
Abstract
17-estradiol (E2) is a neuroprotective hormone with a high anti-inflammatory potential in different neurological disorders. The inflammatory response initiated by spinal cord injury (SCI) involves the processing of interleukin-1beta (IL-1b) and IL-18 mediated by caspase-1 which is under the control of an intracellular multiprotein complex called inflammasome. We recently described in a SCI model that between 24 and 72 h post-injury, most of inflammasome components including IL-18, IL-1b, NLRP3, ASC, and caspase-1 are upregulated. In this study, we investigated the influence of E2 treatment after spinal cord contusion on inflammasome regulation. After contusion of T9 spinal segment, 12-week-old male Wistar rats were treated subcutaneously with E2 immediately after injury and every 12 h for the next 3 days. Behavioral scores were significantly improved in E2-treated animals compared to vehicle-treated groups. Functional improvement in E2-treated animals was paralleled by the attenuated expression of certain inflammasome components such as ASC, NLRP1b, and NLRP3 together with IL1b, IL-18, and caspase-1. On the histopathological level, microgliosis and oligodendrocyte injury was ameliorated. These findings support and extend the knowledge of the E2-mediated neuroprotective function during SCI. The control of the inflammasome machinery by E2 might be a missing piece of the puzzle to understand the anti-inflammatory potency of E2.
Similar content being viewed by others
Avoid common mistakes on your manuscript.
Introduction
Spinal cord injury (SCI) leads to complex cellular and molecular changes within the lesioned site of the central nervous system (CNS) which at the end results in a loss of neural connectivity and, in consequence, functional-behavioral deficits. While significant advances have been made in the early spinal surgery and supportive treatment of SCI [1], the therapeutic control of local cell degeneration and nerve fiber loss either occurring during the acute or chronic stage of the disease are still in its infancy. Local inflammatory processes (i.e., neuroinflammation) presumably have an important impact on the secondary phase of SCI, and it was demonstrated that targeting of the neuroinflammatory responses can improve functional nerve recovery in SCI rats [2, 3]. Such neuroinflammatory cascades can be either beneficial (i.e., support regenerative events) or deleterious (i.e., amplification of local destructive pathways), thus confidentiality being a double-edged sword in the context of SCI outcome. Indeed, it is believed that the balance between pro-inflammatory and intrinsic repair mechanisms may determine the outcome after SCI [4]. A better understanding of the focal neuroinflammatory machinery is therefore urgently needed.
Neuroinflammatory responses include maturation and secretion of the pro-inflammatory cytokines interleukin (IL)-1b and IL-18 which initiate a cascade of physiological events including triggering cell death. The processing of pro-IL-1b and pro-IL-18 requires the activation of the proteolytic enzyme caspase-1 which is mediated by the regulation of nucleotide-binding domain-like receptor proteins (NLRP) termed inflammasomes. The NLRP inflammasomes consist of the NLRP domain, apoptosis-associated speck-like protein containing a caspase activation and recruitment domain (CARD) (ASC), and caspase-1. The formation of inflammasomes activates caspase-1 and subsequently leads to plasma-membrane pore formation and cleavage of chromosomal DNA [5]. In addition, caspase-1 plays a decisive role during cleavage of pro-IL-1b and pro-IL-18 into their mature forms [6]. IL-1b can be produced and released by neurons and glia cells following distinct stimulation, suggesting that different neural cells take part in the brain-intrinsic inflammatory scenario [7]. Inflammasome activation also occurs in the spinal cord after CNS injury [7].
Gonadal steroids have been shown to transmit beneficial effects to the brain in a variety of acute and chronic degenerative CNS processes including multiple sclerosis, ischemic stroke, and a variety of diseases with mitochondrial failure [8–11]. Preclinical studies have clearly demonstrated that gender and sex steroids, i.e., estrogen and progesterone or its metabolites, affect the outcome after SCI and other traumatic brain injuries [12]. This is supported by clinical evidence and the use of progesterone derivatives and glucocorticoids [13, 14]. Mechanistically, sex steroids affect a wide variety of cell and physiological functions which many of them contributing to protect neurons from death [15]. A set of studies have also presented evidence that the anti-inflammatory potency of these steroids is implicated in neuroprotection [16, 17]. In support of this, sex steroids influence the devastating function of microglia in the brain [18–20, 11]. 17b-estradiol (E2) administered to male rats immediately after SCI caused a significant decrease in the number of infiltrating cells and activity of macrophages/microglia in the injured spinal cord [21, 22] and improve locomotor function [23]. Further, E2 reduced the number of TUNEL-positive cells and secondary damage at the epicenter of SCI [24]. In male rats, E2 reduced oligodendrocyte apoptosis [25].
Only recently, sex steroids were shown also to act at the level of the inflammasome platform and to regulate their assembly and function. In a stroke model, both female sexual hormones reduced the inflammasome activity [26–28]. These findings spurred our interest to analyze in detail the efficacy of E2 in the control of inflammasome activation after traumatic spinal cord contusion in adult male rats.
