary injuries [132]. Primary injury after TBI includes direct mechanical damage to brain tissue and blood vessels, causing neuronal loss and necrotic cell death [133]. Following the primary injury, the secondary injury further damages the brain tissue, which occurs seconds to minutes following the primary injury, involving various biochemical processes, including oxidative stress, inflammation, mitochondrial dysfunction, and blood–brain barrier (BBB) disruption [133].
Mitochondrial dysfunction and oxidative stress are typical changes contributing to secondary cell injury in TBI [7]. Excessive release of ROS and RNS following TBI causes oxidative damage to the cell, including lipoperoxidation of the cell membranes, various organelles, and microstructures within neurons. These changes result in widespread neuronal injury and death [134]. Notably, the oxidative stress-induced lipid peroxidation of mitochondrial membranes led to the disruption of mitochondrial function [134]. In addition to serving as the powerhouse of the neurons, mitochondria are essential in maintaining calcium homeostasis [135]. The impaired mitochondrial function and disrupted intracellular calcium homeostasis contribute significantly to the pathophysiology of delayed neuronal damage and death following TBI [136]. Therefore, mitochondria-targeted antioxidants or antioxidant therapies have attracted considerable efforts in TBI studies [7, 137].
Several studies have reported that hydrogen’s anti-inflammatory and antioxidative effects are implicated in protecting against TBI [45, 47]. In a controlled cortical impact model, 2% hydrogen therapy was employed from 30 min to 5 h following TBI [47]. Interestingly, TBI animals with hydrogen therapy exhibited a better neurological outcome, improving BBB integrity, and attenuating cerebral lesion volume and brain edema [47]. Further analysis found that hydrogen significantly enhanced the activities of antioxidant enzymes and alleviated the release of oxidative products [47]. Notably, the microRNA-21 inhibitor inhibited these beneficial changes, indicating that miR-21 is critical in hydrogen therapy following TBI [47]. Another study demonstrated the beneficial effects of hydrogen-rich saline in ameliorating early brain injury in a TBI animal model [57]. The hydrogen-rich saline is administered for 72 h following TBI. Intriguingly, neurological deficits, brain edema, neuronal necroptosis, neuroinflammation, and oxidative stress are significantly alleviated in the hydrogen-treated groups [57]. Notably, hydrogen modulated necroptosis via the regulation of the ROS/HO-1 signaling pathway, suggesting the neuroprotective effects of hydrogen partly depend on ROS/heme oxygenase 1 (HO-1) signaling pathway-regulated necroptosis [57]. The HO-1 is an antioxidant enzyme downstream of the Nrf2 pathway [138]. In the TBI animal model, hydrogen-rich water promotes the disassociation of Nrf2 from Keap1, resulting in the Nrf2 nuclear translocation followed by binding to the antioxidant response element (ARE) and producing endogenous antioxidant enzyme, including HO-1 [55]. This finding suggests that the modulation of the Keap1/Nrf2/ARE signaling pathway contributes to the antioxidative effects of hydrogen and is imperative to protect against TBI [55]. Similar to the findings in other brain diseases, the beneficial effects of hydrogen on TBI are also concentration-dependent [139]. For example, hydrogen therapy alleviated cerebral lesions following TBI in a concentration-dependent manner, wherein 4% hydrogen exhibites better effects in preserving brain tissue than 1% and 2% hydrogen [139], suggesting hydrogen therapy exists the optimal dosage, which needs more study.
Remaining Issues and Challenges
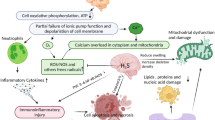
As mentioned previously, hydrogen therapy with various administration routes has shown great potential for neuroprotection in brain disorders, including neurodegenerative diseases [66, 67, 140], brain injury [32, 57, 82], and mental diseases [58, 94] (Fig. 1). However, several unresolved issues still need to be addressed.

Primary mechanisms of molecular hydrogen therapies in brain disorders. Nearly all neurodegenerative diseases, brain injuries, and mood disorders are characterized by mitochondrial function and neuroinflammation. The dysfunction of mitochondria induces excessive production of ROS and the excessively released inflammatory cytokines, which further damage mitochondria and active glial cells. All these changes induce oxidative damage to DNA, protein, and lipids, which finally induce cellular damage, neuronal degeneration, and neuronal loss. However, hydrogen works as an electron donor to selectively scavenge the excessive hydroxyl radical to reduce oxidative stress directly. Additionally, hydrogen enhances the activity of antioxidant enzymes, reduces the release of proinflammatory factors, and activates other neuroprotective pathways
The Exact Molecular Targets and Mechanisms Remain Unclear
Although hydrogen’s antioxidant and anti-inflammatory properties have been widely studied in various brain disorders [32, 57, 66, 67, 82, 140], the crosstalk among anti-inflammation, anti-oxidation, and other pathways are still unclear. Additionally, although studies have found the protective effects of hydrogen on mitochondria and the regulation of various genes and proteins, further investigation is needed to determine whether these modulations are the cause or the results of the hydrogen’s anti-inflammatory and antioxidant effects [141].
No Consensus on Hydrogen Administration and Dose Regimes
As mentioned previously, various routes of hydrogen administration have shown their preventive and therapeutic effects in various brain diseases [32, 57, 66, 67, 82, 140]. The neuroprotective effects of hydrogen therapy in brain disorders are displayed in a dose-dependent, gender-dependent, and time-dependent manner [69, 80, 104]. However, there is no consensus on the optimal hydrogen administration and dose regimes [142]. Therefore, more studies are needed to further our understanding of the optimal routes for hydrogen administration, dosage, pharmacokinetics, toxicity, and biology in animal and clinical studies [142].
New Routes for Hydrogen Administration Are Needed
Although hydrogen administration via drinking or injection has shown its advantages in various brain diseases [32, 57, 66, 67, 82, 140], the solubility of molecular hydrogen in water and saline is relatively low [37]. Additionally, the storage of hydrogen-rich water or saline needs a specific container. For example, aluminum containers retain hydrogen longer than glass and plastic containers [37]. Moreover, the daily hydrogen-rich saline injection has the risk of infection and would be very dangerous if hydrogen-dissolved saline were injected directly into the vein [42]. Therefore, new routes for hydrogen administration are needed. For example, in previous studies, a Si-based agent that continues generating molecule hydrogen has been developed and exerts neuroprotective effects in a mouse model of Parkinson’s disease [120]. The new routes for hydrogen administration should have the following advantages: portable, easy to store, and reach a specific target area with expected concentration and release hydrogen continuously. Nanoparticles for hydrogen generation may hold great potential for hydrogen utilization in preventing or treating brain diseases [143].
