
Abstract
Traumatic brain injury (TBI) is defined as an alteration in brain function or other evidence of brain pathology caused by an external force. When epilepsy develops following TBI, it is known as post-traumatic epilepsy (PTE). PTE occurs in a subset of patients suffering from different types and severities of TBI, occurs more commonly following severe injury, and greatly impacts the quality of life for patients recovering from TBI. Similar to other types of epilepsy, PTE is often refractory to drug treatment with standard anti-seizure drugs. No therapeutic approaches have proven successful in the clinic to prevent the development of PTE. Therefore, novel treatment strategies are needed to stop the development of PTE and improve the quality of life for patients after TBI. Interestingly, TBI represents an excellent clinical opportunity for intervention to prevent epileptogenesis as typically the time of initiation of epileptogenesis (i.e., TBI) is known, the population of at-risk patients is large, and animal models for preclinical studies of mechanisms and treatment targets are available. If properly identified and treated, there is a true opportunity to prevent epileptogenesis after TBI and stop seizures from ever happening. With that goal in mind, here we review previous attempts to prevent PTE both in animal studies and in humans, we examine how biomarkers could enable better-targeted therapeutics, and we discuss how genetic variation may predispose individuals to PTE. Finally, we highlight exciting new advances in the field that suggest that there may be novel approaches to prevent PTE that should be considered for further clinical development.
Similar content being viewed by others

Avoid common mistakes on your manuscript.
Introduction
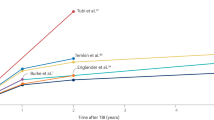
Globally, an estimated 2.4 million people are diagnosed with epilepsy each year. Thus, a new person is diagnosed with epilepsy every 13 s [1]. Epileptogenesis refers to the development and extension of tissue capable of generating spontaneous seizures, resulting in (a) the development of an epileptic condition and/or (b) progression of epilepsy after it is established [2]. In 60% of those affected, epileptogenesis is initiated by structural causes such as traumatic brain injury (TBI) [3, 4]. Recent epidemiologic data indicate that approximately 2.5 million people experience TBI annually, both in Europe and the USA. The risk of epileptogenesis increases according to the severity of TBI: about two- to four-fold after mild, eight-fold after moderate, and 16-fold after severe TBI [5, 6]. Up to 53% of patients with penetrating TBI develop epilepsy [7, 8]. Post-traumatic epilepsy (PTE) is estimated to account for approximately 5% of all epilepsies and 20% of structural epilepsies [9]. Mild TBI comprises over 90% of all TBI [10], and thus the total number of patients developing epilepsy after mild TBI can be expected to be greater than that of patients developing epilepsy after severe TBI, which has been the focus of experimental and clinical PTE studies (Fig. 1).

Potential mechanisms and therapeutic strategies to treat post-traumatic epilepsy. A schematic of the healthy, injured, and epileptic brain. Mechanisms that have been implicated in post-traumatic epileptogenesis are indicated with a red question mark. Mechanisms that may be targeted to reduce brain injury are indicated in blue. Mechanisms that may be targeted to reverse or prevent epileptogenesis are indicated in green. We also point out that small molecule and genetic screens may be able to identify novel mechanisms and treatment strategies
TBI refers to an alteration in brain function, or other evidence of brain pathology, caused by an external force [11]. Seizures after TBI have been classically categorized into immediate (seizures ≤ 24 h post-TBI), early (≤ 7 days), or late (> 7 days) seizures. A person with PTE suffers repeated unprovoked seizures that result from TBI and occur more than a week after the initial injury [12]. According to the current International league Against Epilepsy definitions, PTE belongs to structural epilepsies, and is diagnosed if the subject experiences one unprovoked seizure > 7 days post-TBI [3, 12, 13]. Approximately 80% of TBI patients who eventually develop epilepsy will receive a PTE diagnosis within 2 years after the TBI [5, 15]. Both clinical and basic science studies have investigated a wide range of biological processes that may be involved in the transition from an injured brain to an epileptic brain. Changes occurring with TBI, including neuroinflammation, neuronal cell death, and changes in synaptic abundance and function, just to name a few, have been studied for their role in PTE [16,17,18]. While there are diverse cellular, molecular, metabolic, and circuit-level changes, we have not been able to identify a causative molecular or cellular event and its temporal relation to the occurrence of PTE. Innovation in both clinical care and basic science suggests that we may be closer than ever to identifying novel therapeutic approaches to prevent PTE. In this review, we examine past failures in preventing PTE, consider diagnostic and genetic information that could guide targeted interventions, and highlight exciting new approaches with may help reduce the prevalence and impact of PTE.
Old and New Strategies for Prevention of Epilepsy After TBI—Still No Treatments in Clinic
In a review of pharmacologic prophylaxis for PTE, Rapport II and Penry [19] cite the first anti-epileptogenesis studies conducted in head-injured patients using diphenylhydantoin [20, 21]. Since then, the concept of using compounds designed to suppress epileptic seizures (anti-seizure drugs) to prevent the complex molecular and cellular processes that drive epileptogenesis [anti-epileptogenic drugs (AEGs)] was expanded to carbamazepine, phenobarbital and valproic acid (for review, see [22]). According to ClinicalTrials.gov, initial studies on PTE are also planned using the third-generation anti-seizure drugs lacosamide (NCT01110187), levetiracetam (NCT01463033, NCT02631759, NCT00566046), and topiramate (NCT00598923). In addition, a study using acetylcholinesterase inhibitor, huperzine A (NCT01676311) was planned but is now discontinued. Biperiden, a cholinergic antagonist acting in the muscarinic receptor, is still under investigation (biperiden (NCT01048138)) [23]. So far, the use of anti-seizure drugs has not resulted in favorable anti-epileptogenic effects, and their use has been recommended only for the first post-injury week to suppress immediate and early seizures (Brain Trauma Foundation Guidelines) [24].
The recent rapid progress in modeling PTE provides an opportunity to vigorously assess preclinical candidate treatments in different clinically relevant epileptogenic injuries, mimicking the heterogeneity of TBI in humans with PTE (Table 1). Long-term video-electroencephalogram (EEG) monitoring studies have shown that epileptogenesis can be triggered in different strains of rats and mice by various injury types, including focal (e.g., controlled cortical impact-induced TBI, CCI), diffuse (repetitive weight-drop), mixed type injury (e.g., lateral-fluid percussion TBI), and blast injury. Although gaps in animal models remain, including epileptogenesis in females and younger animals, currently available animal models have identified candidate molecular, cellular, and network epileptogenic mechanisms that will pave the way for the discovery of novel treatments for PTE.