Material and Methods
Animals and Surgery
Research and animal care procedures were approved by the Review Board for the Care of Animal Subjects of the district government (Tehran, Iran). In vivo experiments were performed with 14-week-old male Wistar rats (350–400 g, Pasteur, Iran). Animals were maintained in a pathogen-free and climate control environment with access to water and food and alternate light and dark cycles, 12 h each. SCI was performed as previously described [29, 30]. Briefly, after shaving the surgical area, rats were deeply anesthetized with 2.0% isoflurane in O2 (Abbott, Germany) and placed in a modified stereotactic frame. Skin incision and blunt dissection of the muscle layers over the area of the vertebral T10 level (spinal T9) were performed. Afterwards, an adjustable forceps was applied to the spinous processes of the second vertebra rostral the Th8 vertebra to stabilize the spinal cord. In addition, transverse processes of Th7 and Th9 were secured by transverse process clamps of the spinal cord adaptor (Stoelting Co, USA). Under a surgical microscope, dorsal laminectomy of T8 vertebra was performed using a fine rongeur. After laminectomy, the spinal cord was compressed by placing a 50 g weight on the exposed spinal cord column for 5 min using a rectangular plate which is longitudinally oriented over the spinal cord. The plate had an area of 11.0 mm2 (2.2 × 5.0 mm) and a concave shape that ensured equal distribution of the pressure on the spinal cord. During the surgery, to avoid local hypothermia of the exposed tissue, a warm plate was used. Postoperative aftercare included saline rehydration (2 mL) and Baytril (0.3 mL, subcutaneously, twice daily) to prevent urinary tract infection. Additionally, surgery-induced bladder dysfunction was compensated by manual emptying twice a day. The animals were sacrificed at different time points, 12, 24, and 72 h post SCI, and after intracardial perfusion, the spinal cords were harvested and proceeded for molecular and biochemical analysis. Sham-operated animals underwent the same anesthesia and surgical procedures with the exception of contusion. For immunohistochemistry staining, 12 animals were assigned randomly to experimental groups with a size of four animals per group. For molecular and biochemical studies, the numbers of animals in the experimental groups were as follows: sham (n = 5), SCI 12 h (n = 5), SCI 24 h (n = 6), SCI 72 h (n = 7), and SCI 72 h plus E2 (n = 7).
Hormone Treatment
17β-estradiol (E2) was purchased from Sigma–Aldrich (Munich, Germany). Rats subjected to SCI were randomly assigned to receive E2 or vehicle (sesame oil plus ethanol as solvent) treatment. E2 was initially dissolved in ethanol and further diluted in sesame oil to obtain final steroid concentrations in the experiment (25 μg/kg body weight) [31]. E2 or vehicle was administrated subcutaneously as neck depots (500 μL) immediately after the SCI and every 12 h thereafter up to 72 h after injury.
Locomotion Testing
Locomotion deficits of rats was evaluated using the Basso, Beattie, and Bresnahan (BBB) scale which is one of the most commonly used assessment methods for rating the motor function of rats after SCI. This score is categorized into 21 levels (0 complete paralysis to 21 normal) as published previously by our group [29]. The scale assesses hind limb movements, body weight support, forelimb to hind limb coordination, and whole body movements. All evaluations were performed by an investigator blinded for the treatment groups. This score was performed before surgery (0), 12, 24, and 72 h after injury in all groups.
RNA Isolation and Real-Time PCR
Gene expression studies were performed with tissues corresponding to the epicenter of injury. Total RNA was extracted using peqGold RNA TriFast (PeqLab, Germany) as previously described [32]. RNA concentration was measured using a NanoDrop 1000 device (PeqLab, Germany). Complementary DNA of 1 μg of total RNA was synthesized using the M-MLV reverse transcription (RT)-kit and random hexanucleotide primers (Invitrogen, Germany). Quantitative real-time PCR (qrtPCR) analysis was performed using the MyIQ detection system (Biorad, Germany). Relative quantification was calculated by the ΔΔCt-method using the qbase + software (Biogazelle, Belgium). Data were expressed as relative amount of the target gene to the amount of a reference gene (CycloA). Previous studies verified CycloA to be stably expressed under all given experimental conditions. Values of sham animals were set to one. Data of interest are given as relative expression. A list of used primers and analyzed genes is given in Table 1. The inflammasome complexes which were evaluated in this study are as follows: IL-1b, IL-18, ASC, NLRP3, NLRP1b, and NLRC4.
Immunohistochemistry
For immunohistochemistry, rats were transcardially perfused with 4% formaldehyde (Sigma, Germany) and were embedded in paraffin (Merck, Germany). Paraffin-embedded sections (5 μm) were rehydrated, unmasked by Tris/EDTA pH 9.0 buffer, and processed for immunohistochemistry using a Vectastain-DAB Kit (Vector Laboratories, Burlingame, CA). After heat-induced antigen retrieval (HIER), sections were incubated with 10% goat serum (Sigma, Germany) for 30 min. Then, slices were incubated overnight at 4 °C with the respective primary antibodies (for details see Table 2). For blocking endogenous peroxidase, sections were incubated with H2O2/PBS (0.3%) (Roth, Germany). Afterwards, sections were incubated with the appropriate secondary antibodies followed by the ABC complex. diaminobenzidine (DAB) was used as chromogen substrate. Finally, sections were counterstained with hematoxylin, dehydrated in graded alcohols, and mounted.
For cell parameter quantification, the whole spinal cord region of interest (complete white and gray matter) was digitally recorded using a Nikon Eclipse 55i (Nikon, Germany) and a ×20 objective for image acquisition. Quantification of cell numbers was performed by manual counting the number of positive cells using the ImageJ 3 software. Cell numbers are expressed as cells/mm2, and only those cells with a clearly visible nucleus (hematoxylin staining) were counted. For each animal, four slices were analyzed with a distance of 50 μm in between.
Immunofluorescence Double-Labeling
In brief, after rehydration, the 5-μm formalin-fixed sections were unmasked with Citrate/EDTA (pH 9.0) buffer and blocked in PBS containing 2% FCS and 1% BSA. Afterwards, the slices were incubated overnight with the primary antibodies at the given concentrations diluted in blocking solution (see Table 2). The following antibodies were used: anti-olig2 antibody (pan-oligodendrocytes) and anti-APC antigen (mature oligodendrocytes) were combined either with anti-ASC or anti-NLRP3. Fluorescent anti-rabbit antibody (1:500, Alexa Fluor 488, Invitrogen, Germany) and anti-mouse/anti-goat antibodies (1:500, Alexa Fluor 598, Invitrogen, Germany) were used as secondary antibodies.