The Effects of Hydrogen on Neurovascular Coupling Remains Unclear, and the Half-Life of Hydrogen in Bio-fluids Deserves Further Investigation
Neurovascular coupling is defined as a tight coupling between cerebral blood flow and neural activity, which is intrinsically regulated by a complex interplay between neurophysiological and hemodynamic signals [144]. The impaired neurovascular coupling has been detected in various brain disorders, including neurodegenerative disease, brain injury, and psychiatric disorders [145,146,147]. However, the effects of hydrogen on neurovascular coupling in physiology and pathological conditions remain unclear. Additionally, to investigate the effects of hydrogen in physiology and pathological conditions, we should know the half-life of hydrogen in the bio-fluids, including blood, plasma, and cerebrospinal fluid. Therefore, this issue should also be addressed in future studies.
Conclusions
As mentioned previously, the biological properties of hydrogen, especially the antioxidant and anti-inflammatory effects, make it a promising candidate for various neurodegenerative diseases and brain injuries (Table 1) [45,46,47, 114, 127]. However, the short biological half-life and the low saturation make the administration complex [37, 42]. Additionally, the mechanisms underlying the neuroprotection of hydrogen, especially the exact molecular targets and mechanism, remain unclear [141]. Therefore, more preclinical studies are needed to understand the pathways and the biological changes impacted by molecular hydrogen therapy. Moreover, the optimal concentration, the mode of administration, pharmacokinetics, biology, and the clinical application of hydrogen are still lacking [45]. Nevertheless, it has been suggested that hydrogen has great therapeutic potential in treating various brain disorders [141].
Data Availability
Not applicable.
Abbreviations
- AD :
-
Alzheimer’s disease
- PD :
-
Parkinson’s disease
- TBI :
-
Traumatic brain injury
- HIE :
-
Neonatal hypoxic-ischemic encephalopathy
- ROS :
-
Reactive oxygen species
- RNS :
-
Reactive nitrogen species
- EAE :
-
Experimental autoimmune encephalomyelitis
- MPTP :
-
1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine
- OGD/R :
-
Oxygen-glucose deprivation (OGD)/reperfusion
- BBB :
-
Blood-brain barrier
References
Tan X, Shen F, Dong WL, Yang Y, Chen G (2018) The role of hydrogen in Alzheimer’s disease. Med Gas Res 8(4):176–180. https://doi.org/10.4103/2045-9912.248270
Dole M, Wilson FR, Fife WP (1975) Hyperbaric hydrogen therapy: a possible treatment for cancer. Science 190(4210):152–154. https://doi.org/10.1126/science.1166304
Ohsawa I, Ishikawa M, Takahashi K, Watanabe M, Nishimaki K, Yamagata K, Katsura K, Katayama Y et al (2007) Hydrogen acts as a therapeutic antioxidant by selectively reducing cytotoxic oxygen radicals. Nat Med 13(6):688–694. https://doi.org/10.1038/nm1577
Singh A, Kukreti R, Saso L, Kukreti S (2019) Oxidative stress: a key modulator in neurodegenerative diseases. Molecules 24 (8). https://doi.org/10.3390/molecules24081583
Wu C, Yang L, Tucker D, Dong Y, Zhu L, Duan R, Liu TC, Zhang Q (2018) Beneficial effects of exercise pretreatment in a sporadic Alzheimer’s rat model. Med Sci Sports Exerc 50(5):945–956. https://doi.org/10.1249/MSS.0000000000001519
Yang L, Wu C, Parker E, Li Y, Dong Y, Tucker L, Brann DW, Lin HW et al (2022) Non-invasive photobiomodulation treatment in an Alzheimer disease-like transgenic rat model. Theranostics 12(5):2205–2231. https://doi.org/10.7150/thno.70756
Yang L, Youngblood H, Wu C, Zhang Q (2020) Mitochondria as a target for neuroprotection: role of methylene blue and photobiomodulation. Transl Neurodegener 9(1):19. https://doi.org/10.1186/s40035-020-00197-z
Dias V, Junn E, Mouradian MM (2013) The role of oxidative stress in Parkinson’s disease. J Parkinsons Dis 3(4):461–491. https://doi.org/10.3233/JPD-130230
Guo JD, Zhao X, Li Y, Li GR, Liu XL (2018) Damage to dopaminergic neurons by oxidative stress in Parkinson’s disease (Review). Int J Mol Med 41(4):1817–1825. https://doi.org/10.3892/ijmm.2018.3406
Yang L, Dong Y, Wu C, Youngblood H, Li Y, Zong X, Li L, Xu T et al (2021) Effects of prenatal photobiomodulation treatment on neonatal hypoxic ischemia in rat offspring. Theranostics 11(3):1269–1294. https://doi.org/10.7150/thno.49672
Yang L, Tucker D, Dong Y, Wu C, Lu Y, Li Y, Zhang J, Liu TC et al (2018) Photobiomodulation therapy promotes neurogenesis by improving post-stroke local microenvironment and stimulating neuroprogenitor cells. Exp Neurol 299(Pt A):86–96. https://doi.org/10.1016/j.expneurol.2017.10.013
Eastman CL, D’Ambrosio R, Ganesh T (2020) Modulating neuroinflammation and oxidative stress to prevent epilepsy and improve outcomes after traumatic brain injury. Neuropharmacology 172:107907. https://doi.org/10.1016/j.neuropharm.2019.107907
Talley Watts L, Long JA, Chemello J, Van Koughnet S, Fernandez A, Huang S, Shen Q, Duong TQ (2014) Methylene blue is neuroprotective against mild traumatic brain injury. J Neurotrauma 31(11):1063–1071. https://doi.org/10.1089/neu.2013.3193
El Khashab IH, Abdelsalam RM, Elbrairy AI, Attia AS (2019) Chrysin attenuates global cerebral ischemic reperfusion injury via suppression of oxidative stress, inflammation and apoptosis. Biomed Pharmacother 112:108619. https://doi.org/10.1016/j.biopha.2019.108619
