
Abstract
Docosahexaenoic acid (DHA) and neuroprotectin D1 (NPD1) are neuroprotective after experimental ischemic stroke. To explore underlying mechanisms, SD rats underwent 2 h of middle cerebral artery occlusion (MCAo) and treated with DHA (5 mg/kg, IV) or NPD1 (5 μg/per rat, ICV) and vehicles 1 h after. Neuro-behavioral assessments was conducted on days 1, 2, and 3, and on week 1, 2, 3, or 4. BrdU was injected on days 4, 5, and 6, immunohistochemistry was performed on week 2 or 4, MRI on day 7, and lipidomic analysis at 4 and 5 h after onset of stroke. DHA improved short- and long-term behavioral functions and reduced cortical, subcortical, and total infarct volumes (by 42, 47, and 31%, respectively) after 2 weeks and reduced tissue loss by 50% after 4 weeks. DHA increased the number of BrdU+/Ki-67+, BrdU+/DCX+, and BrdU+/NeuN+ cells in the cortex, subventricular zone, and dentate gyrus and potentiated NPD1 synthesis in the penumbra at 5 h after MCAo. NPD1 improved behavior, reduced lesion volumes, protected ischemic penumbra, increased NeuN, GFAP, SMI-71-positive cells and vessels, axonal regeneration in the penumbra, and attenuated blood-brain barrier (BBB) after MCAo. We conclude that docosanoid administration increases neurogenesis and angiogenesis, activates NPD1 synthesis in the penumbra, and diminishes BBB permeability, which correlates to long-term neurobehavioral recovery after experimental ischemic stroke.
Similar content being viewed by others


Avoid common mistakes on your manuscript.
Introduction
Stroke is the fifth leading cause of death in the USA and the primary cause of long-term disability [1]. Only one FDA-approved therapy exists for treatment of acute ischemic stroke, the thrombolytic tissue plasminogen activator (tPA), but unfortunately, only 3–5% of patients can be elected to undergo this treatment [2, 3]. Therefore, an effective treatment for stroke remains an unmet health care need.
Currently, translational enabling research strategies, in addition to neuroprotection, include neurorestoration to enhance post-stroke tissue repair [4, 5]. A mechanism for neurorestoration is neurogenesis activation, which occurs in the subgranular zone of the dentate gyrus (DG) and the subventricular zone of the lateral ventricles [5]. Newborn neurons are generated from neural stem/progenitor cells in the subgranular zone (SGZ) of the DG through differentiation and maturation. Ischemic injury induces neurogenesis and promotes migration of neuroblasts along the blood vessels into the damaged area [6]. Thus, neurogenesis may lead to functional recovery of the ischemic area through proliferation, migration, differentiation, and integration of newly generated neural cells into circuits [7]. Moreover, neurogenesis and angiogenesis leading to repair implement structural and functional restoration of the injured brain and represent targets for potential mediators [4].
A candidate for neuro-reparative modulation is docosahexaenoic acid (DHA; 22:6, n-3), a member of the omega-3 (n-3) polyunsaturated fatty acids (PUFAs) family [8], which promotes neuronal differentiation and neurite growth [9]. Several signaling pathways might be involved in the induction of neural differentiation by DHA [10], and dietary supplementation of n-3 PUFA boosts adult hippocampal neurogenesis associated with cognitive and behavioral processes, promotes synaptic plasticity, increases long-term potentiation, modulates synaptic protein expression, and stimulates dendritic arborization and new spines formation [11]. Moreover, DHA increases the abundance of growth-associated protein-43 (GAP-43), a protein associated with growth cone formation as a marker of axonal growth [12]. Although neurogenesis is a key in long-term cognitive and functional recovery after brain injury [13,14,15,16], there are no effective targeted strategies to enhance neurogenesis. There have been limitations to apply this knowledge to clinical purposes, which include lack of effective targeted strategies to enhance neurogenesis, difficulty crossing BBB, and unwanted or intolerable side effects. Focal cerebral ischemic injury triggers molecular and cellular repair mechanisms, which contribute to recovery and activate neural progenitor proliferation and cell migration [13, 17, 18].
The morbidity after ischemic stroke is lower in populations with high intakes of n-3 PUFA [19]. A diet rich in n-3 PUFA, like DHA, also lowers the risk of several diseases, including heart disease and age-related macular degeneration [20, 21]. DHA is an n-3 PUFA that is involved in neurological development, vision, and cognition [20]. Supplementation with the n-3 PUFA eicosapentaenoic acid and DHA increased hippocampal neurogenesis in aged rats and stimulated immediate early neurogenesis events that promoted neuronal differentiation by increasing the population of neurons with longer neurites [22,23,24,25]. DHA fosters neurite growth in hippocampal neurons [26], upregulates maintenance of both neurogenic and gliogenic neural stem/progenitor cells [27], and promotes neurite outgrowth of embryonic hippocampal cells and embryonic cortical cells [28].
DHA improves behavioral function, decreases infarct volume, attenuates BBB permeability and promotes cell survival in the ischemic penumbra and attenuates cerebral edema in 1 week of survival after focal cerebral ischemia in rats [29,30,31]. DHA is the precursor of bioactive mediators, the docosanoids [8, 32]. Neuroprotectin D1 (NPD1; 10R,17S-dihydroxy-docosa-4Z,7Z,11E,13E,15Z,19Z-hexaenoic acid) is produced at the onset of brain ischemia-reperfusion [33]. When given to the third ventricle after middle cerebral artery occlusion (MCAo), it inhibits polymorphonuclear neutrophil (PMN) infiltration, downregulates ischemia-induced gene expression, and reduces stroke-mediated tissue damage [33]. The neuroprotective bioactivity of NPD1 includes inflammatory modulation, induction of cell survival, and restoring disrupted homeostasis [8, 34,35,36,37].
Although neurorestorative and neuroprotective actions of DHA have been demonstrated after experimental stroke, the effect of NPD1 administration has not been fully characterized so far. We used non-invasive magnetic resonance imaging (MRI) and LC-MS/MS-mediator lipidomic analysis in conjunction with behavioral, histological, and immunostaining methods to define mechanisms of DHA action after stroke in rats.
Material and Methods
Animals
All studies were in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the US Department of Health and Human Services and were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center (LSUHSC), New Orleans. Male Sprague-Dawley (SD) rats (3–4 months old; Charles River Laboratory, Wilmington, MA) were fasted overnight with free access to water prior to the surgical procedure. Anesthesia was induced by the inhalation of 3% isoflurane in 70% NO and 30% O2 mixed gases, and then maintained with 1% isoflurane in the same mixed gases during the procedure. Orally intubated animals were mechanically ventilated after the immobilization by injection of pancuronium bromide (0.6 mg/kg, IV). The catheters were implanted into the right femoral artery and vein for the blood sampling and drug infusion. Serial analyses of arterial blood gases, plasma glucose, arterial blood pressure, and heart rate were conducted before, during, and after surgical procedure. Rectal (CMA/150 Temperature Controller, CMA/Microdialysis AB, Stockholm, Sweden) and cranial (temporalis muscle; Omega Engineering, Stamford, CT) temperatures were closely monitored before, during, and after MCAo. Rectal temperature and body weight were monitored daily until sacrifice.