Biochemical Analysis
Protein levels of active IL-1b and IL-18 were determined using enzyme-linked immunoabsorbent assay (ELISA) kits (900-K91, Peprotech, Rocky Hill, USA, for IL-1b and ABIN416245, antibodies-online.com for IL-18). Briefly, spinal cord samples were harvested at 72 h after SCI and homogenized in PBS (0.02 mol/L, pH 7.0–7.2). Afterwards, supernatants were analyzed by ELISA. According to the manufacturer’s instructions, samples were assayed in duplicate at absorbance rates for IL-1b at 405 nm and IL-18 at 450 nm. Concentrations were calculated from the respective standard curves and expressed as pg per mg total protein.
Measurement of Plasma Sex Hormone Levels
Two milliliters of blood were collected in EDTA-coated tubes via the right external jugular vein catheter in various groups before operation and at 12, 24, and 72 h post-injury. Plasma was separated immediately by centrifugation for 5 min at 8000 rmp. Samples were then stored at −80 °C until assayed for sex hormone levels. Plasma levels of E2 were determined by an EIA kit (Cayman Chemical, Ann Arbor, MI) according to the manufacturer’s instructions.
SDS PAGE and Western Blot
Spinal cord tissue was lysed in ice-cold RIPA buffer consisting of 50 mM Tris-HCl, pH 8.0, 1% (v/v) Nonidet P-40 (Sigma, Igepal, CA), 0.1% SDS (sodium dodecyl sulfate), 0.5% sodium deoxycholate and protease inhibitor cocktail (Complete Mini, Roche, Mannheim, Germany). Protein concentrations were determined using the BCA™ Protein Assay Kit (Pierce, Bonn, Germany) according to the manufacturer’s protocol. Same amounts of protein samples (approx. 30 μg per lane) were loaded, separated by 8–12% (v/v) discontinuous sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE), and transferred onto a PVDF membranes (Roche, Manheim, Germany). After blocking with 5% skimmed milk in Tris-buffered saline containing 0.05% Tween 20 (TBS-T) for 1 h at room temperature, PVDF membranes were incubated with primary antibodies (Table 2) over night at 4 °C. After washing with TBS-T, membranes were incubated with a peroxidase-conjugated goat anti-rabbit (Bio-Rad, USA) secondary antibody for 2 h at room temperature. Visualization was achieved using the enhanced chemiluminescence method (ECL plus, Pierce Scientific, Waltham, MA, USA) according to the manufacturer’s protocol. For densitometric quantification, intensities of the specific bands were normalized to GAPDH in the same blot using ImageJ software (free Java software provided by the National Institute of Health, Bethesda, MD, USA).
Data Analysis
All data are given as means ± SEM. Statistical differences between various groups were analyzed by one-way analysis of variance (ANOVA) followed by Tukey’s post hoc test using GraphPad Prism 5 (GraphPad Software Inc., USA). The BBB test was evaluated using two-way ANOVA.
Results
Behavioral Assessment
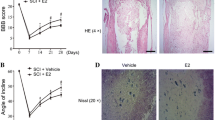
We used the BBB scoring criteria to evaluate the lower limb motor function at time point 0 (before SCI), 12, 24, and 72 h after SCI. Our results show that locomotion of all SCI-induced rats (including the vehicle group) exhibited partial recovery after 72 h. Rats treated with E2 displayed a significantly better locomotor activity compared to vehicle rats after 72 h (Fig. 1a). Notably, differences in functional recovery between vehicle- and E2-treated animals became evident between 24 and 72 h post SCI, indicating that not the initial tissue injury but rather neuroinflammation-mediated secondary tissue damage was under the control of sex steroid treatment.

a BBB locomotion assessment at 12, 24, and 72 h after SCI onset. a 72 h post SCI, the BBB scores decreased to zero in the SCI vehicle and SCI + E2 groups and remained until 24 h at this level. Thereafter, BBB in the SCI + E2 group recovered significantly better compared to SCI vehicle. b, c Expression of IL-1b and IL-18 mRNAs was significantly induced after SCI (72 h). These effects were significantly dampened in the SCI + E2 group. d, e PCR data are supported by ELISA analysis. Again, SCI induced IL-1b and IL-18 levels and E2 administration abolished partially these effects at 72 h. f Presents a selective Western blot for active caspase-1 and the housekeeping marker GAPDH, as well as the corresponding semi-quantitative densitometric quantification. SCI increased and E2 inhibited the transformation into the active caspase-1. §§ p ≤ 0.01 sham vs. SCI vehicle, §§§ p ≤ 0.001 sham vs. SCI vehicle, *p ≤ 0.05 SCI vehicle vs. SCI + E2, **p ≤ 0.01 SCI vehicle vs. SCI + E2, **p ≤ 0.001 SCI vehicle vs. SCI + E2. Data represent means ± SEM
E2 Ameliorates SCI-Induced Inflammatory Parameters
Both IL-1b and IL-18 are synthesized as inactive cytoplasmic precursors that are processed as biologically active forms in response to pro-inflammatory stimuli by caspase-1 [33]. One possibility of processing these two cytokines involves the activation of inflammasomes [34]. PCR studies showed a massive (>10 fold) induction of IL-1b and IL-18 messenger RNAs (mRNAs) in the SCI vehicle groups (Fig. 1b, c). In both cases, E2 reduced the IL-induction by more than 50%. Similarly, protein values of both active ILs determined by ELISA rose after SCI and revealed significant lower levels in the E2 group compared to SCI vehicle (Fig. 1d, e). Figure 1f shows the corresponding Western blots for active caspase-1. As can be seen, SCI significantly increased caspase-1 enzyme which was blocked by E2 treatment.