Solleiro-Villavicencio H, Rivas-Arancibia S (2018) Effect of chronic oxidative stress on neuroinflammatory response mediated by CD4(+)T cells in neurodegenerative diseases. Front Cell Neurosci 12:114. https://doi.org/10.3389/fncel.2018.00114
Pawate S, Shen Q, Fan F, Bhat NR (2004) Redox regulation of glial inflammatory response to lipopolysaccharide and interferongamma. J Neurosci Res 77(4):540–551. https://doi.org/10.1002/jnr.20180
Hsieh HL, Yang CM (2013) Role of redox signaling in neuroinflammation and neurodegenerative diseases. Biomed Res Int 2013:484613. https://doi.org/10.1155/2013/484613
Chan PH (2001) Reactive oxygen radicals in signaling and damage in the ischemic brain. J Cereb Blood Flow Metab 21(1):2–14. https://doi.org/10.1097/00004647-200101000-00002
Leng F, Edison P (2021) Neuroinflammation and microglial activation in Alzheimer disease: where do we go from here? Nat Rev Neurol 17(3):157–172. https://doi.org/10.1038/s41582-020-00435-y
Yang B, Xu J, Li Y, Dong Y, Li Y, Tucker L, Yang L, Zong X et al (2020) Photobiomodulation therapy for repeated closed head injury in rats. J Biophotonics 13(2):e201960117. https://doi.org/10.1002/jbio.201960117
Yang L, Feng S, Wu C, Yang L (2022) Microglia-mediated Abeta propagation in Alzheimer’s disease. Neurosci Bull. https://doi.org/10.1007/s12264-022-00907-9
d’Errico P, Ziegler-Waldkirch S, Aires V, Hoffmann P, Mezo C, Erny D, Monasor LS, Liebscher S et al (2022) Microglia contribute to the propagation of Abeta into unaffected brain tissue. Nat Neurosci 25(1):20–25. https://doi.org/10.1038/s41593-021-00951-0
Merlo S, Spampinato SF, Caruso GI, Sortino MA (2020) The ambiguous role of microglia in Abeta toxicity: chances for therapeutic intervention. Curr Neuropharmacol 18(5):446–455. https://doi.org/10.2174/1570159X18666200131105418
Shen XY, Gao ZK, Han Y, Yuan M, Guo YS, Bi X (2021) Activation and role of astrocytes in ischemic stroke. Front Cell Neurosci 15:755955. https://doi.org/10.3389/fncel.2021.755955
Li M, Li Z, Yao Y, Jin WN, Wood K, Liu Q, Shi FD, Hao J (2017) Astrocyte-derived interleukin-15 exacerbates ischemic brain injury via propagation of cellular immunity. Proc Natl Acad Sci U S A 114(3):E396–E405. https://doi.org/10.1073/pnas.1612930114
Takano T, Oberheim N, Cotrina ML, Nedergaard M (2009) Astrocytes and ischemic injury. Stroke 40(3 Suppl):S8-12. https://doi.org/10.1161/STROKEAHA.108.533166
Zong X, Dong Y, Li Y, Yang L, Li Y, Yang B, Tucker L, Zhao N et al (2020) Beneficial effects of theta-burst transcranial magnetic stimulation on stroke injury via improving neuronal microenvironment and mitochondrial integrity. Transl Stroke Res 11(3):450–467. https://doi.org/10.1007/s12975-019-00731-w
Kanwar JR (2005) Anti-inflammatory immunotherapy for multiple sclerosis/experimental autoimmune encephalomyelitis (EAE) disease. Curr Med Chem 12(25):2947–2962. https://doi.org/10.2174/092986705774462833
Kwon HS, Koh SH (2020) Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes. Transl Neurodegener 9(1):42. https://doi.org/10.1186/s40035-020-00221-2
Chen X, Guo C, Kong J (2012) Oxidative stress in neurodegenerative diseases. Neural Regen Res 7(5):376–385. https://doi.org/10.3969/j.issn.1673-5374.2012.05.009
Ishibashi T (2019) Therapeutic efficacy of molecular hydrogen: a new mechanistic insight. Curr Pharm Des 25(9):946–955. https://doi.org/10.2174/1381612825666190506123038
Nagatani K, Wada K, Takeuchi S, Kobayashi H, Uozumi Y, Otani N, Fujita M, Tachibana S et al (2012) Effect of hydrogen gas on the survival rate of mice following global cerebral ischemia. Shock 37(6):645–652. https://doi.org/10.1097/SHK.0b013e31824ed57c
Hayashida K, Sano M, Kamimura N, Yokota T, Suzuki M, Maekawa Y, Kawamura A, Abe T et al (2012) H(2) gas improves functional outcome after cardiac arrest to an extent comparable to therapeutic hypothermia in a rat model. J Am Heart Assoc 1(5):e003459. https://doi.org/10.1161/JAHA.112.003459
Hugyecz M, Mracsko E, Hertelendy P, Farkas E, Domoki F, Bari F (2011) Hydrogen supplemented air inhalation reduces changes of prooxidant enzyme and gap junction protein levels after transient global cerebral ischemia in the rat hippocampus. Brain Res 1404:31–38. https://doi.org/10.1016/j.brainres.2011.05.068
Cui J, Chen X, Zhai X, Shi D, Zhang R, Zhi X, Li X, Gu Z et al (2016) Inhalation of water electrolysis-derived hydrogen ameliorates cerebral ischemia-reperfusion injury in rats - a possible new hydrogen resource for clinical use. Neuroscience 335:232–241. https://doi.org/10.1016/j.neuroscience.2016.08.021
Cole AR, Sperotto F, DiNardo JA, Carlisle S, Rivkin MJ, Sleeper LA, Kheir JN (2021) Safety of prolonged inhalation of hydrogen gas in air in healthy adults. Crit Care Explor 3(10):e543. https://doi.org/10.1097/CCE.0000000000000543
Ohta S (2014) Molecular hydrogen as a preventive and therapeutic medical gas: initiation, development and potential of hydrogen medicine. Pharmacol Ther 144(1):1–11. https://doi.org/10.1016/j.pharmthera.2014.04.006
Tian R, Hou Z, Hao S, Wu W, Mao X, Tao X, Lu T, Liu B (2016) Hydrogen-rich water attenuates brain damage and inflammation after traumatic brain injury in rats. Brain Res 1637:1–13. https://doi.org/10.1016/j.brainres.2016.01.029
Kamimura N, Nishimaki K, Ohsawa I, Ohta S (2011) Molecular hydrogen improves obesity and diabetes by inducing hepatic FGF21 and stimulating energy metabolism in db/db mice. Obesity (Silver Spring) 19(7):1396–1403. https://doi.org/10.1038/oby.2011.6