Transient Middle Cerebral Artery Occlusion
The right middle cerebral artery (MCA) was occluded by poly-l-lysine coated filament [38]. A 4-cm of 3-0 monofilament nylon suture coated with poly-lysine was introduced via the proximal ECA into the internal carotid artery and MCA. The correct suture position was confirmed by feeling a certain resistance during filament forwarding or by advancing the suture a defined distance (21–22 mm) from the common carotid artery bifurcation. Then, the animals were allowed to awaken from anesthesia and returned to their cages. After 2 h of MCAo, rats were re-anesthetized with the same anesthetic combination and intraluminal sutures were removed. The neck incision was closed with silk sutures, and the animals were allowed to survive with free access to food and water. Confirmation of MCAo: Animals were allowed to awaken from anesthesia, and after 60 min, they were tested on a standardized neurobehavioral battery (0 = normal and 12 = maximal deficit) to confirm the presence of a high-grade neurologic deficit. Only rats with a high-grade neurological deficit (10 or greater) were used. Thirty minutes after suture removal, a brain infusion cannula was implanted into the right lateral ventricle for NPD1 administration in each rat. Briefly, rats were anesthetized with 3% isoflurane and were secured to a stereotaxic apparatus with skull leveled between the bregma and lambda. A sterile stainless-steel cannula (5 mm long) was implanted into the lateral ventricle using the stereotaxic coordinates (0.2 mm caudal to bregma, 2 mm lateral to midline, and 5 mm below the dura). Cannulas were removed after treatment was completed.
Treatments
Animals were randomly assigned to four treatment groups: DHA, Saline, NPD1, and CSF. DHA (5 mg/kg, IV, Cayman, Ann Arbor, MI) was dissolved in saline and administered IV 1 h after 2 h of MCAo at a constant rate over 3 min using an infusion pump. Vehicle-treated rats received an IV infusion of a comparable volume of 0.9% saline. NPD1 (5 μg/per rat, ICV) was dissolved in artificial cerebrospinal fluid (CSF), and vehicle (CSF) was administered into the right lateral ventricle at a constant rate over 3 min using an infusion pump at 3 h after onset of MCAo. Highly neuroprotective DHA and NPD1 doses in transient MCAo were chosen from previously published studies [39, 40]. All treatments and analysis were administered by researchers blinded to the treatment groups.
Experimental Design
In Series 1
Effect of DHA on neurogenesis during 2 weeks survival: DHA or saline (n = 10–12 per group), behavioral testing (1, 2, 3, and 14 days), 5-bromo-2′-deoxyuridine (BrdU) in vivo labeling was performed on days 4, 5, and 6, histopathology and immunohistochemistry on day 14. In series 2: Effect of DHA on NPD1 synthesis and long-term survival: DHA or saline (n = 6–8 per group), lipidomic analysis at 4 and 5 h, behavioral testing (on days 1, 2, 3, and weeks 1, 2, 3, and 4), histopathology and immunohistochemistry were extended to 4-week duration. In series 3: Effect of NPD1 on ischemic penumbra: NPD1 or CSF (n = 7–10 per group), behavioral testing (on days 1, 2, 3, and 7), MRI, histopathology, and immunohistochemistry on day 7.
Behavioral Tests
A composite neurological battery consisted of two components: (1) a postural reflex test, designed to examine forelimb and upper-body posture in response to tail-suspension and lateral displacement, regarded as being sensitive to both cortical and striatal lesions; and (2) an elicited forelimb placing test, which examines sensorimotor integration by assessing placing reactions to visual, tactile, and proprioceptive stimuli. The total neurologic score was graded on a scale from 0 (normal) to 12 (maximal deficit) [38].
5-Bromo-2′-Deoxyuridine (BrdU) In Vivo Labeling
BrdU was used to label recently proliferated cells. BrdU (50 mg/kg, in saline, Sigma, St Louis, MO) was administered IP on days 4, 5, and 6 after MCAo [41,42,43]. Rats were perfused with 0.9% saline followed by 4% paraformaldehyde on week 2 after onset of MCAo. The brains were removed and then subjected to immunohistochemistry. The sections were incubated in 2 N HCl to denature DNA at RT for 1 h then neutralized with 0.1 M boric acid (pH 8.5) for 5 min twice. After these additional steps, immunohistochemistry was performed as described below.
Histopathology and Immunohistochemistry
Rats were perfused with 4% paraformaldehyde on week 1, 2, or 4 after MCAo. Brains were then removed and embedded in a gelatin matrix using MultiBrain™ Technology (NeuroScience Associates, Knoxville, TN) as previously described [44]. To quantify infarct volume, histological sections were digitized (MCID core imaging software; InterFocus Imaging Ltd., Cambridge, England) at nine standardized coronal levels (bregma levels + 5.2, + 2.7, + 1.2, − 0.3, − 1.3, − 1.8, − 3.8, − 5.0, and − 7.3 mm) using a CCD camera (QICAM Fast 1394, QIMAGING, British Columbia, Canada) [38]. Brain sections were imaged on a motorized microscope BX61VS (Olympus, Japan) at ×10 objective.
An investigator blinded to the experimental groups then outlined the zone of the cortical and subcortical infarct as well as the left and right hemispheres of each section. Infarct volume was calculated as the integrated product of the cross-sectional area and inter-sectional distance and corrected for brain swelling [22]. Brain edema was measured by the differences of ipsi- and contra-lateral hemispheres [22]. Residual (normal) tissue in the right hemisphere, tissue loss, and percent [relative to unlesioned (left) hemisphere volume] was calculated [45].
Immunohistochemistry was performed on adjacent sections at week 2 or 4 after MCAo. The following antibodies were used: NeuN (Chemicon Inc., Billerica, MA, USA) for mature neurons; anti-BrdU (AbD Serotec, Raleigh, NC, USA) for newly proliferated cells; Ki-67 (BD PharMingen, San Jose, CA, USA) as a marker for mitotic cells; Doublecortin (DCX, Santa Cruz Biotechnology, Santa Cruz, CA) as a marker for immature and early post-mitotic neurons; Glial fibrillary acidic protein (GFAP, Dako, Denmark) to label reactive astrocytes; Iba-1 (Wako, Richmond, VA, USA) for activated microglia/microphages; SMI-71 (BioLegend, San Diego, CA), as a rat BBB vascular marker; Hoechst 33342 (Sigma, Oakville, ON, Canada) as a marker for neuronal apoptosis; biotinylated anti-rat immunoglobulin (IgG) antibody (BioLegend, San Diego, CA) to detect neurovascular unit (NVU) breakdown; and anti-phosphorylated growth-associated protein (pGAP-43) (Merck Millipore, Billerica, MA) to detect axonal sprouting density.
The number of positive cells and immunopositive vessels were counted in dentate gyrus (DG), which is composed of the following: crest, lateral blade (suprapyramidal portion) and medial blade (infrapyramidal portion), sub-ventricular zone (at bregma level − 3.8 mm), and in the cortex and striatum at the level of the central lesion (bregma level − 0.3 mm). Data were expressed as numbers of positive cells and vessels per high-power microscopic field (magnification ×40). The images of sections were obtained using confocal laser microscope (LSM510, Carl Zeiss MicroImaging, Irvine, CA) following specific experimental protocols. The images were acquired with dimension 212.3 μm × 212.3 μm using Zen software (Carl Zeiss MicroImaging). Image analysis was conducted using ImageJ software (National Institute of Health, Bethesda, MD). BrdU/Ki-67+, BrdU/DCX+, and BrdU/NeuN+ cells were counted within the image field in the region of DG, sub-ventricular zone, and cortical peri-infarct zone. NeuN+ and GFAP+ cells and Iba-1 fluorescent intensity were analyzed and measured in ischemic penumbra (cortex) and core (subcortex).