Table 2 summarizes the current in vivo treatment studies on epileptogenesis in relevant animal models. Most of them have administered small molecules with a variety of mechanisms of action as a monotherapy. Commonly targeted mechanisms include oxidative stress, neuroinflammation, neuroprotection, and restoration of inhibitory GABAergic function. Unlike in status epilepticus-induced epileptogenesis models, gene therapy, administration of monoclonal antibodies, or treatments targeting DNA or RNA have not yet been tested in PTE models. Some laboratories have applied ketogenic diet, hypothermia, focal passive cooling, treadmill exercise, or transplantation of GABAergic progenitors (Table 2). These pharmacological treatments have typically been initiated within hours after the injury. Studies delivering anti-epileptogenic interventions at later time points would also greatly inform future clinical studies and would complement the development of biomarkers to identify high-risk patients at later timepoints. Ex vivo analysis of tissue excitability or in vivo analysis of seizure susceptibility, incidence of epilepsy, or characteristics of epilepsy phenotype (seizure frequency, duration or behavioral severity of seizures) have been used as outcome measures. Assessment of a 50% responder rate has been challenging as it would require large animal numbers as typically 25–50% of animals with TBI develop epilepsy within the 4–6 months follow-up (Table 2). So far, none of the treatments except the transplantation of GABAergic progenitors has been able to prevent the development of epilepsy.
Advancing Biomarkers from Basic and Clinical Studies
One of the biggest challenges in preventing post-traumatic epilepsy is identifying patients who are at the greatest risk following injury. Only a fraction of those who suffer a traumatic brain injury will go on to develop epilepsy. Clinicians are often hesitant to use aggressive or experimental interventions when there is little certainty that a patient is at significant risk of developing PTE. Therefore, developing biomarkers that are predictive of PTE would be extremely useful in stratifying patients into those that have a low probability of developing epilepsy from those that are at significantly higher risk. For patients at high risk, targeted interventions may slow or prevent the development of PTE. With this goal in mind, much work has been devoted, both in clinical and basic epilepsy research, to identifying useful biomarkers predictive of the later development of PTE (Table 3).
A biomarker is a characteristic that can be objectively measured as an indicator of normal biologic processes, pathogenic processes, or responses to an exposure or intervention, including therapeutic interventions. Biomarker modalities include molecular, histologic, radiographic, or physiologic characteristics. To improve the understanding and use of biomarker terminology in biomedical research, clinical practice, and medical product development, the FDA-NIH Joint Leadership Council developed the BEST Resource (Biomarkers, EndpointS, and other Tools). The seven BEST biomarker categories include (a) susceptibility/risk biomarkers, (b) diagnostic biomarkers, (c) monitoring biomarkers, (d) prognostic biomarkers, (e) predictive biomarkers, (f) pharmacodynamic/response biomarkers, and (g) safety biomarkers.
The current status of biomarker development in PTE field is summarized in Table 3. Many types of biomarkers have been examined to predict the development of epilepsy after different epileptogenic etiologies (for recent review see [88]), including molecular biomarkers (serum proteins, non-coding RNAs, cerebrospinal fluid (CSF)), imaging biomarkers (MRI, PET), and EEG based biomarkers. Unfortunately, no useful clinical biomarkers have been rigorously validated using sufficient patient populations. That said, there are a few exciting studies that suggest clinical biomarkers of PTE may be not that far down the road. A study of 256 Caucasian adults showed that high levels of the inflammatory cytokine IL-1β in the CSF, relative to levels in the serum, were associated with an increased risk of developing PTE over time [73]. While quite promising, this study had a small sample size and did not include statistical analysis of the predictive value of IL-1β CSF/serum ratios in PTE. None the less, this is an exciting line of research worthy of further development. In two other studies, EEG-based biomarkers suggest that epileptiform activity (epileptiform discharges, lateralized periodic discharges, generalized periodic discharges, or lateralized rhythmic delta activity) [79] and early seizures [80] are predictive of later development of PTE. These studies underscore the importance of robust, quantitative analysis in defining biomarkers of PTE and in applying statistical modeling to pinpoint the most predictive parameters in the data matrix as biomarkers. Moreover, analyses of the sensitivity, specificity, quantitative cut-off values for PTE prediction are needed to rigorously apply a biomarker in an individual subject [e.g., receiver operating characteristic analysis (ROC)]. While increased abnormal EEG and early post-injury seizures seem like straightforward potential biomarkers, the heterogeneity in the human population, in the injury itself, and in the type of clinical data collected has made it extremely challenging to pinpoint a specific metric useful in predicting PTE. More work is needed to develop these important clinical tools.
Many promising predictive biomarkers of PTE have also been identified in basic science studies and have promise as potential clinical biomarkers. For example, in the fluid-percussion model of PTE, MRI and FDG-PET imaging at 1 week and 3 months post-TBI time points were able to predict which animals would go onto develop PTE [43]. Multiple EEG biomarkers, which may eventually be useful in treating human PTE, have also been identified as predictive in animal models. These include shortening of sleep spindles [81] and the presence of pathological high frequency oscillations (HFOs) [82]. In these preclinical studies, the predictive power of these metrics was confirmed using robust statistical approaches. In the near future, we hope that these metrics can be investigated in relevant clinical populations to determine if their utility applies to human TBI/PTE.
Identifying and validating biomarkers of PTE is of critical importance for three main reasons. First, aggressive interventions for those at the highest risk of developing PTE could prevent the development of epilepsy. If those interventions have adverse effects, however, clinicians may hesitate to treat unless the risk of developing PTE is known to be high. Validated biomarkers can give clinical confidence, on a case-by-case basis, that using interventions are worthwhile based on patient risk. Second, it is extremely challenging and costly to carry out clinical trials for PTE prevention. Because only a subset of patients with TBI develops PTE, a significant number of patients must be used to conclusively ascertain if a treatment is effective. If a validated biomarker could show which patients were at high-risk, clinical studies could be enriched for patients with the greatest likelihood of developing PTE. This would dramatically reduce the cost and number of patients needed to test new PTE treatments. This is particularly important for mild TBI, where patients often do not seek immediate clinical care after injury, but are still at risk of developing PTE. Perhaps most importantly, biomarkers enable patients and care givers to understand their risk and plan accordingly. Clearly, the development and validation of a clinical biomarker for PTE would greatly impact the field and patients who suffer from TBI.