Asada R, Tazawa K, Sato S, Miwa N (2020) Effects of hydrogen-rich water prepared by alternating-current-electrolysis on antioxidant activity, DNA oxidative injuries, and diabetes-related markers. Med Gas Res 10(3):114–121. https://doi.org/10.4103/2045-9912.296041
Shimouchi A, Nose K, Shirai M, Kondo T (2012) Estimation of molecular hydrogen consumption in the human whole body after the ingestion of hydrogen-rich water. Adv Exp Med Biol 737:245–250. https://doi.org/10.1007/978-1-4614-1566-4_36
Tian Y, Zhang Y, Wang Y, Chen Y, Fan W, Zhou J, Qiao J, Wei Y (2021) Hydrogen, a novel therapeutic molecule, regulates oxidative stress, inflammation, and apoptosis. Front Physiol 12:789507. https://doi.org/10.3389/fphys.2021.789507
Jiang B, Li Y, Dai W, Wu A, Wu H, Mao D (2021) Hydrogen-rich saline alleviates early brain injury through regulating of ER stress and autophagy after experimental subarachnoid hemorrhage. Acta Cir Bras 36(8):e360804. https://doi.org/10.1590/ACB360804
Chu X, Cao L, Yu Z, Xin D, Li T, Ma W, Zhou X, Chen W et al (2019) Hydrogen-rich saline promotes microglia M2 polarization and complement-mediated synapse loss to restore behavioral deficits following hypoxia-ischemic in neonatal mice via AMPK activation. J Neuroinflammation 16(1):104. https://doi.org/10.1186/s12974-019-1488-2
Hu HW, Chen ZG, Liu JG, Chen G (2021) Role of hydrogen in traumatic brain injury: a narrative review. Med Gas Res 11(3):114–120. https://doi.org/10.4103/2045-9912.314331
Liu Y, Dong F, Guo R, Zhang Y, Qu X, Wu X, Yao R (2019) Hydrogen-Rich Saline Ameliorates Experimental Autoimmune Encephalomyelitis in C57BL/6 mice via the Nrf2-ARE signaling pathway. Inflammation 42(2):586–597. https://doi.org/10.1007/s10753-018-0915-3
Wang L, Zhao C, Wu S, Xiao G, Zhuge X, Lei P, Xie K (2018) Hydrogen gas treatment improves the neurological outcome after traumatic brain injury via increasing miR-21 expression. Shock 50(3):308–315. https://doi.org/10.1097/SHK.0000000000001018
Cramer T, Kisliouk T, Yeshurun S, Meiri N (2015) The balance between stress resilience and vulnerability is regulated by corticotropin-releasing hormone during the critical postnatal period for sensory development. Dev Neurobiol 75(8):842–853. https://doi.org/10.1002/dneu.22252
Liu FT, Xu SM, Xiang ZH, Li XN, Li J, Yuan HB, Sun XJ (2014) Molecular hydrogen suppresses reactive astrogliosis related to oxidative injury during spinal cord injury in rats. CNS Neurosci Ther 20(8):778–786. https://doi.org/10.1111/cns.12258
Huang JL, Liu WW, Manaenko A, Sun XJ, Mei QY, Hu Q (2019) Hydrogen inhibits microglial activation and regulates microglial phenotype in a mouse middle cerebral artery occlusion model. Med Gas Res 9(3):127–132. https://doi.org/10.4103/2045-9912.266987
Li T, Liu T, Chen X, Li L, Feng M, Zhang Y, Wan L, Zhang C et al (2020) Microglia induce the transformation of A1/A2 reactive astrocytes via the CXCR7/PI3K/Akt pathway in chronic post-surgical pain. J Neuroinflammation 17(1):211. https://doi.org/10.1186/s12974-020-01891-5
Iketani M, Ohsawa I (2017) Molecular hydrogen as a neuroprotective agent. Curr Neuropharmacol 15(2):324–331. https://doi.org/10.2174/1570159x14666160607205417
Huang L (2016) Molecular hydrogen: a therapeutic antioxidant and beyond. Med Gas Res 6(4):219–222. https://doi.org/10.4103/2045-9912.196904
Wu Y, Yuan M, Song J, Chen X, Yang H (2019) Hydrogen gas from inflammation treatment to cancer therapy. ACS Nano 13(8):8505–8511. https://doi.org/10.1021/acsnano.9b05124
Yuan J, Wang D, Liu Y, Chen X, Zhang H, Shen F, Liu X, Fu J (2018) Hydrogen-rich water attenuates oxidative stress in rats with traumatic brain injury via Nrf2 pathway. J Surg Res 228:238–246. https://doi.org/10.1016/j.jss.2018.03.024
Duleh S, Wang X, Komirenko A, Margeta M (2016) Activation of the Keap1/Nrf2 stress response pathway in autophagic vacuolar myopathies. Acta Neuropathol Commun 4(1):115. https://doi.org/10.1186/s40478-016-0384-6
Hu Y, Feng X, Chen J, Wu Y, Shen L (2022) Hydrogen-rich saline alleviates early brain injury through inhibition of necroptosis and neuroinflammation via the ROS/HO-1 signaling pathway after traumatic brain injury. Exp Ther Med 23(2):126. https://doi.org/10.3892/etm.2021.11049
Gao Q, Song H, Wang XT, Liang Y, Xi YJ, Gao Y, Guo QJ, LeBaron T et al (2017) Molecular hydrogen increases resilience to stress in mice. Sci Rep 7(1):9625. https://doi.org/10.1038/s41598-017-10362-6
Ostojic SM (2018) Inadequate production of H(2) by gut microbiota and Parkinson disease. Trends Endocrinol Metab 29(5):286–288. https://doi.org/10.1016/j.tem.2018.02.006
Scheperjans F, Aho V, Pereira PA, Koskinen K, Paulin L, Pekkonen E, Haapaniemi E, Kaakkola S et al (2015) Gut microbiota are related to Parkinson’s disease and clinical phenotype. Mov Disord 30(3):350–358. https://doi.org/10.1002/mds.26069
Varesi A, Pierella E, Romeo M, Piccini GB, Alfano C, Bjorklund G, Oppong A, Ricevuti G, Esposito C, Chirumbolo S et al. (2022) The potential role of gut microbiota in Alzheimer’s disease: from diagnosis to treatment. Nutrients 14 (3). https://doi.org/10.3390/nu14030668
Meyer M, Tebbe U, Piiper J (1980) Solubility of inert gases in dog blood and skeletal muscle. Pflugers Arch 384(2):131–134. https://doi.org/10.1007/BF00584428