IgG staining intensity was calculated and averaged at the same levels as assessed for ischemic damage as previously described [46, 47]. To calculate the intensity of IgG staining, the images were converted to grayscale, and the mean gray values were recorded and compared. ImageJ software assigns black pixels for the numerical value of “0” and white pixels for the numerical value of “1.” Gradations of gray are assigned the numerical values in between, increasing with pixel lightness and decreasing with pixel darkness. As such, IgG intensity values were expressed as the reciprocal of mean gray for graphical clarity. All sections were imaged at the same time with the same settings and with no adjustment to brightness or contrast. IgG stain intensity was measured in the entire contralateral and ipsilateral hemispheres, as well as the cortex and striatum.
Magnetic Resonance Imaging (MRI) Acquisition and Analysis of Total Lesion, Core, and Penumbra Volumes
High-resolution ex vivo MRI was performed on 4% paraformaldehyde-fixed brains on day 7 using an 11.7 T Bruker Advance 8.9-cm horizontal bore instrument equipped with an 89-mm (ID) receiver coil (Bruker Biospin, Billerica, MA). T2WI, diffusion-weighted images (DWI), 3D volumes, and apparent diffusion coefficient (ADC) maps were collected as we previously described [48]. T2 and ADC maps were computed from T2WI and DWI, respectively. We used Hierarchical Region Splitting (HRS) to automatically identify core and penumbra volumes (total lesion = core + penumbra) from T2 relaxation and water mobility (ADC), as we have published previously [32]. Our penumbral tissue determination by HRS (implemented in MATLAB) was validated previously by use of perfusion-weighted imaging (PWI)/DWI subtractions at each brain level [32]. The penumbra from HRS was defined using T2 and ADC values, which were between normal appearing brain tissue and ischemic core. Data from each modality were summarized per group.
Brain Sampling and Lipidomic Analysis
Rats were sacrificed at 4 and 5 h after MCAo to investigate the effect of DHA on NPD1 synthesis in the ischemic penumbra. The brains were removed, divided into right and left hemispheres, and penumbra (cortex) were dissected at bregma level − 0.3 mm for lipidomic analysis (see diagram in Fig. 6). NPD1, 17-hydroxydocosahexaenoic acid (17-HDHA; a stable derivative of 17H(p)DHA, the NPD1 precursor), and 14-hydroxydocosahexaenoic acid (14-HDHA; a precursor of maresin 1) were characterized from the penumbra by liquid chromatography-photodiode array detection-electrospray ionization-tandem mass spectrometry (LC-PDA-ESI-MS/MS) using a TSQ Quantum Ultra UPLC-MS/MS triple stage tandem mass spectrometer from Thermo Electronic Company, Inc., at SRM mode [49].
Lipid Extraction
Brain samples were homogenized with glass homogenizer on ice in such a way that the total volume of homogenate was 3 ml in cold MeOH. Five microliters of internal standard mixture (5 ng of LTB4-d4, 15-HETE-d8, PGD2-d4, EPA-d5 and 25 ng of AA-d8, all purchased from Cayman Chemical, Ann Arbor, MI) was sonicated in the water bath with ice for 30 min. Sample was stored at − 80 °C overnight for the extraction. The following day, C18 column (BondElut 500 mg, Agilent Technologies) was equilibrated with 20 ml cold MeOH followed by 20 ml cold H2O in a vacuum manifold for Solid Phase Extraction. The sample was diluted with cold pH 3.5 H2O such that MeOH concentration is 10~15% and pH of the sample became 3.5~4.0 before loading it to the column. The sample was loaded into the column with 1~2 drops per minute. The column was washed with H2O and 2 ml hexane, and lipid was eluted with 10 ml methylformate. Sample was dried under N2 flow and re-suspended with 10 μl MeOH followed by 5 μl H2O.
Liquid Chromatography/Mass Spectrometry
Xevo TQ-S equipped with Acquity I Class UPLC (Waters) was used for lipidomics. Acquity UPLC HSS T3 1.8 μm 2.1 × 50 mm column was used for fatty acids and their derivatives. Seventy-five percent of solvent A (H2O + 0.1% acetic acid) and 25% of solvent B (90% acetonitrile, 10% isopropanol) with 0.6 ml/min flow were used for the first 1 min, then gradient to 100% of solvent B at 8.5 min followed 100% B until 11 min. Then, the column was re-equilibrated to 75% A and 25% B for 2 min. The capillary voltage was − 2.5 kV, desolvation temperature at 600 °C, desolvation gas flow at 1100 L/h, cone gas at 150 L/h, and nebulizer pressure at 7.0 bar with the source temperature at 150 °C. MassLynx 4.1 software was used for operation and recording the data.
Statistical Analysis
Values are presented as means ± SEM. Repeated-measures analysis of variance (ANOVA), followed by Bonferroni tests, was used for multiple-group comparisons. Two-tailed Student’s t test was used for two-group comparisons. A value of p < 0.05 was regarded as statistically significant. Statistical analyses were conducted by Prism 5 (Graph Pad Software, Inc., La Jolla, CA).
Results
Physiological Variables
Rectal and cranial (temporalis muscle) temperatures, arterial blood gases, plasma glucose, hematocrit, blood pressure, and heart beating rates showed no significant differences among groups. There were no adverse behavioral side effects observed after DHA or NPD1 administration to rats in all groups. No animals died during this study.
DHA Increases Neurogenesis During 2-Week Survival
A single DHA dose, administered at 1 h after 2 h of MCAo, improved composite neurologic score and substantial improvement of postural reflex, tactile (lateral and dorsal), visual (sideways), and proprioceptive contralateral forelimb placing reactions (Fig. 1a). Saline-treated rats exhibited consistent pan-necrotic lesions involving both cortical and subcortical regions of the right hemisphere, characterized microscopically by disruption of neuronal, glial, and vascular elements (Fig. 1b). By contrast, infarct size was dramatically reduced by DHA and was mostly localized in the subcortical areas.

DHA improves behavioral and histopathological outcomes 2 weeks after stroke. Rats were subjected to 2 h of MCAo and treated with DHA or saline iv at 3 h after onset of stroke. a Total neurological score (normal score = 0, maximal score = 12) and time course of recovery of postural reflex, tactile (dorsal and lateral), visual, and proprioceptive contralateral forelimb reactions (normal score = 0, maximal deficit = 2) following MCAo in rats. b Representative Nissl stained coronal sections (bregma − 0.3 mm) from rats treated with saline or DHA. c Total, cortical, and subcortical infarct areas measured at nine coronal levels. d Integrated infarct volumes, residual and relative tissue loss. Values are mean ± SEM; n = 10–11 per group. *p < 0.05 vs. saline group (repeated measured ANOVA followed by Bonferroni test)
Total, cortical, and subcortical infarct areas in the DHA-treated rats were markedly reduced at multiple bregma levels (Fig. 1c) as well as total, cortical, and subcortical infarct volumes by 42%, by 48%, and by 31% (Fig. 1d). DHA preserved more residual tissue (by 24%) and relative tissue loss (by 42%) (Fig. 1d).
Representative fluorescent images of brains from DHA- or vehicle-treated rats are shown in Fig. 2. DHA increased BrdU/Ki-67+, BrdU/DCX+, and BrdU/NeuN+ cells in the cortical infarct border, a wall of ventricle in the SVZ, and a granular cell layer (GCL) in the DG after MCAo (Fig. 2a–c). BrdU/DCX+ cells from DG granular cell layer from DHA-treated animals showed the most robust and elongated dendrites toward the molecular layer of DG (Fig. 2b).