Genetic Modifiers of PTE
It is not well understood why some people develop PTE after TBI, while others do not. Many factors are likely at play including injury severity, inflammatory response, age at time of injury, time after injury, secondary “hits,” including other TBIs, stress, disruption of sleep, and many others. Of course, one must also consider that underlying genetic variations may contribute to the progression of secondary injury and manifestation of PTE. The effect may not be linear but rather depend, for example, on injury severity. A number of human and animal studies suggest that there is reason to believe that genetic variability that is not pathological under normal circumstances predisposes individuals to developing PTE.
Human Studies
While only preliminary analysis of genetic modifiers of PTE have been identified, a number of candidate gene variants have been suggested [89]. Methylenetetrahydrofolate reductase (MTHFR) is an enzyme involved in amino acid metabolism. The C677T MTHFR variant has been examined as genetic risk factor for epilepsy, may be over-represented in epilepsy patients, and is suggested to be linked to migraine and alcohol withdrawal seizures. In a study of 800 epileptic patients and 800 controls, the C677T variant was enriched in patients who had documented PTE [90]. While exciting, larger studies need to be done to determine if C677T MTHFR is strongly linked to PTE. Glutamic acid decarboxylase (GAD) is an enzyme critical to generating the inhibitory neurotransmitter GABA. Single-nucleotide polymorphisms (SNPs) in GAD1, one of the two GAD isoforms, were shown to be linked to post-traumatic seizures occurring shortly after TBI (< 1 week post-TBI) and PTE (1 week–6 months post-TBI) [91]. Again, these preliminary studies are intriguing but require further investigation to confirm their functional significance and strength of linkage to PTE. Changes in GABAergic inhibition are likely associated with PTE as GABAergic interneurons are lost after TBI [16, 92,93,94] and restoring GABAergic inhibition after TBI can prevent PTE [72, 95] in animal models. The adenosine A1 receptor (A1R) is a G-protein coupled receptor that is powerfully anti-convulsant and neuroprotective due to its ability to activate G-protein coupled inwardly rectifying K (GIRK) channels and inhibit presynaptic Ca2+ channels. In a study of over 200 patients with a severe TBI, SNPs in the A1R gene were linked to post-traumatic seizures occurring within one week following injury [96]. Again, A1Rs are closely linked to epilepsy as adenosine acting at A1Rs is thought to play a significant role in terminating seizures [97, 98] and genetic deletion of the A1R gene results in increased mortality in a rodent model of TBI [99]. Perhaps most interestingly, a SNP in the interleukin-1beta (IL-1b) gene, an inflammatory cytokine, has been shown to be linked to PTE risk over time [73]. The functional effects of this IL-1b SNP are unknown, but as mentioned above, the CSF/serum IL-1b ratio may also serve as a biomarker of PTE, strongly tying IL-1b to TBI/PTE. Finally, while not linked to PTE, there are several genetic polymorphisms, including APOE4, that are linked to poor outcomes after TBI [100, 101]. By combining genetic modifiers that affect TBI outcomes and early seizures, with those that affect PTE, we can build a more comprehensive understanding of how underlying genetic variation contributes to epileptogenesis. Properly powered clinical studies are critical to this goal.
Animal Studies
Only five studies have used genetically modified mice in PTE studies (Table 2). APP/PS1 mice showed increased prevalence of epilepsy [102]. Pijet et al. [103] showed increased prevalence of epilepsy, increased seizure frequency and susceptibility to PTZ-induced seizures in Mmp-9 over-expressing mice. Interestingly, deficiency in another extracellular matrix system (urokinase-type plasminogen activator) had minor if any effect on epileptogenesis [59, 60]. Adenosine A1R knockout mice show increased susceptibility to acute post-TBI seizures, but no longer-term risk of epileptogenesis was reported [99].
Overall, understanding the contribution of genetics on the evolution of epileptogenesis is at its infancy. Perhaps the most convincing evidence is the observation of higher incidence of epileptogenesis in CD1 mouse strain as compared to B6 strain [14] (Table 1). Even less is known about the contribution of genetics to therapy response. Further studies to replicate the current observations in larger study populations are warranted. It remains to be studies whether genetic markers can be used in stratification of patient populations in epileptogenesis clinical studies.
In Vitro Approaches to Understand Epileptogenesis and Develop Anti-epileptogenic Treatments
The development of PTE is likely driven by multiple factors which over time transform healthy brain networks in vivo into networks that generate seizures. Because of the in vivo nature of PTE, relatively few purely in vitro approaches exist to model epileptogenesis. One exception to this rule is the organotypic slice culture model of epilepsy developed by the Staley Lab. In this approach, hippocampal brain slices are prepared from neonatal rodents and cultured in vitro for weeks to months. Interestingly, over this time window, organotypic slices undergo dynamic changes in neuronal activities that eventually evolve into in vitro seizure-like events. In the first week of culture, only spike-like activity occurs. By 3 weeks in culture, > 50% of slices display prolonged, ictal-like activity [104, 105]. This evolution of ictal-like activity has been harnessed to investigate candidate mechanisms of epileptogenesis, like PI3K-Akt signaling [106, 107] as well as to perform drug screening aimed at identifying novel anti-convulsant and anti-epileptogenic therapies [108]. When combined with in vivo validation, this approach may hold promise as an alternative to purely in vivo–based drug discovery for anti-convulsant and anti-epileptogenic therapies.
As novel approaches are developed to grow cerebral organoids in vitro from induced pluripotent stem cells (iPSCs), new ways to model TBI and PTE will emerge. Intriguing recent work examines how contusional injury, similar to the controlled cortical impact model, affects neuronal viability, neurotransmission, and cell signaling in cerebral organoids [109]. This study shows that neurons die, that glutamate is released, and that pAKT and GSK3b signaling are reduced after physical injury in cerebral organoids. Whether cerebral organoids go onto develop something like PTE remains to be seen, but this is an exciting step towards developing additional in vitro assays for TBI and later PTE.