Neff RA, Wang M, Vatansever S, Guo L, Ming C, Wang Q, Wang E, Horgusluoglu-Moloch E, Song WM, Li A et al. (2021) Molecular subtyping of Alzheimer’s disease using RNA sequencing data reveals novel mechanisms and targets. Sci Adv 7 (2). https://doi.org/10.1126/sciadv.abb5398
Sinyor B, Mineo J, Ochner C (2020) Alzheimer’s disease, inflammation, and the role of antioxidants. J Alzheimers Dis Rep 4(1):175–183. https://doi.org/10.3233/ADR-200171
Cui Y, Zhang H, Ji M, Jia M, Chen H, Yang J, Duan M (2014) Hydrogen-rich saline attenuates neuronal ischemia–reperfusion injury by protecting mitochondrial function in rats. J Surg Res 192(2):564–572. https://doi.org/10.1016/j.jss.2014.05.060
Li J, Wang C, Zhang JH, Cai JM, Cao YP, Sun XJ (2010) Hydrogen-rich saline improves memory function in a rat model of amyloid-beta-induced Alzheimer’s disease by reduction of oxidative stress. Brain Res 1328:152–161. https://doi.org/10.1016/j.brainres.2010.02.046
Wang C, Li J, Liu Q, Yang R, Zhang JH, Cao YP, Sun XJ (2011) Hydrogen-rich saline reduces oxidative stress and inflammation by inhibit of JNK and NF-kappaB activation in a rat model of amyloid-beta-induced Alzheimer’s disease. Neurosci Lett 491(2):127–132. https://doi.org/10.1016/j.neulet.2011.01.022
Yarza R, Vela S, Solas M, Ramirez MJ (2015) c-Jun N-terminal Kinase (JNK) Signaling as a therapeutic target for Alzheimer’s disease. Front Pharmacol 6:321. https://doi.org/10.3389/fphar.2015.00321
Hou C, Peng Y, Qin C, Fan F, Liu J, Long J (2018) Hydrogen-rich water improves cognitive impairment gender-dependently in APP/PS1 mice without affecting Abeta clearance. Free Radic Res 52(11–12):1311–1322. https://doi.org/10.1080/10715762.2018.1460749
Choi SH, Bylykbashi E, Chatila ZK, Lee SW, Pulli B, Clemenson GD, Kim E, Rompala A, Oram MK, Asselin C et al. (2018) Combined adult neurogenesis and BDNF mimic exercise effects on cognition in an Alzheimer’s mouse model. Science 361 (6406). https://doi.org/10.1126/science.aan8821
Jiao SS, Shen LL, Zhu C, Bu XL, Liu YH, Liu CH, Yao XQ, Zhang LL et al (2016) Brain-derived neurotrophic factor protects against tau-related neurodegeneration of Alzheimer’s disease. Transl Psychiatry 6(10):e907. https://doi.org/10.1038/tp.2016.186
Lin CL, Huang WN, Li HH, Huang CN, Hsieh S, Lai C, Lu FJ (2015) Hydrogen-rich water attenuates amyloid beta-induced cytotoxicity through upregulation of Sirt1-FoxO3a by stimulation of AMP-activated protein kinase in SK-N-MC cells. Chem Biol Interact 240:12–21. https://doi.org/10.1016/j.cbi.2015.07.013
van der Horst A, Burgering BM (2007) Stressing the role of FoxO proteins in lifespan and disease. Nat Rev Mol Cell Biol 8(6):440–450. https://doi.org/10.1038/nrm2190
Rodriguez M, Valez V, Cimarra C, Blasina F, Radi R (2020) Hypoxic-ischemic encephalopathy and mitochondrial dysfunction: facts, unknowns, and challenges. Antioxid Redox Signal 33(4):247–262. https://doi.org/10.1089/ars.2020.8093
Allen KA, Brandon DH (2011) Hypoxic ischemic encephalopathy: pathophysiology and experimental treatments. Newborn Infant Nurs Rev 11(3):125–133. https://doi.org/10.1053/j.nainr.2011.07.004
Arteaga O, Alvarez A, Revuelta M, Santaolalla F, Urtasun A, Hilario E (2017) Role of antioxidants in neonatal hypoxic-ischemic brain injury: new therapeutic approaches. Int J Mol Sci 18 (2). https://doi.org/10.3390/ijms18020265
Qin X, Cheng J, Zhong Y, Mahgoub OK, Akter F, Fan Y, Aldughaim M, Xie Q et al (2019) Mechanism and treatment related to oxidative stress in neonatal hypoxic-ischemic encephalopathy. Front Mol Neurosci 12:88. https://doi.org/10.3389/fnmol.2019.00088
Millar LJ, Shi L, Hoerder-Suabedissen A, Molnar Z (2017) Neonatal hypoxia ischaemia: mechanisms, models, and therapeutic challenges. Front Cell Neurosci 11:78. https://doi.org/10.3389/fncel.2017.00078
Htun Y, Nakamura S, Kusaka T (2021) Hydrogen and therapeutic gases for neonatal hypoxic-ischemic encephalopathy: potential neuroprotective adjuncts in translational research. Pediatr Res 89(4):753–759. https://doi.org/10.1038/s41390-020-0998-z
Cai J, Kang Z, Liu WW, Luo X, Qiang S, Zhang JH, Ohta S, Sun X et al (2008) Hydrogen therapy reduces apoptosis in neonatal hypoxia-ischemia rat model. Neurosci Lett 441(2):167–172. https://doi.org/10.1016/j.neulet.2008.05.077
Shiraishi H, Okamoto H, Yoshimura A, Yoshida H (2006) ER stress-induced apoptosis and caspase-12 activation occurs downstream of mitochondrial apoptosis involving Apaf-1. J Cell Sci 119(Pt 19):3958–3966. https://doi.org/10.1242/jcs.03160
Wang P, Zhao M, Chen Z, Wu G, Fujino M, Zhang C, Zhou W, Zhao M et al (2020) Hydrogen gas attenuates hypoxic-ischemic brain injury via regulation of the MAPK/HO-1/PGC-1a pathway in neonatal rats. Oxid Med Cell Longev 2020:6978784. https://doi.org/10.1155/2020/6978784
Consoli V, Sorrenti V, Grosso S, Vanella L (2021) Heme oxygenase-1 signaling and redox homeostasis in physiopathological conditions. Biomolecules 11 (4). https://doi.org/10.3390/biom11040589
Htun Y, Nakamura S, Nakao Y, Mitsuie T, Nakamura M, Yamato S, Jinnai W, Koyano K et al (2019) Hydrogen ventilation combined with mild hypothermia improves short-term neurological outcomes in a 5-day neonatal hypoxia-ischaemia piglet model. Sci Rep 9(1):4088. https://doi.org/10.1038/s41598-019-40674-8