Effect of Steroids on Inflammasome Expression
In a previous study, we have shown that NLRP3 inflammasome complexes were significantly upregulated in the epicenter 72 h after SCI [29]. Therefore, we analyzed the effect of E2 on inflammasome expression at this time point. As shown in Fig. 2, SCI induced the expression of ASC (Fig. 2a), NLRP3 (Fig. 2b), and NLRP1b (Fig. 2c) but not NLRC4 mRNA (Fig. 2d). The application of E2 significantly diminished the SCI effect with respect to ASC, NLRP3, and NLRP1b. Analysis of the corresponding protein values by Western blotting showed an increase in ASC (Fig. 2e, f) and NLRP3 (Fig. 2 e, g) protein after SCI which ameliorate with E2 treatment. Interestingly, NLRP1b levels seemed to decrease (although not significantly) and E2 to restore the levels to basic values (Fig. 2 e, h). At the transcriptional level, NLRC4 was not affected by E2 by SCI or E2 substitution (data not shown).

Regulation of inflammasome components after SCI or E2 substitution. mRNA expression levels (a–d) and the corresponding protein levels (e–h). SCI significantly induced the transcription of ASC, NLRP3, NLRP1b, and NLRC4 genes. The administration of E2 only reduced the increase of ASC, NLRP3, and NLRP1b but not NLRC4. Corresponding Western blots (e) and the semi-quantitative assessment of protein values are shown (f–h). Note that only ASC (f) and NLRP3 (g) proteins were induced by SCI, whereas NLRP1 (h) seems to be slightly decreased. Again, E2 abolished to a large extent the induction of ASC and NLRP3 and restored NLRP1b levels to baseline values. No regulatory changes in NLRC4 protein was observed after SCI and E2 (data not shown). §§ p ≤ 0.01 sham vs. SCI vehicle, §§§ p ≤ 0.001 sham vs. SCI vehicle, *p ≤ 0.05 SCI vehicle vs. SCI + E2, **p ≤ 0.01 SCI vehicle vs. SCI + E2, **p ≤ 0.001 SCI vehicle vs. SCI + E2. Data represent means ± SEM
Regulation of Estrogen Receptor Expression and Serum E2 Plasma Levels after of SCI
We have further analyzed the effect of SCI and E2 treatment on local expression of nuclear estrogen receptor (ER)-alpha and ER-beta. In the intact spinal cord, nuclear ERs are found in neurons and different glial subtypes [35]. As shown in Fig. 3a, b, mRNA expression of ER-alpha was reduced (P = 0.056), although not reaching significant values, during the first 72 h after SCI. E2 substitution did not affect ER-alpha expression at 72 h. In contrast, ER-beta expression levels were significantly elevated 12 h after SCI but then decreased again to basal values (Fig. 3c, d).

PCR analysis of ER-alpha and ER-beta mRNA expression 12, 24, and 72 h after SCI and after E2 substitution. ER-alpha and ER-beta expression levels were decreased or increased (only at 12 h) after SCI, respectively (a, c). Hormone substitution had no effect on these levels (b, d). E2 plasma levels increased at 24 and 72 h after SCI (e), and E2 substitution significantly boosted this effect (f). § p ≤ 0.05 sham vs. SCI vehicle. §§ p ≤ 0.01 sham vs. SCI vehicle, ***p ≤ 0.001 SCI vehicle vs. SCI + E2. Data represent means ± SEM
Endogenous E2 plasma levels were analyzed by ELISA 12, 24, and 72 h post-injury and after E2 substitution at 72 h. E2 values significantly increased 24 and 72 h after SCI (Fig. 3e). E2 substitution increased plasma E2 levels by approx. 50-fold (Fig. 3f) compared to vehicle at 72 h.
E2 Effects on Oligodendrocytes and Microgliosis After SCI
Using Iba-1 staining to identify microglia/macrophages, we found in sham-operated animals low numbers of Iba-1-positive cells which appeared equally distributed throughout the gray matter (Fig. 4a). Seventy-two hours after SCI, Iba-1 cell numbers significantly increased. On the morphological level, Iba-1-positive cells displayed a resting, ramified morphology in sham-operated animals, whereas after SCI, these cells clearly showed to be activated (swollen and retracted cell processes). Notably, E2 application reduced this effect.

Assessment of Iba-1-positive (microglia/macrophage, (a) the insert shows the region of interest, i.e., the ventral horn), Olig2-positive (pan-oligodendrocyte, (b) the insert shows the region of interest, i.e., the ventral funiculus) and APC-positive (mature oligodendrocyte, (c) the insert shows the region of interest, i.e., the ventral funiculus) cell numbers in the epicenter of spinal cord 72 h after SCI. Representative microphotographs of the different experimental groups are also included (sham, SCI vehicle, and SCI+E2). Note that SCI induced microgliosis and reduced oligodendrocyte numbers which were both partially antagonized by E2. §§ p ≤ 0.01 sham vs. SCI vehicle. §§§ p ≤ 0.001 sham vs. SCI vehicle. **p ≤ 0.01 SCI vehicle vs. SCI + E2, ***p ≤ 0.001 SCI vehicle vs. SCI + E2. Data represent means ± SEM. Scale bar: 100 μm
Besides microglia, astroglia, and neurons [35], ERs are also expressed in oligodendrocyte progenitors and mature oligodendrocytes [36]. Further, we therefore analyzed whether these cell populations are (direct or indirect) E2 targets in our experimental study. SCI induced a significant loss of Olig2- and APC-positive cells in the gray and white matter (Fig. 4b, c), and E2 substitution partially reversed this effect.
Co-Localization of ASC and NLRP3 with Olig2 (Oligodendrocytes) and APC (Mature Oligodendrocytes)
In our previous study, we have shown that neurons, microglia, and astrocytes co-express NLRP3 and ASC in the spinal cord after injury model. Since these cells were protected under E2 treating, we attempted in this study to attribute the different inflammasome components to oligodendrocyte subtypes in the spinal cord. Therefore, double-immunofluorescence staining of olig2 and APC as pan-oligodendrocyte and mature oligodendrocyte markers respectively with anti-NLRP3 and ASC antibodies were performed. Double-immunofluorescence staining revealed that ASC and NLRP3 are associated with olig2 (Fig. 5a), whereas APC+ cells did not obviously express inflammasome markers (Fig. 5b).