DHA enhances neurogenesis at 2 weeks after MCAo. Representative images of double-labeling immunostaining of a BrdU/ki-67+ (BrdU-green and ki-67-red), b BrdU/DCX+ (BrdU-red and DCX-green), and c BrdU/NeuN+ (BrdU-green and NeuN-red) in peri-infarct cortex, SVZ, and DG 2 weeks after MCAo. Scale bar 50 μm. Arrows indicate dendrites of BrdU/DCX+ cells. SVZ subventricular zone, DG dentate gyrus
Cellular counts for BrdU/Ki-67+, BrdU/DCX+, and BrdU/NeuN+ cells are presented in Fig. 3b. DHA increased the number of BrdU/Ki-67+ cells in the cortex, SVZ and DG total by 88, 40, and 138%, respectively, and the number of BrdU/DCX+ cells (65, 29, and 53%) and BrdU/NeuN+ cells (49, 28, and 42%, respectively) (Fig. 3b).

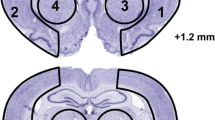
DHA increases neurogenesis, and improves neurobehavioral and histopathological outcome after stroke. a Coronal brain diagrams showing locations of regions for cell count in peri-infarct cortex (C), SVZ at bregma level + 1.2 mm, and three regions of DG at bregma level—3.8 mm (1 DG crest, 2 infrapyramidal DG, 3 suprapyramidal DG). b 2 weeks survival: Quantification of double-labeling BrdU/ki-67+, BrdU/DCX+, BrdU/NeuN+ cells in peri-infarct cortex, SVZ, and DG after MCAo. Values are mean ± SEM; n = 10–11 per group. *p < 0.05 vs. saline group (repeated measured ANOVA followed by Bonferroni test). c 4 weeks survival: Total neurological scores. d Representative mosaic images of Nissl stained brain sections from saline- and DHA-treated rats. e Residual tissue, total and relative tissue loss. Values are mean ± SEM; n = 7–9 per group. *p < 0.05 vs. saline group (repeated measured ANOVA followed by Bonferroni test)
DHA Activates NPD1 Synthesis in the Ischemic Penumbra
The saline-treated group showed a continuous severe composite neurologic score despite moderate improvement throughout the 4-week survival period. Rats treated with DHA displayed improved neurologic score (Fig. 3c). Large zones of infarct with development of cavities in the cortex and subcortex were observed in the saline-treated rats (Fig. 3d). In contrast, the DHA-treated rat has much smaller infarction, mostly in the subcortical area. DHA preserved residual tissue volume by 31%, reduced total tissue loss by 42%, and incurred relative tissue loss by 48% (Fig. 3e).
Representative fluorescent images of brains from DHA- or vehicle-treated rats are presented in Fig. 4a, b. Saline-treated rats showed extensive neuronal loss and increased GFAP-positive reactive astrocytes outlining the infarct border and massive Iba-1-positive microglia/macrophage infiltration. In contrast, DHA attenuated neuronal damage and decreased Iba-1 microglial infiltration and GFAP-positive reactive astrocytes. Figure 4c, d present NeuN+ and GFAP+ cell counts and Iba-1+ fluorescent intensity. DHA significantly increased expression of NeuN and decreased GFAP-positive cell counts and Iba-1+ fluorescent intensity.

DHA attenuates cell damage 4 weeks after MCAo. a, b Representative images of NeuN+ (green), GFAP+ (red), Iba-1+ (green), and Hoechst staining in the cortical and subcortical regions. Scale bar 50 μm. c Coronal brain diagram showing locations of regions for cell count in peri-infarct cortex (C) and subcortex (S). d Quantification of NeuN, GFAP, and Iba-1 in peri-infarct cortex and subcortex. Values are mean ± SEM; n = 7–9 per group. *p < 0.05 vs. saline group (repeated measured ANOVA followed by Bonferroni test)
Figure 5a shows deconvolution microscopy images double labeled with DCX and NeuN in the molecular layer of the dentate gyrus. In the DHA-treated animals, DCX immunoreactive cells were increased and displayed extensive processes extending throughout the cellular layers of the DG, and these DCX-positive neurons had significant dendritic growth (Fig. 5a, b). Quantification of DCX-positive neurons in the granular layer of DG demonstrated that DHA-treated animals had a higher ipsilateral/contralateral hemisphere ratio of DCX-positive cells (Fig. 5c).

DHA enhances neurogenesis after stroke. a Deconvolution microscopy images double-labeled with DCX (green) and NeuN (red) from saline- and DHA-treated rats 4 weeks after MCAo. DCX immunopositive cells are located in granular layer of the dentate gyrus. Arrows show DCX+ cells and arrowheads indicate the dendritic projections. b Representative brightfield images of DCX from saline- and DHA-treated animals. Arrows indicate DCX+ cells and arrowheads show projections in DG (bregma level − 2.16 mm). c Ratio of DCX+ cells in ipsilateral side compared to contralateral side. Data are means ± SEM; n = 4–5 per group. *p < 0.05 vs. saline group (repeated measured ANOVA followed by Bonferroni test)
Figure 6a, b presents total neurological score and lipidomic analysis, conducted at 4 and 5 h after MCAo. DHA-treated rats exhibited improved neurological scores at 4 and 5 h after reperfusion (Fig. 6a). LC-MS/MS-based mediator lipidomic analysis of ipsilateral cortical region demonstrated synthesis of NPD1 following DHA treatment but not with saline treatment in the ischemic penumbra at 5 h after MCAo (Fig. 6b). We ascertained NPD1 synthesis in the penumbra by isolating and characterizing 17-HDHA, a stable derivative of 17H(p)DHA, the short-lived NPD1 precursor. Since 17-HDHA and NPD1 were enhanced in the penumbra by DHA, we conclude that active NPD1 synthesis occurs under these conditions. In addition, increased expression of 14-HDHA, a precursor to the anti-inflammatory, pro-resolving maresin 1, was detected in the penumbra as well (Fig. 6b). Figure 6c–f presents the daughter scan of m/z 343, 17-HDHA and 14-HDHA fragment pattern, and full fragment spectra from the same sample, which indicates that MRM used for quantification was correctly assigned.