In addition to this purely in vitro approach, by coupling in vivo injury with in vitro assays, many studies have identified potential mechanisms of PTE and assayed the effects of various drugs. These include neuronal cell death, excitatory neuron sprouting, neuroinflammation, changes in metabolic activity, glial cell dysfunction, and more.
Conclusions
Recent successes in modeling various epileptogenic human brain injuries in rodents and larger animals [110], in identifying subjects at risk for PTE (high CSF/serum Il-1β, N3-REM spindles), and in pinpointing epileptogenic regions in the injured brain (HFOs) provide promise that novel approaches to identify and treat PTE are closer than ever before. Recent emphasis on clinically relevant study designs and outcome measures, combined with statistically powered preclinical multicenter studies can be expected to advance the field remarkably over the next years [111]. There is hope on the horizon.
References
https://www.who.int/news-room/fact-sheets/detail/epilepsy
Pitkänen A, Engel Jr. J. Past and Present Definitions of Epileptogenesis and Its Biomarkers. Neurotherapeutics. 2014;11(2).
Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935-1984. Epilepsia. 1993;34(3):453–68.
Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia [Internet]. 2017;58(4):512–21. Available from: http://www.ncbi.nlm.nih.gov/pubmed/28276062
Annegers JF, Hauser WA, Coan SP, Rocca WA. A population-based study of seizures after traumatic brain injuries. N Engl J Med [Internet]. 1998 Jan 1 [cited 2016 Feb 22];338(1):20–4. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9414327
Pitkänen A, Immonen R. Epilepsy related to traumatic brain injury. Neurotherapeutics [Internet]. 2014 Apr;11(2):286–96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24554454
Frey LC. Epidemiology of posttraumatic epilepsy: a critical review. Epilepsia [Internet]. 2003;44 Suppl 1:11–7. Available from: http://www.ncbi.nlm.nih.gov/pubmed/14511389
Salazar AM, Grafman J. Post-traumatic epilepsy. clinical clues to pathogenesis and paths to prevention. Handb Clin Neurol. 2015;128:525–38.
Herman ST. Epilepsy after brain insult: targeting epileptogenesis. Neurology. 2002;59:S21–6.
Maas AIR, Menon DK, Adelson PD, Andelic N, Bell MJ, Belli A, et al. Traumatic brain injury: integrated approaches to improve prevention, clinical care, and research. Lancet Neurol [Internet]. 2017;16(12):987–1048. Available from: http://www.ncbi.nlm.nih.gov/pubmed/29122524
Menon DK, Schwab K, Wright DW, Maas AI, Demographics and Clinical Assessment Working Group of the International and Interagency Initiative toward Common Data Elements for Research on Traumatic Brain Injury and Psychological Health. Position statement: definition of traumatic brain injury. Arch Phys Med Rehabil [Internet]. 2010 Nov;91(11):1637–40. Available from: http://www.ncbi.nlm.nih.gov/pubmed/21044706
Jennett WB. Early traumatic epilepsy. Definition and identity. Lancet (London, England) [Internet]. 1969 May 24;1(7604):1023–5. Available from: http://www.ncbi.nlm.nih.gov/pubmed/4181257
Fisher RS, Acevedo C, Arzimanoglou A, Bogacz A, Cross JH, Elger CE, et al. ILAE official report: a practical clinical definition of epilepsy. Epilepsia. 2014 Apr;55(4):475–82.
Reid WM, Rolfe A, Register D, Levasseur JE, Churn SB, Sun D. Strain-related differences after experimental traumatic brain injury in rats. J Neurotrauma. 2010;27(7):1243–53.
Englander J, Bushnik T, Duong TT, Cifu DX, Zafonte R, Wright J, et al. Analyzing risk factors for late posttraumatic seizures: A prospective, multicenter investigation. Arch Phys Med Rehabil. 2003;84(3 SUPPL. 1):365–73.
Koenig JB, Dulla CG. Dysregulated glucose metabolism as a therapeutic target to reduce post-traumatic epilepsy. Front Cell Neurosci. 2018;12(October):1–19.
Engel J, Pitkänen A. Biomarkers for epileptogenesis and its treatment. Neuropharmacology [Internet]. 2020 May;167:107735. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0028390819302941
Sharma S, Tiarks G, Haight J, Bassuk AG. Neuropathophysiological Mechanisms and Treatment Strategies for Post-traumatic Epilepsy. Front Mol Neurosci. 2021;14(February).
II RLR, PENRY JK. Pharmacologic Prophylaxis of Posttraumatic Epilepsy A Review. Epilepsia. 1972;13(2):295–304.
Hoff H, Hoff H. Fortschritte in der Behandlung der Epilepsie. Mschr Psychiat Neurol. 1947;(114):105–18.
Birkmayer W. Die Behandlung der tramatischen Epilepsie. Wen klin Wschr. 1951;(63):606–9.
Temkin NR. Antiepileptogenesis and seizure prevention trials with antiepileptic drugs: Meta-analysis of controlled trials. Epilepsia. 2001;42(4):515–24.
Benassi SK, Alves JGSM, Guidoreni CG, Massant CG, Queiroz CM, Garrido-Sanabria E, et al. Two decades of research towards a potential first anti-epileptic drug. Seizure [Internet]. 2021;(February). Available from: https://doi.org/10.1016/j.seizure.2021.02.031
Carney N, Totten AM, O’Reilly C, Ullman JS, Hawryluk GWJ, Bell MJ, et al. Guidelines for the Management of Severe Traumatic Brain Injury, Fourth Edition. Neurosurgery. 2017;80(1):6–15.
Golarai G, Greenwood a C, Feeney DM, Connor J a. Physiological and structural evidence for hippocampal involvement in persistent seizure susceptibility after traumatic brain injury. J Neurosci. 2001;21(21):8523–37.
Chrzaszcz M, Venkatesan C, Dragisic T, Watterson DM, Wainwright MS. Minozac treatment prevents increased seizure susceptibility in a mouse “two-hit” model of closed skull traumatic brain injury and electroconvulsive shock-induced seizures. J Neurotrauma. 2010;27(7):1283–95.