Matchett GA, Fathali N, Hasegawa Y, Jadhav V, Ostrowski RP, Martin RD, Dorotta IR, Sun X et al (2009) Hydrogen gas is ineffective in moderate and severe neonatal hypoxia-ischemia rat models. Brain Res 1259:90–97. https://doi.org/10.1016/j.brainres.2008.12.066
Geng C, Guo Y, Wang C, Liao D, Han W, Zhang J, Jiang P (2020) Systematic impacts of chronic unpredictable mild stress on metabolomics in rats. Sci Rep 10(1):700. https://doi.org/10.1038/s41598-020-57566-x
Patki G, Solanki N, Atrooz F, Allam F, Salim S (2013) Depression, anxiety-like behavior and memory impairment are associated with increased oxidative stress and inflammation in a rat model of social stress. Brain Res 1539:73–86. https://doi.org/10.1016/j.brainres.2013.09.033
Carrasco M, Hong C, Nienhuis JK, Harbin SM, Fitzgerald KD, Gehring WJ, Hanna GL (2013) Increased error-related brain activity in youth with obsessive-compulsive disorder and other anxiety disorders. Neurosci Lett 541:214–218. https://doi.org/10.1016/j.neulet.2013.02.017
Ng F, Berk M, Dean O, Bush AI (2008) Oxidative stress in psychiatric disorders: evidence base and therapeutic implications. Int J Neuropsychopharmacol 11(6):851–876. https://doi.org/10.1017/S1461145707008401
Bajpai A, Verma AK, Srivastava M, Srivastava R (2014) Oxidative stress and major depression. J Clin Diagn Res 8(12):04–07. https://doi.org/10.7860/JCDR/2014/10258.5292
Allam F, Dao AT, Chugh G, Bohat R, Jafri F, Patki G, Mowrey C, Asghar M et al (2013) Grape powder supplementation prevents oxidative stress-induced anxiety-like behavior, memory impairment, and high blood pressure in rats. J Nutr 143(6):835–842. https://doi.org/10.3945/jn.113.174649
Salim S, Sarraj N, Taneja M, Saha K, Tejada-Simon MV, Chugh G (2010) Moderate treadmill exercise prevents oxidative stress-induced anxiety-like behavior in rats. Behav Brain Res 208(2):545–552. https://doi.org/10.1016/j.bbr.2009.12.039
Ji Y, Luo J, Zeng J, Fang Y, Liu R, Luan F, Zeng N (2021) Xiaoyao pills ameliorate depression-like behaviors and oxidative stress induced by olfactory bulbectomy in rats via the activation of the PIK3CA-AKT1-NFE2L2/BDNF signaling pathway. Front Pharmacol 12:643456. https://doi.org/10.3389/fphar.2021.643456
Zhang Y, Su WJ, Chen Y, Wu TY, Gong H, Shen XL, Wang YX, Sun XJ et al (2016) Effects of hydrogen-rich water on depressive-like behavior in mice. Sci Rep 6:23742. https://doi.org/10.1038/srep23742
Franchi L, Eigenbrod T, Munoz-Planillo R, Nunez G (2009) The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol 10(3):241–247. https://doi.org/10.1038/ni.1703
Matovic E, Delibegovic S (2019) Adrenocorticotropic hormone (ACTH) and cortisol monitoring as stress markers during laparoscopic cholecystectomy: standard and low intraabdominal pressure and open cholecystectomy. Med Arch 73(4):257–261. https://doi.org/10.5455/medarh.2019.73.257-261
Perry RE, Rincon-Cortes M, Braren SH, Brandes-Aitken AN, Opendak M, Pollonini G, Chopra D, Raver CC et al (2019) Corticosterone administration targeting a hypo-reactive HPA axis rescues a socially-avoidant phenotype in scarcity-adversity reared rats. Dev Cogn Neurosci 40:100716. https://doi.org/10.1016/j.dcn.2019.100716
Khaibullin T, Ivanova V, Martynova E, Cherepnev G, Khabirov F, Granatov E, Rizvanov A, Khaiboullina S (2017) Elevated levels of proinflammatory cytokines in cerebrospinal fluid of multiple sclerosis patients. Front Immunol 8:531. https://doi.org/10.3389/fimmu.2017.00531
Castro-Borrero W, Graves D, Frohman TC, Flores AB, Hardeman P, Logan D, Orchard M, Greenberg B et al (2012) Current and emerging therapies in multiple sclerosis: a systematic review. Ther Adv Neurol Disord 5(4):205–220. https://doi.org/10.1177/1756285612450936
Ohl K, Tenbrock K, Kipp M (2016) Oxidative stress in multiple sclerosis: central and peripheral mode of action. Exp Neurol 277:58–67. https://doi.org/10.1016/j.expneurol.2015.11.010
Stys PK, Zamponi GW, van Minnen J, Geurts JJ (2012) Will the real multiple sclerosis please stand up? Nat Rev Neurosci 13(7):507–514. https://doi.org/10.1038/nrn3275
Haider L, Fischer MT, Frischer JM, Bauer J, Hoftberger R, Botond G, Esterbauer H, Binder CJ et al (2011) Oxidative damage in multiple sclerosis lesions. Brain 134(Pt 7):1914–1924. https://doi.org/10.1093/brain/awr128
Schluesener HJ, Seid K (2000) Heme oxygenase-1 in lesions of rat experimental autoimmune encephalomyelitis and neuritis. J Neuroimmunol 110(1–2):114–120. https://doi.org/10.1016/s0165-5728(00)00352-0
Zhao M, Liu MD, Pu YY, Wang D, Xie Y, Xue GC, Jiang Y, Yang QQ et al (2016) Hydrogen-rich water improves neurological functional recovery in experimental autoimmune encephalomyelitis mice. J Neuroimmunol 294:6–13. https://doi.org/10.1016/j.jneuroim.2016.03.006
MacMahon Copas AN, McComish SF, Fletcher JM, Caldwell MA (2021) The pathogenesis of Parkinson’s disease: a complex interplay between astrocytes, microglia, and T lymphocytes? Front Neurol 12:666737. https://doi.org/10.3389/fneur.2021.666737
Mahul-Mellier AL, Burtscher J, Maharjan N, Weerens L, Croisier M, Kuttler F, Leleu M, Knott GW et al (2020) The process of Lewy body formation, rather than simply alpha-synuclein fibrillization, is one of the major drivers of neurodegeneration. Proc Natl Acad Sci U S A 117(9):4971–4982. https://doi.org/10.1073/pnas.1913904117
Blesa J, Trigo-Damas I, Quiroga-Varela A, Jackson-Lewis VR (2015) Oxidative stress and Parkinson’s disease. Front Neuroanat 9:91. https://doi.org/10.3389/fnana.2015.00091