Co-localization of NLRP3 and ASC with Olig2 (Pan-oligodendrocyte) (a) after spinal cord contusion. Mature oligodendrocytes which were stained with APC did not reveal ASC and NLRP3 co-staining in the experimental SCI model (b). Scale bar: 50 μm
Discussion
Despite the diversity of SCI paradigms and the dissimilar dosage and treatment protocols used in the various experimental studies, the majority of animal studies reported beneficial effects of E2 on locomotor recovery [37] and tissue sparing [38]. The absence of a clear protective action of E2 in this CNS damage model in some studies [16] might result from known strain differences in the outcome and therapeutic accessibility after SCI [39, 40]. Irrespective of this controversy, there is existing considerable support that E2 is neuroprotective and anti-inflammatory in a variety of CNS injury models either acute or chronic, including SCI [27].
E2 exerts its genomic or nongenomic actions via two traditional estrogen receptors ERa and ERb [41]. The genomic actions of estrogen are initiated upon binding ERa and ERb in the cytoplasm or nucleus. After ligand binding, ERa or ERb can directly regulate transcriptional processes in the nucleus, either by binding specific estrogen response elements (EREs), which are located within the promoters of target genes, or by interacting with other DNA-binding transcription factors that modulate gene expression. The target genes include anti-apoptotic mediators of the bcl-2 family and pro-inflammatory cytokines. In the spinal cord, estrogen receptors are found in both neurons and different glial subtypes [35]. Several spinal cord neuronal populations express low to moderate levels of ERa or ERb, whereas spinal cord astrocytes often express ERa [42] and spinal cord microglia primarily ERb [43]. All estrogen receptor subtypes are found in oligodendrocyte progenitors and oligodendrocytes in vitro [36]. However, ERb is the main isoform expressed in oligodendrocytes of the adult CNS including the spinal cord and is localized to the plasma membrane and myelin [44].
Following SCI, immune system-related cells infiltrate the injury epicenter. These cells could also be the target of estrogen as studies have shown expression of ERs in immune cells infiltrating the inflamed spinal cord in animal models of multiple sclerosis [15]. A high E2 dose administered to male rats immediately after SCI caused a significant decrease in the number of infiltrating cells like monocytes, macrophages, and neutrophils which occur after SCI at the center of injury [21]. Other investigations confirmed the reduction in the spreading of inflammatory cells and the decrease in the number or activity of macrophages/microglia in the injured spinal cord [22]. The attenuation of microglia/macrophage reactivity, assessed qualitatively, appeared to continue even in chronic phases [23]. The anti-inflammatory effects of E2 in the experimental SCI studies showed a time dependency with regard to pro-inflammatory cytokine and chemokine expression. Ritz and Hausmann found that E2 further increased injury-elicited and transient upregulation of IL-1a and IL-1b protein in the spinal cord at 6 h post-injury [45]. On the other hand, E2 treatment decreased SCI-provoked increase in TNF-a, IL-1b and CCL2/MCP-1 mRNA levels in mice at 24 h post-injury [24]. In this study, we demonstrated that E2 dampened IL-1b and IL-18 protein and mRNA levels at 72 h after SCI. Possible mechanisms of E2 to attenuate the inflammatory responses in CNS injuries are as follows: (1) E2 prevents transcription of inflammatory cytokine genes in microglia/macrophages by interfering with the translocation of NFκB from the cytoplasm to the nucleus [46, 43] and (2) E2 dampens pro-inflammatory chemokine gene expression in spinal cord astrocytes, an effect involving direct interaction of ERα with nuclear NFκB [42]. Additionally, we showed using a classical SCI contusion model that E2 exerts its protective and anti-inflammatory function at the intracellular platform of inflammasomes by a partial prevention of the inflammasome-caspase-1 activation including NLRP3 and ASC.
The inflammasomes are cytosolic protein complexes which act as intracellular sensors of disruption of cell homeostasis. Their stimulation leads to the proteolytic cleavage of the pro-inflammatory cytokines pro-IL-1b and pro-IL-18 through the activation of caspase-1. The active complex consists of a central scaffold protein for which it is named (e.g., NLRP1, NLRP3, NLRC4, and AIM2), an ASC containing caspase activation and recruitment domain (CARD), which is mandatory for most inflammasomes, and the precursor form of caspase-1 enzyme, pro-caspase-1. How the inflammasomes are activated is still debated, but in the case of NLRP3, which is the most studied caspase-1 activator [47], several signals related to cell damage and stress such as generation of extracellular ATP, production of reactive oxygen species (ROS), and activators that form crystalline/particulate ligands are likely to be involved [48]. When the inflammasome is activated, pro-caspase-1 undergoes an autolysis leading to the formation of caspase-1, which in turn cleaves pro-IL-1b, generating the mature cytokine that is subsequently released along with caspase-1 [49] via nonclassical secretory pathways [3, 4]. The NLRP3 inflammasome has been linked to acute disorders (from infections [50] to acute brain injury [27, 29]) and chronic diseases exhibiting an inflammatory component such as experimental autoimmune encephalitis [51], Parkinson’s disease [52], Alzheimer’s disease [53], and amyotrophic lateral sclerosis [54]. The NLRP3 inflammasome has been studied in microglia and macrophages but has also been suggested to have functions in neurons [29, 27, 55]. In previous experiments, we analyzed the time course of inflammasome activation after SCI and showed that the protein and mRNA levels of NLRP3 complexes are highly expressed at 72 h after injury [29]. Further using double-immunofluorescence staining, we revealed that most of the NLRP3+ cells were neurons. In addition, microglia and astrocytes also seem to express NLRP3, whereas the adaptor protein ASC was mainly confined to microglia.