DHA increases production of 14 HDHA, 17 HDHA, and NPD1 after stroke. a, b Two hours MCAo, treatment with DHA or saline, behavioral score, and lipidomic analysis were conducted at 4 and 5 h after stroke. a Total neurological score (normal score = 0; maximal deficit = 12). b Quantitative analysis of lipid extracts from the ischemic penumbra after MCAo and coronal brain diagram showing location of region for lipidomic in ischemic penumbra. DHA treatment dramatically increases production of 17 HDHA (precursor of NPD1), NPD1, and 14 HDHA (precursor of maresin 1) at 5 h after stroke. Data are means ± SEM; n = 6 per group. *p < 0.05 vs. saline group (repeated measured ANOVA followed by Bonferroni test). c Present the daughter scan of m/z 343, which is for mono-HDHAs (green line) from a DHA-treated animal. MRM chromatogram of 17-HDHA (blue line) and 14-HDHA (red line) are also plotted to confirm their retention time. d, e Show 17-HDHA and 14-HDHA fragment pattern and full fragment spectra from the same sample. They show very nice match, indicating that MRM used for quantification was correctly assigned. f Demonstrate NPD1 peaks from two MRM transitions (m/z 359–> 206, m/z 359–> 153), matching to the standard in the retention time (data not shown)
NPD1 Enhances Neurogenesis, Improves Axonal Regeneration, and Attenuates BBB Permeability
A significant improvement in neurological score was evident in both NPD1-treated groups compared with CSF-treated rats within 1 day of treatment, and the score was sustained at every observation point throughout the 7-day survival period (Fig. 7a). Ischemic core, penumbra, and total lesion volumes (computed from T2WI) were dramatically reduced by NPD1 treatments (Fig. 7b). T2-weighted imaging (T2WI) revealed large lesions, and T2 hyperintensities were observed in the ischemic core and penumbra of CSF-treated rats, consistent with edema formation (Fig. 7c). In contrast, both NPD1 treatments markedly reduced lesion volumes in the ischemic core and penumbra and total lesion volumes, as shown in the 3D lesion volumes (Fig. 7c). Brains from CSF-treated rats exhibited a pannecrotic lesion involving both cortical and subcortical regions of the right hemisphere (Fig. 7d). By contrast, infarct size in the rats treated with NPD1 showed less extensive damage, mostly in the subcortical area. NPD1 protection was extensive in the frontal-parietal cortex and subcortex (Fig. 7d, e). Total infarct volume, corrected for brain swelling, was significantly reduced in both NPD1-treated groups by 78 and 89% (Fig. 7e). CSF-treated rats exhibited large lesions, characterized by loss of neuronal, glial, and vascular elements (Fig. 7f). In contrast, NPD1-treated rats showed less infarction with an increased number of NeuN, GFAP-positive cells, and SMI-71-positive vessels in the ipsilateral hemisphere (Fig. 7f).

Neuroprotectin D1 (NPD1) promotes motor functional recovery, protects the penumbra, reduces MRI lesion volumes, attenuates cell damage, and diminishes neurovascular unit (NVU) disruption 1 week after ischemic stroke. Rats were subjected to 2 h of MCAo, and NPD1-SS, NPD1-ME (as sodium salts or methyl esters; 5 μg/per rat), or CSF was administered into lateral cerebral ventricle at 3 h after onset of stroke. a Total neurological score during 7 days survival period. b Ischemic core, penumbra, and total lesion volumes computed from T2WI images on day 7. c Representative T2WI, pseudo-images, core/penumbra, and 3D lesion volumes computed from T2WI on day 7. Core and penumbra were extracted from the entire brain. Core (red) and penumbral (blue) tissues were automatically identified in vehicle- and NPD1-treated animals using the computational MRI method, Hierarchal Region Splitting, for penumbra identification. d Representative Nissl stained brain sections from rats treated with CSF, NPD1-SS, and NPD1-ME. e Cortical, subcortical, and total infarct volumes from all groups. f Representative NeuN (blue), SMI-71 (brown), and GFAP (dark green) stained brain sections from all groups. g NVU breakdown was assessed by immunodetection of immunoglobulin G (IgG) within the parenchyma. IgG staining (brown) indicating NVU breakdown. h Bar graph shows that NPD1-SS and NPD1-ME significantly reduced IgG immunoreactivity in cortex, subcortex and whole right hemisphere (total). Values shown are means ± SD (n = 5–6 per group). *p < 0.05, vs. the vehicle group (repeated measures ANOVA followed by Bonferroni tests)
Ischemic disruption of the neurovascular unit was measured by infiltration of endogenous immunoglobulin G (IgG) into the brain parenchyma (Fig. 7g, h). IgG staining intensity was observed in the ipsilateral hemisphere after MCAo (Fig. 7g). In contrast, treatment with both NPD1 showed significantly reduced IgG immunoreactivity in the cortex, subcortex, and whole right hemisphere (total) (Fig. 7g, h). Figure 8a–c displays representative NeuN-, SMI-71-, and GFAP-stained brain sections and cellular counts (regions delineated in diagram b) from all groups. SMI-71, an endothelial barrier antigen, is a marker for BBB integrity. CSF-treated rats showed extensive neuronal loss, reduction of GFAP-reactive astrocytes, and SMI-71-positive vessels. Both NPD1 treatments increased NeuN-positive neurons and GFAP-positive reactive astrocytes and protected blood vessel integrity (Fig. 8a, c). As a result of both NPD1 treatments, blood vessel density (SMI-71) was increased within the penumbral tissues with parallel formation of denser GFAP-rich scar tissue. Thus, enhancement of blood vessel density likely facilitates neurogenesis and synaptogenesis, which, in turn, contribute to improved repair and, ultimately, improved functional recovery.

NPD1 attenuates cellular damage and promotes axonal regeneration after ischemic stroke. a Representative NeuN (brown), SMI-71 (light brown), and GFAP (dark green) stained brain sections from all groups. b Coronal brain diagram (bregma + 1.2 mm) showing locations of regions for NeuN, SMI-71, and GFAP-positive cell counts in the cortex (a, b, and c) and striatum (s). c Quantification of NeuN, SMI-71, and GFAP from all groups. Data are mean ± SD; n = 4/group; *p < 0.05 vs. CSF group; repeated measures ANOVA followed by Bonferroni tests. d–f Axonal sprouting was identified by expression of the phosphorylated growth-associated protein (pGAP-43). d Coronal digram showing locations for pGAP-43 intensity measurement. e Immunoreactivity to pGAP-43 in the infarct boundary. f Bar graph shows optical stain intensity from all groups. Values shown are means ± SD (n = 5–6 per group). *p < 0.05, vs. the vehicle group (repeated measures ANOVA followed by Bonferroni tests)
Axonal sprouting was identified by expression of pGAP-43, which is concentrated in the growth cones of developing neurites (Fig. 8d–f). Treatment with NPD1 increased immunoreactivity to pGAP-43 in the infarct boundary (Fig. 8e). Optical stain intensity was greatly enhanced by NPD1-ME treatment, and there were no differences between CSF and NPD1-SS groups (Fig. 8f).
Discussion
Our results demonstrate that DHA and NPD1 increase neurogenesis and angiogenesis, reduce tissue damage, diminish BBB breakdown, and protect the ischemic penumbra when applied 1 h after 2 h of MCA-o. Overall, decreased infarct volumes, upon treatment, were mirrored by enhanced neurological recovery. Neurogenesis assessed by BrdU labeling associated with specific cell phase markers identifies the proliferation, migration, and differentiation of the neuronal precursor in focal cerebral ischemia. BrdU is used to monitor cell proliferation and differentiation because it is a thymidine analog that incorporates the DNA of dividing cells in the S-phase [50]. However, BrdU can label the cell division of unrelated cells, such as DNA repair and/or the apoptosis process in post-mitotic cells [50]. Therefore, we applied double immunohistochemistry with BrdU and Ki-67 to test the effect of DHA on continuous proliferation of newly born neural precursor cells after ischemic injury, because Ki-67 is expressed in mitotic cell cycling, but it is not detected during the DNA repair process [51]. Our results indicated that DHA treatment increased the number of BrdU/Ki-67+ cells in the crest, lateral blade (suprapyramidal portion), and medial blade (infrapyramidal portion) of DG and total DG, SVZ, and infarct border 2 weeks after MCAo. It suggested that DHA stimulates the proliferation of neural stem cells in neurogenic regions (DG and SVZ) and promotes the continuous proliferation of migrated neural stem cells into the peri-infarct areas.