Segura-Aguilar J, Paris I, Munoz P, Ferrari E, Zecca L, Zucca FA (2014) Protective and toxic roles of dopamine in Parkinson’s disease. J Neurochem 129(6):898–915. https://doi.org/10.1111/jnc.12686
Zucca FA, Basso E, Cupaioli FA, Ferrari E, Sulzer D, Casella L, Zecca L (2014) Neuromelanin of the human substantia nigra: an update. Neurotox Res 25(1):13–23. https://doi.org/10.1007/s12640-013-9435-y
Hattingen E, Magerkurth J, Pilatus U, Mozer A, Seifried C, Steinmetz H, Zanella F, Hilker R (2009) Phosphorus and proton magnetic resonance spectroscopy demonstrates mitochondrial dysfunction in early and advanced Parkinson’s disease. Brain 132(Pt 12):3285–3297. https://doi.org/10.1093/brain/awp293
Zuo L, Motherwell MS (2013) The impact of reactive oxygen species and genetic mitochondrial mutations in Parkinson’s disease. Gene 532(1):18–23. https://doi.org/10.1016/j.gene.2013.07.085
Muftuoglu M, Elibol B, Dalmizrak O, Ercan A, Kulaksiz G, Ogus H, Dalkara T, Ozer N (2004) Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov Disord 19(5):544–548. https://doi.org/10.1002/mds.10695
Bian M, Liu J, Hong X, Yu M, Huang Y, Sheng Z, Fei J, Huang F (2012) Overexpression of parkin ameliorates dopaminergic neurodegeneration induced by 1- methyl-4-phenyl-1,2,3,6-tetrahydropyridine in mice. PLoS One 7(6):e39953. https://doi.org/10.1371/journal.pone.0039953
Filograna R, Beltramini M, Bubacco L, Bisaglia M (2016) Antioxidants in Parkinson’s disease therapy: a critical point of view. Curr Neuropharmacol 14(3):260–271. https://doi.org/10.2174/1570159x13666151030102718
Fujita K, Seike T, Yutsudo N, Ohno M, Yamada H, Yamaguchi H, Sakumi K, Yamakawa Y et al (2009) Hydrogen in drinking water reduces dopaminergic neuronal loss in the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine mouse model of Parkinson’s disease. PLoS ONE 4(9):e7247. https://doi.org/10.1371/journal.pone.0007247
Yanagihara T, Arai K, Miyamae K, Sato B, Shudo T, Yamada M, Aoyama M (2005) Electrolyzed hydrogen-saturated water for drinking use elicits an antioxidative effect: a feeding test with rats. Biosci Biotechnol Biochem 69(10):1985–1987. https://doi.org/10.1271/bbb.69.1985
Yoritaka A, Takanashi M, Hirayama M, Nakahara T, Ohta S, Hattori N (2013) Pilot study of H(2) therapy in Parkinson’s disease: a randomized double-blind placebo-controlled trial. Mov Disord 28(6):836–839. https://doi.org/10.1002/mds.25375
Yoritaka A, Ohtsuka C, Maeda T, Hirayama M, Abe T, Watanabe H, Saiki H, Oyama G et al (2018) Randomized, double-blind, multicenter trial of hydrogen water for Parkinson’s disease. Mov Disord 33(9):1505–1507. https://doi.org/10.1002/mds.27472
Yoritaka A, Abe T, Ohtsuka C, Maeda T, Hirayama M, Watanabe H, Saiki H, Oyama G et al (2016) A randomized double-blind multi-center trial of hydrogen water for Parkinson’s disease: protocol and baseline characteristics. BMC Neurol 16:66. https://doi.org/10.1186/s12883-016-0589-0
Kobayashi Y, Imamura R, Koyama Y, Kondo M, Kobayashi H, Nonomura N, Shimada S (2020) Renoprotective and neuroprotective effects of enteric hydrogen generation from Si-based agent. Sci Rep 10(1):5859. https://doi.org/10.1038/s41598-020-62755-9
Li H, Luo Y, Yang P, Liu J (2019) Hydrogen as a complementary therapy against ischemic stroke: a review of the evidence. J Neurol Sci 396:240–246. https://doi.org/10.1016/j.jns.2018.11.004
Kuriakose D, Xiao Z (2020) Pathophysiology and treatment of stroke: present status and future perspectives. Int J Mol Sci 21 (20). https://doi.org/10.3390/ijms21207609
Lee M, Lim JS, Kim CH, Lee SH, Kim Y, Hun Lee J, Jang MU, Sun OhM et al (2021) High neutrophil-lymphocyte ratio predicts post-stroke cognitive impairment in acute ischemic stroke patients. Front Neurol 12:693318. https://doi.org/10.3389/fneur.2021.693318
Zivancevic K, Lovic D, Andjus PR, Radenovic L (2021) Neuroinflammation in post-ischemic brain. In: Pluta R (ed) Cerebral Ischemia. Brisbane (AU). https://doi.org/10.36255/exonpublications.cerebralischemia.2021.neuroinflammation
Rodrigo R, Fernandez-Gajardo R, Gutierrez R, Matamala JM, Carrasco R, Miranda-Merchak A, Feuerhake W (2013) Oxidative stress and pathophysiology of ischemic stroke: novel therapeutic opportunities. CNS Neurol Disord Drug Targets 12(5):698–714. https://doi.org/10.2174/1871527311312050015
Wu L, Xiong X, Wu X, Ye Y, Jian Z, Zhi Z, Gu L (2020) Targeting oxidative stress and inflammation to prevent ischemia-reperfusion injury. Front Mol Neurosci 13:28. https://doi.org/10.3389/fnmol.2020.00028
Huang L, Lenahan C, Boling W, Tang J, Zhang JH (2021) Molecular hydrogen application in stroke: bench to bedside. Curr Pharm Des 27(5):703–712. https://doi.org/10.2174/1381612826666200917152316
Wu X, Li X, Liu Y, Yuan N, Li C, Kang Z, Zhang X, Xia Y et al (2018) Hydrogen exerts neuroprotective effects on OGD/R damaged neurons in rat hippocampal by protecting mitochondrial function via regulating mitophagy mediated by PINK1/Parkin signaling pathway. Brain Res 1698:89–98. https://doi.org/10.1016/j.brainres.2018.06.028
Popescu C, Anghelescu A, Daia C, Onose G (2015) Actual data on epidemiological evolution and prevention endeavours regarding traumatic brain injury. J Med Life 8(3):272–277
Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, Bragge P, Brazinova A et al (2017) Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol 16(12):987–1048. https://doi.org/10.1016/S1474-4422(17)30371-X