Ghadiri T, Vakilzadeh G, Hajali V, Khodagholi F. Progesterone modulates post-traumatic epileptogenesis through regulation of BDNF-TrkB signaling and cell survival-related pathways in the rat hippocampus. Neurosci Lett [Internet]. 2019;709(June):134384. Available from: https://doi.org/10.1016/j.neulet.2019.134384
HAMM RJ, PIKE BR, TEMPLE MD, ODELL DM, LYETH BG. the Effect of Postinjury Kindled Seizures on Cognitive Performance of Traumatically Brain-Injured Rats. Vol. 136, Experimental Neurology. 1995. p. 143–8.
D’Ambrosio R, Fairbanks JP, Fender JS, Born DE, Doyle DL, Miller JW. Post-traumatic epilepsy following fluid percussion injury in the rat. Brain. 2004;127(2):304–14.
D’Ambrosio R, Fender JS, Fairbanks JP, Simon EA, Born DE, Doyle DL, et al. Progression from frontal-parietal to mesial-temporal epilepsy after fluid percussion injury in the rat. Brain [Internet]. 2005 Jan;128(Pt 1):174–88. Available from: http://www.ncbi.nlm.nih.gov/pubmed/15563512
D’Ambrosio R, Eastman CL, Darvas F, Fender JS, Verley DR, Farin FM, et al. Mild passive focal cooling prevents epileptic seizures after head injury in rats. Ann Neurol [Internet]. 2013 Feb [cited 2016 Feb 22];73(2):199–209. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3608748&tool=pmcentrez&rendertype=abstract
Atkins CM, Truettner JS, Lotocki G, Sanchez-Molano J, Kang Y, Alonso OF, et al. Post-traumatic seizure susceptibility is attenuated by hypothermia therapy. Eur J Neurosci. 2010;32(11):1912–20.
Bao Y-H, Bramlett HM, Atkins CM, Truettner JS, Lotocki G, Alonso OF, et al. Post-traumatic seizures exacerbate histopathological damage after fluid-percussion brain injury. J Neurotrauma [Internet]. 2011;28(1):35–42. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3019585&tool=pmcentrez&rendertype=abstract
Nemes A, Najm IM, Gale JT, Ying Z, Johnson M, Gonzalez-Martinez J. Underlying Cortical Dysplasia as Risk Factor for Traumatic Epilepsy: An Animal Study. J Neurotrauma. 2016;9:1–9.
Lowenstein DH, Thomas MJ, Smith DH, McIntosh TK. Selective vulnerability of dentate hilar neurons following traumatic brain injury: a potential mechanistic link between head trauma and disorders of the hippocampus. J Neurosci [Internet]. 1992;12(12):4846–53. Available from: http://www.ncbi.nlm.nih.gov/pubmed/1464770
Reeves TM, Lyeth BG, Phillips LL, Hamm RJ, Povlishock JT. The effects of traumatic brain injury on inhibition in the hippocampus and dentate gyrus. Brain Res. 1997;757(1):119–32.
Gurkoff GG, Giza CC, Shin D, Auvin S, Sankar R, Hovda DA. Acute neuroprotection to pilocarpine-induced seizures is not sustained after traumatic brain injury in the developing rat. Neuroscience. 2009;164(2):862–76.
Kharatishvili I, Nissinen JP, McIntosh TK, Pitkänen A. A model of posttraumatic epilepsy induced by lateral fluid-percussion brain injury in rats. Neuroscience. 2006;140(2):685–97.
Kharatishvili I, Immonen R, Gröhn O, Pitkänen A. Quantitative diffusion MRI of hippocampus as a surrogate marker for post-traumatic epileptogenesis. Brain. 2007;130(12).
Echegoyen J, Armstrong C, Morgan RJ, Soltesz I. Single application of a CB1 receptor antagonist rapidly following head injury prevents long-term hyperexcitability in a rat model. Epilepsy Res. 2009;85(1):123–7.
Schwartzkroin PA, Wenzel HJ, Lyeth BG, Poon CC, DeLance A, Van KC, et al. Does ketogenic diet alter seizure sensitivity and cell loss following fluid percussion injury? Epilepsy Res [Internet]. 2010;92(1):74–84. Available from: https://doi.org/10.1016/j.eplepsyres.2010.08.009
Bolkvadze T, Pitkänen A. Development of post-traumatic epilepsy after controlled cortical impact and lateral fluid-percussion-induced brain injury in the mouse. J Neurotrauma. 2012;29(5).
Shultz SR, Cardamone L, Liu YR, Edward Hogan R, MacCotta L, Wright DK, et al. Can structural or functional changes following traumatic brain injury in the rat predict epileptic outcome? Epilepsia. 2013;54(7):1240–50.
Mukherjee S, Zeitouni S, Cavarsan CF, Shapiro LA. Increased seizure susceptibility in mice 30 days after fluid percussion injury. Front Neurol. 2013;4 MAR(March):1–11.
Goodrich GS, Kabakov AY, Hameed MQ, Dhamne SC, Rosenberg PA, Rotenberg A. Ceftriaxone treatment after traumatic brain injury restores expression of the glutamate transporter, GLT-1, reduces regional gliosis, and reduces post-traumatic seizures in the rat. J Neurotrauma [Internet]. 2013;30(16):1434–41. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=3741415&tool=pmcentrez&rendertype=abstract
Hameed MQ, Goodrich GS, Dhamne SC, Amandusson A, Hsieh T-H, Mou D, et al. A rapid lateral fluid percussion injury rodent model of traumatic brain injury and post-traumatic epilepsy. Neuroreport [Internet]. 2014;25:532–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24553065
Campbell JN, Gandhi A, Singh B, Churn SB. Traumatic Brain Injury Causes a Tacrolimus-Sensitive Increase in Non-Convulsive Seizures in a Rat Model of Post-Traumatic Epilepsy. Int J Neurol brain Disord [Internet]. 2014;1(1):1–11. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4287390&tool=pmcentrez&rendertype=abstract
Wang X, Wang Y, Zhang C, Liu C, Yang H-F, Hu W-H, et al. Endogenous cannabinoid system alterations and their role in epileptogenesis after brain injury in rat. Epilepsy Res [Internet]. 2016;128:35–42. Available from: http://linkinghub.elsevier.com/retrieve/pii/S0920121116302406
Reid AY, Bragin A, Giza CC, Staba RJ, Engel J. The progression of electrophysiologic abnormalities during epileptogenesis after experimental traumatic brain injury. Epilepsia. 2016;57(10):1558–67.