The effect of DHA on the development of neuronal lineage was evaluated by labeling neuronal precursor cells with BrdU and DCX. DCX is present in migrating neuroblast and young/immature neurons [51, 52]. BrdU/DCX+ cells were shown to have long and robust dendritic processes in the GCL toward the molecular layers in the DG. BrdU/DCX+ cells in DHA-treated rats were more abundant in the crest, infrapyramidal, and suprapyradmial blades of the DG, SVZ, and infarct border. The results indicate that DHA directed the development of neural precursor cells to neuronal lineages following ischemic injury and resulted in persistent development of newly generated cells in ischemic border in a sustained fashion 4 weeks after treatment.
Determination of differentiation and its phenotype is an essential step for neurogenesis. Phenotype of newly generated cells can be determined by double labeling with BrdU and NeuN. NeuN is detected in most neurons of the nervous system and serves as a marker of post-mitotic and newly born mature neurons [50]. Treatment with DHA increased the number of BrdU/NeuN+ cells in crest, infrapyramidal, suprapyramidal of DG and total DG, SVZ, and peri-infarct. In addition, our results indicate that newly generated mature neurons by DHA successfully integrated into existed neuronal circuits in the ischemic border. Although the current study provides evidence that supports DHA augmented neurogenesis followed by ischemic injury, further studies are required to validate whether newly born neurons mediated by DHA are fully functional in neural circuits.
In this study, we showed that DHA reduced the total, cortical, and subcortical infarct volume, relative tissue loss, and preserved the residual (viable) tissue in the ischemic hemispheres. The histologic distribution of molecular markers in the infarcted regions of DHA-treated brains differed from saline-treated brains in that DHA brains showed attenuated cellular death of both astrocytes and neurons within the infracted region. These results indicate that treatment with DHA protects neurons and astrocytes, which are critical for the maintenance and protection of neurons via the secretion of neurotrophic mediators.
Focal ischemic stroke leads to impaired sensorimotor and cognitive functions with 70–80% of patients displaying hemiparesis immediately after stroke [14]. Functional deficits in rodents following MCAo resemble sensorimotor deficits. Since the ultimate goal of any stroke therapy is the restoration of behavioral functions, two tests of the sensorimotor battery were used to detect neurological deficits following experimental stroke. DHA improved overall neurological recovery highlighted by the time course of recovery of postural reflex and proprioceptive and tactile contralateral forelimb reactions through the 2- and 4-week survival period. Both NPD1-SS and NMD1-ME treatments also dramatically improved behavioral deficits during 7-day survival period.
In the present study, we investigated whether intracerebroventricular (ICV) injection of NPD1 is neuroprotective against focal cerebral ischemia. NPD1, a stereospecific derivative of DHA, formed through a lipoxygenase enzyme that acts on free DHA, reduces infarct volume at 48 h after reperfusion [33]. NPD1 is a pro-survival, anti-apoptotic signaling molecule, which acts by inhibiting proinflammatory gene expression induced by oxidative-stress and by promoting cell survival through up-regulation of anti-apoptotic proteins and binding toxic peroxides [20, 33, 53, 54].
Ischemic stroke intervention requires protection of the ischemic penumbra. Neuroimaging of the penumbra is a strategy for detecting tissue at risk [55], especially when up to 44% of patients may still retain penumbral tissue 18 h after stroke [56]. We employed MRI, for the detection of changes in water content and diffusion that characterize acute ischemic stroke. Ischemic core, penumbra, and total lesion volumes (computed from T2WI) were dramatically reduced by both NPD1 treatments. Our lipidomic study demonstrated synthesis of NPD1 and 17-HDHA, following DHA treatment, in the ischemic penumbra at 5 h after MCAo. In addition, increased expression of 14-HDHA, a side product of the precursor to the anti-inflammatory, pro-resolving maresin 1, was detected in the penumbra as well.
Cerebral ischemia initiates a complex cascade of cellular, molecular, and metabolic events that lead to irreversible brain damage [57]. As the infarct progresses, the neurovascular lesions worsen [58]. The complex processes that occur after stroke require the targeting of multiple factors and cells, including glia, vascular, and inflammatory cells. Our novel results demonstrate that NPD1 is endogenously produced in the stroke-injured brain upon DHA treatment. Maximal protection was detected in the cortex (the penumbral area) and also in the subcortical area. Smaller infarcts with less pancellular damage, denser areas of eosinophilic, and shrunken neurons along the infarct margin were detected in both NPD1-treated groups. NPD1 treatments increased the number of surviving neurons, axonal sprouting, and blood vessel density in the penumbra with parallel formation of denser GFAP-rich scar tissue. The integrity of blood vessels facilitates neurogenesis and synaptogenesis, which in turn contributes to improved functional recovery.
Ischemic brain injury disrupts the BBB and then triggers a cascade of events, leading to edema formation, secondary brain injury, and poor neurological outcomes [58,59,60]. Clinically, BBB disruption occurs in more than one third of stroke patients and is associated with poor outcomes and lower survival rates following stroke [61]. Since cerebral ischemia and BBB damage are closely correlated, approaches for protecting BBB integrity and reducing BBB permeability could help elucidate the pathophysiological mechanism in brain ischemia, leading to novel therapies as well as providing a mechanistic roadmap to evaluate efficacy of treatment. No prior studies have evaluated the effect of NPD1 on BBB dysfunction after cerebral ischemia-reperfusion injury. In our study, ischemic disruption of the neurovascular unit was measured by infiltration of endogenous IgG into the brain parenchyma. We found that NPD1 administration was protected against ischemia-induced BBB disruption, as evidenced by reduced IgG immunoreactivity in the cortex, subcortex, and whole right hemisphere.
In summary, our study demonstrates that administration of DHA starting at 1 h after 2 h of stroke increases neurogenesis and angiogenesis, activates NPD1 synthesis in the penumbra, and diminishes BBB, which leads to long-term neurobehavioral recovery after experimental stroke. In addition, we show that NPD1 administration exerts similar effects. This experimental therapy has potential to be translated into clinical applications.