Dulla CG, Pitkanen A (2021) Novel approaches to prevent epileptogenesis after traumatic brain injury. Neurotherapeutics 18(3):1582–1601. https://doi.org/10.1007/s13311-021-01119-1
Yue Y, Shang C, Dong H, Meng K (2019) Interleukin-1 in cerebrospinal fluid for evaluating the neurological outcome in traumatic brain injury. Biosci Rep 39 (4). 10.1042/BSR20181966
Schimmel SJ, Acosta S, Lozano D (2017) Neuroinflammation in traumatic brain injury: a chronic response to an acute injury. Brain Circ 3(3):135–142. https://doi.org/10.4103/bc.bc_18_17
Hill RL, Singh IN, Wang JA, Hall ED (2017) Time courses of post-injury mitochondrial oxidative damage and respiratory dysfunction and neuronal cytoskeletal degradation in a rat model of focal traumatic brain injury. Neurochem Int 111:45–56. https://doi.org/10.1016/j.neuint.2017.03.015
Li X, Liang M, Jiang J, He R, Wang M, Guo X, Shen M, Qin R (2018) Combined inhibition of autophagy and Nrf2 signaling augments bortezomib-induced apoptosis by increasing ROS production and ER stress in pancreatic cancer cells. Int J Biol Sci 14(10):1291–1305. https://doi.org/10.7150/ijbs.26776
Weber JT (2012) Altered calcium signaling following traumatic brain injury. Front Pharmacol 3:60. https://doi.org/10.3389/fphar.2012.00060
Hall ED, Vaishnav RA, Mustafa AG (2010) Antioxidant therapies for traumatic brain injury. Neurotherapeutics 7(1):51–61. https://doi.org/10.1016/j.nurt.2009.10.021
Nappi G, Facchinetti F, Martignoni E, Petraglia F, Manzoni GC, Sances G, Sandrini G, Genazzani AR (1985) Endorphin patterns within the headache spectrum disorders. Cephalalgia 5(Suppl 2):201–210. https://doi.org/10.1177/03331024850050S240
Ji X, Liu W, Xie K, Liu W, Qu Y, Chao X, Chen T, Zhou J et al (2010) Beneficial effects of hydrogen gas in a rat model of traumatic brain injury via reducing oxidative stress. Brain Res 1354:196–205. https://doi.org/10.1016/j.brainres.2010.07.038
Lin YT, Shi QQ, Zhang L, Yue CP, He ZJ, Li XX, He QJ, Liu Q et al (2022) Hydrogen-rich water ameliorates neuropathological impairments in a mouse model of Alzheimer’s disease through reducing neuroinflammation and modulating intestinal microbiota. Neural Regen Res 17(2):409–417. https://doi.org/10.4103/1673-5374.317992
Ge L, Yang M, Yang NN, Yin XX, Song WG (2017) Molecular hydrogen: a preventive and therapeutic medical gas for various diseases. Oncotarget 8(60):102653–102673. https://doi.org/10.18632/oncotarget.21130
Ghanizadeh A, Berk M (2013) Molecular hydrogen: an overview of its neurobiological effects and therapeutic potential for bipolar disorder and schizophrenia. Med Gas Res 3(1):11. https://doi.org/10.1186/2045-9912-3-11
Mao Y, Guo L (2012) Nanomaterials for renewable hydrogen production, storage and utilization. Progress in Nat Sci: Mater Int 22:522–534. https://doi.org/10.1016/j.pnsc.2012.12.003
Lee SH, Jin SH, An J (2019) The difference in cortical activation pattern for complex motor skills: A functional near- infrared spectroscopy study. Sci Rep 9(1):14066. https://doi.org/10.1038/s41598-019-50644-9
Sharp PS, Ameen-Ali KE, Boorman L, Harris S, Wharton S, Howarth C, Shabir O, Redgrave P et al (2020) Neurovascular coupling preserved in a chronic mouse model of Alzheimer’s disease: methodology is critical. J Cereb Blood Flow Metab 40(11):2289–2303. https://doi.org/10.1177/0271678X19890830
Lin WH, Hao Q, Rosengarten B, Leung WH, Wong KS (2011) Impaired neurovascular coupling in ischaemic stroke patients with large or small vessel disease. Eur J Neurol 18(5):731–736. https://doi.org/10.1111/j.1468-1331.2010.03262.x
Piilgaard H, Lauritzen M (2009) Persistent increase in oxygen consumption and impaired neurovascular coupling after spreading depression in rat neocortex. J Cereb Blood Flow Metab 29(9):1517–1527. https://doi.org/10.1038/jcbfm.2009.73
Funding
This study was supported by the National Key Research and Development Program of China (2017YFB0403801), the National Natural Science Foundation of China (31971096, 31771256, 31971099, and 32100918), the project funded by the China Postdoctoral Science Foundation (2021M690060 and 2022T150227), and the Sigma Xi Grants in Aid of Research (GIAR) program (G03152021115804390).
Author information
Authors and Affiliations
Contributions
C. W. reviewed the literature and drafted the manuscript. C. W. and P. Z. drew the figures and tables. L. D. Y. supervised the writing and edited the manuscript. All authors read and approved the final manuscript.
Corresponding author
Ethics declarations
Ethics Approval and Consent to Participate
Not applicable.
Consent for Publication
Not applicable.
Competing Interests
The authors declare no competing interests.
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Rights and permissions
Springer Nature or its licensor (e.g. a society or other partner) holds exclusive rights to this article under a publishing agreement with the author(s) or other rightsholder(s); author self-archiving of the accepted manuscript version of this article is solely governed by the terms of such publishing agreement and applicable law.
About this article

Cite this article
Wu, C., Zou, P., Feng, S. et al. Molecular Hydrogen: an Emerging Therapeutic Medical Gas for Brain Disorders. Mol Neurobiol 60, 1749–1765 (2023). https://doi.org/10.1007/s12035-022-03175-w
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-022-03175-w