Wang X, Wang Y, Zhang C, Liu C, Zhao B, Wei N, et al. CB1 receptor antagonism prevents long-term hyperexcitability after head injury by regulation of dynorphin-KOR system and mGluR5 in rat hippocampus. Brain Res [Internet]. 2016;1646:174–81. Available from: https://doi.org/10.1016/j.brainres.2016.05.055
Statler KD, Scheerlinck P, Pouliot W, Hamilton M, White HS, Dudek FE. A potential model of pediatric posttraumatic epilepsy. Epilepsy Res. 2009;86(2–3):221–3.
Hunt RF, Scheff SW, Smith BN. Posttraumatic epilepsy after controlled cortical impact injury in mice. Exp Neurol [Internet]. 2009;215(2):243–52. Available from: https://doi.org/10.1016/j.expneurol.2008.10.005
Hunt RF, Scheff SW, Smith BN. Regionally localized recurrent excitation in the dentate gyrus of a cortical contusion model of posttraumatic epilepsy. J Neurophysiol [Internet]. 2010 Mar;103(3):1490–500. Available from: http://www.ncbi.nlm.nih.gov/pubmed/20089815
Guo D, Zeng L, Brody DL, Wong M. Rapamycin Attenuates the Development of Posttraumatic Epilepsy in a Mouse Model of Traumatic Brain Injury. PLoS One. 2013;8(5).
Butler CR, Boychuk J a, Smith BN. Effects of Rapamycin Treatment on Neurogenesis and Synaptic Reorganization in the Dentate Gyrus after Controlled Cortical Impact Injury in Mice. Front Syst Neurosci [Internet]. 2015;9(November):163. Available from: http://www.pubmedcentral.nih.gov/articlerender.fcgi?artid=4661228&tool=pmcentrez&rendertype=abstract
Kelly KM, Miller ER, Lepsveridze E, Kharlamov EA, Mchedlishvili Z. Posttraumatic seizures and epilepsy in adult rats after controlled cortical impact. Epilepsy Res [Internet]. 2015;117:104–16. Available from: https://doi.org/10.1016/j.eplepsyres.2015.09.009
Ping X, Jin X. Transition from Initial Hypoactivity to Hyperactivity in Cortical Layer V Pyramidal Neurons after Traumatic Brain Injury In Vivo. J Neurotrauma. 2016;33:354–61.
Eslami M, Ghanbari E, Sayyah M, Etemadi F, Choopani S, Soleimani M, et al. Traumatic brain injury accelerates kindling epileptogenesis in rats. Neurol Res. 2015;6412(7):1743132815Y0000000086.
Bolkvadze T, Rantala J, Puhakka N, Andrade P, Pitkänen A. Epileptogenesis after traumatic brain injury in Plau-deficient mice. Epilepsy Behav [Internet]. 2015 Oct;51:19–27. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26253597
Bolkvadze T, Puhakka N, Pitkänen A. Epileptogenesis after traumatic brain injury in Plaur-deficient mice. Epilepsy Behav [Internet]. 2016 Jul;60:187–96. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27208924
Miszczuk D, Dębski KJ, Tanila H, Lukasiuk K, Pitkänen A. Traumatic Brain Injury Increases the Expression of Nos1, Aβ Clearance, and Epileptogenesis in APP/PS1 Mouse Model of Alzheimer’s Disease. Mol Neurobiol. 2016;53(10).
Bugay V, Bozdemir E, Vigil FA, Chun SH, Holstein DM, Elliott WR, et al. A Mouse Model of Repetitive Blast Traumatic Brain Injury Reveals Post-Trauma Seizures and Increased Neuronal Excitability. J Neurotrauma. 2020;37(2):248–61.
Nissinen J, Andrade P, Natunen T, Hiltunen M, Malm T, Kanninen K, et al. Disease-modifying effect of atipamezole in a model of post-traumatic epilepsy. Epilepsy Res [Internet]. 2017 Oct;136:18–34. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0920121117302061
Saraiva ALL, Ferreira APO, Silva LFA, Hoffmann MS, Dutra FD, Furian AF, et al. Creatine reduces oxidative stress markers but does not protect against seizure susceptibility after severe traumatic brain injury. Brain Res Bull [Internet]. 2012 Feb 10;87(2–3):180–6. Available from: http://www.ncbi.nlm.nih.gov/pubmed/22051612
Gerbatin RR, Silva LFA, Hoffmann MS, Della-Pace ID, do Nascimento PS, Kegler A, et al. Delayed creatine supplementation counteracts reduction of GABAergic function and protects against seizures susceptibility after traumatic brain injury in rats. Prog Neuro-Psychopharmacology Biol Psychiatry. 2019;92(January):328–38.
Raible DJ, Frey LC, Del Angel YC, Carlsen J, Hund D, Russek SJ, et al. JAK/STAT pathway regulation of GABAA receptor expression after differing severities of experimental TBI. Exp Neurol [Internet]. 2015;271:445–56. Available from: https://doi.org/10.1016/j.expneurol.2015.07.001
Liu SJ, Zheng P, Wright DK, Dezsi G, Braine E, Nguyen T, et al. Sodium selenate retards epileptogenesis in acquired epilepsy models reversing changes in protein phosphatase 2A and hyperphosphorylated tau. Brain. 2016;139(7):1919–38.