We provide now for the first time data suggesting a mainly inhibitory role for E2 with respect to inflammasome stimulation and dampening of neuroinflammatory processes after SCI. In parallel, E2 reduced microglia accumulation and decreased the amounts of IL-18 and Il-1b. Since microglia represents a known source of inflammasomes, E2 effects on inflammasomes are related to changes in microglia infiltration and proliferation. After SCI, dying or dead neurons release distinct compounds which can stimulate the resident microglia to proliferate and form specific microglial subtype cells with specific morphology. Activated microglia recruit peripheral blood-derived immune cells to infiltrate the injured area and to initiate and perpetuate local inflammation. Macrophages also are attracted after microglial activation, and their numbers transiently increase within 24 h after injury [56]. In contrast, microglial cell numbers increase gradually during the first week post-injury [57]. Microglia is supposed to be the main cell type in the brain responsible for IL-1b and IL-18 secretion [58]. A recent report showed that macrophages also secrete IL-18 in a NLRP-3/ASC-dependent manner in response to a stimulation with Staphylococcus aureus [59]. Further, it was shown that IL-18 production occurs in microglia in response to the classical activators ATP and is NLRP3- and caspase-1-dependent [58]. This is of particular interest, since IL-18 is currently believed to act as an important set screw for neuroinflammation [60]. Another important observation in this study was that E2 protects oligodendrocytes after SCI. It has been shown before that in male rats sustaining a contusion injury, repeated administration of E2 reduced oligodendrocyte apoptosis [25]. Further using double-immunofluorescence staining, we revealed that NLRP3 and ASC are well-associated with oligodendrocyte, but only with Olig2 (pan-oligodendrocytes) whereas APC+ cells did not express an inflammasome component in our SCI model. In a cuprizone-induced demyelination animal model, E2 together with progesterone prevented partially the demyelination of the corpus callosum [61]. Thus, it is likely that these protective effects are partly exerted through direct actions on oligodendrocytes which is supported by in vitro investigations showing that the death of cortical oligodendrocytes, elicited by a cytotoxic agent, is reduced by E2 [62].
In summary, our study presents data that E2 effectively interferes and reduces the expression of several inflammasome components after SCI and thereby reduces local inflammation in the spinal cord.
References
Agostinello J, Battistuzzo CR, Skeers P, Bernard S, Batchelor PE (2016) Early spinal surgery following thoracolumbar spinal cord injury: process of care from trauma to theatre. Spine
Cox A, Varma A, Banik N (2015) Recent advances in the pharmacologic treatment of spinal cord injury. Metab Brain Dis 30(2):473–482
Allison DJ, Thomas A, Beaudry K, Ditor DS (2016) Targeting inflammation as a treatment modality for neuropathic pain in spinal cord injury: a randomized clinical trial. J Neuroinflammation 13(1):152
DiSabato DJ, Quan N, Godbout JP (2016) Neuroinflammation: the devil is in the details. J Neurochem 139 Suppl 2:136–153
Lin W-P, Xiong G-P, Lin Q, Chen X-W, Zhang L-Q, Shi J-X, Ke Q-F, Lin J-H (2016) Heme oxygenase-1 promotes neuron survival through down-regulation of neuronal NLRP1 expression after spinal cord injury. J Neuroinflammation 13(1):1
Latz E, Xiao TS, Stutz A (2013) Activation and regulation of the inflammasomes. Nat Rev Immunol 13(6):397–411
de Rivero Vaccari JP, Lotocki G, Alonso OF, Bramlett HM, Dietrich WD, Keane RW (2009) Therapeutic neutralization of the NLRP1 inflammasome reduces the innate immune response and improves histopathology after traumatic brain injury. J Cereb Blood Flow Metab 29(7):1251–1261
Ulbrich C, Zendedel A, Habib P, Kipp M, Beyer C, Dang J (2012) Long-term cerebral cortex protection and behavioral stabilization by gonadal steroid hormones after transient focal hypoxia. J Steroid Biochem Mol Biol 131(1):10–16
Habib P, Dang J, Slowik A, Victor M, Beyer C (2014) Hypoxia-induced gene expression of aquaporin-4, cyclooxygenase-2 and hypoxia-inducible factor 1α in rat cortical astroglia is inhibited by 17β-estradiol and progesterone. Neuroendocrinology 99(3–4):156–167
Braun A, Dang J, Johann S, Beyer C, Kipp M (2009) Selective regulation of growth factor expression in cultured cortical astrocytes by neuro-pathological toxins. Neurochem Int 55(7):610–618
Kipp M, Hochstrasser T, Schmitz C, Beyer C (2015) Female sex steroids and glia cells: impact on multiple sclerosis lesion formation and fine tuning of the local neurodegenerative cellular network. Neurosci Biobehav Rev 67:125–136
Olsen ML, Campbell SC, McFerrin MB, Floyd CL, Sontheimer H (2010) Spinal cord injury causes a wide-spread, persistent loss of Kir4. 1 and glutamate transporter 1: benefit of 17β-oestradiol treatment. Brain 133(4):1013–1025
Bracken MB, Shepard MJ, Collins WF, Holford TR, Young W, Baskin DS, Eisenberg HM, Flamm E et al (1990) A randomized, controlled trial of methylprednisolone or naloxone in the treatment of acute spinal-cord injury: results of the Second National Acute Spinal Cord Injury Study. N Engl J Med 322(20):1405–1411