References
Mozaffarian D, Benjamin E, Go A, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Després JP et al (2015) Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 131:e29–e322. https://doi.org/10.1161/CIR.0000000000000152
Hirano T (2015) Evaluation of cerebral perfusion in patients undergoing intravenous recombinant tissue plasminogen activator thrombolysis. Neurol Med Chir 55:789–795. https://doi.org/10.2176/nmc.ra2015-0111
Jauch E, Saver J, Adams HJ et al (2013) Guidelines for the early management of patients with acute ischemic stroke. Stroke 44:870–947. https://doi.org/10.1161/STR.0b013e318284056a
Font M, Arboix A, Krupinski J (2010) Angiogenesis, neurogenesis and neuroplasticity in ischemic stroke. Curr Cardiol Rev 6:238–244. https://doi.org/10.2174/157340310791658802
Van Praaq H, Schinder A, Christie B et al (2002) Functional neurogenesis in the adult hippocampus. Nature 415:1030–1034. https://doi.org/10.1038/4151030a
Zhang R, Xue Y, Lu S et al (2006) Bcl-2 enhances neurogenesis and inhibits apoptosis of newborn neurons in adult rat brain following a transient middle cerebral artery occlusion. Neurobiol Dis 24:345–356. https://doi.org/10.1016/j.nbd.2006.07.012
Tobin M, Bonds J, Minshall R et al (2014) Neurogenesis and inflammation after ischemic stroke: what is known and where we go from here. J Cereb Blood Flow Metab 34:1573–1584. https://doi.org/10.1038/jcbfm.2014.130
Bazan NG, Molina MF, Gordon WC (2011) Docosahexaenoic acid signalolipidomics in nutrition: significance in aging, neuroinflammation, macular degeneration, Alzheimer’s, and other neurodegenerative diseases. Annu Rev Nutr 31:321–351. https://doi.org/10.1146/annurev.nutr.012809.104635
Kan I, Melamed E, Offen D, Green P (2007) Docosahexaenoic acid and arachidonic acid are fundamental supplements for the induction of neuronal differentiation. J Lipid Res 48:513–517. https://doi.org/10.1194/jlr.C600022-JLR200
Kawakita E, Hashimoto M, Shido O (2006) Docosahexaenoic acid promotes neurogenesis in vitro and in vivo. Neuroscience 139:991–997. https://doi.org/10.1016/j.neuroscience.2006.01.021
Crupi R, Marino A, Cuzzocrea S (2013) n-3 fatty acids: role in neurogenesis and neuroplasticity. Curr Med Chem 20:2953–2963. https://doi.org/10.2174/09298673113209990140
Cao D, Kevala K, Kim J, Moon HS, Jun SB, Lovinger D, Kim HY (2009) Docosahexaenoic acid promotes hippocampal neuronal development and synaptic function. J Neurochem 111:510–521. https://doi.org/10.1111/j.1471-4159.2009.06335.x
TD Azad VA, Steinberg G (2016) Neurorestoration after stroke. Neurosurg Focus 40:e2. https://doi.org/10.3171/2016.2.FOCUS15637
Patel R, McMullan P (2017) Neuroprotection in the treatment of acute ischemic stroke. Prog Cardiovasc Dis 59:542–548. https://doi.org/10.1016/j.pcad.2017.04.005
Shapiro L (2017) Altered hippocampal neurogenesis during the first 7 days after a fluid percussion traumatic brain injury. Cell Transplant 26:1314–1318. https://doi.org/10.1177/0963689717714099
Wang X, Gao X, Michaelski S et al (2016) Traumatic brain injury severity affects neurogenesis in adult mouse hippocampus. J Neurotrauma 33:721–733. https://doi.org/10.1089/neu.2015.4097
Molina-Holgado E, Molina-Holgado F (2010) Mending the broken brain: neuroimmune interactions in neurogenesis. J Neurochem 114:1277–1290. https://doi.org/10.1111/j.1471-4159.2010.06849.x
Hermann D, Chopp M (2012) Promoting brain remodelling and plasticity for stroke recovery: therapeutic promise and potential pitfalls of clinical translation. Lancet Neurol 11:369–380. https://doi.org/10.1016/S1474-4422(12)70039-X
He K, Rimm E, Merchant A, Rosner BA, Stampfer MJ, Willett WC, Ascherio A (2002) Fish consumption and risk of stroke in men. JAMA 288:3130–3136. https://doi.org/10.1001/jama.288.24.3130
Bazan N (2009) Neuroprotectin D1-mediated anti-inflammatory and survival signaling in stroke, retinal degenerations, and Alzheimer’s disease. J Lipid Res 50:400–405. https://doi.org/10.1194/jlr.R800068-JLR200
Simopoulos A (2008) The importance of the omega-6/omega-3 fatty acid ratio in cardiovascular disease and other chronic diseases. Exp Biol Med 233:674–688. https://doi.org/10.3181/0711-MR-311
Zhang W, Wang H, Zhang H, Leak RK, Shi Y, Hu X, Gao Y, Chen J (2015) Dietary supplementation with omega-3 polyunsaturated fatty acids robustly promotes neurovascular restorative dynamics and improves neurological functions after stroke. Exp Neurol 272:170–180. https://doi.org/10.1016/j.expneurol.2015.03.005
Pu H, Jiang X, Hu X, Xia J, Hong D, Zhang W, Gao Y, Chen J et al (2016) Delayed docosahexaenoic acid treatment combined with dietary supplementation of omega-3 fatty acids promotes long-term neurovascular restoration after ischemic stroke. Transl Stroke Res 7:521–534. https://doi.org/10.1007/s12975-016-0498-y
Dyall S (2015) Long-chain omega-3 fatty acids and the brain: a review of the independent and shared effects of EPA, DPA and DHA. Front Aging Neurosci 7:52. https://doi.org/10.3389/fnagi.2015.00052
Dagai L, Peri-Naor R, Birk R (2009) Docosahexaenoic acid significantly stimulates immediate early response genes and neurite outgrowth. Neurochem Res 34:867–875. https://doi.org/10.1007/s11064-008-9845-z
Calderon F, Kim H (2004) Docosahexaenoic acid promotes neurite growth in hippocampal neurons. J Neurochem 90:979–988. https://doi.org/10.1111/j.1471-4159.2004.02520.x
Sakayori N, Maekawa M, Numayama-Tsuruta K, Katura T, Moriya T, Osumi N (2011) Distinctive effects of arachidonic acid and docosahexaenoic acid on neural stem/progenitor cells. Genes Cells 16:778–790. https://doi.org/10.1111/j.1365-2443.2011.01527.x
Hashimoto M, Hossain S, Al Mamun A et al (2017) Docosahexaenoic acid: one molecule diverse functions. Crit Rev Biotechnol 37:579–597. https://doi.org/10.1080/07388551.2016.1207153
Belayev L, Khoutorova L, Atkins K et al (2011) Docosahexaenoic acid therapy of experimental ischemic stroke. Transl Stroke Res 2:33–41. https://doi.org/10.1007/s12975-010-0046-0
Eady T, Belayev L, Khoutorova L et al (2012) Docosahexaenoic acid signaling modulates cell survival in experimental ischemic stroke penumbra and initiates long-term repair in young and aged rats. PLoS One 7:e46151. https://doi.org/10.1371/journal.pone.0046151
Hong S, Khoutorova L, Bazan N, Belayev L (2015) Docosahexaenoic acid improves behavior and attenuates blood-brain barrier injury induced by focal cerebral ischemia in rats. Exp Transl Stroke Med 7:3. https://doi.org/10.1186/s13231-014-0012-0
Serhan C, Dalli J, Colas R et al (2015) Protectins and maresins: new pro-resolving families of mediators in acute inflammation and resolution bioactive metabolome. Biochim Biophys Acta 1851:397–413. https://doi.org/10.1016/j.bbalip.2014.08.006
Marcheselli V, Hong S, Lukiw W et al (2003) Novel docosanoids inhibit brain ischemia-reperfusion-mediated leukocyte infiltration and pro-inflammatory gene expression. J Biol Chem 278:43807–43817. https://doi.org/10.1074/jbc.M305841200
Mukherjee P, Marcheselli V, Serhan C, Bazan N (2004) Neuroprotectin D1: a docosahexaenoic acid-derived docosatriene protects human retinal pigment epithelial cells from oxidative stress. Proc Natl Acad Sci U S A 101:8491–8496. https://doi.org/10.1073/pnas.0402531101