Neuberger EJ, Swietek B, Corrubia L, Prasanna A, Santhakumar V. Enhanced Dentate Neurogenesis after Brain Injury Undermines Long-Term Neurogenic Potential and Promotes Seizure Susceptibility. Stem Cell Reports [Internet]. 2017;9(3):972–84. Available from: https://doi.org/10.1016/j.stemcr.2017.07.015
Cantu D, Croker D, Shacham S, Tamir S, Dulla C. In vivo KPT-350 treatment decreases cortical hyperexcitability following traumatic brain injury. Brain Inj [Internet]. 2020;34(11):1489–96. Available from: https://doi.org/10.1080/02699052.2020.1807056
Yang L, Afroz S, Valsamis HA, Michelson HB, Goodman JH, Ling DSF. Early intervention with levetiracetam prevents the development of cortical hyperexcitability and spontaneous epileptiform activity in two models of neurotrauma in rats. Exp Neurol [Internet]. 2021;337(December 2020):113571. Available from: https://doi.org/10.1016/j.expneurol.2020.113571
Silva LFA, Hoffmann MS, Gerbatin R da R, Fiorin F da S, Dobrachinski F, Mota BC, et al. Treadmill exercise protects against pentylenetetrazol-induced seizures and oxidative stress after traumatic brain injury. J Neurotrauma [Internet]. 2013 Jul 15;30(14):1278–87. Available from: http://www.ncbi.nlm.nih.gov/pubmed/23530735
Zhu B, Eom J, Hunt RF. Transplanted interneurons improve memory precision after traumatic brain injury. Nat Commun [Internet]. 2019;10(1):1–12. Available from: https://doi.org/10.1038/s41467-019-13170-w
Diamond ML, Ritter AC, Failla MD, Boles JA, Conley YP, Kochanek PM, et al. IL-1β associations with posttraumatic epilepsy development: A genetics and biomarker cohort study. Epilepsia [Internet]. 2015 Jul;56(7):991–1001. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26149793
Mazarati A, Medel-Matus J-S, Shin D, Jacobs JP, Sankar R. Disruption of intestinal barrier and endotoxemia after traumatic brain injury: Implications for post-traumatic epilepsy. Epilepsia. 2021 Apr;
Pitkänen A, Immonen R. Epilepsy Related to Traumatic Brain Injury. Neurotherapeutics. 2014;11(2).
Manninen E, Chary K, Lapinlampi N, Andrade P, Paananen T, Sierra A, et al. Acute thalamic damage as a prognostic biomarker for post‐traumatic epileptogenesis. Epilepsia [Internet]. 2021 Aug 9;62(8):1852–64. Available from: https://doi.org/10.1111/epi.16986
Messori A, Polonara G, Carle F, Gesuita R, Salvolini U. Predicting posttraumatic epilepsy with MRI: prospective longitudinal morphologic study in adults. Epilepsia [Internet]. 2005 Sep [cited 2016 Feb 22];46(9):1472–81. Available from: http://www.ncbi.nlm.nih.gov/pubmed/16146443
Pitkänen A, Löscher W, Vezzani A, Becker AJ, Simonato M, Lukasiuk K, et al. Advances in the development of biomarkers for epilepsy. Lancet Neurol [Internet]. 2016;15(8):843–56. Available from: https://doi.org/10.1016/S1474-4422(16)00112-5
Kim JA, Boyle EJ, Wu AC, Cole AJ, Staley KJ, Zafar S, et al. Epileptiform activity in traumatic brain injury predicts post-traumatic epilepsy. Ann Neurol. 2018;83(4):858–62.
Tubi MA, Lutkenhoff E, Blanco MB, McArthur D, Villablanca P, Ellingson B, et al. Early seizures and temporal lobe trauma predict post-traumatic epilepsy: A longitudinal study. Neurobiol Dis [Internet]. 2019 Mar;123:115–21. Available from: https://linkinghub.elsevier.com/retrieve/pii/S0969996118301529
Andrade P, Nissinen J, Pitkänen A. Generalized Seizures after Experimental Traumatic Brain Injury Occur at the Transition from Slow-Wave to Rapid Eye Movement Sleep. J Neurotrauma. 2017;34(7).
Bragin A, Li L, Almajano J, Alvarado-Rojas C, Reid AY, Staba RJ, et al. Pathologic electrographic changes after experimental traumatic brain injury. Epilepsia [Internet]. 2016 May;57(5):735–45. Available from: http://www.ncbi.nlm.nih.gov/pubmed/27012461
Lapinlampi N, Andrade P, Paananen T, Hämäläinen E, Ekolle Ndode‐Ekane X, Puhakka N, et al. Postinjury weight rather than cognitive or behavioral impairment predicts development of posttraumatic epilepsy after lateral fluid‐percussion injury in rats. Epilepsia [Internet]. 2020 Sep 12;61(9):2035–52. Available from: https://doi.org/10.1111/epi.16632
Di Sapia R, Moro F, Montanarella M, Iori V, Micotti E, Tolomeo D, et al. In-depth characterization of a mouse model of post-traumatic epilepsy for biomarker and drug discovery. Acta Neuropathol Commun. 2021 Apr;9(1):76.
Bragin A, Engel J, Wilson CL, Fried I, Mathern GW. Hippocampal and entorhinal cortex high-frequency oscillations (100--500 Hz) in human epileptic brain and in kainic acid--treated rats with chronic seizures. Epilepsia [Internet]. 1999 Feb;40(2):127–37. Available from: http://www.ncbi.nlm.nih.gov/pubmed/9952257
Liu S, Gurses C, Sha Z, Quach MM, Sencer A, Bebek N, et al. Stereotyped high-frequency oscillations discriminate seizure onset zones and critical functional cortex in focal epilepsy. Brain [Internet]. 2018 Jan 30; Available from: http://www.ncbi.nlm.nih.gov/pubmed/29394328
Roehri N, Pizzo F, Lagarde S, Lambert I, Nica A, McGonigal A, et al. High-frequency oscillations are not better biomarkers of epileptogenic tissues than spikes. Ann Neurol [Internet]. 2017 Dec 15; Available from: http://www.ncbi.nlm.nih.gov/pubmed/29244226
Simonato M, Agoston D V., Brooks-Kayal A, Dulla C, Fureman B, Henshall DC, et al. Identification of clinically relevant biomarkers of epileptogenesis — a strategic roadmap. Nat Rev Neurol. 2021;17(4):231–42.
Cotter D, Kelso A, Neligan A. Genetic biomarkers of posttraumatic epilepsy: A systematic review. Seizure [Internet]. 2017;46:53–8. Available from: https://doi.org/10.1016/j.seizure.2017.02.002
Scher AI, Wu H, Tsao JW, Blom HJ, Feit P, Nevin RL, et al. MTHFR C677T genotype as a risk factor for epilepsy including post-traumatic epilepsy in a representative military cohort. J Neurotrauma. 2011;28(9):1739–45.
Darrah SD, Miller MA, Ren D, Hoh NZ, Scanlon JM, Conley YP, et al. Genetic variability in glutamic acid decarboxylase genes: Associations with post-traumatic seizures after severe TBI. Epilepsy Res [Internet]. 2013;103(2–3):180–94. Available from: https://doi.org/10.1016/j.eplepsyres.2012.07.006
Cantu D, Walker K, Andresen L, Taylor-Weiner A, Hampton D, Tesco G, et al. Traumatic Brain Injury Increases Cortical Glutamate Network Activity by Compromising GABAergic Control. Cereb Cortex. 2015;25(8):2306–20.