Gonzalez SL, Saravia F, Deniselle MCG, Lima AE, De Nicola AF (1999) Glucocorticoid regulation of motoneuronal parameters in rats with spinal cord injury. Cell Mol Neurobiol 19(5):597–611
Kipp M, Beyer C (2009) Impact of sex steroids on neuroinflammatory processes and experimental multiple sclerosis. Front Neuroendocrinol 30(2):188–200
Elkabes S, Nicot AB (2014) Sex steroids and neuroprotection in spinal cord injury: a review of preclinical investigations. Exp Neurol 259:28–37
Giatti S, Melcangi RC, Pesaresi M (2016) The other side of progestins: effects in the brain. J Mol Endocrinol 57(2):R109–R126
Johann S, Beyer C (2013) Neuroprotection by gonadal steroid hormones in acute brain damage requires cooperation with astroglia and microglia. J Steroid Biochem Mol Biol 137:71–81
Arevalo M-A, Santos-Galindo M, Bellini M-J, Azcoitia I, Garcia-Segura LM (2010) Actions of estrogens on glial cells: implications for neuroprotection. Biochimica et Biophysica Acta (BBA)-General Subjects 1800(10):1106–1112
Habib P, Dreymueller D, Ludwig A, Beyer C, Dang J (2013) Sex steroid hormone-mediated functional regulation of microglia-like BV-2 cells during hypoxia. J Steroid Biochem Mol Biol 138:195–205
Sribnick EA, Wingrave JM, Matzelle DD, Wilford GG, Ray SK, Banik NL (2005) Estrogen attenuated markers of inflammation and decreased lesion volume in acute spinal cord injury in rats. J Neurosci Res 82(2):283–293
Siriphorn A, Dunham KA, Chompoopong S, Floyd CL (2012) Postinjury administration of 17β-estradiol induces protection in the gray and white matter with associated functional recovery after cervical spinal cord injury in male rats. J Comp Neurol 520(12):2630–2646
Sribnick EA, Samantaray S, Das A, Smith J, Matzelle DD, Ray SK, Banik NL (2010) Postinjury estrogen treatment of chronic spinal cord injury improves locomotor function in rats. J Neurosci Res 88(8):1738–1750
Cuzzocrea S, Genovese T, Mazzon E, Esposito E, Di Paola R, Muia C, Crisafulli C, Peli A et al (2008) Effect of 17β-estradiol on signal transduction pathways and secondary damage in experimental spinal cord trauma. Shock 29(3):362–371
Lee JY, Choi SY, Oh TH, Yune TY (2012) 17β-Estradiol inhibits apoptotic cell death of oligodendrocytes by inhibiting RhoA-JNK3 activation after spinal cord injury. Endocrinology 153(8):3815–3827
Slowik A, Beyer C (2015) Inflammasomes are neuroprotective targets for sex steroids. J Steroid Biochem Mol Biol 153:135–143
Lammerding L, Slowik A, Johann S, Beyer C, Zendedel A (2015) Poststroke inflammasome expression and regulation in the peri-infarct area by gonadal steroids after transient focal ischemia in the rat brain. Neuroendocrinology 103(5):460–475
Xu Y, Sheng H, Bao Q, Wang Y, Lu J, Ni X (2016) NLRP3 inflammasome activation mediates estrogen deficiency-induced depression-and anxiety-like behavior and hippocampal inflammation in mice. Brain Behav Immun 56:175–186
Zendedel A, Johann S, Mehrabi S, Joghataei M-T, Hassanzadeh G, Kipp M, Beyer C (2016) Activation and regulation of NLRP3 inflammasome by intrathecal application of SDF-1a in a spinal cord injury model. Mol Neurobiol 53(5):3063–3075
Zendedel A, Nobakht M, Bakhtiyari M, Beyer C, Kipp M, Baazm M, Joghataie MT (2012) Stromal cell-derived factor-1 alpha (SDF-1α) improves neural recovery after spinal cord contusion in rats. Brain Res 1473:214–226
Dang J, Mitkari B, Kipp M, Beyer C (2011) Gonadal steroids prevent cell damage and stimulate behavioral recovery after transient middle cerebral artery occlusion in male and female rats. Brain Behav Immun 25(4):715–726
Zendedel A, Habib P, Dang J, Lammerding L, Hoffmann S, Beyer C, Slowik A (2015) Omega-3 polyunsaturated fatty acids ameliorate neuroinflammation and mitigate ischemic stroke damage through interactions with astrocytes and microglia. J Neuroimmunol 278:200–211
Martinon F, Tschopp J (2007) Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ 14(1):10–22
Ogura Y, Sutterwala FS, Flavell RA (2006) The inflammasome: first line of the immune response to cell stress. Cell 126(4):659–662
Papka R, Storey-Workley M, Shughrue P, Merchenthaler I, Collins J, Usip S, Saunders P, Shupnik M (2001) Estrogen receptor-α and-β immunoreactivity and mRNA in neurons of sensory and autonomic ganglia and spinal cord. Cell Tissue Res 304(2):193–214
Jung-Testas I, Renoir M, Bugnard H, Greene GL, Baulieu E-E (1992) Demonstration of steroid hormone receptors and steroid action in primary cultures of rat glial cells. J Steroid Biochem Mol Biol 41(3):621–631
Kachadroka S, Hall AM, Niedzielko TL, Chongthammakun S, Floyd CL (2010) Effect of endogenous androgens on 17 β-estradiol-mediated protection after spinal cord injury in male rats. J Neurotrauma 27(3):611–626
Hubscher CH, Fell JD, Gupta DS (2010) Sex and hormonal variations in the development of at-level allodynia in a rat chronic spinal cord injury model. Neurosci Lett 477(3):153–156
Kjell J, Sandor K, Josephson A, Svensson CI, Abrams MB (2013) Rat substrains differ in the magnitude of spontaneous locomotor recovery and in the development of mechanical hypersensitivity after experimental spinal cord injury. J Neurotrauma 30(21):1805–1811
Schmitt C, Miranpuri GS, Dhodda VK, Isaacson J, Vemuganti R, Resnick DK (2006) Changes in spinal cord injury–induced gene expression in rat are strain-dependent. Spine J 6(2):113–119