Asatryan A, Bazan N (2017) Molecular mechanisms of signaling via the docosanoid neuroprotection D1 for cellular homeostasis and neuroprotection. J Biol Chem 292:12390–12397. https://doi.org/10.1074/jbc.R117.783076
Belayev L, Mukherjee P, Balaszczuk V et al (2017) Neuroprotectin D1 upregulates Iduna expression and provides protection in cellular uncompensated oxidative stress and in experimental ischemic stroke. Cell Death Differ 24:1091–1099. https://doi.org/10.1038/cdd.2017.55
Calandria J, Asatryan A, Balaszczuk V et al (2015) NPD1-mediated stereoselective regulation of BIRC3 expression through cREL is decisive for neuronal cell survival. Cell Death Differ 22:1363–1377. https://doi.org/10.1038/cdd.2014.233
Belayev L, Alonso O, Busto R et al (1996) Middle cerebral artery occlusion in the rat by intraluminal suture: neurological and pathological evaluation of an improved model. Stroke 27:1616–1622. https://doi.org/10.1161/01.STR.27.9.1616
Belayev L, Khoutorova L, Atkins K, Bazan N (2009) Robust docosahexaenoic acid-mediated neuroprotection in a rat model of transient, focal cerebral ischemia. Stroke 40:3121–3126. https://doi.org/10.1161/STROKEAHA.109.555979
Yao C, Zhang J, Chen F, Lin Y (2013) Neuroprotectin D1 attenuates brain damage induced by transient middle cerebral artery occlusion in rats through TRPC6/CREB pathways. Mol Med Rep 8:543–550. https://doi.org/10.3892/mmr.2013.1543
Arvidsson A, Collin T, Kirik D, Kokaia Z, Lindvall O (2002) Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat Med 8:963–970. https://doi.org/10.1038/nm747
Thiyagarajan M, Fernandez J, Lane S et al (2008) Activated protein C promotes neovascularization and neurogenesis in postischemic brain via protease-activated receptor 1. J Neurosci 28:12788–12797. https://doi.org/10.1523/JNEUROSCI.3485-08.2008
Zhang Y, Pardridge W (2006) Blood–brain barrier targeting of BDNF improves motor function in rats with middle cerebral artery occlusion. Brain Res 1111:227–229. https://doi.org/10.1016/j.brainres.2006.07.005
Thompson S, Gibson T, Thompson B et al (2006) Relationship of calpain-mediated proteolysis to the expression of axonal and synaptic plasticity markers following traumatic brain injury in mice. Exp Neurol 201:253–265. https://doi.org/10.1016/j.expneurol.2006.04.01
Belayev L, Khoutorova L, Zhao K et al (2009) A novel neurotrophic therapeutic strategy for experimental stroke. Brain Res 1280:117–123. https://doi.org/10.1016/j.brainres.2009.05.030
Haley M, Lawrence C (2017) The blood–brain barrier after stroke: structural studies and the role of transcytotic vesicles. J Cereb Blood Flow Metab 37:456–470. https://doi.org/10.1177/0271678X16629976
Readnower R, Chavko M, Adeeb S et al (2010) Increase in blood–brain barrier permeability, oxidative stress, and activated microglia in a rat model of blast-induced traumatic brain injury. J Neurosci Res 88:3530–3539. https://doi.org/10.1002/jnr.22510
Obenaus A, Dilmac N, Tone B, Tian HR, Hartman R, Digicaylioglu M, Snyder EY, Ashwal S (2011) Long-term magnetic resonance imaging of stem cells in neonatal ischemic injury. Ann Neurol 69:282–291. https://doi.org/10.1002/ana.22168
Bazan N, Eady T, Khoutorova L et al (2012) Novel aspirin-triggered neuroprotectin D1 attenuates cerebral ischemic injury after experimental stroke. Exp Neurol 236:122–130. https://doi.org/10.1016/j.expneurol.2012.04.007
von Bohlen Und Halbach O (2007) Immunohistological markers for staging neurogenesis in adult hippocampus. Cell Tissue Res 329:409–420 . doi: https://doi.org/10.1007/s00441-007-0432-4
von Bohlen Und Halbach O (2011) Immunohistological markers for proliferative events, gliogenesis, and neurogenesis within the adult hippocampus. Cell Tissue Res 345:1–19 . doi: https://doi.org/10.1007/s00441-011-1196-4
Rao M, Shetty A (2004) Efficacy of doublecortin as a marker to analyse the absolute number and dendritic growth of newly generated neurons in the adult dentate gyrus. Eur J Neurosci 19:234–246. https://doi.org/10.1111/j.0953-816X.2003.03123.x
Bazan N (2005) Neuroprotectin D1 (NPD1): a DHA-derived mediator that protects brain and retina against cell injury-induced oxidative stress. Brain Pathol 15:159–166. https://doi.org/10.1111/j.1750-3639.2005.tb00513.x
Bazan N (2007) Homeostatic regulation of photoreceptor cell integrity: significance of the potent mediator neuroprotectin D1 biosynthesized from docosahexaenoic acid: The proctor lecture. Invest Ophthalmol Vis Sci 48:4866–4881. https://doi.org/10.1167/iovs.07-0918
Donnan G, Baron J, Ma H, Davis S (2009) Penumbral selection of patients for trials of acute stroke therapy. Lancet Neurol 8:261–269. https://doi.org/10.1016/S1474-4422(09)70041-9
Ebinger M, De Silva D, Christensen S et al (2009) Imaging the penumbra - strategies to detect tissue at risk after ischemic stroke. J Clin Neurosci 16:178–187. https://doi.org/10.1016/j.jocn.2008.04.002
Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808. https://doi.org/10.1038/nm.2399
Mogoantă L, Pirici D, Pop O, Bălşeanu AT, Rolea E, Dahnovici RM (2010) Study of vascular microdensity in areas of cerebral ischemia on experimental model. Romanian J Morphol Embryol 51:725–731
Belayev L, Busto R, Zhao W, Ginsberg M (1996) Quantitative evaluation of blood-brain barrier permeability following middle cerebral artery occlusion in rats. Brain Res 739:88–96. https://doi.org/10.1016/S0006-8993(96)00815-3
Yang Y, Rosenberg G (2011) Blood–brain barrier breakdown in acute and chronic cerebrovascular disease. Stroke 42:3323–3328. https://doi.org/10.1161/STROKEAHA.110.608257
Rosenberg G (2012) Neurological diseases in relation to the blood–brain barrier. J Cereb Blood Flow Metab 32:1139–1151. https://doi.org/10.1038/jcbfm.2011.197
Acknowledgments
We also would like to thank NeuroScience Associates, Inc., for performing immunohistochemical staining.
Funding
This work was supported by the National Institute of Neurological Disorders and Stroke grant NS046741 (NGB) and National Institute of General Medical Sciences grant GM103340 (NGB).
Author information
Authors and Affiliations
Contributions
LB and NGB conceived the project, and LB, NGB, and HM wrote the manuscript. LB, NGB, S-HH, and RBO designed the experiments. S-HH, HM, SJM, RF, LK, VB, and RSF performed the experiments, collected, and analyzed data. AO conducted and analyzed MRI experiments. All authors discussed the results and edited the manuscript.
Corresponding author
Ethics declarations
All studies were in compliance with the guidelines outlined in the Guide for the Care and Use of Laboratory Animals from the US Department of Health and Human Services and were approved by the Institutional Animal Care and Use Committee of the Louisiana State University Health Sciences Center (LSUHSC), New Orleans.
Conflict of Interest
The authors declare that they have no conflicts of interest.
Rights and permissions
About this article

Cite this article
Belayev, L., Hong, SH., Menghani, H. et al. Docosanoids Promote Neurogenesis and Angiogenesis, Blood-Brain Barrier Integrity, Penumbra Protection, and Neurobehavioral Recovery After Experimental Ischemic Stroke. Mol Neurobiol 55, 7090–7106 (2018). https://doi.org/10.1007/s12035-018-1136-3
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-018-1136-3