Huusko N, Pitkänen A. Parvalbumin immunoreactivity and expression of GABAA receptor subunits in the thalamus after experimental TBI. Neuroscience [Internet]. 2014 May 16;267:30–45. Available from: http://www.ncbi.nlm.nih.gov/pubmed/24607347
Huusko N, Römer C, Ndode-Ekane XE, Lukasiuk K, Pitkänen A. Loss of hippocampal interneurons and epileptogenesis: a comparison of two animal models of acquired epilepsy. Brain Struct Funct. 2013;220(1).
Hunt RF, Baraban SC. Interneuron transplantation as a treatment for epilepsy. Cold Spring Harb Perspect Med. 2015;5(12).
Wagner AK, Miller MA, Scanlon J, Ren D, Kochanek PM, Conley YP. Adenosine A1 receptor gene variants associated with post-traumatic seizures after severe TBI. Epilepsy Res [Internet]. 2010;90(3):259–72. Available from: https://doi.org/10.1016/j.eplepsyres.2010.06.001
Etherington LA V., Frenguelli BG. Endogenous adenosine modulates epileptiform activity in rat hippocampus in a receptor subtype-dependent manner. Eur J Neurosci. 2004;19(9):2539–50.
Dale N, Frenguelli B. Release of Adenosine and ATP During Ischemia and Epilepsy. Curr Neuropharmacol. 2009;7(3):160–79.
Kochanek PM, Vagni VA, Janesko KL, Washington CB, Crumrine PK, Garman RH, et al. Adenosine A1 receptor knockout mice develop lethal status epilepticus after experimental traumatic brain injury. J Cereb Blood Flow Metab. 2006;26(4):565–75.
Teasdale G, Jennett B. Assessment of coma and impaired consciousness. A practical scale. Lancet (London, England). 1974 Jul;2(7872):81–4.
Jordan BD, Relkin NR, Ravdin LD, Jacobs AR, Bennett A, Gandy S. Apolipoprotein E ε4 associated with chronic traumatic brain injury in boxing. J Am Med Assoc. 1997;278(2):136–40.
Miszczuk D, Dębski KJ, Tanila H, Lukasiuk K, Pitkänen A. Traumatic Brain Injury Increases the Expression of Nos1, Aβ Clearance, and Epileptogenesis in APP/PS1 Mouse Model of Alzheimer’s Disease. Mol Neurobiol [Internet]. 2016 Dec;53(10):7010–27. Available from: http://www.ncbi.nlm.nih.gov/pubmed/26671618
Pijet B, Stefaniuk M, Kostrzewska-Ksiezyk A, Tsilibary PE, Tzinia A, Kaczmarek L. Elevation of MMP-9 Levels Promotes Epileptogenesis After Traumatic Brain Injury. Mol Neurobiol. 2018;55(12):9294–306.
Dyhrfjeld-Johnsen J, Berdichevsky Y, Swiercz W, Sabolek H, Staley KJ. Interictal spikes precede ictal discharges in an organotypic hippocampal slice culture model of epileptogenesis. J Clin Neurophysiol. 2010;27(6):418–24.
Lillis KP, Wang Z, Mail M, Zhao GQ, Berdichevsky Y, Bacskai B, et al. Evolution of network synchronization during early epileptogenesis parallels synaptic circuit alterations. J Neurosci. 2015;35(27):9920–34.
Berdichevsky Y, Dryer AM, Saponjian Y, Mahoney MM, Pimentel CA, Lucini CA, et al. PI3K-Akt signaling activates mTOR-mediated epileptogenesis in organotypic hippocampal culture model of post- Traumatic epilepsy. J Neurosci. 2013;33(21):9056–67.
Song Y, Pimentel C, Walters K, Boller L, Ghiasvand S, Liu J, et al. Neuroprotective levels of IGF-1 exacerbate epileptogenesis after brain injury. Sci Rep [Internet]. 2016;6:1–12. Available from: https://doi.org/10.1038/srep32095
Berdichevsky Y, Saponjian Y, Park K Il, Roach B, Pouliot W, Lu K, et al. Staged anticonvulsant screening for chronic epilepsy. Ann Clin Transl Neurol. 2016;3(12):908–23.
Liaudanskaya V, Chung JY, Mizzoni C, Rouleau N, Berk AN, Wu L, et al. Modeling Controlled Cortical Impact Injury in 3D Brain-Like Tissue Cultures. Adv Healthc Mater. 2020;9(12):1–14.
Ulyanova A, Cottone C, Litt B, Chen I, Johnson V, Wolf J. A translational model of post-traumatic epileptogenesis. J Neurotrauma. 2019;36(13):B08-06.
Pitkänen A, Paananen T, Kyyriäinen J, Das Gupta S, Heiskanen M, Vuokila N, et al. Biomarkers for posttraumatic epilepsy. Epilepsy Behav [Internet]. 2020 Apr 18;107080. Available from: http://www.ncbi.nlm.nih.gov/pubmed/32317161
Funding
This study was supported by the Medical Research Council of the Academy of Finland (Grants 272249, 273909, and 2285733–9) (AP), the European Union’s Seventh Framework Programme (FP7/2007–2013) under grant agreement n°602102 (EPITARGET) (AP), the National Institute of Neurological Disorders and Stroke (NINDS) (Centres without Walls (U54 NS100064) (AP), R33 NS096948 (CD), R01 NS113499 (CD), R01 NS100706 (CD)), and the Department of Defense (W81XWH-18–1-0669 (CD) and W81XWH-17–1-0531 (CD)).
Funding
Disclosure forms provided by the authors are available with the online version of this article.
Author information
Authors and Affiliations
Corresponding authors
Additional information
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary Information
Below is the link to the electronic supplementary material.
Rights and permissions
About this article

Cite this article
Dulla, C. ., Pitkänen, A. Novel Approaches to Prevent Epileptogenesis After Traumatic Brain Injury. Neurotherapeutics 18, 1582–1601 (2021). https://doi.org/10.1007/s13311-021-01119-1
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s13311-021-01119-1