Nilsson S, Gustafsson J-Å (2002) Biological role of estrogen and estrogen receptors. Crit Rev Biochem Mol Biol 37(1):1–28
Giraud SN, Caron CM, Pham-Dinh D, Kitabgi P, Nicot AB (2010) Estradiol inhibits ongoing autoimmune neuroinflammation and NFκB-dependent CCL2 expression in reactive astrocytes. Proc Natl Acad Sci 107(18):8416–8421
Wu W-f, Tan X-j, Dai Y-B, Krishnan V, Warner M, Gustafsson J-Å (2013) Targeting estrogen receptor β in microglia and T cells to treat experimental autoimmune encephalomyelitis. Proc Natl Acad Sci 110(9):3543–3548
Arvanitis DN, Wang H, Bagshaw RD, Callahan JW, Boggs JM (2004) Membrane-associated estrogen receptor and caveolin-1 are present in central nervous system myelin and oligodendrocyte plasma membranes. J Neurosci Res 75(5):603–613
Ritz M-F, Hausmann ON (2008) Effect of 17β-estradiol on functional outcome, release of cytokines, astrocyte reactivity and inflammatory spreading after spinal cord injury in male rats. Brain Res 1203:177–188
Hu R, Sun H, Zhang Q, Chen J, Wu N, Meng H, Cui G, Hu S et al (2012) G-protein coupled estrogen receptor 1 mediated estrogenic neuroprotection against spinal cord injury. Crit Care Med 40(12):3230–3237
Walsh JG, Muruve DA, Power C (2014) Inflammasomes in the CNS. Nat Rev Neurosci 15(2):84–97
Schroder K, Tschopp J (2010) The inflammasomes. Cell 140(6):821–832
Clark AK, D’Aquisto F, Gentry C, Marchand F, McMahon SB, Malcangio M (2006) Rapid co-release of interleukin 1β and caspase 1 in spinal cord inflammation. J Neurochem 99(3):868–880
Ramos HJ, Lanteri MC, Blahnik G, Negash A, Suthar MS, Brassil MM, Sodhi K, Treuting PM et al (2012) IL-1β signaling promotes CNS-intrinsic immune control of West Nile virus infection. PLoS Pathog 8(11):e1003039
Hoegen T, Tremel N, Klein M, Angele B, Wagner H, Kirschning C, Pfister H-W, Fontana A et al (2011) The NLRP3 inflammasome contributes to brain injury in pneumococcal meningitis and is activated through ATP-dependent lysosomal cathepsin B release. J Immunol 187(10):5440–5451
Codolo G, Plotegher N, Pozzobon T, Brucale M, Tessari I, Bubacco L, de Bernard M (2013) Triggering of inflammasome by aggregated α–synuclein, an inflammatory response in Synucleinopathies. PLoS One 8(1):e55375
Heneka MT, Kummer MP, Stutz A, Delekate A, Schwartz S, Vieira-Saecker A, Griep A, Axt D et al (2013) NLRP3 is activated in Alzheimer/’s disease and contributes to pathology in APP/PS1 mice. Nature 493(7434):674–678
Johann S, Heitzer M, Kanagaratnam M, Goswami A, Rizo T, Weis J, Troost D, Beyer C (2015) NLRP3 inflammasome is expressed by astrocytes in the SOD1 mouse model of ALS and in human sporadic ALS patients. Glia 63(12):2260–2273
Compan V, Baroja-Mazo A, López-Castejón G, Gomez AI, Martínez CM, Angosto D, Montero MT, Herranz AS et al (2012) Cell volume regulation modulates NLRP3 inflammasome activation. Immunity 37(3):487–500
Nguyen HX, Beck KD, Anderson AJ (2011) Quantitative assessment of immune cells in the injured spinal cord tissue by flow cytometry: a novel use for a cell purification method. JoVE (Journal of Visualized Experiments) 50:e2698–e2698
Jin X, Ishii H, Bai Z, Itokazu T, Yamashita T (2012) Temporal changes in cell marker expression and cellular infiltration in a controlled cortical impact model in adult male C57BL/6 mice. PLoS One 7(7):e41892
Gustin A, Kirchmeyer M, Koncina E, Felten P, Losciuto S, Heurtaux T, Tardivel A, Heuschling P et al (2015) NLRP3 inflammasome is expressed and functional in mouse brain microglia but not in astrocytes. PLoS One 10(6):e0130624
Mariathasan S, Weiss DS, Newton K, McBride J, O’Rourke K, Roose-Girma M, Lee WP, Weinrauch Y et al (2006) Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 440(7081):228–232
Felderhoff-Mueser U, Schmidt OI, Oberholzer A, Bührer C, Stahel PF (2005) IL-18: a key player in neuroinflammation and neurodegeneration? Trends Neurosci 28(9):487–493
Acs P, Kipp M, Norkute A, Johann S, Clarner T, Braun A, Berente Z, Komoly S et al (2009) 17β-estradiol and progesterone prevent cuprizone provoked demyelination of corpus callosum in male mice. Glia 57(8):807–814
Takao T, Flint N, Lee L, Ying X, Merrill J, Chandross KJ (2004) 17beta-estradiol protects oligodendrocytes from cytotoxicity induced cell death. J Neurochem 89(3):660–673
Acknowledgements
This work was supported by two internal grants from the Medical Clinic of the RWTH Aachen University START (A.Z. and A.S.). We appreciate the discussions with and advice from Prof. J. Bernhagen, formerly head of the Institute of Biochemistry at the RWTH Aachen University.
Author information
Authors and Affiliations
Corresponding author
Ethics declarations
Research and animal care procedures were approved by the Review Board for the Care of Animal Subjects of the district government (Tehran, Iran).
Additional information
Adib Zendedel and Fabian Mönnink contributed equally
Rights and permissions
About this article

Cite this article
Zendedel, A., Mönnink, F., Hassanzadeh, G. et al. Estrogen Attenuates Local Inflammasome Expression and Activation after Spinal Cord Injury. Mol Neurobiol 55, 1364–1375 (2018). https://doi.org/10.1007/s12035-017-0400-2
Received:
Accepted:
Published:
Issue Date:
DOI: https://doi.org/10.1007/s12035-017-0400-2